
Abstract
Background
Tumor-induced osteomalacia is a rare, acquired paraneoplastic syndrome, including hypophosphatemia, high serum alkaline phosphatase, reduced active vitamin D, suboptimal bone mineral density, bone pain, fragility fractures, and muscle weakness.
Case presentation
We report a case of 74–year–old male of mixed ancestry with hypophosphatemia resistant to treatment despite optimal compliance, associated with profound reduction of bone mineral density and multiple nontraumatic fractures, including bilateral rib fractures, lower-thoracic (T11, T12) vertebrae, and two fractures involving the surgical and anatomical neck of the right humerus. We discuss an approach to identifying the underlying cause of hypophosphatemia associated with fragility fractures, and options for management of this rare condition.
Conclusion
Although rare, tumor-induced osteomalacia can be diagnosed if a logical stepwise approach is implemented. Surgery could be curative if the tumor is properly located and is resectable.
Similar content being viewed by others


Background
Osteomalacia usually manifests with a predictable pattern of biochemical derangements, including hypophosphatemia, elevated serum alkaline phosphatase, and low or inappropriately normal active vitamin D, associated with reduced bone mineral density [1]. Additional clinical manifestations of osteomalacia include bone pain, fragility fractures, and muscle weakness, and in children, rickets may lead to severe stunting of their longitudinal growth, if not managed optimally [2,3,4]. Several disorders resulting in hypophosphatemia can cause osteomalacia by directly limiting the mineralization process [5, 6]. Vitamin D deficiency, resulting in malabsorption, for example from celiac disease, defective hydroxylation of vitamin D, or resistance to vitamin D, may result in deficient plasma calcium and phosphate concentrations, and in the long term, can cause osteomalacia [7, 8]. Phosphate deficiency may also result from poor dietary intake or renal losses associated with Fanconi syndrome, hypophosphatemic osteomalacia, and in tumor-induced osteomalacia (TIO) [1, 5]. The pathogenic mechanism of TIO is the excessive secretion of fibroblast growth factor 23 (FGF-23), a phosphatonin secreted by the tumor arising from mesenchymal tissue, which can result in reduced renal phosphate reabsorption and impaired vitamin D 1α-hydroxylase activity [1, 9].
Laboratory workup to elucidate the cause of hypophosphatemia includes confirmation of reduced renal tubular reabsorption of phosphate, serum phosphate and calcium, alkaline phosphatase, 25-hydroxy vitamin D, 1–25-hydroxy vitamin D, genetic testing for hypophosphatemic osteomalacia, and, if available, FGF-23 concentrations [3, 4]. Treatment includes the combination of vitamin D (calcitriol and alfacalcidol) and phosphate supplementation. If a proven mesenchymal tumor is identified consistent with TIO, surgical resection is recommended and is potentially curative [2, 9, 10].
Case presentation
A 74-year–old man of mixed ancestry, married father and pensioner, with neither significant past medical history nor a relevant family history, was referred to the endocrine service because of severe osteoporosis. The diagnosis of osteoporosis was made based on the findings of a dual x-ray absorptiometry (DEXA) scan, performed by a private radiology practice, which showed severely low bone mineral density, with T-scores of the lumbar spine (L1–L4) of −3.1 standard deviations (SD), left femoral neck −4.3 SD, left total hip −3.4 SD, right femoral neck −4.7 SD, right total hip −4.1 SD, and right forearm −1.3 SD.
His history is noteworthy for long-standing generalized body pain and muscle weakness for about 7 years. His muscle weakness progressed in severity rendering him bed bound for 2 years prior to presentation. At the time of his review by the endocrine service, he was only taking calciferol and calcium supplementation but did not demonstrate any improvement in mobility. Significant proximal muscle weakness (4/5) was elicited at both the shoulder and pelvic girdles, with generalized muscle wasting. His family observed progressive loss of longitudinal height and noted that he tired easily. There were no apparent symptoms suggestive of an occult malignancy. He had neither a suggestive family history of hypophosphatemic osteomalacia, nor Fanconi syndrome. Moreover, he was a life-long nonsmoker and consumed no alcohol.
Additional clinical examination revealed a bed-bound elderly man, with a barrel chest, and a flail segment of his chest, consistent with rib fractures. Hemodynamically, he was stable, blood pressure was 130/85 mmHg and pulse rate 82 per minute in sinus rhythm. He used accessory respiratory muscles in keeping with respiratory distress. Chest auscultation revealed bilateral scattered crackles. Importantly, he was severely kyphotic, and the rest of the systemic examination was noncontributory.
Initial laboratory workup showed that his serum creatinine was 59 (64–104) µmol/L, full blood count, liver function tests, thyroid function tests, uric acid, and lipogram were all within normal limits. Moreover, additional tests revealed a serum phosphate of 0.39 (0.78–1.4) mmol/L, calcium 2.01 (2.2–2.56) mmol/L, albumin 46 (35–52) g/L, alkaline phosphatase 302 (53–128) U/L, and a simultaneous measure of parathyroid hormone of 16.8 (1.6–6.9) pmol/L. We confirmed that he was deficient in serum 25–hydroxy-vitamin D [45.2 (replete > 72.5) nmol/L]. His fractional excretion of phosphate was 29% (< 5%) and the ratio of tubular maximum reabsorption of phosphate to glomerular filtration rate (TmP/GFR) was 0.33 mmol/L (normal range 0.8–1.35 mmol/L), taken contemporaneously with a serum phosphate of 0.44 (0.78–1.4) mmol/L, indicates profound phosphaturia. Venous blood gas showed a pH of 7.38 (7.35–7.45), pCO2 5.6 (4.66–6.38) kPa, HCO3 25 (19–24) mmol/L, and the erythrocyte sedimentation rate was 4 (0–20) mm/hour. Additional tests to identify the underlying cause for his fragility fractures included a normal serum protein electrophoresis, absence of Bence–Jones proteins, and an 08:00 serum testosterone of 13.4 (6.7–25.7) nmol/L. Antitissue transglutaminase antibodies, antigliadin, and endomysial antibodies were additionally negative. In view of his particularly high phosphaturia, exclusion of Fanconi syndrome was undertaken, which demonstrated normal urinary excretion of amino acids, absence of glycosuria, and sodium and potassium excretion in the urine were entirely within normal limits.
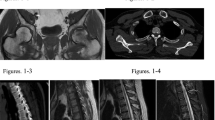
He was continued on calciferol and alfacalcidol and commenced on phosphate supplementation. Despite regular use of these medicines and normalization of his serum calcium and vitamin D levels, low serum phosphate and elevated alkaline phosphatase and parathyroid hormone persisted. Oncogenic osteomalacia was considered having excluded a genetic form of hypophosphatemic osteomalacia. A Gallium Ga 68-DOTANOC PET/CT showed a focus of avid uptake in the soft tissue of the posterior aspect of the right humeral head, and a representative magnetic resonance imaging (MRI) scan showed an ill-defined nodular lesion corresponding with the initial area of uptake, measuring 12 × 12 mm, related to the distal teres minor muscle fibers (Fig. 1). Associated with the defined lesion on MRI scan, two fractures of the right head of humerus were seen, with one of the fractures being recently sustained. Multiple rib fractures were also seen. The lesion in the right shoulder was considered to be the likely culprit. Due to our resource limitations, it was deemed too costly to initiate somatostatin analog therapy, and surgery was postponed until the end of the COVID-19 pandemic.

Demonstration of a tumor in the right humeral head by Gallium 68-DOTANOC PET/CT (A) and magnetic resonance imaging (MRI) (B)
Discussion
We present a rare case of hypophosphatemic osteomalacia, resulting in severe osteoporosis and multiple fractures from tumor-inducing osteomalacia complicated by severe vitamin D deficiency. Although we did not have histological evidence of a mesenchymal tumor, we feel that there is sufficient indirect evidence for this, based on the positive Gallium Ga 68-DOTANOC PET/CT results and confirmation of a well-defined lesion on MRI.
Severe reduction in bone mineral density and fragility fractures can, rarely, be caused by hypophosphatemia [11]. Specifically, tumor-induced osteomalacia (TIO) is a rare, frequently missed condition, which is challenging to diagnose. The symptoms and biochemical derangements are often vague and mirror other conditions, for example, osteoporosis. It is important to know that inadvertent use of bisphosphonates could have aggravated the condition considerably [12]. Until 2018, only 500 cases were reported worldwide [13], with a peak age distribution of between 40 and 45 years [14].
TIO is a rarely acquired, paraneoplastic syndrome, resulting in hypophosphatemia, low levels of active vitamin D, severe osteomalacia, bone pain, fragility fractures, and muscle weakness. Patients with TIO are generally symptomatic for bone pain, muscle weakness, and gait disturbance, likely a consequence of the pelvic girdle myopathy [2]. A Chinese retrospective analysis of 144 cases revealed that bone pain was the most common reported symptom, followed by height loss in more than 50% of patients [15]. Pathological fractures of both thoracic and lumbar vertebrae, ribs, femurs, and the pelvis were the major cause of morbidity associated with TIO [13].
The typical biochemical pattern includes low serum phosphate, normal to low calcium, normal parathyroid hormone (PTH), low or inappropriately normal 1,25-dihydroxy vitamin D, normal 25-hydroxyvitamin D, elevated alkaline phosphatase, and elevated fibroblast growth factor 23 (FGF-23) concentrations in the serum. Increased phosphate excretion in urine is demonstrated by a low tubular reabsorption of phosphate [16]. The usual presentation is generally through investigation for fragility fractures, bone pain, muscle weakness, and height loss [15].
The principal pathogenic mechanism of TIO is over-production of fibroblast growth factor 23 (FGF23), a phosphatonin that causes renal phosphate wasting, secondary to it is role in regulating renal inorganic phosphate reabsorption and modifying 25-hydroxyvitamin D 1α-hydroxylase activity [17]. We did not have access to measurement of serum FGF-23.
The differential diagnosis of TIO includes genetic causes of hypophosphatemic osteomalacia, in particular, X-linked hypophosphatemic rickets (XLH), autosomal dominant hypophosphatemic rickets (ADHR), and autosomal recessive hypophosphatemic rickets (ARHR) [18]. These aforementioned conditions may manifest with a similar phenotype to TIO, but usually a positive family history is elicited, with early onset gait and skeletal deformities. Both TIO and these inherited forms of hypophosphatemic rickets are associated with elevated FGF23. The positive family history may corroborate a genetic cause for hypophosphatemia, which is further proven by genetic testing, including PHEX mutation for XLH, mutations in the FGF23 gene causing ADHR (including R176Q, R176W, R179Q, R179W), and mutations for autosomal recessive hypophosphatemic rickets (ARHR), including inactivating mutations in the DMP1 gene, ENPP1 gene, and mutations in the FAM20C gene encoding a protein kinase [19,20,21]. FGF-23 is presumed to reduce the expression of both 1-α-hydroxylase enzyme and two phosphate channels namely [Na-Pi 2a) and (Na-Pi 2c)], leading to phosphate wasting in the intestine and kidneys [22,23,24,25].
A logical stepwise process has been proposed to locate the offending tumor, by combining functional and anatomical tests, using PET/CT scan, CT scan, and MRI [14]. These tumors sometimes express somatostatin receptors [26, 27], which facilitates location, using somatostatin-receptor (SSTR) imaging, but they lack specificity, necessitating a pathological diagnosis [28] Following functional imaging, anatomical imaging is necessary to confirm a pathological lesion using ultrasound, CT, and MRI, depending on the location [13]. Despite all the advances in the investigations for diagnosis, there is a proportion of patients who fail to demonstrate a positive result. Imaging may be repeated every 1–2 years in the hope that the tumor may declare itself [13].
Medical therapy is indicated during workup of these patients, when tumors cannot be located or a complete excision cannot be performed [29]. The basic regimen for TIO includes phosphate and active vitamin D (calcitriol and alfacalcidol) supplements [30] Side effects include nephrolithiasis, nephrocalcinosis, impaired renal function, and either secondary or tertiary hyperparathyroidism [29], necessitating close surveillance. Somatostatin receptor analogues have been used with some success [29, 31,32,33]. Inconsistent results are reported regarding the efficacy of somatostatin receptor-based therapy with octreotide in this setting [31,32,33]. FGF-23 antibodies, for example, burosumab, a fully human IgG1 monoclonal antibody directed at fibroblast growth factor 23, is now licensed to be used in the treatment of TIO [33].
Conclusions
TIO remains a rare, but potentially treatable, paraneoplastic syndrome, requiring a high index of suspicion. Careful attention in assessing patients for osteoporosis who also have a low serum phosphate, should alert the clinician to either a hereditary form of hypophosphatemia or an oncogenic condition, such as tumor-induced osteomalacia.
Availability of data and materials
All laboratory data are available through the repository held by the National Health Laboratory Services (NHLS), South Africa and radiology is available from the Department of Radiology, Groote Schuur Hospital, Cape Town.
Abbreviations
- TIO:
-
Tumor-induced osteomalacia
- FGF-23:
-
Fibroblast growth factor 23
- DEXA:
-
Dual energy x-ray absorptiometry
- SD:
-
Standard deviations
- Ga 68-DOTANOC PET/CT:
-
Gallium 68 (68Ga) 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA)–octreotate (DOTATATE, GaTate) positron emission tomography/computed tomography
- MRI:
-
Magnetic resonance imaging
- XLH:
-
X-linked hypophosphatemic rickets
- ADHR:
-
Autosomal dominant hypophosphatemic rickets
- ARHR:
-
Autosomal recessive hypophosphatemic rickets
- PHEX:
-
Phosphate regulating gene with homologies to endopeptidases on the X-chromosome
- DMP1:
-
Dentin matrix acidic phosphoprotein 1
- ENPP1:
-
Ectonucleotide pyrophosphatase/phosphodiesterase-1
- FAM20C:
-
Family with sequence similarity 20, member C
- PET/CT:
-
Positron emission tomography/computed tomography
- CT:
-
Computed tomography
- SSTR:
-
Somatostatin receptor
- IgG1:
-
Immunoglobulin G1
Reference
Minisola S, et al. Tumour-induced osteomalacia. Nat Rev Dis Primers. 2017;3(1):1–15.
Lewiecki EM, Urig EJ Jr, Williams RC Jr. Tumor-induced osteomalacia: lessons learned. Arthrit Rheum. 2008;58(3):773–7.
Reginato AJ, Coquia JA. Musculoskeletal manifestations of osteomalacia and rickets. Best Pract Res Clin Rheumatol. 2003;17(6):1063–80.
Hazzazi MA, et al. Clinical presentation and etiology of osteomalacia/rickets in adolescents. Saudi J Kidney Dis Transplant. 2013;24(5):938.
Penido MGM, Alon US. Phosphate homeostasis and its role in bone health. Pediatr Nephrol. 2012;27(11):2039–48.
Thalayasingam B. Coeliac disease as a cause of osteomalacia and rickets in the Asian immigrant population. Br Med J (Clin Res Ed). 1985;290(6475):1146.
Hughes MR, et al. Regulation of serum 1alpha, 25-dihydroxyvitamin D3 by calcium and phosphate in the rat. Science. 1975;190(4214):578–80.
Hutchison FN, Bell NH. Osteomalacia and rickets. In: Seminars in nephrology; Amsterdam: Elsevier. 1992.
Kumar R, Folpe AL, Mullan BP. Tumor-induced osteomalacia. Transl Endocrinol Metab. 2015;7(3):1871.
Peacock M, et al. The action of 1αhydroxy vitamin D 3 and phosphate supplements in hypophosphataemic osteomalacia. In: Phosphate metabolism. Springer; 1977. p. 431–40.
Yin Z, et al. Tumor-induced osteomalacia. Osteoporos Sarcopenia. 2018;4(4):119–27.
Cundy T, et al. Bisphosphonate-induced deterioration of osteomalacia in undiagnosed adult Fanconi syndrome. JBMR Plus. 2020;4(8):e10374.
Jung G-H, et al. A 9-month-old phosphaturic mesenchymal tumor mimicking the intractable rickets. J Pediatric Orthop B. 2010;19(1):127–32.
Feng J, et al. The diagnostic dilemma of tumor induced osteomalacia: a retrospective analysis of 144 cases. Endocrine J. 2017; EJ16-0587.
White KE, et al. The autosomal dominant hypophosphatemic rickets (ADHR) gene is a secreted polypeptide overexpressed by tumors that cause phosphate wasting. J Clin Endocrinol Metab. 2001;86(2):497–500.
Pal R, et al. Tumor-induced osteomalacia: experience from three tertiary care centers in India. Endocr Connect. 2019;8(3):266–76.
Beygi S, Denio A, Sharma TS. The foot that broke both hips: a case report and literature review of tumor-induced osteomalacia. Case Rep Rheumatol. 2017;2017:1.
White KE, et al. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet. 2000;26(3):345–8.
Bergwitz C, Miyamoto K-I. Hereditary hypophosphatemic rickets with hypercalciuria: pathophysiology, clinical presentation, diagnosis and therapy. Pflügers Archiv-Eur J Physiol. 2019;471(1):149–63.
Sommer S, et al. The phosphatonins and the regulation of phosphate transport and vitamin D metabolism. J Steroid Biochem Mol Biol. 2007;103(3–5):497–503.
González-Lamuño, D., Hypophosphataemic rickets: diagnosis algorithm—how not to make a mistake. Adv Therapy. 2020; 1–10.
Urakawa I, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444(7120):770–4.
Shimada T, et al. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004;19(3):429–35.
Sabbagh Y, et al. Intestinal phosphate transport. Adv Chronic Kidney Dis. 2011;18(2):85–90.
Yamazaki Y, et al. Increased circulatory level of biologically active full-length FGF-23 in patients with hypophosphatemic rickets/osteomalacia. J Clin Endocrinol Metab. 2002;87(11):4957–60.
Reubi JC, et al. Somatostatin and vasoactive intestinal peptide receptors in human mesenchymal tumors: in vitro identification. Can Res. 1996;56(8):1922–31.
El-Maouche D, et al. 68Ga-DOTATATE for tumor localization in tumor-induced osteomalacia. J Clin Endocrinol Metab. 2016;101(10):3575–81.
van Boekel G, et al. Tumor producing fibroblast growth factor 23 localized by two-staged venous sampling. Eur J Endocrinol. 2008;158(3):431–8.
Seufert J, et al. Octreotide therapy for tumor-induced osteomalacia. N Engl J Med. 2001;345(26):1883–8.
Kim DH, Sung DH, Min YK. Hypophosphatemic osteomalacia induced by low-dose adefovir therapy: focus on manifestations in the skeletal system and literature review. J Bone Miner Metab. 2013;31(2):240–6.
Ovejero D, et al. Octreotide is ineffective in treating tumor-induced osteomalacia: results of a short-term therapy. J Bone Miner Res. 2017;32(8):1667–71.
Ishii A, et al. The levels of somatostatin receptors in causative tumors of oncogenic osteomalacia are insufficient for their agonist to normalize serum phosphate levels. Calcif Tissue Int. 2010;86(6):455–62.
Fukumoto S. FGF23-FGF receptor/Klotho pathway as a new drug target for disorders of bone and mineral metabolism. Calcif Tissue Int. 2016;98(4):334–40.
Acknowledgements
Khalid Aligail thanks the patient for consenting to publication of his case, and the co-authors for their support and comments during the preparation of this manuscript.
Funding
No funding was received for this case report.
Author information
Authors and Affiliations
Contributions
The authors contributed equally to investigation and management of this patient. The manuscript was co-authored jointly, but ILR supervised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The faculty of Health Sciences, Human Research and Ethics Committee University of Cape Town waives the need for ethics approval for case studies, but individual patient consent is required for publication. Patient consent was obtained.
Consent for publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interests
We have no conflict of interest, whatsoever.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Aligail, K., Dave, J.A. & Ross, I.L. Tumor-induced osteomalacia: a case report. J Med Case Reports 16, 22 (2022). https://doi.org/10.1186/s13256-021-03220-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13256-021-03220-7