
Abstract
The open reading frame YGL157w in the genome of the yeast Kluyveromyces marxianus strain DMB1 encodes a putative uncharacterized oxidoreductase. However, this protein shows 46% identity with the Saccharomyces cerevisiae S288c NADPH-dependent methylglyoxal reductase, which exhibits broad substrate specificity for aldehydes. In the present study, the YGL157w gene product (KmGRE2) was purified to homogeneity from overexpressing Escherichia coli cells and found to be a monomer. The enzyme was strictly specific for NADPH and was active with a wide variety of substrates, including aliphatic (branched-chain and linear) and aromatic aldehydes. The optimal pH for methylglyoxal reduction was 5.5. With methylglyoxal as a substrate, the optimal temperature for enzyme activity at pH 5.5 was 45°C. The enzyme retained more than 70% of its activity after incubation for 30 min at temperatures below 35°C or at pHs between 5.5 and 9.0. In addition, the KmGRE2-overexpressing E. coli showed improved growth when cultivated in cedar hydrolysate, as compared to cells not expressing the enzyme. Taken together, these results indicate that KmGRE2 is potentially useful as an inhibit decomposer in E. coli cells.
Similar content being viewed by others


Introduction
The NADPH-dependent methylglyoxal reductase (EC 1.1.1.283) in Saccharomyces cerevisiae is termed GRE2. Using NADPH as a coenzyme, GRE2 catalyzes the stereoselective reduction of a broad range of substrates, including aldehydes and diketones, as well as aliphatic and aromatic ketones (Chen et al. 2003; Murata et al. 1985). In S. cerevisiae, this enzyme functions within the high osmolarity glycerol pathway (Garay-Arroyo and Covarrubias 1999), and its expression is induced by environmental conditions, including ionic, osmotic, oxidative, heat shock and heavy metal-related stresses (Garay-Arroyo and Covarrubias 1999; Krantz et al. 2004; Liu et al. 2008; Rep et al. 2001; Rutherford and Bird 2004). GRE2 also shows isovaleraldehyde reductase activity and so acts as a suppressor of filamentation (Chen et al. 2003; Hauser et al. 2007). To date, GRE2 and homologues have been purified to homogeneity from S. cerevisiae (Chen et al. 2003; Murata et al. 1985), Aspergillus niger (Inoue et al. 1988) and goat liver (Ray and Ray 1984), and their enzymatic properties have been characterized. In addition, the three-dimensional structures of the S. cerevisiae S288c GRE2 apo enzyme and the enzyme-NADP+ complex expressed in Escherichia coli have been solved (Guo et al. 2014). Based on its structural features, GRE2 is classified as a member of the extended short-chain-dehydrogenase/reductase superfamily (Müller et al. 2010).
S. cerevisiae GRE2 is currently being used as a versatile biocatalyst for the stereoselective synthesis of hydroxy compounds, which serve as building blocks in the production of pharmaceuticals and other fine chemicals (Choi et al. 2010; Ema et al. 2008; Müller et al. 2010; Park et al. 2010). Another advantageous feature of GRE2 is a decomposer in bacteria. For example, GRE2 is used for glycolaldehyde degradation during bioethanol production in S. cerevisiae (Jayakody et al. 2013). In addition, a S. cerevisiae strain overexpressing a GRE2 with site-directed mutagenesis exhibited enhanced furfural and 5-hydroxymethylfurfural (HMF) detoxification (Moon and Liu 2012). Conversely, in a S. cerevisiae GRE2 knockout strain growth was suppressed by environmental stress (Warringer and Blomberg 2006), and filament formation was increased in the presence of isoamyl alcohol (Hauser et al. 2007). Hence, GRE2 is regarded as a key enzyme necessary for inhibitor and stress tolerance in S. cerevisiae.
We recently isolated Kluyveromyces marxianus strain DMB1, a thermotolerant yeast, from sugarcane bagasse hydrolysate and determined its genomic sequence (Suzuki et al. 2014). Within the sequence, we identified open reading frame YGL157w, which shows 46% identity with S. cerevisiae S288c GRE2 (Figure 1). In the hope of identifying a more stable GRE2 homologue, in the present study, we purified and characterized the enzyme from K. marxianus strain DMB1 after its overexpression in E. coli cells. In addition, we examined its ability to improve growth of cells cultured in cedar hydrolysate.

Multiple sequence alignment of KmGRE2 and GRE2. The sequence alignment was prepared using ClustalW (Thompson et al. 1994). Residues involved in the substrate-binding site in GRE2 are boxed.
Materials and Methods
Construction of expression vectors
The plasmid pET-16b/YGL157w was constructed for production of K. marxianus YGL157w protein with a N-terminal hexahistidine tag. After preparation of genomic DNA from K. marxianus strain DMB1 (strain number: HUT7412), the YGL157w gene (accession number: LC016711) was amplified using PCR with KOD -plus- DNA polymerase (Toyobo, Osaka, Japan) and the primers 5′-CAT ATGACGTACGTTGTGGTTACTGGTGC-3′ (the NdeI site is in bold and the initiation codon is in italics) and 5′-GGATCC TTAGTTGTTAGCCTTTAGTATTTG-3′ (the BamHI site is in bold and termination codon is in italics). The PCR product was cloned into pTA2 (Toyobo, Osaka, Japan) and sequenced to check for PCR errors. The YGL157w gene was then excised from the resulting plasmid using NdeI and BamHI and subcloned into pET-16b (Novagen, Hessen, Germany) to give pET-16b/YGL157w.
Expression of proteins
YGL157w protein was expressed in E. coli BL21 (DE3) cells harboring pET-16b/YGL157w and then purified to homogeneity. The cells were grown at 37°C for 3 h in Luria-Bertani (LB) medium (1 L) containing 100 mg/L ampicillin. After inducing expression by addition of isopropyl β-D-1-thiogalactopyranoside (IPTG) to a final concentration of 1.0 mM, the culture was incubated for an additional 3 h. The cells were then harvested, suspended in 20 mM Tris–HCl buffer (pH 7.9) containing 500 mM NaCl (buffer A) and 5 mM imidazole, and disrupted by ultrasonication. The resultant lysate was clarified by centrifugation (27,500 × g for 15 min at 4°C), after which the supernatant was applied to a Chelating Sepharose Fast Flow column (20 mL; GE Healthcare, Buckinghamshire, UK) charged with Ni2+ and equilibrated with buffer A containing 5 mM imidazole. After washing the column with buffer A containing 5 mM imidazole (40 mL) and then 60 mM imidazole (60 mL), the recombinant YGL157w protein was eluted with buffer A containing 500 mM imidazole. The active fractions were pooled, concentrated using a Vivaspin 20 concentrator (10,000 MWCO, Sartorius AG, Goettingen, Germany) and loaded onto a HiLoad 26/60 Superdex 200 pg column (GE Healthcare) equilibrated with 20 mM Tris–HCl buffer (pH 8.0) containing 50 mM NaCl. The active fractions were pooled and dialyzed against 20 mM Tris–HCl buffer (pH 7.2). Finally, the dialysate was concentrated and the resultant solution was used for biochemical experiments.
Protein concentrations were determined using the Bradford method with bovine serum albumin (BSA) serving as the standard (Bradford 1976).
Molecular mass determination
SDS-PAGE was carried out on a 10% polyacrylamide gel using the method of Laemmli (1970). EzStandard PrestainBlue (ATTO, Tokyo, Japan) was used as the molecular mass standards. The protein sample was boiled for 5 min in EzApply (ATTO). Protein bands were visualized by staining with EzStainAqua (ATTO).
The molecular mass of the native enzyme was determined by gel filtration column chromatography using a Superdex 200 Increase 10/300 GL column. Conalbumin (75 kDa), ovalbumin (43 kDa), carbonic anhydrase (29 kDa), ribonuclease A (13.7 kDa) and aprotinin (6.5 kDa) served as molecular standards (GE Healthcare).
Assay of enzyme activity
KmGRE2 activity was measured by monitoring the decreases in the absorbance at 340 nm caused by the reduction of aldehyde, or the increases in the absorbance caused by the oxidation of alcohol. The mixture (1 mL) used for the reductive reaction contained 100 mM acetate buffer (pH 5.5), 5 mM aldehyde, 0.2 mM NADPH and YGL157w protein. The mixture (1 mL) used for the oxidative reaction contained 100 mM bicarbonate-NaOH (pH 10.0), 5 mM alcohol, 1.25 mM NADP+ and YGL157w protein. The reaction was started by the addition of coenzymes, and the absorbance at 340 nm was monitored at 25°C using a Shimadzu UV-2450 (Kyoto, Japan). The extinction coefficient of NADPH was 6.22 mM−1 cm−1. One unit of enzyme was defined as the amount of enzyme producing 1 μmol of NADPH per min at 25°C in the reductive reaction of methylglyoxal.
Effects of pH and temperature on enzyme activity
The pH dependence of the reduction catalyzed by YGL157w protein was determined at 25°C using 100 mM concentrations of acetate (pH 4.0–5.5) and citrate (pH 5.5–6.5). The temperature dependence was evaluated by measuring the reductive reaction at temperatures ranging from 25 to 45°C.
Effects of pH and temperature on enzyme stability
The effect of pH on enzyme stability was evaluated by incubating 100 nM YGL157w protein for 30 min at 35°C with 50 mM concentrations of acetate (pH 5.0–5.5), citrate (pH 5.5–6.5), phosphate (pH 6.5–8.0), borate-NaOH (pH 8.0–9.0) and bicarbonate-NaOH (pH 9.0–11.0). The enzyme solution was then rapidly cooled on ice, and the remaining activity was determined using the standard reduction assay. The thermal stability was determined by incubating YGL157w protein in 20 mM Tris–HCl buffer (pH 7.2) for 30 min at temperatures ranging from 25–45°C. The enzyme solution was then rapidly cooled on ice, and the remaining activity was determined using the standard reduction assay.
Determination of kinetic parameters
The initial velocity of the reductive reaction was analyzed using the standard assay conditions. To determine the kinetic constants for methylglyoxal and NADPH, several concentrations of methylglyoxal (0.05–15 mM) or NADPH (0.01–0.15 mM) were used. The initial velocity was then plotted against the substrate concentration, and the K m and k cat values were determined by curve fitting using Igor Pro ver. 3.14 (WaveMetrics, Tigard, OR, USA).
Preparation of hydrolysate
Lignocellulosic biomass material (Japanese cedar) was milled using a cutter mill (MKCM-3; Masuko Sangyo, Saitama, Japan), after which the resulting particles were used as the initial raw material. According to Lee et al. (2010), mechanochemical and hydrothermal pretreatment was carried out. The resulting sample was hydrolyzed using 20 FPU/g of Acremonium cellulase (Meiji Seika Pharma, Nagoya, Japan) and 40 μL/g of Optimash BG (Genencor International, Rochester, NY, USA) in 50 mM citrate buffer (pH 5.0) at 50°C and 150 rpm. After incubation for 48 h, the reaction mixture was harvested by centrifugation, and the supernatant was filtered through a 0.2 μm filter (Merck Millipore, Billerica, MA, USA). The pH of the mixture was then adjusted to 6.5, the mixture was diluted, and the resulting solution was used as the hydrolysate. Further details of the procedure are provided elsewhere (Akita et al. 2015).
Effect of KmGRE2 expression on cell growth
The effect of KmGRE2 expression was evaluated by cultivation in a test tube using 3 mL of hydrolysate containing 0.5 mM IPTG, which was incubated at 37°C and 180 rpm. E. coli BL21 (DE3) cells harboring pET-16b/YGL157w or pET-16b were pregrown overnight and then diluted 1:100 with fresh hydrolysate. Cultures were monitored for cell growth at OD600 using an Eppendorf BioSpectrometer (Eppendorf, Hamburg, Germany).
Quantification of sugars and aldehydes
After clarifying the culture by centrifugation and filtration, the supernatant was subjected to high performance liquid chromatography (HPLC). Quantification was performed using an Aminex HPX-87H cationic exchange column connected to an Aminex 85H Micro-Guard Column (Bio-Rad Labs, Richmond, CA, USA). The chromatographic conditions for sugars and aldehydes were as follows: mobile phase, 4.5 mM H2SO4 or 8 mM H2SO4; flow rate, 0.6 mL · min−1; and the column oven temperature, 65°C or 35°C. Sugars and aldehydes were detected using a Jasco RI-2031 Plus Intelligent Refractive Index Detector (Jasco, Tokyo, Japan) or a Jasco UV-2070 Plus Intelligent UV/VIS Detector at 278 nm (Jasco).
Results
Purification and molecular mass determination of KmGRE2
After expression in 4.86 g (wet weight) of E. coli cells harboring pET-16b/YGL157w, YGL157w protein was purified using two successive purification steps: Chelating Sepharose Fast Flow column chromatography and gel filtration chromatography (Figure 2A). Ultimately, a pure protein was obtained with an overall yield of 82.4% (Table 1).

Purification of YGL157w protein. (A) Purification steps followed by SDS-PAGE. Proteins were separated by SDS-PAGE and visualized by EzStainAqua staining: lane 1, protein molecular size markers; lane 2, crude extract; lane 3, Chelating Sepharose Fast Flow column chromatography pool; lane 4, HiLoad 26/60 Superdex 200 pg column pool. (B) Determination of molecular mass using gel filtration chromatography.
The apparent molecular mass of the YGL157w protein was determined to be about 36 kDa using Superdex 200 Increase 10/300 GL column gel filtration chromatography (Figure 2B). SDS-PAGE of the enzyme showed one major band of 40 kDa (Figure 2A), suggesting the native protein exists as a monomer.
Substrate specificity and kinetic properties of KmGRE2
When assessed the enzymatic activity of recombinant YGL157w protein, we found that it catalyzed the reduction of linear, branched-chain and aromatic aldehydes using NADPH as the coenzyme (Table 2). High levels of activity were observed with isovaleraldehyde (C5), methylglyoxal (C3) and valeraldehyde (C5), while the lower levels were observed with octanal (C8), benzaldehyde (C7) and HMF (C6). YGL157w protein showed no activity toward p-anisaldehyde (C8), p-hydroxy benzaldehyde (C7), D-alanine, l-alanine, D-lactate, L-lactate and pyruvate. Only NADPH was utilized as the cofactor for reduction of methylglyoxal by the enzyme; NADH was not inactive. In addition, using NADP+ as the coenzyme, no activity was observed for oxidative reactions toward alcohol-analogs such as isoamyl alcohol, isobutanol, 2-propanol, 1-hexanol, 1-heptanol and 1-octanol under the described conditions. These results demonstrate that YGL157w gene encodes a NADPH-dependent GRE2, which we are calling KmGRE2.
After measuring the initial rates at various methylglyoxal or NADPH concentrations, regression analyses were used to fit the data to the Michaelis-Menten equation (data not shown). The K m and k cat values for methylglyoxal were calculated as 0.30 ± 0.018 mM and 1.3 × 103 ± 15 min−1, respectively. The kinetic parameters for NADPH were 0.028 ± 0.0012 mM and 1.4 × 103 ± 22 min−1 mM−1, respectively. In addition, the k cat/K m for methylglyoxal and NADPH were 4.4 × 103 and 5.1 × 104 min−1 mM−1, respectively. These results are similar to those of S. cerevisiae (Murata et al. 1985).
Effects of pH and temperature on enzyme activity and stability
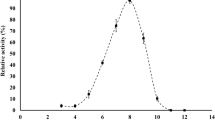
The effect of pH on the reduction of methylglyoxal was determined by assessing the enzyme activity at several pHs. At a temperature of 25°C, the optimum pH was about 5.5 (Figure 3A). When the temperature dependence at pH 5.5 was examined, maximum activity was observed at around 45°C (Figure 3B). Moreover, when KmGRE2 was incubated for 30 min at various temperatures in 20 mM Tris–HCl buffer (pH 7.2), KmGRE2 retained more than 80% of its activity at temperatures below 35°C (Figure 3C). On the other hand, there was a complete loss of activity when the enzyme was incubated at temperatures above 45°C. When the effect of pH on the stability of the enzyme was evaluated based on the activity remaining after incubation at 35°C for 30 min, we found that KmGRE2 retained more than 70% of its activity at pHs between 5.5 and 9.0 (Figure 3D). Somewhat disappointingly, however, KmGRE2 showed nearly the same temperature and pH stability as S. cerevisiae GRE2 (Park et al. 2010).

Effects of pH and temperature on KmGRE2 activity and stability. The markers of buffer were indicated following: circles, acetate; squares, citrate; diamonds, phosphate, isosceles triangles, borate-NaOH and right triangles, bicarbonate-NaOH. (A) Effect of pH on KmGRE2 activity. (B) Effect of temperature on KmGRE2 activity. (C) KmGRE2 activity after incubation for 30 min at various temperatures in the 20 mM Tris–HCl buffer (pH 7.2). (D) KmGRE2 activity after incubation for 30 min at 35°C in buffer solutions of various pHs. Error bars indicate SE (n = 3).
Cell growth in hydrolysate from cedar
To assess the ability of KmGRE2 as decomposer, the effect of KmGRE2 expression on the growth of cell in cedar hydrolysate was determined by monitoring the E. coli growth. When cedar hydrolysate was prepared, glucose and xylose were mainly included as the sugars, whereas aldehyde inhibitors such as furfural and HMF were mainly generated. Thus, these sugars and aldehydes were detected by HPLC. The pH of hydrolysate was decided based on the optimal pH for KmGRE2 activity and the possible growth pH of E. coli. When cultivated in the hydrolysate under these conditions, KmGRE2-overexpressing E. coli showed more rapid growth than E. coli not expressing the enzyme (Figure 4), with enhanced furfural degradation (Table 3).

Growth of KmGRE2-overexpressing E. coli cells (circles) and cells not expressing KmGRE2 (squares). Error bars indicate SE (n = 3).
Discussion
In the present study, we succeeded in expressing the YGL157w gene from K. marxianus strain DMB1 in E. coli cells and purifying the product. Characterization of the purified enzyme showed that KmGRE2 harbored strong NADPH-dependent reductive activities toward at least 10 aldehyde substrates (Tables 2). The higher activities were observed on C3 branched-chain and C3 to C7 linear aldehydes, whereas lower or no activities were detected for C8 linear aldehyde and C6 to C8 aromatic aldehydes. Conversely, S. cerevisiae GRE2 showed the highest activity for phenyglyoxal (C8) in the presence of NADPH (Murata et al. 1985). When we compared the amino acid sequences of KmGRE2 and S. cerevisiae S288c GRE2, we found that Ser127, Tyr165 and Lys169 in GRE2 were completely conserved in KmGRE2 as Ser127, Tyr165 and Lys169 (Figure 1). The three aforementioned residues in GRE2 are considered the crucial roles for the substrate dehydrogenation: Ser127 stabilizes the substrate, Tyr165 acts on a catalytic base and Lys169 facilitates the catalysis at neutral pH (Guo et al. 2014). However, two residues responsible for the substrate binding differ between the two enzymes: Phe85 and Tyr128 in GRE2 are respectively replaced by Cys85 and Val128 in KmGRE2 (Guo et al. 2014) (Figure 1). These substitutions may reduce the hydrophobic interactions for aromatic aldehydes in KmGRE2, which suggests that the molecular mechanism for substrate recognition differs between KmGRE2 and GRE2. To assess the molecular mechanism, we are now trying to obtain crystals of cofactor and/or substrate-bound KmGRE2.
The utilization of biofuel from lignocellulosic biomass holds promise as a means of abating global warming. This has prompted the development of a number of bioconversion methods for biofuel production (Akita et al. 2015; Lan and Liao 2013; Nakashima et al. 2014; da Silva et al. 2014). But while those methods produced several kinds of biofuels from hydrolysate derived of lignocellulosic biomass, the productivities and yields were often low (Akita et al. 2015; Lan and Liao 2013; Nakashima et al. 2014; da Silva et al. 2014). One of the mentioned causes of the low productivity is microbial growth inhibition by aldehyde inhibitors (Mills et al. 2009). Because aldehyde inhibitors such as furfural, HMF, glycolaldehyde, methylglyoxal and vanillin are generated mainly during the biomass hybridization process (Jarboe and Chi 2013; Jayakody et al. 2011), they are able to inhibit microbial growth and interfere with subsequent fermentation (Jayakody et al. 2011; Liu et al. 2008; Mills et al. 2009; Moon and Liu 2012). Consequently, we proposed that KmGRE2 utilizes as inhibitor decomposer. To confirm the ability of KmGRE2 to play decomposer, we assessed the effect of KmGRE2 expression on cell growth in cedar hydrolysate, production of which led to the formation of both furfural and HMF. As anticipated, the KmGRE2-overexpressing E. coli showed substantial growth improvement (Figure 4). We think that the growth improvement was achieved by enhanced furfural degradation, which provided for the preferable culture conditions at early culture phase. In fact, the OD600 of KmGRE2-overexpressing E. coli at 6 to 12 h were 1.3–1.6-fold higher than these of not expressing E. coli. On the other hand, the less activity toward HMF in KmGRE2-overexpressing E. coli remained unclear. The omics analysis on the metabolic response of KmGRE2-overexpressing E. coli may reveal this phenomenon. Recently, we developed a simple and efficient method involving biomass-inducible chromosome-based expression system (BICES) for expressing foreign genes without the use of plasmids or expensive inducers (Akita et al. 2015; Nakashima et al. 2014). This method can also be used to produce biofuels, but the productivity and yield were markedly diminished when hydrolysate from Japanese cedar as the carbon source for isobutanol production (Akita et al. 2015). We are now planning to integrate KmGRE2 gene into the genome of the E. coli strain involving BICES. We anticipate that this will improve growth rates, thereby increasing the productivity and yield.
References
Akita H, Nakashima N, Hoshino T (2015) Bacterial production of isobutanol without expensive reagents. Appl Microbiol Biotechnol 99:991–999
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Chen CN, Porubleva L, Shearer G, Svrakic M, Holden LG, Dover JL, Johnston M, Chitnis PR, Kohl DH (2003) Associating protein activities with their genes: rapid identification of a gene encoding a methylglyoxal reductase in the yeast Saccharomyces cerevisiae. Yeast 20:545–554
Choi YH, Choi HJ, Kim D, Uhm KN, Kim HK (2010) Asymmetric synthesis of (S)-3-chloro-1-phenyl-1-propanol using Saccharomyces cerevisiae reductase with high enantioselectivity. Appl Microbiol Biotechnol 87:185–193
da Silva TL, Gouveia L, Reis A (2014) Integrated microbial processes for biofuels and high value-added products: the way to improve the cost effectiveness of biofuel production. Appl Microbiol Biotechnol 98:1043–1053
Ema T, Ide S, Okita N, Sakai T (2008) Highly efficient chemoenzymatic synthesis of methyl (R)-o-chloromandelate, a key intermediate for clopidogrel, via asymmetric reduction with recombinant Escherichia coli. Adv Synth Catal 350:2039–2044
Garay-Arroyo A, Covarrubias AA (1999) Three genes whose expression is induced by stress in Saccharomyces cerevisiae. Yeast 22:879–892
Guo PC, Bao ZZ, Ma XX, Xia Q, Li WF (2014) Structural insights into the cofactor-assisted substrate recognition of yeast methylglyoxal/isovaleraldehyde reductase Gre2. Biochim Biophys Acta 1844:1486–1492
Hauser M, Horn P, Tournu H, Hauser NC, Hoheisel JD, Brown AJ, Dickinson JR (2007) A transcriptome analysis of isoamyl alcohol-induced filamentation in yeast reveals a novel role for Gre2p as isovaleraldehyde reductase. FEMS Yeast Res 7:84–92
Inoue Y, Rhee H, Watanabe K, Murata K, Kimura A (1988) Metabolism of 2-oxoaldehyde in mold. Purification and characterization of two methylglyoxal reductases from Aspergillus niger. Eur J Biochem 171:213–218
Jarboe LR, Chi Z (2013) Inhibition of microbial biocatalysts by biomass-derived aldehydes and methods for engineering tolerance. In: Torrioni L, Pescasseroli E (eds) New Developments in Aldehydes Research. Nova Science Publishers, New York
Jayakody LN, Hayashi N, Kitagaki H (2011) Identification of glycolaldehyde as the key inhibitor of bioethanol fermentation by yeast and genome-wide analysis of its toxicity. Biotechnol Lett 33:285–292
Jayakody LN, Horie K, Hayashi N, Kitagaki H (2013) Engineering redox cofactor utilization for detoxification of glycolaldehyde, a key inhibitor of bioethanol production, in yeast Saccharomyces cerevisiae. Appl Microbiol Biotechnol 97:6589–6600
Krantz M, Nordlander B, Valadi H, Johansson M, Gustafsson L, Hohmann S (2004) Anaerobicity prepares Saccharomyces cerevisiae cells for faster adaptation to osmotic shock. Eukaryot Cell 3:1381–1390
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Lan EI, Liao JC (2013) Microbial synthesis of n-butanol, isobutanol, and other higher alcohols from diverse resources. Bioresour Technol 135:339–349
Lee SH, Chang F, Inoue S, Endo T (2010) Increase in enzyme accessibility by generation of nanospace in cell wall supramolecular structure. Bioresour Technol 101:7218–7223
Liu ZL, Moon J, Andersh BJ, Slininger PJ, Weber S (2008) Multiple gene-mediated NAD(P)H-dependent aldehyde reduction is a mechanism of in situ detoxification of furfural and 5-hydroxymethylfurfural by Saccharomyces cerevisiae. Appl Microbiol Biotechnol 81:743–753
Mills TY, Sandoval NR, Gill RT (2009) Cellulosic hydrolysate toxicity and tolerance mechanisms in Escherichia coli. Biotechnol Biofuels 2:26
Moon J, Liu ZL (2012) Engineered NADH-dependent GRE2 from Saccharomyces cerevisiae by directed enzyme evolution enhances HMF reduction using additional cofactor NADPH. Enzyme Microb Technol 50:115–120
Müller M, Katzberg M, Bertau M, Hummel W (2010) Highly efficient and stereoselective biosynthesis of (2S,5S)-hexanediol with a dehydrogenase from Saccharomyces cerevisiae. Org Biomol Chem 8:1540–1550
Murata K, Fukuda Y, Simosaka M, Watanabe K, Saikusa T, Kimura A (1985) Metabolism of 2-oxoaldehyde in yeasts. Purification and characterization of NADPH-dependent methylglyoxal-reducing enzyme from Saccharomyces cerevisiae. Eur J Biochem 151:631–636
Nakashima N, Akita H, Hoshino T (2014) Establishment of a novel gene expression method, BICES (biomass-inducible chromosome-based expression system), and its application for production of 2,3-butanediol and acetoin. Metab Eng 25:204–214
Park HJ, Jung J, Choi H, Uhm KN, Kim HK (2010) Enantioselective bioconversion using Escherichia coli cells expressing Saccharomyces cerevisiae reductase and Bacillus subtilis glucose dehydrogenase. J Microbiol Biotechnol 20:1300–1306
Ray M, Ray S (1984) Purification and partial characterization of a methylglyoxal reductase from goat liver. Biochim Biophys Acta 802:119–127
Rep M, Proft M, Remize F, Tamás M, Serrano R, Thevelein JM, Hohmann S (2001) The Saccharomyces cerevisiae Sko1p transcription factor mediates HOG pathway-dependent osmotic regulation of a set of genes encoding enzymes implicated in protection from oxidative damage. Mol Microbiol 40:1067–1083
Rutherford JC, Bird AJ (2004) Metal-responsive transcription factors that regulate iron, zinc, and copper homeostasis in eukaryotic cells. Eukaryot Cell 3:1–13
Suzuki T, Hoshino T, Matsushika A (2014) Draft genome sequence of Kluyveromyces marxianus strain DMB1, isolated from sugarcane bagasse hydrolysate. Genome Announc 2:e00733-14
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Warringer J, Blomberg A (2006) Involvement of yeast YOL151W/GRE2 in ergosterol metabolism. Yeast 23:389–398
Acknowledgment
We are grateful to all members of the Bio-conversion Research Team at our Institute [Biomass Refinery Research Center, National Institute of Advanced Industrial Sciences and Technology (AIST)] for their technical assistance and valuable discussion.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contribution
HA designed this study, performed experiments, participated in the interpretation of the results and drafted the manuscript. MW and TS participated in the design and coordination of this study and helped to revise the manuscript. NN and TH conceived and designed this study, coordinated the experiments, interpreted the results and revised the manuscript for important intellectual content. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Akita, H., Watanabe, M., Suzuki, T. et al. Characterization of the Kluyveromyces marxianus strain DMB1 YGL157w gene product as a broad specificity NADPH-dependent aldehyde reductase. AMB Expr 5, 17 (2015). https://doi.org/10.1186/s13568-015-0104-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13568-015-0104-9