
Abstract
Apolipoprotein E ε4 allele (APOE4) has been shown to associate with increased susceptibility to SARS-CoV-2 infection and COVID-19 mortality in some previous genetic studies, but information on the role of APOE4 on the underlying pathology and parallel clinical manifestations is scarce. Here we studied the genetic association between APOE and COVID-19 in Finnish biobank, autopsy and prospective clinical cohort datasets. In line with previous work, our data on 2611 cases showed that APOE4 carriership associates with severe COVID-19 in intensive care patients compared with non-infected population controls after matching for age, sex and cardiovascular disease status. Histopathological examination of brain autopsy material of 21 COVID-19 cases provided evidence that perivascular microhaemorrhages are more prevalent in APOE4 carriers. Finally, our analysis of post-COVID fatigue in a prospective clinical cohort of 156 subjects revealed that APOE4 carriership independently associates with higher mental fatigue compared to non-carriers at six months after initial illness. In conclusion, the present data on Finns suggests that APOE4 is a risk factor for severe COVID-19 and post-COVID mental fatigue and provides the first indication that some of this effect could be mediated via increased cerebrovascular damage. Further studies in larger cohorts and animal models are warranted.
Similar content being viewed by others


Introduction
Apolipoprotein E ε4 allele (APOE4) is the strongest genetic risk factor for sporadic Alzheimer’s disease (AD), and it has also been linked to increased risk of other neurodegenerative conditions, such as dementia with Lewy bodies and Parkinson disease dementia [1, 2]. In addition, APOE4 has been shown to associate with increased susceptibility to SARS-CoV-2 infection and COVID-19 mortality in the UK Biobank Cohort [3, 4], and associations between APOE4 and COVID-19 have also been reported in some other candidate gene studies [5,6,7,8]. Interestingly, recent studies have revealed that similar genetic pathways are involved in AD and severe COVID-19 [9], also in direct linkage to APOE4 [10]. Furthermore, these diseases share many co-morbidities and risk factors, such as age, gender, hypertension, diabetes and obesity [11].
Damage to the cerebral microvasculature has been suggested to be a central mediator of the neurological complications often seen in hospitalised COVID-19 patients [11]. Similarly, blood–brain barrier (BBB) dysfunction and cerebrovascular pathology have been shown to be involved in APOE4-associated neurodegenerative conditions [12]. However, information on the role of APOE4 in COVID-19-associated pathology and parallel clinical manifestations remains scarce.
Here we studied the association between APOE and COVID-19 in FinnGen (https://www.finngen.fi/en) utilising the COVID-19 susceptibility GWAS release, and extended the work to include autopsy brain tissues and a prospective clinical cohort of Finnish COVID-19 patients. The Finnish population is a genetically advantageous target to further assess initial genetic discoveries because the frequency of APOE4 is high, and its associations with related morbidity, such as AD and vascular pathology are exceptionally strong in Finland [13, 14].
Materials and methods
Ethics statement
Patients and controls in FinnGen provided informed consent for biobank research, based on the Finnish Biobank Act (https://www.finngen.fi/fi). All DNA samples and data were pseudonymised. Participants of the RECOVID-study [protocol approved by Ethical board of Helsinki University Hospital (HUS/1949/2020)] provided written informed consent. Clinical autopsies were performed at the Department of Pathology, HUS Diagnostic Center, Helsinki University Hospital according to Finnish legislation, with consent granted by the next of kin. The protocol covering COVID-19 autopsies was approved by the Ethics Committee of Helsinki University Hospital (HUS/1238/ 2020).
FinnGen biobank resource
We extracted COVID-19 cases and controls from FinnGen (Release (R) 7, cutoff date March 27th 2021), which comprises prospective epidemiological cohorts (initiated in 1992), disease-based cohorts, and hospital biobank samples. All cases were extracted based on the same criteria: cases were FinnGen samples who according to the National Infectious Disease Register had had COVID-19 and from whom information on disease severity (as per level of care: home, hospital ward or intensive care unit) was available. The genotype data is integrated via a unique national personal identification number with national registries such as the hospital discharge registry, national death registry and medication reimbursement registry. Information on COVID-19 is piped into FinnGen via the national register of infectious diseases. FinnGen R7 included 3469 individuals who had a diagnosis of COVID-19. We excluded 858 cases lacking information on possible hospitalisation. We divided the remaining 2,611 cases into three groups based on the level of care as a surrogate for the severity of disease: home-isolation, hospitalisation but no intensive care, and intensive care. We selected controls from the large nationwide population-based cohorts (FINRISK1992-2012 [15], FINHEALTH2017 [16], HEALTH2000/2012 [17]) included in FinnGen. These cohorts included 43,884 individuals from whom we excluded five individuals without phenotype data and 340 individuals who had had COVID-19, leaving 43,539 individuals as controls. Then we used the MatchIt v.4.1.0 package [18] as implemented in R [19] to match all three case groups (at-home-treatment, hospitalisation, intensive care) separately to controls. Matching was performed based on age (age at death/loss-of-follow up or current age) and sex using default parameters, except that five controls were matched to every case. In a complementary analysis, we added a composite phenotype variable of any cardiovascular disease (FinnGen R7 phenotype code FG_CVD that includes hypertensive diseases, ischaemic and other heart diseases, pulmonary embolism, cerebrovascular diseases, diseases of arteries/arterioles/capillaries, diseases of veins/lymphatic vessels and lymph nodes, and other cardiovascular diseases) as an additional matching term.
APOE genotype was derived from the single-nucleotide polymorphisms rs429358 and rs7412 that were genotyped in FinnGen using a microarray or imputed using a Finnish reference panel. The APOE ε1/ε3 genotype is indistinguishable from the ε2/ε4 genotype but since the ε1 is exceedingly rare, the ε2/ε4 genotype was used for all individuals with the above allele combination.
Recovery after critical COVID-19 infection (RECOVID) study design and subjects
Participants of the RECOVID-study were asked for separate written informed consent for APOE analysis. All participants were adult (≥ 18 years of age) native Finnish-speakers. Prior major neurological disease (e.g., traumatic brain injury, dementia, stroke or Parkinson´s disease) or developmental disability were exclusion criteria. All hospitalised COVID-19 patients with a diagnosis between March 1 and December 31, 2020 were recruited within three months from hospital discharge by mailed invitation or personal contact during a follow-up visit. Home-isolated patients with confirmed SARS-CoV-2 infection (RT-PCR or antibody test) from the same time period were recruited within three months from diagnosis by press and online announcements. Non-infected controls were recruited by online announcements in early 2021. Demographic data and blood samples were collected from all participants, and clinical data of the inpatient period was recorded from hospital-treated cohorts. One-hundred and fifty-six study participants (of whom 108 were COVID-19 patients and 48 controls) completed written questionnaires including the multi-dimensional fatigue scale (MFI-20) at six months post-discharge from hospital or recovery from acute COVID-19 (Additional file 1: Supplementary Fig. 1).
Autopsies
Clinical autopsies were performed at the Department of Pathology, HUS Diagnostic Center according to Finnish legislation, with consent granted by the next of kin. The modified full autopsies were carried out in an autopsy room appropriate for handling infectious decedents, with appropriate personal protective equipment. All autopsies included the complete exploration of the visceral cavity and craniotomy. Modifications included extended neuropathological sampling (samples from 3–12 brain areas) in those 10 cases where a full neuropathological examination was not performed.
Neuropathological examination
Full neuropathological examination of the brain was performed on 11 cases. Each brain was dissected after at least 10 days of formaldehyde-fixation and at least 15 samples from different brain areas were collected. H&E stainings were performed using standardised protocols at the Department of Pathology, Helsinki University Hospital.
Immunohistochemistry (IHC)
Beta-amyloid and CD68 immunostainings were performed at the Department of Pathology, Helsinki University Hospital using standard protocols (details in Additional file 1: Supplementary Table 1).
Histopathological analysis of perivascular haemorrhage (“Bleed Grade” and “Bleed Score”)
Bleed Grade
The extent of perivascular haemorrhage was graded in brain samples from midbrain, pons, medulla, cerebellum, frontal cortex and basal ganglia by two pathologists (JK, HP), reaching consensus using a consultation microscope (Nikon Eclipse i80; eye-pieces CFI 10x/22; objectives: Nikon Plan Fluor 20x/0.50, Nikon Plan Fluor 10x/0.30, Nikon Plan Fluor 4x/0.13, Nikon Plan UW 2x/0.06). Grades were defined as follows: 3 = unequivocal perivascular haemorrhage in two or more foci as seen in one field-of-view (FOV) using a 2 × objective, 2 = unequivocal perivascular haemorrhage as seen in one FOV using a 2 × objective, 1 = unequivocal perivascular haemorrhage as seen in one FOV using a 4 × objective, 0 = no unequivocal perivascular haemorrhage in any FOV as seen using a 4 × objective. The pathologists graded the samples blinded to the information on genotype.
Bleed Score
The number of haemorrhagic foci were determined in brain samples from midbrain, pons, medulla, cerebellum, frontal cortex and basal ganglia by one pathologist (JK) using the same microscope as in the Bleed Grade. Scoring was performed as follows: four parenchymal hot spots for haemorrhage (total FOV 95 mm2) were scored using the 4 × objective and counting the number of haemorrhagic foci. Perivascular haemorrhage was defined as an enlarged perivascular space with red blood cells or enlarged perivascular space with debris and pigmented macrophages. The border of the perivascular space also had to show evidence of neuropil degeneration or vacuolation. Meningeal haemorrhage was not counted. The pathologist scored the samples blinded to information on genotype.
Analysis of cerebral amyloid angiopathy (CAA)
CAA was determined by a neuropathologist (LM) based on beta-amyloid-immunopositivity in at least one vessel on sections from frontal cortex and cerebellum.
Histopathological analysis of microglial reactivity
CD68-stained sections from midbrain, pons and medulla were assessed by two pathologists (JK, HP) using a modified version of the method described by Poloni and colleagues [20]. Briefly, the CD68-stained slide was assessed for representative areas using a 4 × objective, and five FOVs were scored according to the 4-grade scheme using a 10 × objective (FOV 3.8 mm2), with confirmatory use of higher magnification where needed. The 4-point scale (0–3) used was identical to the work by Poloni: 0 = absence of both perivascular infiltrate and microglial nodules and < 20 amoeboid cells/reactive microglial cells; 1 = presence of at least one perivascular infiltrate or 1 micronodule or > 20 amoeboid cells/reactive microglial cells; 2 = presence of 2–4 microglial nodules; and 3 = presence of > 4 microglial nodules. The pathologists graded the samples blinded to information on genotype, and grade was agreed upon by consensus at the consulting microscope described above.
In addition, a count of stained foci was determined by one pathologist (JK) in 3 hot spot areas using a 20 × objective (FOV 0.95 mm2). Where a single cell could be identified, the stained cells were counted. The pathologist scored the samples blinded to information on genotype.
Sanger sequencing
We genotyped APOE using Sanger sequencing. All consented participants of the RECOVID study gave venous blood samples from which we extracted DNA using standard methods. In the AUTOPSY cohort, DNA was isolated from frozen tissues collected at autopsy using standard protocols. We amplified DNA fragments containing rs429358 and rs7412 using previously published primers [21] that amplify rs429358 and rs7412 on the same DNA fragment. The amplification reaction contained 1X DreamTaq Buffer, 0.35 mM of dNTP mix, 0.6 μM of forward and reverse primers, 1 M of Betaine, 1.25U of DreamTaq DNA Polymerase (Applied Biosystems, ThermoFisher Scientific, Waltham, MA, USA), approximately 50 ng of template DNA and nuclease-free water to attain a total volume of 20 μl. The cycling conditions have been previously published [21]. We then purified the amplicons using Exonuclease I—Shrimp Alkaline Phosphatase Clean Up (Applied Biosystems) according to the manufacturer’s protocol. We then sequenced the purified amplicons using BigDye Terminator v3.1 Cycle sequencing kit (Applied Biosystems) according to the manufacturer’s protocol with the forward primer. Sequences were analysed on a ABI3730XL DNA Analyzer (Applied Biosystems) at the Finnish Institute of Molecular Medicine (FIMM). We determined the genotypes of rs429358 and rs7412 using Sequencher (Sequencher® version 5.1 DNA sequence analysis software, Gene Codes Corporation, Ann Arbor, MI USA).
Fatigue testing (Multidimensional Fatigue Inventory MFI-20)
We evaluated chronic fatigue at a 6-month follow-up using the Multidimensional Fatigue Inventory (MFI-20) score [22]. The questionnaire was translated into Finnish and was a part of a larger set of questions concerning neuropsychological recovery and mental health. MFI-20 measures five subscales of fatigue (General Fatigue, Physical Fatigue, Mental Fatigue, Reduced Motivation, Reduced Activity), each of which is probed with four questions. The subject specifies the extent to which each statement relates to her/him on a 5-point Likert scale, ranging from “Yes, that is true” to “No, that is not true”. The range of possible scores is 4–20 in each fatigue subscale, and 20–100 in total fatigue, with a higher score signifying a higher level of fatigue.
Statistical analysis of the AUTOPSY and RECOVID cohorts
All statistical analyses were performed using GraphPad Prism (version 8.4.2 for Windows, GraphPad Software, San Diego, California USA, www.graphpad.com), SPSS (IBM Corp. Released 2020. IBM SPSS Statistics for Windows, Version 27.0. Armonk, NY: IBM Corp), and the statistical software R [19].
We used Fisher’s exact test as implemented in R’s stats package to test if carrying at least one APOE ε4 allele associated with each COVID-19 case group. We used linear mixed-effect model as implemented in R’s nlme (Pinheiro J, Bates D, DebRoy S, Sarkar D, R Core Team (2021). nlme: Linear and Nonlinear Mixed Effects Models. R package version 3.1–153, https://CRAN.R-project.org/package=nlme) and cumulative link mixed model as implemented in R’s ordinal (Christensen, R. H. B. (2019). ordinal—Regression Models for Ordinal Data. R package version 2019.12–10, https://CRAN.R-project.org/package=ordinal) package to determine the statistical significances of perivascular bleed score and bleed grade depending on APOE4 carriership, respectively. Additionally, we used exact Pearson’s chi-square test and Mann–Whitney U test as implemented in SPSS to determine the statistical significance of perivascular bleed grade and bleed score depending on APOE4 carriership specifically in different brain areas, respectively. All patients who filled in the MFI-20 questionnaire were included in the analysis. We used a negative binomial regression model as implemented in the MASS [23] package in R to analyse factors associated with the MFI-20 fatigue score in both univariate and multivariate analyses (Tables 2 and 3). Negative binomial regression was chosen over Poisson regression to compensate for the overdispersion in data. COVID-19 severity was classified into three categories, as done in a previously published analysis of fatigue associated with long-COVID [24]. The categories were: 0 – noninfected controls, 1 – home-isolated COVID-19, 2 – hospitalised COVID-19. As COVID-19 severity was the only non-binary categorical variable in multivariate binomial regression model and reached statistical significance in the model in one but not all factor levels, its independence was further analysed by comparison (ANOVA) with the same model with severity excluded.
Results
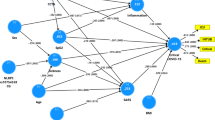
To assess the association between APOE and COVID-19, we used data from FinnGen release 7, which integrates genome-wide genotype data of over 300,000 subjects with phenotype data derived from national registries. First, we identified 2611 individuals with a COVID-19 diagnosis in the National Registry of Infectious diseases by March 27th 2021 and with known disease-severity: home-isolated (n = 2259, 31.8% APOE4 carriers); hospitalised but no intensive care (n = 304, 36.2% APOE4 carriers); and intensive care (n = 48, 50.0% APOE4 carriers). In a complementary analysis, we matched five controls from nationwide population-based cohorts to every case by age and sex, and also by diagnosis of cardiovascular disease (Fig. 1). Compared to the matched controls, APOE4 carriership was not significantly more frequent in the home-isolated (p = 0.11, odds ratio (OR) = 0.92, 95% confidence interval (CI) 0.84–1.02, Fisher’s exact test) nor hospital treated COVID-19 patients (p = 0.64, OR = 1.07, 95% CI 0.82–1.39, Fisher’s exact test). However, APOE4 carriership was more prevalent among COVID-19 patients treated in the intensive care unit (ICU) (p = 0.0064, OR = 2.47, 95% CI 1.25–4.89, Fisher’s exact test), and the association persisted also after matching for cardiovascular disease (p = 0.020, OR = 2.15, 95% CI 1.09 – 4.24, Fisher’s exact test).

Flow chart and results of FinnGen Release 7 data analysis
Next, we looked into the prevalence of APOE4 in two COVID-19 cohorts, which we sequenced for APOE genotype status, and matched the genotypes with histological results from neuropathological autopsy material, or clinical records, respectively. The cohorts were (1) a series of autopsies comprising 21 cases with COVID-19 infection confirmed by RT-PCR (group “AUTOPSY”, see Methods and Additional file 1: Supplementary Table 2), and (2) a prospective cohort study of Recovery After Critical COVID-19 Infection (RECOVID). The RECOVID cohort comprised 58 patients treated for critical COVID-19 in the intensive care unit (ICU) in Helsinki University Hospital, and 33 COVID-19 patients admitted at ward-level care (WARD). In line with our main aim to study the impact of COVID-19 severity, the ICU and WARD cohorts were compared to 37 COVID-19 patients who did not need hospitalisation during recovery (HOME), and 53 non-infected individuals (NOCOV; see Additional file 1: Supplementary Fig. 1 and Methods for more details on the study design). The demographic and APOE4 data of both cohorts are shown in Table 1.
APOE4 carriership was not associated with severity of COVID-19 in the small AUTOPSY or RECOVID cohorts (Table 1). For the AUTOPSY group, we used the FinnGen population as controls (p = 0.49, Fisher’s exact test). In the RECOVID cohort, we compared the various severity grades of the disease to the non-infected group (ICU vs NOCOV, p = 0.69; WARD vs NOCOV p = 0.82, HOME vs NOCOV p = 1, Fisher’s exact test in all comparisons).
Interestingly, we found that AUTOPSY and RECOVID cohorts included APOE4 homozygotes (cases RX and A14; Fig. 2) sharing a striking pathological phenotype: the presence of abundant petechial microvascular haemorrhages in multiple brain areas confirmed by either brain MRI or neuropathological autopsy. The clinical features of RX are described in detail in the Supplementary Information. The neuropathological and clinical features of A14 have been described in detail previously [25]. Here, we also performed whole-genome sequencing of this patient (findings of the main COVID-19-associated variants are shown in Additional file 1: Supplementary Table 3), in addition to the APOE analysis. Microvascular brain injury has been reported to associate with COVID-19 [26], and autopsied COVID-19 patients have been reported to exhibit micro- and perivascular haemorrhages [27, 28] (but see also [29]). However, we are not aware of any reports associating it with the APOE genotype status. Therefore, we set out to look closer at brain microvascular damage in the AUTOPSY cohort.

Microvascular brain haemorrhages in RECOVID and AUTOPSY cohorts. a MRI showing cortical microhaemorrhages in a case with APOE ε4/ε4. b MRI showing cerebellopontine microhaemorrhages in the same case as (a). c Brain subarachnoidal microhaemorrhages at autopsy in a case with APOE ε3/ε4. d Brain pontine microhaemorrhages at neuropathological examination in a case with APOE ε4/ε4. e Histological H&E section showing pontine microhaemorrhages in the same case as (c). f Histological H&E section showing pontine microhaemorrhages in the same case as (d). Red arrows indicate microhaemorrhages. Scale bars represent 100 μm in (e) and (f)
We examined histopathological sections from different brain areas of all 21 subjects of the AUTOPSY cohort, using haematoxylin–eosin (H&E) staining and IHC stainings. The severity of perivascular haemorrhage was assessed with a 4-grade scoring system (see Bleed Grade in Methods) and a continuous bleed score for the frequency of perforating microvascular damage (see Bleed Score in Methods) in six brain areas (frontal cortex, mesencephalon, pons, basal ganglia, cerebellum and medulla). APOE4 carriers had a significantly higher bleed grade than non-carriers overall (p = 0.0024, cumulative link mixed model), with the most pronounced effect in cerebellum (nominal p = 0.024, Pearson’s chi-square test). Basal ganglia showed a similar trend (nominal p = 0.062) with all of the APOE4 carriers having the most severe grade. With respect to the bleed score, there was no statistically significant effect of APOE4 carriership (p = 0.201, linear mixed-effect model) over the whole dataset. In both analyses, and in each brain area, the mean value for bleed grade/score was higher in APOE4 carriers (Fig. 3 and Additional file 1: Supplementary Table 4).

Presence of microvascular haemorrhages across brain areas. a Bleed Grades of all autopsied cases (n = 21). b Bleed Grades with APOE2 carriers excluded (n = 17). c Bleed Scores of all autopsied cases. d Bleed Scores with APOE2 carriers excluded. a,c,d,eMissing sample from 1 patient; bMissing samples from 2 patients. * stands for nominal p < 0.05 (non-significant after Bonferroni’s correction for multiple testing), ** stands for nominal p < 0.01 (significant after Bonferroni’s correction for multiple testing)
A recent mechanistic study on APOE4-related cerebrovascular disorders [30] did not include APOE ε2 (APOE2) -containing genotypes, and the role of APOE2 as a protective versus predisposing factor is under debate and seems to vary according to condition [31]. Therefore, we further analysed the microhaemorrhage data excluding APOE2 carriers. In this analysis, the effect of APOE4 to the bleed grade persisted (p = 0.0053). In bleed scores, there was still no statistically significant difference when assessing the data as a whole (p = 0.114), but in brain area -specific comparisons microhaemorrhages in pons were significantly more prevalent in APOE4 carriers (p = 0.007, significant after correction with Bonferroni’s method for multiple testing) (Fig. 3).
Perivascular activated (CD68-positive) microglia in the brainstem have previously been reported as a common finding in COVID-19 patients [32]. As APOE4-associated neuroinflammatory effects are known to be partially mediated by microglia [33], we also tested whether microglial activation was associated with APOE4 carriership in COVID-19; however, there was no such association (Fig. 4, p > 0.05 in all studied brain areas).

Abundance and pro-inflammatory reactivity of CD68 + microglial cells across brain areas. a The number of CD68-positive cells in evaluated brain areas; three FOVs evaluated per area from each autopsied case (n = 21). b Grade of microglial activation in evaluated brain areas, as described in [20]; five FOVs scored per area from each autopsied case (n = 21). P values calculated with unpaired t-test. aMissing sample from 1 patient; b,cMissing sample from 2 patients
Chronic fatigue has been recently reported [24, 34, 35] as a prevalent symptom in post-acute COVID-19 syndrome (PACS, or “long COVID”). Similar observations were also published after the severe acute respiratory syndrome (SARS) epidemic in 2003 [36]. As APOE4 strongly associates with various kinds of cognitive dysfunctions, we also analysed its potential role on the long-term neuropsychological recovery after COVID-19, using MFI-20 fatigue scores (validated for post-viral syndrome in adults [37]) six months post-discharge. In a previous report, which showed increased post-COVID fatigue after severe initial illness at a 6-month follow-up [24], the analysis was stratified to hospitalised and home-isolated patients. To enable a direct comparison with the above study, we combined the ICU and WARD groups into one cohort of hospitalised patients. However, we found that APOE4 carriership was not associated with total MFI-20 scores, nor with the subcategories of general fatigue, physical fatigue, reduced activity or reduced motivation (Table 2).
Notably, the subcategory of mental fatigue was associated with a higher score in hospitalised (ICU + WARD) APOE4 carriers compared to non-carriers (9.9 vs 8.0, p = 0.029, negative binomial regression, Table 2), and this association remained independently significant after factoring in other plausibly contributing patient features in a multivariate model (p = 0.038, negative binomial regression, Table 3). APOE4 carriership similarly elevated mental fatigue scores also in the NOCOV group (9.8 vs 7.4, p = 0.0062), whereas it did not have an effect in the HOME group (10.8 vs 11.2, p = 0.82).
We further analysed several factors (sex, age, COVID-19 severity, APOE4 carriership and key comorbidities) in relation to mental fatigue in all RECOVID participants (Table 3), and APOE4 remained a significant contributor (p = 0.015 in multivariate model). The severity of COVID-19 was not associated with mental fatigue after controlling for other factors (p = 0.11 in multivariate model, Table 3).
Discussion
Previous studies [3, 4] showed a genetic association between APOE4 carriership and COVID-19 positivity, as well as mortality. Using Finnish biobank (FinnGen study), autopsy and clinical evidence, we confirm and extend these findings. First, we show an association between APOE4 carriership and COVID-19 patients requiring critical care. Second, our data suggests that APOE4 carriership increases the incidence of cerebral microhaemorrhages in COVID-19 patients, and that APOE4-associated COVID-19 neuropathology might be driven more by the perivascular damage than by microglial activation. Finally, our findings in the RECOVID cohort suggest that APOE4 carriership may be a risk factor for prolonged mental fatigue after severe COVID-19.
Several candidate-gene studies (Additional file 1: Supplementary Table 5a) have found an association between APOE4 and SARS-CoV-2 infection or the severity of COVID-19, whereas genome-wide association studies on COVID-19 (Additional file 1: Supplementary Table 5b) have failed to find evidence for an association between the APOE locus and COVID-19. There are several reasons for the discrepant results, such as different definitions for cases and controls, different age distributions and different ancestries. Candidate-gene studies may be more susceptible to false positive findings and publication bias. In genome-wide association analyses, reaching the genome-wide significance threshold requires a very large sample size or a very large effect size. The largest genome-wide association studies published were multinational and it is known that APOE4 frequency varies in different populations. Moreover, genome-wide association studies test the association of individual variants. Thus, they did not directly test the association of APOE4, just the association of APOE4 proxy variants, which may decrease power.
The populations with APOE4 carrier frequency above the global average (like the Finns or in the UK) are ideal for studying the genetic association reported here, because the effect size of APOE4 carriership for increased susceptibility to severe COVID-19 is modest, and therefore likely to be masked in more heterogeneous transethnic populations, such as in a recent global genetic mapping of COVID-19 [38].
The key limitations of this study are the low number of ICU-treated subjects in our biobank data and subjects overall in the RECOVID study, which was not adequately powered to detect an effect of APOE4 on COVID-19 severity. Additionally, the association of APOE4 with mental fatigue was not consistent, as the control group also showed it, while the HOME group did not. Selection bias could account for the finding in the NOCOV group, since the cohort was recruited via open invitation and, thus, individuals experiencing fatigue-like cognitive symptoms may have been more eager to participate. The HOME group on the other hand showed higher mental fatigue scores in general, which suggests that factors unrelated to COVID-19 could be masking an underlying effect of APOE4. Moreover, only the subcategory of mental fatigue (which comprises questions probing the subjects’ ability to direct attention and keep focused) differed significantly between genotypes, whereas the other dimensions of the MFI-20 questionnaire did not. Therefore, further studies with more complete test batteries are warranted to study the link of APOE genotype and cognitive symptoms. Limitations of our AUTOPSY cohort include the small number of patients with variable comorbidities, COVID-19 disease courses and mechanisms of death. When assessing haemorrhages, even small bleeds were taken into account. It is noteworthy that in Finland anticoagulation treatment was routinely used in COVID-19 patients even in the early stages of the pandemic, and the majority of the autopsied patients were treated with anticoagulants. Many of them suffered from hypoxia and had been treated in ICU for varying lengths of times, affecting the bleeding tendency in variable ways. Most importantly, APOE4 is per se associated with early mortality and cognitive decline, and if and how this is modified by COVID-19 is a question that cannot be conclusively answered by this study alone.
APOE4 is the strongest common genetic risk factor for late-onset AD, and it has also been shown to associate with cognitive decline independently of AD pathology [39]. These effects have been mechanistically connected to the APOE4-induced compromised cerebrovascular integrity including the breakdown of the BBB in both humans and animal models, mediated by inflammatory pathways in BBB-forming pericytes, as well as in neurons [30]. COVID-19 has also been shown to damage the microvasculature of the brain [26] and lead to enhanced expression of APOE at the barriers of the CNS: the choroid plexus and in astrocytes, which participate in formation of the BBB [40]. Interestingly, an in vitro study based on human-induced pluripotent stem cell (hiPSC) models has suggested that astrocytes and neurons expressing APOE4 homozygously are more vulnerable to SARS-CoV-2 infection than those with APOE3, and APOE4 astrocytes also exhibit a more severe cytopathogenic effect to SARS-CoV-2 infection, which might mediate the severity of COVID-19 [41]. However, direct viral involvement and neurotropism seem to be rare events in human subjects [26]. Furthermore, the effects of APOE in the context of COVID-19 are likely not restricted to the brain—for instance, APOE has a pro-inflammatory role also in the lungs [42], and APOE4 is a well-known risk factor for atherosclerosis and organ damage outside the brain predisposing to life-threatening vicious cycles. Thus, we hypothesise that the severe multi-organ inflammatory and coagulopathic manifestations of COVID-19 are potentiated by the effects of APOE4, and therefore APOE4 carriers might be prone to developing a more severe course of disease. Studies in larger cohorts and animal models are warranted to investigate this hypothesis.
Conclusions
The present data on Finns suggests that APOE4 is a risk factor for severe COVID-19 and post-COVID mental fatigue and provides the first indication that some of this effect could be mediated via increased cerebrovascular damage. Further studies in larger cohorts and animal models are warranted.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding authors upon reasonable request.
Abbreviations
- AD:
-
Alzheimer’s disease
- APOE2 :
-
Apolipoprotein E ε2 allele
- APOE4 :
-
Apolipoprotein E ε4 allele
- AUTOPSY:
-
Cohort of autopsied cases
- BBB:
-
Blood–brain barrier
- CAA:
-
Cerebral amyloid angiopathy
- FOV:
-
Field-of-view
- HOME:
-
Cohort of COVID-19 patients who did not need hospitalisation during recovery
- ICU:
-
Intensive care unit; cohort of COVID-19 patients admitted to an intensive care unit
- IHC:
-
Immunohistochemistry
- MFI-20:
-
Multi-dimensional fatigue scale—20
- NOCOV:
-
Cohort of non-COVID-19-infected individuals
- PACS:
-
Post-acute COVID-19 syndrome
- RECOVID:
-
Recovery After Critical COVID-19 Infection
- WARD:
-
Cohort of COVID-19 patients admitted at ward-level care
References
Yamazaki Y, Zhao N, Caulfield TR, Liu C-C, Bu G (2019) Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurol 15:501–518
Tsuang D, Leverenz JB, Lopez OL, Hamilton RL, Bennett DA, Schneider JA et al (2013) APOE ϵ4 Increases risk for dementia in pure synucleinopathies. JAMA Neurol 70:223–228
Kuo C-L, Pilling LC, Atkins JL, Masoli JAH, Delgado J, Kuchel GA et al (2020) ApoE e4e4 genotype and mortality with COVID-19 in UK Biobank. J Gerontol Ser A 75:1801–1803
Kuo C-L, Pilling LC, Atkins JL, Masoli JAH, Delgado J, Kuchel GA et al (2020) APOE e4 genotype predicts severe COVID-19 in the UK Biobank community cohort. J Gerontol Ser A 75:2231–2232
Hubacek JA, Dlouha L, Dusek L, Majek O, Adamkova V (2021) Apolipoprotein E4 allele in subjects with COVID-19. Gerontology 67:320–322
Al-Jaf SMA, Niranji SS, Ali HN, Mohammed OA (2021) Association of Apolipoprotein e polymorphism with SARS-CoV-2 infection. Infect Genet Evol 95:105043
Del Ser T, Fernández-Blázquez MA, Valentí M, Zea-Sevilla MA, Frades B, Alfayate E et al (2021) Residence, clinical features, and genetic risk factors associated with symptoms of COVID-19 in a cohort of older people in madrid. Gerontology 67:281–289
Kuo C-L, Pilling LC, Atkins JL, Fortinsky RH, Kuchel GA, Melzer D (2021) APOE e4 genotypes increase risk of delirium during COVID-19-related hospitalizations: evidence from a large UK Cohort. J Gerontol Ser A. https://doi.org/10.1093/gerona/glab184/6309638
Magusali N, Graham AC, Piers TM, Panichnantakul P, Yaman U, Shoai M et al (2021) A genetic link between risk for Alzheimer’s disease and severe COVID-19 outcomes via the OAS1 gene. Brain. https://doi.org/10.1093/brain/awab337/6382473
Xiong N, Schiller MR, Li J, Chen X, Lin Z (2021) Severe COVID-19 in Alzheimer’s disease: APOE4’s fault again? Alzheimer’s Res Ther 13:1–5
Miners S, Kehoe PG, Love S (2020) Cognitive impact of COVID-19: looking beyond the short term. Alzheimer’s Res Ther 12:1–16
Sweeney MD, Kisler K, Montagne A, Toga AW, Zlokovic BV (2018) The role of brain vasculature in neurodegenerative disorders. Nat Neurosci 21:1318–1331
Eichner JE, Dunn ST, Perveen G, Thompson DM, Stewart KE, Stroehla BC (2002) Apolipoprotein E polymorphism and cardiovascular disease: A HuGE review. Am J Epidemiol 155:487–495
Peuralinna T, Tanskanen M, Mäkelä M, Polvikoski T, Paetau A, Kalimo H et al (2011) APOE and AβPP gene variation in cortical and cerebrovascular amyloid-β pathology and Alzheimer’s disease: a population-based analysis. J Alzheimer’s Dis 26:377–385
Borodulin K, Tolonen H, Jousilahti P, Jula A, Juolevi A, Koskinen S et al (2018) Cohort profile: the national FINRISK study. Int J Epidemiol 47:696–696i
Borodoulin K, Sääksjärvi K. FinHealth 2017 Study – Methods. Finnish Inst. Heal. Welfare. Rep. 17/2019.
Heistaro S. Methodology report : Health 2000 survey. Kansanterveyslaitos; 2008 [cited 2021 Sep 27]; Available from: https://www.julkari.fi/handle/10024/78185
Ho D, Imai K, King G, Stuart EA (2011) MatchIt: nonparametric preprocessing for parametric causal inference. J Stat Softw 42:1–28
R Core Team (2020). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/.
Poloni TE, Medici V, Moretti M, Visonà SD, Cirrincione A, Carlos AF et al (2021) COVID-19-related neuropathology and microglial activation in elderly with and without dementia. Brain Pathol 31:e12997
Zhong L, Xie Y-Z, Cao T-T, Wang Z, Wang T, Li X et al (2016) A rapid and cost-effective method for genotyping apolipoprotein E gene polymorphism. Mol Neurodegener 11:1–8
Smets EMA, Garssen B, Bonke B, De Haes JCJM (1995) The multidimensional Fatigue Inventory (MFI) psychometric qualities of an instrument to assess fatigue. J Psychosom Res Elsevier 39:315–325
Venables WN, Ripley BD (2002) Modern Applied Statistics with S. Fourth Edition. Springer, New York. ISBN 0-387-95457-0.
Blomberg B, Mohn KG-I, Brokstad KA, Zhou F, Linchausen DW, Hansen B-A et al (2021) Long COVID in a prospective cohort of home-isolated patients. Nat Med 27:1607–1613
Kantonen J, Mahzabin S, Mäyränpää MI, Tynninen O, Paetau A, Andersson N et al (2020) Neuropathologic features of four autopsied COVID-19 patients. Brain Pathol 30:1012–1016
Lee M-H, Perl DP, Nair G, Li W, Maric D, Murray H et al (2020) Microvascular injury in the brains of patients with Covid-19. N Engl J Med 384:481–483
Maiese A, Manetti AC, Bosetti C, Del DF, La RR, Frati P et al (2021) SARS-CoV-2 and the brain: a review of the current knowledge on neuropathology in COVID-19. Brain Pathol. https://doi.org/10.1111/bpa.13013
Mukerji SS, Solomon IH (2021) What can we learn from brain autopsies in COVID-19? Neurosci Lett 742:135528
Glatzel M, Hagel C, Matschke J, Sperhake J, Deigendesch N, Tzankov A et al (2021) Neuropathology associated with SARS-CoV-2 infection. Lancet 397:276
Montagne A, Nikolakopoulou AM, Huuskonen MT, Sagare AP, Lawson EJ, Lazic D et al (2021) APOE4 accelerates advanced-stage vascular and neurodegenerative disorder in old Alzheimer’s mice via cyclophilin A independently of amyloid-β. Nat Aging 1:506–520
Li Z, Shue F, Zhao N, Shinohara M, Bu G (2020) APOE2: protective mechanism and therapeutic implications for Alzheimer’s disease. Mol Neurodegener 15:1–19
Matschke J, Lütgehetmann M, Hagel C, Sperhake JP, Schröder AS, Edler C et al (2020) Neuropathology of patients with COVID-19 in Germany: a post-mortem case series. Lancet Neurol 19:919–929
Shi Y, Manis M, Long J, Wang K, Sullivan PM, Remolina Serrano J et al (2019) Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J Exp Med 216:2546–2561
Taquet M, Dercon Q, Luciano S, Geddes JR, Husain M, Harrison PJ (2021) Incidence, co-occurrence, and evolution of long-COVID features: A 6-month retrospective cohort study of 273,618 survivors of COVID-19. PLOS Med 18:e1003773
Xie Y, Bowe B, Al-Aly Z (2021) Burdens of post-acute sequelae of COVID-19 by severity of acute infection, demographics and health status. Nat Commun 12:1–12
Lam MH-B, Wing Y-K, Yu MW-M, Leung C-M, Ma RCW, Kong APS et al (2009) Mental morbidities and chronic fatigue in severe acute respiratory syndrome survivors: long-term follow-up. Arch Int Med 169:2142–2147
Dencker A, Sunnerhagen KS, Taft C, Lundgren-Nilsson Å (2015) Multidimensional fatigue inventory and post-polio syndrome – a Rasch analysis. Heal Qual Life Outcomes 13:1–6
Mapping the human genetic architecture of COVID-19. Nat 2021 . Nature Publishing Group; 2021 [cited 2021 Sep 25];1–8. Available from: https://www.nature.com/articles/s41586-021-03767-x
Montagne A, Nation DA, Sagare AP, Barisano G, Sweeney MD, Chakhoyan A et al (2020) APOE4 leads to blood–brain barrier dysfunction predicting cognitive decline. Nature 581:71–76
Yang AC, Kern F, Losada PM, Agam MR, Maat CA, Schmartz GP et al (2021) Dysregulation of brain and choroid plexus cell types in severe COVID-19. Nature 595:565–571
Wang C, Zhang M, Garcia G, Tian E, Cui Q, Chen X et al (2021) ApoE-isoform-dependent SARS-CoV-2 neurotropism and cellular response. Cell Stem Cell 28:331-342.e5
Gordon EM, Yao X, Xu H, Karkowsky W, Kaler M, Kalchiem-Dekel O et al (2019) Apolipoprotein E is a concentration-dependent pulmonary danger signal that activates the NLRP3 inflammasome and IL-1β secretion by bronchoalveolar fluid macrophages from asthmatic subjects. J Allergy Clin Immunol 144:426-441.e3
Acknowledgements
We thank Prof. Anders Paetau for his advice on interpretation of the neuropathological findings; Kristiina Nokelainen, Lilja Jansson, Susanna Saarinen, Anne Nummisto, Katja Lindström, Kirsi Kallioheimo and Pirjo Niiniketo for excellent technical help; autopsy technicians Marko Kariniemi, Seppo Kolari, Kirpo Kakkonen, Esa Ihalainen, Sari Lyytikäinen and Piia Muikku for their valuable assistance; Eija Koponen, Sari Renholm and Tarja Suhonen for all practical help; Anna Hästbacka, Erik Mäkitalo and Inari Taskila for acquisition of the clinical data; Sanna Koskinen, Annamari Tuulio-Henriksson for design of the clinical work and acquisition of the clinical data; Henriikka Ollila and Salla Kattainen for interviewing patients and acquisition of the clinical data. We also want to acknowledge the participants and investigators of the FinnGen study. Sequencing of FinnGen samples was performed by the Sequencing unit of Institute for Molecular Medicine Finland FIMM Technology Centre, University of Helsinki. Sequencing unit is supported by Biocenter Finland. Lastly, we would like to offer our condolences to the next of kin of the deceased, and express our gratitude for the trust they have bestowed upon us.
Funding
This work was funded by Academy of Finland (341007, LM; 336439 and 335527, AK), Juho Vainio Foundation (OC), The Finnish Medical Foundation (MIM, AK), Helsinki University Hospital grant (PJT), University of Helsinki Brain profiling project funds (UHBrain, Karri Kaivola), Helsinki University Hospital grant (Finnish government TYH2021310, JH), Nordforsk (JH), Sigrid Jusélius Foundation (SNK, Kai Kaila), Finnish Brain Foundation (SNK).
Author information
Authors and Affiliations
Consortia
Contributions
SNK: design of the work, acquisition of the genetic data, analysis and interpretation of the genetic, pathological and clinical data, drafting and revision of the manuscript. JK: design of the work, acquisition of the genetic and pathological data, analysis and interpretation of the pathological and genetic data, revision of the manuscript. Karri Kaivola: design of the work, acquisition of the genetic data, analysis and interpretation of the genetic data, revision of the manuscript. LH: design of the work, acquisition of the clinical data, revision of the manuscript. MIM: design of the work, analysis and interpretation of the pathological data, revision of the manuscript. HP: acquisition of the pathological data, analysis and interpretation of the pathological data, revision of the manuscript. JM: acquisition of the clinical data, revision of the manuscript. MP: acquisition of the genetic data, analysis and interpretation of the genetic data, revision of the manuscript. MK: acquisition of the pathological data, analysis and interpretation of the pathological data, revision of the manuscript. JT: analysis and interpretation of the genetic, pathological and clinical data, revision of the manuscript. OC: acquisition of the pathological data, funding, revision of the manuscript. AK: acquisition of the pathological data, funding, revision of the manuscript. OV: acquisition of the pathological data, funding, revision of the manuscript. MJ: design of the work, acquisition of the clinical data, revision of the manuscript. PJT: design of the work, acquisition of the genetic data, funding, revision of the manuscript. KK: funding, drafting and revision of the manuscript. JH: design of the work, acquisition of the genetic and clinical data, analysis and interpretation of the genetic and clinical data, funding, revision of the manuscript. LM: design of the work, acquisition of the genetic and pathological data, analysis and interpretation of the genetic and pathological data, funding, revision of the manuscript. All authors read and approved the final manuscript
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Patients and controls in FinnGen provided informed consent for biobank research, based on the Finnish Biobank Act (https://www.finngen.fi/fi). All DNA samples and data were pseudonymised. Participants of the RECOVID-study [protocol approved by Ethical board of Helsinki University Hospital (HUS/1949/2020)] provided written informed consent. Clinical autopsies were performed at the Department of Pathology, HUS Diagnostic Center, Helsinki University Hospital according to Finnish legislation, with consent granted by the next of kin. The protocol covering COVID-19 autopsies was approved by the Ethics Committee of Helsinki University Hospital (HUS/1238/2020).
Consent for publication
A consent to publish individual pictures/details was obtained from the subjects or their next-of-kin.
Competing interests
Pentti J. Tienari holds a patent on C9orf72 in diagnostics and treatment of ALS/FTD and has made paid consultations to Roche, Biogen, Merck, Teva, Sanofi-Genzyme and Novartis. Other authors declare no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Supplementary Information.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kurki, S.N., Kantonen, J., Kaivola, K. et al. APOE ε4 associates with increased risk of severe COVID-19, cerebral microhaemorrhages and post-COVID mental fatigue: a Finnish biobank, autopsy and clinical study. acta neuropathol commun 9, 199 (2021). https://doi.org/10.1186/s40478-021-01302-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-021-01302-7