
Abstract
Background
Alpha-synuclein (α-syn) is considered the main pathophysiological protein component of Lewy bodies in synucleinopathies. α-Syn is an intrinsically disordered protein (IDP), and several types of structural conformations have been reported, depending on environmental factors. Since IDPs may have distinctive functions depending on their structures, α-syn can play different roles and interact with several proteins, including amyloid-beta (Aβ) and tau, in Alzheimer’s disease (AD) and other neurodegenerative disorders.
Main body
In previous studies, α-syn aggregates in AD brains suggested a close relationship between AD and α-syn. In addition, α-syn directly interacts with Aβ and tau, promoting mutual aggregation and exacerbating the cognitive decline. The interaction of α-syn with Aβ and tau presented different consequences depending on the structural forms of the proteins. In AD, α-syn and tau levels in CSF were both elevated and revealed a high positive correlation. Especially, the CSF α-syn concentration was significantly elevated in the early stages of AD. Therefore, it could be a diagnostic marker of AD and help distinguish AD from other neurodegenerative disorders by incorporating other biomarkers.
Conclusion
The overall physiological and pathophysiological functions, structures, and genetics of α-syn in AD are reviewed and summarized. The numerous associations of α-syn with Aβ and tau suggested the significance of α-syn, as a partner of the pathophysiological roles in AD. Understanding the involvements of α-syn in the pathology of Aβ and tau could help address the unresolved issues of AD. In particular, the current status of the CSF α-syn in AD recommends it as an additional biomarker in the panel for AD diagnosis.
Similar content being viewed by others

Introduction
Alzheimer’s disease (AD) is characterized by extracellular amyloid-beta (Aβ) plaques and intracellular neurofibrillary or extraneuronal ghost tau tangles in the brain. Recently, the pathophysiology of other proteins, including the triggering receptor expressed on myeloid cells 2, transactive response DNA-binding protein 43, and α-synuclein (α-syn), has drawn attention for their direct and indirect associations in AD. α-Syn is the major constituent protein of Lewy bodies, the hallmark of Parkinson’s disease (PD) [1]. Accumulation of α-syn has been found in the brain of patients with AD, as well as in patients with synucleinopathies, such as PD, dementia with Lewy bodies (DLB), and multiple system atrophy [2]. However, the physiological and pathophysiological structures or functions of α-syn in AD are not fully understood. Furthermore, whether altered α-syn levels are a causal factor or consequential result of AD is unknown.
In 1993, Uéda et al. proposed an amyloidosis mechanism of α-syn and its involvement in the pathogenesis of AD by identifying non-Aβ components (NACs), now known as fragments of α-syn, in Aβ plaques [3]. Immunolabeling using an α-syn antibody verified abundant α-syn at the center of Aβ plaques, confirming the contribution of α-syn in the formation of Aβ plaques [4]. In addition, α-syn-positive inclusions were co-localized with neurofibrillary tangles, suggesting the influence of tau aggregation in Lewy body formation [5]. Previous studies showd that over half of the patients with autopsy-confirmed familial or sporadic AD had comorbid α-syn pathology in addition to Aβ and tau [5,6,7]. AD patients with autopsy-diagnosed Lewy body variants presented with more rapid cognitive deterioration and a higher mortality rate than patients with pure AD [8]. In contrast, a study showed no correlation of concurrent Lewy body abnormalities in AD with variability in clinical features, including cognitive decline, disease duration, or the presence of hallucinations or extrapyramidal signs [9]. This was supported by a recent autopsy study in which age-associated clinical and cognitive heterogeneities were mediated by the mid-frontal/hippocampal neurofibrillary tangle ratio, not by non-AD pathologies, such as the α-syn co-pathology [10]. On the other hand, patients with autopsy-confirmed quadruple pathologies (Aβ, tau, α-syn, and transactive response DNA-binding protein 43) were associated with a probability of aggressive progression in disease [11].
Although Lewy bodies were most often found in the amygdala and hippocampus of patients with AD, α-syn-positive inclusions, which differ from typical Lewy bodies, were also found [5, 7]. Regional distributions of α-syn and spreads of its pathological patterns differed between typical Lewy body diseases and AD. While the identified pathology of α-syn in DLB and PD spread from the brainstem to the limbic area and the neocortex [12, 13], α-syn in AD with amygdala-predominant Lewy bodies (AD/ALB) may show a spread to lower regions from the upper neuraxis [14]. In addition, immunohistochemical and biochemical analyses of DLB have revealed unique strain-like variations in α-syn pathologies in the amygdala, which are less in AD/ALB [15]. These close associations of α-syn in AD pathology and distinctions from synucleinopathies suggested its possible differential classification of the disease groups depending on the presence of α-syn aggregations [5]. In contrast, despite the absence of Lewy body-related pathology, levels of monomers and oligomers of intracellular soluble α-syn were elevated in the inferior temporal lobe of the AD brain, indicating the importance of the soluble form of α-syn in AD [16, 17]. Furthermore, α-syn levels were elevated in the cerebrospinal fluid (CSF) of patients with AD and are strongly correlated with tau levels [18]. Neuropathologically diagnosed AD patients with α-syn pathology had lower CSF total tau, phosphorylated tau 181, and neurogranin levels, which correlated with elevated α-syn levels [19]. Although the pathophysiological roles of α-syn in AD are unclear, growing evidence suggests that α-syn is directly involved in the pathophysiology of AD.
Variability in the α-syn structure
α-syn is a small protein composed of 140 amino acids (aa) and classified into three major domains (Fig. 1) [20]. The hydrophilic N-terminal domain (1–60 aa) is an amphipathic domain with an alpha-helical structure that includes the repeated consensus sequence KTKEGV. A characteristic property of this region is its involvement in binding to the lipid membrane [21]. Several missense mutations in this domain, such as A53T, A30P, and E46K, are involved in neurodegenerative disorders, particularly PD [22]. The central domain (61–95 aa) is referred to as the NAC, which can aggregate by forming a beta-sheet structure through its hydrophobic amino acids [3, 23]. The C-terminal domain (96–140 aa) contains abundant proline and strongly negatively charged amino acids, termed acidic tail, without a specific structure [12]. Alternative splicing of the SNCA gene transcript results in four protein isoforms; however, their roles under physiological and pathological conditions have not been well elucidated [24].

Characteristics of the three regions in α-synuclein. α-syn, alpha-synuclein; NAC, non-amyloid-beta component
α-Syn, considered to be an intrinsically disordered protein, can remain in a natively unfolded protein. Purified recombinant α-syn exists as a non-compact mixture of conformers with little secondary structure [25]. Interestingly, α-syn is stabilized by forming an alpha-helix secondary structure when the N-terminal domain is bound to the lipid membrane [23, 26]. The α-helical secondary structure of α-syn reverted to its unfolded conformation upon dissociation from the membrane [27]. However, purified native α-syn from human erythrocytes exists in the form of a tetramer with an α-helical structure, which could resist self-aggregation into protofibril/fibril conformations [28]. In addition, a stable multimeric state of α-syn is formed in the absence of a lipid membrane under non-denaturing conditions on electron micrograph reconstruction or nuclear magnetic resonance (NMR) studies [29]. However, a study showed that identified α-syn in the mouse brain had an unfolded monomeric structure that was prone to aggregation [30]. A review suggested that α-syn could exist in the monomeric structure of the transient state in the cytoplasm and be multimerized through interactions with the membrane until it resumed its physiological functions in the cell [31]. Evidence remains limited for the helical tetramer of α-syn, and further biochemical studies should be conducted to fully understand the physiological and pathophysiological conformations of α-syn in cells. Cryo-electron microscopy (cryo-EM) presented a high-resolution image of α-syn in the fibril state. With full-length recombinant human α-syn, cryo-EM on negative staining showed helical reconstructions, containing two polymorphic fibrils, i.e., rod and twister [32], supporting α-syn fibrils with different lengths extracted from the brain tissue of patients with PD or multiple system atrophy (MSA) [33, 34]. Compared to α-syn fibrils derived from the final product of the protein misfolding cyclic amplification reaction, a technique for amplifying α-syn aggregates, patients with MSA revealed an average shorter twisting distance of α-syn fibrils than those with PD [35]. Brain-derived α-syn fibrils from patients with DLB showed less twisted and thinner structures than those from patients with MSA [36]. Additionally, Peng et al. demonstrated different seeding activities of α-syn fibril strains in the brains with different synucleinopathies [37]. Additional advanced technologies would allow us to demonstrate that the structure of α-syn fibrils could be disease-specific, which could help understand the importance of structural heterogeneity of α-syn in understanding the pathogenicity of the disease.
Comprehensively, α-syn could be transformed into various conformations depending on the surrounding environment owing to the unstable nature of the structure (Fig. 2). Imbalanced physiological conditions, such as overexpression or mutation, may induce abnormal accumulation and aggregation of α-syn, leading to disease. These conformational diversities of α-syn imply that each α-syn structure may play different roles and/or be involved differently in various neurodegenerative disorders.

Conformational diversity of α-syn. Modeling of α-synuclein monomer (PDB: 1XQ8), tetramer, and fibril from cryo-electron microscopy for both the twister (PDB: 6CU8) and rod polymorph (PDB: 6CU7). α-syn, alpha-synuclein; cryo-EM, cryo-electron microscopy
Interactions with AD-related proteins
Aβ
Aβ, a crucial peptide in the pathophysiology of AD, is derived from the proteolytic process of the amyloid precursor protein by β- and γ-secretase complexes [38, 39]. Interactions of Aβ with other proteins have been suggested to play a pivotal role in the Aβ pathomechanisms of AD [40]. α-Syn has been investigated as a protein that can directly interact with Aβ [41, 42] (Table 1). Direct interactions between Aβ and α-syn have been shown to promote heterotrophic aggregation and intraneuronal accumulation of α-syn, which may exacerbate neuronal pathologies [43]. Based on co-immunoprecipitation in postmortem samples from the AD and DLB brains, α-syn monomers, dimers, trimers, and pentamers formed complexes with Aβ through interactions between the N-terminus of Aβ and the N- and C-terminus of α-syn [44]. A recent in vitro study confirmed that the interaction of Aβ with α-syn could increase the aggregation rate of α-syn, leading to accelerated fibril formation [45]. In several animal studies, transgenic mice with both hSYN and hAPP exhibited accelerated cognitive decline, motor deficits, and formation of α-syn inclusions in the brain, in which the structures of inclusions were partially fibrillar in a double transgenic mouse model and amorphous in single α-syn transgenic mice [43, 46]. Analogously, α-syn can promote Aβ aggregation, but the exact effects of each conformer are still obscure. For instance, α-syn oligomers could induce the formation of Aβ oligomers and stabilize their cross-β structures, resulting in Aβ fibril-like conformations [47]. Fibril forms of α-syn could accelerate the heterogeneous nucleation pathway of Aβ aggregates, whereas α-syn monomers suppressed Aβ aggregation in the secondary step by binding to Aβ fibrils, indicating that different structural forms of α-syn had distinct effects on Aβ aggregation [48]. Recently, α-syn monomers and oligomers were shown to hamper Aβ fibrillization, enhance oligomerization of Aβ monomers, and stabilize Aβ oligomers [49]. Direct interactions between α-syn and Aβ appear to have different consequences, depending on the structural species/stain. Therefore, subsequent studies should clarify the role of α-syn by investigating the defined components in interactions with AD pathophysiology.
Tau
Tau is a protein constituting neurofibrillary tangles, a hallmark of AD, and closely related to α-syn and Aβ (Table 2). Tau and α-syn were co-localized in the axons of cells, and their direct interaction in the nerve cell was demonstrated by affinity chromatography of the human brain cytosol [50]. The binding sites were the C-terminus of α-syn (55–140 aa) and the microtubule-binding region of tau (192–383 aa) [50]. This binding led to increased insoluble high-molecular-weight α-syn species and colocalization of tau and α-syn aggregates [51]. Colocalization of tau and α-syn aggregates could be described by prion-like properties of the two proteins that facilitated mutual homogenous/heterogeneous aggregations (Fig. 3). For instance, α-syn could induce tau aggregation; in turn, tau could synergistically accelerate the fibrillization of α-syn [52]. NMR imaging revealed that the monomeric form of tau selectively interacted with the C-terminal region of the α-syn monomer and accelerates α-syn oligomerization and subsequent fibril formation [53]. In addition, α-syn monomers and fibrils promoted tau aggregations [53]. Oikawa et al. suggested different conformational effects of α-syn; only α-syn fibrils, not monomer α-syn, interacted with tau and hampered microtubule assembly by inhibiting the binding of tau to microtubules [54]. α-Syn mutation hampered or enhanced interactions with tau, but the results remain controversial. In in vitro binding assays, α-syn mutations (A30P and A53T) appeared to have no effect on the tau-binding activity [50]. In Förster resonance energy transfer-based analysis, the A30P mutant exhibited a reduced interaction with tau but no effect of A53T or E46K mutants [55]. In cells co-transfected with tau and each mutant α-syn, all mutations (A30P, A53T, and E46K) increased the binding of α-syn with tau and exacerbated the stability of microtubules [56]. In tau mutation (P301L) related to frontotemporal dementia, the reduced binding affinity between the tau mutant and α-syn may promote tau aggregation and higher α-syn fibrils [57]. Treatment with increasing α-syn fibrils also increased tau aggregation by over 50% in P301L-mutant tau cells in comparison with wild-type tau cells, supporting that tau aggregation could result from the interaction with α-syn fibrils, which were accelerated in the P301L-mutant model [58]. Although the direct interaction between α-syn and tau promoted mutual aggregation, additional evidence is required to determine changes in each conformer and mutation determinant.

Schematic diagram of the possible relationship of α-synuclein with amyloid-beta, tau, and tubulin α-syn, alpha-synuclein; Aβ, amyloid-beta
The interaction between these two proteins also promotes tau phosphorylation. The regulation of tau phosphorylation could be accelerated by α-syn, along with several kinases (Fig. 3). Jensen et al. revealed that α-syn increased tau phosphorylation on S262 and S356 residues by protein kinase A up to 66%, depending on the protein concentration ratio [50]. In α-syn-overexpressing mouse models, the existence of phosphorylated tau was associated with the activation of the extracellular signal-regulated and c-Jun N-terminal kinases, which phosphorylated S396 and S404 residues of tau [59]. Hyperphosphorylations of tau at T181, S396, and S404 residues were also induced by activating the tau kinase glycogen synthase kinase-3β (GSK-3β) [60,61,62]. In the MPP + neurotoxin-induced PD model, α-syn increased the phosphorylation of tau at Ser262, 396, and 404 of tau by forming a heterotrimeric complex with tau and GSK-3β [60, 61]. Moreover, α-syn-deficient cells and α-syn knockout mice showed no change in tau phosphorylation due to the absence of phosphorylated GSK-3β, indicating that tau phosphorylation depended on the presence of α-syn [60, 61]. Under in vitro conditions, α-syn-mediated tau phosphorylation occurred via triple complex formation by the binding of tau to the acidic C-terminus of α-syn and by the interaction between GSK-3β and the NAC and KTEGV domains of α-syn [62]. Tau phosphorylation by this complex was gradually promoted as the α-syn/tau ratio increased, and the molar ratio of tau to α-syn at the maximum point was 1:20 [62]. Consistently, the percentages of phosphorylated tau were increased in the CSF of patients with AD, following increased α-syn/total tau ratio [63]. This accumulated evidence of α-syn-mediated tau phosphorylation suggests that elevated α-syn in AD may promote tau phosphorylation with other kinases under pathophysiological conditions, leading to tau pathology through significantly elevated phosphorylated tau.
Tubulin
Tubulin is an essential cytoskeleton component responsible for fundamental processes, including structural support, organelle transport, and cell division. Its assembly into microtubules is facilitated and stabilized by interactions with the neuronal tau protein. α-Syn is also known to bind tubulin, and the colocalization of these two proteins has been identified in the human and rat brains [64] (Table 3). Although a study reported the effects of α-syn on induction of tubulin polymerization [65], a contradictory result indicated that α-syn could destabilize microtubule assembly by blocking physiological interactions between tau and tubulin [66]. Residues 60–100 of α-syn were identified as the binding site for tubulin, which could contribute to inhibiting microtubule formation [67]. Even α-syn interactions with tubulin exerted different conformational effects on microtubule polymerization. Monomeric α-syn had no effect on microtubule polymerization, but tau-promoted microtubule assembly was inhibited by both protofibrils and α-syn fibrils [54]. In addition, treatment of oligomeric α-syn in dopaminergic neurons hampered tubulin polymerization and decreased mitochondrial function [68]. Taken together, interactions of α-syn with tubulin mainly affect the inhibition of microtubule assembly, and α-syn may be involved in pathogenic mechanisms rather than in normal physiological functions.
The overall functions of α-syn, as a partner of Aβ, tau, and tubulin, were depicted in Fig. 3. This schematic figure focused on the AD pathogenesis induced by involvements with α-syn. On the other hand, the interactions between α-syn and these proteins could be expanded to concomitant pathology. Converse to the accelerated pathology of Aβ and tau by α-syn, synucleinopathies in patients with AD could be initiated by α-syn aggregation due to interaction with proteins. For instance, patients with DLB and PD with dementia would be involved with aberrant aggregations of Aβ and α-syn, leading to the co-existence of senile plaques and Lewy bodies in the brain [69]. Various mixed types of neurodegenerative disorders could be interpreted as causing result in the specific pathology by promoting mutual aggregations of Aβ, tau, and α-syn.
Genetic association of α-synuclein in AD
SNCA, which encodes α-syn, was the first gene discovered in a patient with PD [70]. Most SNCA mutations are associated with PD pathology, and the involvement of AD-related genes as a genetic risk factor is limited. Interestingly, SNCA polymorphism of the GG frequency (rs10516846) was significantly increased in patients with AD compared to healthy control (HC) [71]. In addition, α-syn levels in CSF of patients with early-onset AD (EOAD) were higher in GG (rs10516846) carriers than in AA carriers [71], suggesting an association between SNCA gene polymorphisms and elevated α-syn levels in AD pathophysiology. Peripheral leukocytes in patients with AD showed elevated mRNA expression of SNCA with a reduced methylation rate in the intron 1 part of SNCA, one of the methylation regulatory regions of SNCA [72].
Previous studies have suggested associations between α-syn and representative genes in AD, such as amyloid precursor protein (APP), presenilin 1 (PSEN1), and apolipoprotein E (APOE). α-Syn and phosphorylated α-syn-positive dystrophic neurites have been observed in the brains of APP transgenic mice [73]. Additional overexpression of mutated PSEN1 in APP transgenic mice accelerated Aβ-induced synucleinopathies and further promoted phosphorylation of α-syn [73]. Human studies also showed a high frequency (50–60%) of Lewy body pathology in familial AD groups, supporting an association between AD-related genes and α-syn [6, 74]. Intriguingly, α-syn pathology has been observed in the amygdala in over 90% of patients with autosomal dominant AD in PSEN1 [75]. Direct interactions of α-syn with the presenilin 1 protein were identified in the brain tissues of cognitively normal individuals. They were significantly increased in the tissues of patients with AD and DLB with PSEN1 mutations [76]. Increased interactions of the presenilin 1 protein with α-syn in PSEN1-mutated cell lines were associated with increased membrane binding and α-syn accumulation [76]. Additionally, a study revealed that cells with PSEN1 mutations associated with AD and DLB had exacerbated phosphorylation and accumulation of α-syn [77]. In a recent CSF biomarker study, the tau/α-syn ratio was altered in patients with AD, and in particular, changes in EOAD were statistically higher than those in late-onset AD [63]. Furthermore, CSF α-syn levels were higher in symptomatic autosomal dominant AD mutation carriers than in non-mutation carriers [78]. Considering the high contribution of genetic factors in EOAD, changes in CSF α-syn levels could be influenced by AD-related genes, resulting in greater changes in autosomal dominant AD. APOE encodes the apolipoprotein E protein, an important lipid-binding protein for intercellular lipid redistribution in the CNS, which is a major risk factor for late-onset AD (after the age of 65 years). The possible pathophysiological roles of APOE and α-syn have been investigated, mainly in relation to PD. The apolipoprotein E protein level was elevated by over fourfold in transgenic mice with the α-syn A30P mutation, and APOE knockout in α-syn A30P transgenic mice increased the survival rate, delayed behavioral symptoms, and decreased neuronal degeneration and Aβ aggregates [79]. In particular, the APOEε4-expressing PD mouse model, but not ε2 and ε3, showed increased α-syn pathology and astrogliosis and impaired behavioral ability with worsened neuronal and synaptic loss [80]. In patients with PD, apolipoprotein E was elevated in the CSF with an abundance in dopaminergic neurons of the substantia nigra from postmortem brain tissues [81]. Although the association between the apolipoprotein E protein and α-syn has been focused on in patients with PD, it may be involved in AD pathophysiological conditions. For instance, laboratory studies have shown elevated CSF α-syn levels in APOEε4-carrier patients with AD [63, 78]. Additionally, elevated CSF α-syn levels were significantly associated with Aβ plaque burdens in APOEε4-positive individuals with autosomal dominant AD [78]. Thus, these studies described the associations of the APOEε4 risk allele with CSF α-syn levels and Aβ deposition in AD. Although the exact mechanism has not been clearly elucidated, accumulating evidence suggests that α-syn is strongly associated with AD-related genes and contributes to disease progression at the genetic level.
CSF α-syn in AD
Meta-analysis of CSF α-syn in AD
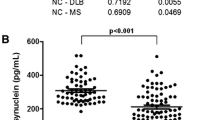
α-Syn is considered to be a biomarker of synucleinopathies, including PD and DLB, as it is the main component of Lewy bodies. However, several studies have investigated the diagnostic performance of the CSF α-syn level in AD. In the meta-analysis, only the total α-syn level was considered, and the phosphorylated or oligomeric forms were excluded. We searched the PubMed and Web of Science databases to extract 38 articles related to the total α-syn level in the CSF of patients of AD. Contrary to the total α-syn level, CSF oligomeric and phosphorylated α-syn levels at serin 129 were consistently elevated in patients with PD [82, 83] but unchanged in those with AD [84]. Since two α-syn species may dominantly affect synucleinopathies rather than AD-related pathology, we speculated that total α-syn mostly of the monomeric form would be suitable for the AD diagnosis. Sixteen studies revealed statistically significant increases α-syn levels in the CSF of patients with AD [78, 84,85,86,87,88,89,90,91,92,93,94,95,96,97,98], whereas a few reports showed reduced levels [99,100,101]. In particular, α-syn levels in the CSF were significantly elevated in patients with AD with all positive CSF triple markers (Aβ42, total tau, and phosphorylated tau) [63, 84]. However, no difference in CSF α-syn concentrations between AD and HC were found in a few studies [63, 102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119]. Inconsistencies across studies may have resulted from misdiagnosis, co-existence of other neurodegenerative disorders, hemolysis, anticoagulant upon collection in plasma, pre-analytic sample handling, technical errors, and particularly platform differences of measurements [119]. To verify changes in CSF α-syn levels in AD, a meta-analysis was conducted with 25 studies in which a normal distribution could be obtained (Fig. 4). In addition, we excluded articles, which were sub-classified groups through additional parameters, such as CSF analysis, positron emission tomography (PET), or longitudinal observation. A total of 25 studies reported that CSF α-syn levels were statistically higher in patients with AD than in HC (Z = 2.94, p = 0.003).

Meta-analysis of the cerebrospinal fluid α-synuclein level in Alzheimer’s disease and healthy controls CSF, cerebrospinal fluid; α-syn, alpha-synuclein; AD, Alzheimer’s disease; HC, healthy controls
Most studies revealed strong positive correlations of CSF α-syn with tau and phosphorylated tau [63, 84, 92,93,94,95, 115,116,117], whereas one study revealed a negative correlation [100]. Whether elevated α-syn levels in the CSF are associated with a causal or protective mechanism in AD is unknown. Considering the high correlation between tau and α-syn levels, synaptic destruction might increase the release of α-syn into the CSF, similar to tau released by neuronal death [18]. In contrast, studies showing reduced CSF α-syn levels in AD suggested lower α-syn secretion from synaptic loss [101]. The correlation between tau and α-syn levels may be associated with the causative mechanisms of AD pathology rather than the consequence of synaptic disruptions or neuronal death. NFL is a representative biomarker released into biofluids as a result of neuron destructions. It is significantly elevated in most neurodegenerative disorders, even in tauopathies and synucleinopathies [120]. In contrast, CSF α-syn levels in patients with tauopathies and synucleinopathies revealed no difference or reduced concentrations in comparison with the HC group [121,122,123,124,125]. A longitudinal study showed that α-syn levels in CSF were reduced in manifested and prodromal patients with PD and slightly declined after 36 months, reflecting no association of reduced CSF α-syn levels with dopaminergic neurodegeneration [126]. These studies supported that elevated α-syn levels in AD were not simply a consequence of synaptic degeneration or neuronal deaths. According to the aforementioned in vitro study, tau phosphorylation increased as the α-syn/tau ratio increased in CSF, which was proven in a previous clinical CSF study [63]. Tau and α-syn levels showed strong positive correlations in the CSF of all groups, including AD, PD, and HC. Furthermore, as the tau/α-syn ratio in the CSF had a strong correlation with the phosphorylated tau rate, tau phosphorylation was modulated according to the ratio of the two proteins. Further studies on different equilibrium states in tau and α-syn in the CSF according to the status of AD pathophysiology could provide important implications for understanding the role of α-syn in AD.
CSF α-syn in the early stage of AD
In previous studies, the highest α-syn levels in CSF were measured in patients with mild cognitive impairment (MCI), showing its involvement in the early stages of AD [91, 96, 117, 127]. The α-syn levels in CSF gradually increased from normal to MCI stages and then decreased in the proceeding stage in AD [78]. In addition, the α-syn levels in CSF were elevated in patients who converted from MCI to AD and with a shorter duration of AD progression [78, 95, 115]. CSF α-syn levels in patients with MCI with stable symptoms not progressing to AD were similar to those of the HC group [78]. These results suggested that α-syn perform pathological functions in AD by rapidly increasing in the period of progression from MCI to symptomatic stages. Similar to tau, CSF α-syn levels were positively associated with brain Aβ plaque deposition in the early stages of AD [78, 118]. Among individuals with subjective complaints of memory dysfunction, CSF α-syn levels in the amyloid PET-positive group tended to be higher than those in the PET-negative group [118]. In addition, the total tau/α-syn ratio in the CSF was highly concordant with CSF Aβ42 and amyloid PET in our study [63]. These clinical study results are consistent with the aforementioned evidence that α-syn could synergistically and directly induce Aβ aggregation in animal models. Taken together, it could assume that α-syn is associated with Aβ-related pathophysiological mechanisms in the early stages of AD.
Incorporation of CSF α-syn with other biomarkers
Incorporation of α-syn in CSF with other biomarkers would have better diagnostic performances. Toledo et al. proposed that patients with elevated phosphorylated tau181 and reduced α-syn levels in CSF could be classified as AD with concomitant Lewy body pathology [92]. Another study reported a differential diagnosis for patients with DLB and AD using combined α-syn and phosphorylated tau181, since the levels of both proteins were reduced in patients with DLB but elevated in patients with AD [93]. The total tau/α-syn ratio was the highest in the CSF of patients with AD [63, 100, 101] and the lowest in the CSF of patients with PD [100]. The incorporation of α-syn with triple CSF markers (Aβ, total tau, and phosphorylated tau181) revealed the best discrimination value between AD and HC and improved differential diagnosis with other neurodegenerative diseases [63, 117]. Although the utility of α-syn as a biomarker for the diagnosis of neurodegenerative diseases remains controversial, the incorporation of CSF α-syn may improve the diagnostic performance in AD and aid in the discrimination of AD from other neurodegenerative diseases. α-Syn may offer an opportunity to overcome the limitations of triple CSF biomarkers.
α-Syn seed amplification assay (SAA) in AD
SAA has been used to detect minimal amounts of misfolded prion in Creutzfeldt-Jakob disease. SAA has recently been expanded to the field of synucleinopathies by detecting misfolded α-syn in the CSF, olfactory mucosa, submandibular gland, skin, and brain [35, 128,129,130,131,132,133,134]. α-Syn SAA distinguished synucleinopathies (PD and MSA) remarkably from other pathological diseases, such as AD, amyotrophic lateral sclerosis, Pick’s disease, corticobasal degeneration, and progressive supranuclear palsy [130, 132, 135]. In particular, among patients with Lewy body-related pathology, limbic/neocortical pathology cases had high positivity in CSF and frontal cortex brain homogenate but lower positivity in AD/ALB, indicating the possibility of the discriminating ability of mixed pathologies with α-syn SAA [136]. Similar to the total α-syn level in CSF, α-syn SAA could be applied to the differential diagnosis of patients with AD with Lewy body-related pathology (particularly AD/ALB cases).
Blood α-syn in AD
Several studies have reported attempts to diagnose AD by measuring total α-syn levels in the blood of patients with AD. Serum levels of α-syn in patients with AD showed no significant difference from those in HC [137]. In addition, there was no discernible intergroup variation in the plasma α-syn levels between AD and HC [96]. In patients with amnestic MCI, plasma α-syn levels increased throughout disease progression and had a discriminatory capacity to indicate the risk of cognitive deterioration [138]. In a recent study, AD and HC had significantly different plasma α-syn levels [139]. Elevated plasma α-syn levels in AD were positively correlated with urinary AD-associated neuronal thread protein but not with serum lipids [139]. Inconsistent results might be owing to various factors, including technical protocols, pre-analytic processes, medications, and particularly hemolysis. Since erythrocytes were the major source of α-syn, the analyzed α-syn levels could be influenced from the released of α-syn from cytosol upon hemolysis [91, 140]. Oligomer or phosphorylated α-syn levels in serum, plasma, and red blood cells were elevated and could be used to diagnose PD or MSA. However, no study has reported the two α-syn species in AD [141,142,143,144]. Although the possible changes of α-syn levels would be present in patients with AD, blood α-syn as a biomarker may not be sufficient in the current stage and should be studied further.
Conclusions
Numerous reports have shown that α-syn is deeply involved in the pathophysiology of AD. Nevertheless, it has not been recognized in the fields of its involvement in AD mechanisms, inter-related biomarkers, or drug development. Similar to other intrinsically disordered proteins, such as Aβ, tau, and prion, α-syn can easily adapt to its diverse structures depending on the environment, wherein each conformation may influence different mechanisms. Elevated α-syn levels in AD could facilitate Aβ oligomerization, tau phosphorylation, activation of kinases, dissociations of tau and tubulin, and tau aggregation. Furthermore, the association of α-syn with genetic factors, such as APP, PSEN1, and APOE, could accelerate AD pathology. As a biomarker, the CSF α-syn levels were the highest in MCI, particularly in rapidly progressing patients to AD. Elevated CSF α-syn levels were also correlated with Aβ depositions in the asymptomatic stage, indicating potential applications in the early diagnosis, as a sensitive indicator of disease progression along with changes in Aβ species, including oligomeric forms. Remarkably, the incorporation of CSF α-syn with other biomarkers had strong potential for the accurate diagnosis of AD and its discrimination from other similar neurodegenerative disorders. Combined CSF α-syn with triple biomarkers could improve diagnostic and prognostic performances. Accurate early identification of AD progression using α-syn and Aβ species may help develop novel therapeutics or better treatments for patients, considering the α-syn mechanism in AD. To date, α-syn has mainly been investigated in synucleinopathies. Further studies on α-syn in AD pathophysiology would contribute to the understanding of its mechanism in AD and other neurodegenerative diseases.
Availability of data and materials
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- AD:
-
Alzheimer’s disease
- Aβ:
-
Amyloid-beta
- α-Syn:
-
α-Synuclein
- PD:
-
Parkinson’s disease
- DLB:
-
Dementia with Lewy bodies
- CSF:
-
Cerebrospinal fluid
- NAC:
-
Non-amyloid-beta component
- Cryo-EM:
-
Cryo-electron microscopy
- GSK-3β:
-
Glycogen synthase kinase-3β
- HC:
-
Healthy control
- EOAD:
-
Early-onset AD
- MCI:
-
Mild cognitive impairment
- PET:
-
Positron emission tomography
References
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388(6645):839–40.
Kim WS, Kågedal K, Halliday GM. Alpha-synuclein biology in Lewy body diseases. Alzheimer’s Research & Therapy. 2014;6(5):73.
Ueda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, et al. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90(23):11282–6.
Masliah E, Iwai A, Mallory M, Ueda K, Saitoh T. Altered presynaptic protein NACP is associated with plaque formation and neurodegeneration in Alzheimer’s disease. Am J Pathol. 1996;148(1):201–10.
Arai Y, Yamazaki M, Mori O, Muramatsu H, Asano G, Katayama Y. Alpha-synuclein-positive structures in cases with sporadic Alzheimer’s disease: morphology and its relationship to tau aggregation. Brain Res. 2001;888(2):287–96.
Lippa CF, Fujiwara H, Mann DM, Giasson B, Baba M, Schmidt ML, et al. Lewy bodies contain altered alpha-synuclein in brains of many familial Alzheimer’s disease patients with mutations in presenilin and amyloid precursor protein genes. Am J Pathol. 1998;153(5):1365–70.
Hamilton RL. Lewy bodies in Alzheimer’s disease: a neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol. 2000;10(3):378–84.
Olichney JM, Galasko D, Salmon DP, Hofstetter CR, Hansen LA, Katzman R, et al. Cognitive decline is faster in Lewy body variant than in Alzheimer’s disease. Neurology. 1998;51(2):351–7.
Holtzer R, Irizarry MC, Sanders J, Hyman BT, Wegesin DJ, Riba A, et al. Relation of quantitative indexes of concurrent α-synuclein abnormalities to clinical outcome in autopsy-proven Alzheimer disease. Arch Neurol. 2006;63(2):226–30.
Smirnov DS, Salmon DP, Galasko D, Goodwill VS, Hansen LA, Zhao Y, et al. Association of neurofibrillary tangle distribution with age at onset–related clinical heterogeneity in Alzheimer disease. An autopsy Study. 2022;98(5):e506–17.
Karanth S, Nelson PT, Katsumata Y, Kryscio RJ, Schmitt FA, Fardo DW, et al. Prevalence and clinical phenotype of quadruple misfolded proteins in older adults. JAMA Neurol. 2020;77(10):1299–307.
Stefanis L. α-Synuclein in Parkinson’s disease. Cold Spring Harbor Perspectives in Medicine. 2012;2(2):a009399.
Sanderson JB, De S, Jiang H, Rovere M, Jin M, Zaccagnini L, et al. Analysis of α-synuclein species enriched from cerebral cortex of humans with sporadic dementia with Lewy bodies. Brain Communications. 2020;2(1):fcaa010.
Uchikado H, Lin W-L, DeLucia MW, Dickson DW. Alzheimer disease with amygdala Lewy bodies: a distinct form of α-synucleinopathy. J Neuropathol Exp Neurol. 2006;65(7):685–97.
Sorrentino ZA, Goodwin MS, Riffe CJ, Dhillon J-KS, Xia Y, Gorion K-M, et al. Unique α-synuclein pathology within the amygdala in Lewy body dementia: implications for disease initiation and progression. Acta Neuropathologica Communications. 2019;7(1):142.
Larson ME, Sherman MA, Greimel S, Kuskowski M, Schneider JA, Bennett DA, et al. Soluble alpha-synuclein is a novel modulator of Alzheimer’s disease pathophysiology. J Neurosci. 2012;32(30):10253–66.
Larson ME, Greimel SJ, Amar F, LaCroix M, Boyle G, Sherman MA, et al. Selective lowering of synapsins induced by oligomeric alpha-synuclein exacerbates memory deficits. Proc Natl Acad Sci U S A. 2017;114(23):E4648–57.
Twohig D, Nielsen HM. alpha-synuclein in the pathophysiology of Alzheimer’s disease. Mol Neurodegener. 2019;14(1):23.
Cousins KAQ, Arezoumandan S, Shellikeri S, Ohm D, Shaw LM, Grossman M, et al. CSF biomarkers of Alzheimer disease in patients with concomitant α-synuclein pathology. Neurology. 2022;99(20):e2303–12.
Breydo L, Wu JW, Uversky VN. Alpha-synuclein misfolding and Parkinson’s disease. Biochim Biophys Acta. 2012;1822(2):261–85.
Vamvaca K, Volles MJ, Lansbury PT Jr. The first N-terminal amino acids of alpha-synuclein are essential for alpha-helical structure formation in vitro and membrane binding in yeast. J Mol Biol. 2009;389(2):413–24.
Miraglia F, Ricci A, Rota L, Colla E. Subcellular localization of alpha-synuclein aggregates and their interaction with membranes. Neural Regen Res. 2018;13(7):1136–44.
Bendor JT, Logan TP, Edwards RH. The function of alpha-synuclein. Neuron. 2013;79(6):1044–66.
Tseng E, Rowell WJ, Glenn O-C, Hon T, Barrera J, Kujawa S, et al. The landscape of SNCA transcripts across synucleinopathies: new insights from long reads sequencing analysis. Front Genet. 2019;10:584.
Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT Jr. NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry. 1996;35(43):13709–15.
Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem. 1998;273(16):9443–9.
Fauvet B, Mbefo MK, Fares MB, Desobry C, Michael S, Ardah MT, et al. alpha-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J Biol Chem. 2012;287(19):15345–64.
Bartels T, Choi JG, Selkoe DJ. alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011;477(7362):107–10.
Wang W, Perovic I, Chittuluru J, Kaganovich A, Nguyen LT, Liao J, et al. A soluble alpha-synuclein construct forms a dynamic tetramer. Proc Natl Acad Sci U S A. 2011;108(43):17797–802.
Burre J, Vivona S, Diao J, Sharma M, Brunger AT, Sudhof TC. Properties of native brain alpha-synuclein. Nature. 2013;498(7453):E4–6; discussion E6-7.
Villar-Pique A, Lopes da Fonseca T, Outeiro TF. Structure, function and toxicity of alpha-synuclein: the Bermuda triangle in synucleinopathies. J Neurochem. 2016;139 Suppl 1:240–55.
Li B, Ge P, Murray KA, Sheth P, Zhang M, Nair G, et al. Cryo-EM of full-length α-synuclein reveals fibril polymorphs with a common structural kernel. Nat Commun. 2018;9(1):3609.
Grazia Spillantini M, Anthony Crowther R, Jakes R, Cairns NJ, Lantos PL, Goedert M. Filamentous α-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett. 1998;251(3):205–8.
Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci. 1998;95(11):6469–73.
Shahnawaz M, Mukherjee A, Pritzkow S, Mendez N, Rabadia P, Liu X, et al. Discriminating α-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature. 2020;578(7794):273–7.
Schweighauser M, Shi Y, Tarutani A, Kametani F, Murzin AG, Ghetti B, et al. Structures of α-synuclein filaments from multiple system atrophy. Nature. 2020;585(7825):464–9.
Peng C, Gathagan RJ, Covell DJ, Medellin C, Stieber A, Robinson JL, et al. Cellular milieu imparts distinct pathological α-synuclein strains in α-synucleinopathies. Nature. 2018;557(7706):558–63.
Selkoe DJ. Amyloid β-protein and the genetics of Alzheimer’s disease*. J Biol Chem. 1996;271(31):18295–8.
Haass C. Take five—BACE and the γ-secretase quartet conduct Alzheimer’s amyloid β-peptide generation. EMBO J. 2004;23(3):483–8.
Rahman MM, Zetterberg H, Lendel C, Härd T. Binding of human proteins to amyloid-β protofibrils. ACS Chem Biol. 2015;10(3):766–74.
Yoshimoto M, Iwai A, Kang D, Otero DA, Xia Y, Saitoh T. NACP, the precursor protein of the non-amyloid beta/A4 protein (A beta) component of Alzheimer disease amyloid, binds A beta and stimulates A beta aggregation. Proc Natl Acad Sci U S A. 1995;92(20):9141–5.
Jensen PH, Højrup P, Hager H, Nielsen MS, Jacobsen L, Olesen OF, et al. Binding of Aβ to α- and β-synucleins: identification of segments in α-synuclein/NAC precursor that bind Aβ and NAC. Biochemical Journal. 1997;323(2):539–46.
Masliah E, Rockenstein E, Veinbergs I, Sagara Y, Mallory M, Hashimoto M, et al. β-Amyloid peptides enhance α-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer’s disease and Parkinson’s disease. Proc Natl Acad Sci. 2001;98(21):12245–50.
Tsigelny IF, Crews L, Desplats P, Shaked GM, Sharikov Y, Mizuno H, et al. Mechanisms of hybrid oligomer formation in the pathogenesis of combined Alzheimer’s and Parkinson’s diseases. PLoS ONE. 2008;3(9): e3135.
Köppen J, Schulze A, Machner L, Wermann M, Eichentopf R, Guthardt M, et al. Amyloid-beta peptides trigger aggregation of alpha-synuclein in vitro. Molecules. 2020;25(3):580.
Clinton LK, Blurton-Jones M, Myczek K, Trojanowski JQ, LaFerla FM. Synergistic interactions between Aβ, tau, and α-synuclein: acceleration of neuropathology and cognitive decline. J Neurosci. 2010;30(21):7281–9.
Atsmon-Raz Y, Miller Y. Non-amyloid-β component of human α-synuclein oligomers induces formation of new Aβ oligomers: insight into the mechanisms that link Parkinson’s and Alzheimer’s diseases. ACS Chem Neurosci. 2016;7(1):46–55.
Chia S, Flagmeier P, Habchi J, Lattanzi V, Linse S, Dobson CM, et al. Monomeric and fibrillar α-synuclein exert opposite effects on the catalytic cycle that promotes the proliferation of Aβ42 aggregates. Proc Natl Acad Sci. 2017;114(30):8005–10.
Candreva J, Chau E, Rice ME, Kim JR. Interactions between soluble species of β-amyloid and α-synuclein promote oligomerization while inhibiting fibrillization. Biochemistry. 2020;59(4):425–35.
Jensen PH, Hager H, Nielsen MS, Hojrup P, Gliemann J, Jakes R. alpha-synuclein binds to Tau and stimulates the protein kinase A-catalyzed tau phosphorylation of serine residues 262 and 356. J Biol Chem. 1999;274(36):25481–9.
Badiola N, de Oliveira RM, Herrera F, Guardia-Laguarta C, Goncalves SA, Pera M, et al. Tau enhances alpha-synuclein aggregation and toxicity in cellular models of synucleinopathy. PLoS ONE. 2011;6(10): e26609.
Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, et al. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science. 2003;300(5619):636–40.
Dasari AKR, Kayed R, Wi S, Lim KH. Tau interacts with the C-terminal region of α-synuclein, promoting formation of toxic aggregates with distinct molecular conformations. Biochemistry. 2019;58(25):2814–21.
Oikawa T, Nonaka T, Terada M, Tamaoka A, Hisanaga S, Hasegawa M. alpha-synuclein fibrils exhibit gain of toxic function, promoting tau aggregation and inhibiting microtubule assembly. J Biol Chem. 2016;291(29):15046–56.
Esposito A, Dohm CP, Kermer P, Bahr M, Wouters FS. alpha-Synuclein and its disease-related mutants interact differentially with the microtubule protein tau and associate with the actin cytoskeleton. Neurobiol Dis. 2007;26(3):521–31.
Qureshi HY, Paudel HK. Parkinsonian neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and alpha-synuclein mutations promote Tau protein phosphorylation at Ser262 and destabilize microtubule cytoskeleton in vitro. J Biol Chem. 2011;286(7):5055–68.
Benussi L, Ghidoni R, Paterlini A, Nicosia F, Alberici AC, Signorini S, et al. Interaction between tau and alpha-synuclein proteins is impaired in the presence of P301L tau mutation. Exp Cell Res. 2005;308(1):78–84.
Waxman EA, Giasson BI. Induction of intracellular tau aggregation is promoted by alpha-synuclein seeds and provides novel insights into the hyperphosphorylation of tau. J Neurosci. 2011;31(21):7604–18.
Kaul T, Credle J, Haggerty T, Oaks AW, Masliah E, Sidhu A. Region-specific tauopathy and synucleinopathy in brain of the alpha-synuclein overexpressing mouse model of Parkinson’s disease. BMC Neurosci. 2011;12:79.
Duka T, Rusnak M, Drolet RE, Duka V, Wersinger C, Goudreau JL, et al. Alpha-synuclein induces hyperphosphorylation of Tau in the MPTP model of parkinsonism. Faseb j. 2006;20(13):2302–12.
Duka T, Duka V, Joyce JN, Sidhu A. Alpha-Synuclein contributes to GSK-3beta-catalyzed Tau phosphorylation in Parkinson’s disease models. Faseb j. 2009;23(9):2820–30.
Kawakami F, Suzuki M, Shimada N, Kagiya G, Ohta E, Tamura K, et al. Stimulatory effect of alpha-synuclein on the tau-phosphorylation by GSK-3beta. Febs j. 2011;278(24):4895–904.
Shim KH, Kang MJ, Suh JW, Pyun J-M, Ryoo N, Park YH, et al. CSF total tau/α-synuclein ratio improved the diagnostic performance for Alzheimer’s disease as an indicator of tau phosphorylation. Alzheimer’s Research & Therapy. 2020;12(1):83.
Alim MA, Hossain MS, Arima K, Takeda K, Izumiyama Y, Nakamura M, et al. Tubulin seeds alpha-synuclein fibril formation. J Biol Chem. 2002;277(3):2112–7.
Alim MA, Ma QL, Takeda K, Aizawa T, Matsubara M, Nakamura M, et al. Demonstration of a role for alpha-synuclein as a functional microtubule-associated protein. J Alzheimers Dis. 2004;6(4):435–42; discussion 43–9.
Moussaud S, Jones DR, Moussaud-Lamodière EL, Delenclos M, Ross OA, McLean PJ. Alpha-synuclein and tau: teammates in neurodegeneration? Mol Neurodegener. 2014;9(1):43.
Zhou RM, Huang YX, Li XL, Chen C, Shi Q, Wang GR, et al. Molecular interaction of alpha-synuclein with tubulin influences on the polymerization of microtubule in vitro and structure of microtubule in cells. Mol Biol Rep. 2010;37(7):3183–92.
Chen L, Jin J, Davis J, Zhou Y, Wang Y, Liu J, et al. Oligomeric alpha-synuclein inhibits tubulin polymerization. Biochem Biophys Res Commun. 2007;356(3):548–53.
Jellinger KA. Interaction between α-synuclein and other proteins in neurodegenerative disorders. ScientificWorldJournal. 2011;11:1893–907.
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276(5321):2045–7.
Wang Q, Tian Q, Song X, Liu Y, Li W. SNCA Gene Polymorphism may contribute to an increased risk of Alzheimer’s disease. J Clin Lab Anal. 2016;30(6):1092–9.
Yoshino Y, Mori T, Yoshida T, Yamazaki K, Ozaki Y, Sao T, et al. Elevated mRNA expression and low methylation of SNCA in Japanese Alzheimer’s disease subjects. J Alzheimers Dis. 2016;54(4):1349–57.
Kurata T, Kawarabayashi T, Murakami T, Miyazaki K, Morimoto N, Ohta Y, et al. Enhanced accumulation of phosphorylated alpha-synuclein in double transgenic mice expressing mutant beta-amyloid precursor protein and presenilin-1. J Neurosci Res. 2007;85(10):2246–52.
Rosenberg CK, Pericak-Vance MA, Saunders AM, Gilbert JR, Gaskell PC, Hulette CM. Lewy body and Alzheimer pathology in a family with the amyloid-β precursor protein APP717 gene mutation. Acta Neuropathol. 2000;100(2):145–52.
Leverenz JB, Fishel MA, Peskind ER, Montine TJ, Nochlin D, Steinbart E, et al. Lewy body pathology in familial Alzheimer disease: evidence for disease- and mutation-specific pathologic phenotype. Arch Neurol. 2006;63(3):370–6.
Winslow AR, Moussaud S, Zhu L, Post KL, Dickson DW, Berezovska O, et al. Convergence of pathology in dementia with Lewy bodies and Alzheimer’s disease: a role for the novel interaction of alpha-synuclein and presenilin 1 in disease. Brain. 2014;137(Pt 7):1958–70.
Kaneko H, Kakita A, Kasuga K, Nozaki H, Ishikawa A, Miyashita A, et al. Enhanced accumulation of phosphorylated alpha-synuclein and elevated beta-amyloid 42/40 ratio caused by expression of the presenilin-1 deltaT440 mutant associated with familial Lewy body disease and variant Alzheimer’s disease. J Neurosci. 2007;27(48):13092–7.
Twohig D, Rodriguez-Vieitez E, Sando SB, Berge G, Lauridsen C, Moller I, et al. The relevance of cerebrospinal fluid alpha-synuclein levels to sporadic and familial Alzheimer’s disease. Acta Neuropathol Commun. 2018;6(1):130.
Gallardo G, Schluter OM, Sudhof TC. A molecular pathway of neurodegeneration linking alpha-synuclein to ApoE and Abeta peptides. Nat Neurosci. 2008;11(3):301–8.
Zhao N, Attrebi ON, Ren Y, Qiao W, Sonustun B, Martens YA, et al. APOE4 exacerbates α-synuclein pathology and related toxicity independent of amyloid. Science Translational Medicine. 2020;12(529):eaay1809.
Paslawski W, Zareba-Paslawska J, Zhang X, Hölzl K, Wadensten H, Shariatgorji M, et al. α-synuclein-lipoprotein interactions and elevated ApoE level in cerebrospinal fluid from Parkinson’s disease patients. Proc Natl Acad Sci. 2019;116(30):15226–35.
Majbour NK, Vaikath NN, van Dijk KD, Ardah MT, Varghese S, Vesterager LB, et al. Oligomeric and phosphorylated alpha-synuclein as potential CSF biomarkers for Parkinson’s disease. Mol Neurodegener. 2016;11(1):7.
Majbour NK, Abdi IY, Dakna M, Wicke T, Lang E, Ali Moussa HY, et al. Cerebrospinal α-synuclein oligomers reflect disease motor severity in DeNoPa longitudinal cohort. Mov Disord. 2021;36(9):2048–56.
Majbour NK, Chiasserini D, Vaikath NN, Eusebi P, Tokuda T, van de Berg W, et al. Increased levels of CSF total but not oligomeric or phosphorylated forms of alpha-synuclein in patients diagnosed with probable Alzheimer’s disease. Sci Rep. 2017;7:40263.
Mukaetova-Ladinska EB, Milne J, Andras A, Abdel-All Z, Cerejeira J, Greally E, et al. Alpha- and gamma-synuclein proteins are present in cerebrospinal fluid and are increased in aged subjects with neurodegenerative and vascular changes. Dement Geriatr Cogn Disord. 2008;26(1):32–42.
Tateno F, Sakakibara R, Kawai T, Kishi M, Murano T. Alpha-synuclein in the cerebrospinal fluid differentiates synucleinopathies (Parkinson disease, dementia with Lewy bodies, multiple system atrophy) from Alzheimer disease. Alzheimer Dis Assoc Disord. 2012;26(3):213–6.
Hall S, Ohrfelt A, Constantinescu R, Andreasson U, Surova Y, Bostrom F, et al. Accuracy of a panel of 5 cerebrospinal fluid biomarkers in the differential diagnosis of patients with dementia and/or parkinsonian disorders. Arch Neurol. 2012;69(11):1445–52.
Viode A, Epelbaum S, Benyounes I, Verny M, Dubois B, Junot C, et al. Simultaneous quantification of tau and alpha-synuclein in cerebrospinal fluid by high-resolution mass spectrometry for differentiation of Lewy body dementia from Alzheimer’s disease and controls. Analyst. 2019;144(21):6342–51.
Hansson O, Hall S, Ohrfelt A, Zetterberg H, Blennow K, Minthon L, et al. Levels of cerebrospinal fluid alpha-synuclein oligomers are increased in Parkinson’s disease with dementia and dementia with Lewy bodies compared to Alzheimer’s disease. Alzheimers Res Ther. 2014;6(3):25.
Chiasserini D, Biscetti L, Eusebi P, Salvadori N, Frattini G, Simoni S, et al. Differential role of CSF fatty acid binding protein 3, alpha-synuclein, and Alzheimer’s disease core biomarkers in Lewy body disorders and Alzheimer’s dementia. Alzheimers Res Ther. 2017;9(1):52.
Korff A, Liu C, Ginghina C, Shi M, Zhang J. alpha-Synuclein in cerebrospinal fluid of Alzheimer’s disease and mild cognitive impairment. J Alzheimers Dis. 2013;36(4):679–88.
Toledo JB, Korff A, Shaw LM, Trojanowski JQ, Zhang J. CSF alpha-synuclein improves diagnostic and prognostic performance of CSF tau and Abeta in Alzheimer’s disease. Acta Neuropathol. 2013;126(5):683–97.
Slaets S, Vanmechelen E, Le Bastard N, Decraemer H, Vandijck M, Martin JJ, et al. Increased CSF alpha-synuclein levels in Alzheimer’s disease: correlation with tau levels. Alzheimers Dement. 2014;10(5 Suppl):S290–8.
Garcia-Ayllon MS, Monge-Argiles JA, Monge-Garcia V, Navarrete F, Cortes-Gomez MA, Sanchez-Paya J, et al. Measurement of CSF alpha-synuclein improves early differential diagnosis of mild cognitive impairment due to Alzheimer’s disease. J Neurochem. 2019;150(2):218–30.
Wang H, Stewart T, Toledo JB, Ginghina C, Tang L, Atik A, et al. A longitudinal study of total and phosphorylated alpha-synuclein with other biomarkers in cerebrospinal fluid of Alzheimer’s disease and mild cognitive impairment. J Alzheimers Dis. 2018;61(4):1541–53.
Seino Y, Nakamura T, Kawarabayashi T, Hirohata M, Narita S, Wakasaya Y, et al. Cerebrospinal fluid and plasma biomarkers in neurodegenerative diseases. J Alzheimers Dis. 2019;68(1):395–404.
Mattsson N, Insel P, Tosun D, Zhang J, Jack CR Jr, Galasko D, et al. Effects of baseline CSF alpha-synuclein on regional brain atrophy rates in healthy elders, mild cognitive impairment and Alzheimer’s disease. PLoS ONE. 2013;8(12): e85443.
Van Hulle C, Jonaitis EM, Betthauser TJ, Batrla R, Wild N, Kollmorgen G, et al. An examination of a novel multipanel of CSF biomarkers in the Alzheimer’s disease clinical and pathological continuum. Alzheimers Dement. 2021;17(3):431–45.
Ohrfelt A, Grognet P, Andreasen N, Wallin A, Vanmechelen E, Blennow K, et al. Cerebrospinal fluid alpha-synuclein in neurodegenerative disorders-a marker of synapse loss? Neurosci Lett. 2009;450(3):332–5.
Parnetti L, Chiasserini D, Bellomo G, Giannandrea D, De Carlo C, Qureshi MM, et al. Cerebrospinal fluid Tau/α-synuclein ratio in Parkinson’s disease and degenerative dementias. Mov Disord. 2011;26(8):1428–35.
Forland MG, Tysnes OB, Aarsland D, Maple-Grodem J, Pedersen KF, Alves G, et al. The value of cerebrospinal fluid alpha-synuclein and the tau/alpha-synuclein ratio for diagnosis of neurodegenerative disorders with Lewy pathology. Eur J Neurol. 2020;27(1):43–50.
Reesink FE, Lemstra AW, van Dijk KD, Berendse HW, van de Berg WD, Klein M, et al. CSF alpha-synuclein does not discriminate dementia with Lewy bodies from Alzheimer’s disease. J Alzheimers Dis. 2010;22(1):87–95.
Mollenhauer B, Cullen V, Kahn I, Krastins B, Outeiro TF, Pepivani I, et al. Direct quantification of CSF alpha-synuclein by ELISA and first cross-sectional study in patients with neurodegeneration. Exp Neurol. 2008;213(2):315–25.
Hong Z, Shi M, Chung KA, Quinn JF, Peskind ER, Galasko D, et al. DJ-1 and alpha-synuclein in human cerebrospinal fluid as biomarkers of Parkinson’s disease. Brain. 2010;133(Pt 3):713–26.
Mollenhauer B, Locascio JJ, Schulz-Schaeffer W, Sixel-Doring F, Trenkwalder C, Schlossmacher MG. alpha-Synuclein and tau concentrations in cerebrospinal fluid of patients presenting with parkinsonism: a cohort study. Lancet Neurol. 2011;10(3):230–40.
Shi M, Bradner J, Hancock AM, Chung KA, Quinn JF, Peskind ER, et al. Cerebrospinal fluid biomarkers for Parkinson disease diagnosis and progression. Ann Neurol. 2011;69(3):570–80.
Wennstrom M, Surova Y, Hall S, Nilsson C, Minthon L, Bostrom F, et al. Low CSF levels of both alpha-synuclein and the alpha-synuclein cleaving enzyme neurosin in patients with synucleinopathy. PLoS ONE. 2013;8(1): e53250.
Magdalinou NK, Paterson RW, Schott JM, Fox NC, Mummery C, Blennow K, et al. A panel of nine cerebrospinal fluid biomarkers may identify patients with atypical parkinsonian syndromes. J Neurol Neurosurg Psychiatry. 2015;86(11):1240–7.
van Steenoven I, Majbour NK, Vaikath NN, Berendse HW, van der Flier WM, van de Berg WDJ, et al. alpha-Synuclein species as potential cerebrospinal fluid biomarkers for dementia with Lewy bodies. Mov Disord. 2018;33(11):1724–33.
Luo X, Hou L, Shi H, Zhong X, Zhang Y, Zheng D, et al. CSF levels of the neuronal injury biomarker visinin-like protein-1 in Alzheimer’s disease and dementia with Lewy bodies. J Neurochem. 2013;127(5):681–90.
Wennstrom M, Londos E, Minthon L, Nielsen HM. Altered CSF orexin and alpha-synuclein levels in dementia patients. J Alzheimers Dis. 2012;29(1):125–32.
Wang Y, Shi M, Chung KA, Zabetian CP, Leverenz JB, Berg D, et al. Phosphorylated alpha-synuclein in Parkinson’s disease. Sci Transl Med. 2012;4(121):121ra20.
Kapaki E, Paraskevas GP, Emmanouilidou E, Vekrellis K. The diagnostic value of CSF alpha-synuclein in the differential diagnosis of dementia with Lewy bodies vs. normal subjects and patients with Alzheimer’s disease. PLoS One. 2013;8(11):e81654.
Oeckl P, Metzger F, Nagl M, von Arnim CA, Halbgebauer S, Steinacker P, et al. Alpha-, beta-, and gamma-synuclein quantification in cerebrospinal fluid by multiple reaction monitoring reveals increased concentrations in Alzheimer’s and Creutzfeldt-Jakob disease but no alteration in synucleinopathies. Mol Cell Proteomics. 2016;15(10):3126–38.
Berge G, Sando SB, Albrektsen G, Lauridsen C, Moller I, Grontvedt GR, et al. Alpha-synuclein measured in cerebrospinal fluid from patients with Alzheimer’s disease, mild cognitive impairment, or healthy controls: a two year follow-up study. BMC Neurol. 2016;16(1):180.
Llorens F, Kruse N, Schmitz M, Shafiq M, da Cunha JE, Gotzman N, et al. Quantification of CSF biomarkers using an electrochemiluminescence-based detection system in the differential diagnosis of AD and sCJD. J Neurol. 2015;262(10):2305–11.
Shi M, Tang L, Toledo JB, Ginghina C, Wang H, Aro P, et al. Cerebrospinal fluid alpha-synuclein contributes to the differential diagnosis of Alzheimer’s disease. Alzheimers Dement. 2018;14(8):1052–62.
Vergallo A, Bun RS, Toschi N, Baldacci F, Zetterberg H, Blennow K, et al. Association of cerebrospinal fluid alpha-synuclein with total and phospho-tau181 protein concentrations and brain amyloid load in cognitively normal subjective memory complainers stratified by Alzheimer’s disease biomarkers. Alzheimers Dement. 2018;14(12):1623–31.
Stewart T, Shi M, Mehrotra A, Aro P, Soltys D, Kerr KF, et al. Impact of pre-analytical differences on biomarkers in the ADNI and PPMI studies: implications in the era of classifying disease based on biomarkers. J Alzheimers Dis. 2019;69(1):263–76.
Bridel C, van Wieringen WN, Zetterberg H, Tijms BM, Teunissen CE, Group atN. Diagnostic value of cerebrospinal fluid neurofilament light protein in neurology: a systematic review and meta-analysis. JAMA Neurology. 2019;76(9):1035–48.
Tokuda T, Qureshi MM, Ardah MT, Varghese S, Shehab SAS, Kasai T, et al. Detection of elevated levels of α-synuclein oligomers in CSF from patients with Parkinson disease. Neurology. 2010;75(20):1766–70.
Heegaard NH, Tanassi JT, Bech S, Salvesen L, Jensen PL, Montezinho LC, et al. Cerebrospinal fluid α-synuclein in the differential diagnosis of parkinsonian syndromes. Future Neurol. 2014;9(5):525–32.
Bousiges O, Philippi N, Lavaux T, Perret-Liaudet A, Lachmann I, Schaeffer-Agalède C, et al. Differential diagnostic value of total alpha-synuclein assay in the cerebrospinal fluid between Alzheimer’s disease and dementia with Lewy bodies from the prodromal stage. Alzheimer’s Research & Therapy. 2020;12(1):120.
Tokutake T, Kasuga K, Tsukie T, Ishiguro T, Shimohata T, Onodera O, et al. Clinical correlations of cerebrospinal fluid biomarkers including neuron-glia 2 and neurofilament light chain in patients with multiple system atrophy. Parkinsonism Relat Disord. 2022;102:30–5.
Schmitz M, Villar-Piqué A, Llorens F, Gmitterová K, Hermann P, Varges D, et al. Cerebrospinal fluid total and phosphorylated α-synuclein in patients with Creutzfeldt-Jakob disease and synucleinopathy. Mol Neurobiol. 2019;56(5):3476–83.
Mollenhauer B, Caspell-Garcia CJ, Coffey CS, Taylor P, Singleton A, Shaw LM, et al. Longitudinal analyses of cerebrospinal fluid α-Synuclein in prodromal and early Parkinson’s disease. Mov Disord. 2019;34(9):1354–64.
Shi M, Tang L, Toledo JB, Ginghina C, Wang H, Aro P, et al. Cerebrospinal fluid α-synuclein contributes to the differential diagnosis of Alzheimer’s disease. Alzheimers Dement. 2018;14(8):1052–62.
Groveman BR, Orrù CD, Hughson AG, Raymond LD, Zanusso G, Ghetti B, et al. Rapid and ultra-sensitive quantitation of disease-associated α-synuclein seeds in brain and cerebrospinal fluid by αSyn RT-QuIC. Acta Neuropathol Commun. 2018;6(1):7.
Shahnawaz M, Tokuda T, Waragai M, Mendez N, Ishii R, Trenkwalder C, et al. Development of a biochemical diagnosis of Parkinson disease by detection of α-synuclein misfolded aggregates in cerebrospinal fluid. JAMA Neurol. 2017;74(2):163–72.
De Luca CMG, Elia AE, Portaleone SM, Cazzaniga FA, Rossi M, Bistaffa E, et al. Efficient RT-QuIC seeding activity for α-synuclein in olfactory mucosa samples of patients with Parkinson’s disease and multiple system atrophy. Translational Neurodegeneration. 2019;8(1):24.
Manne S, Kondru N, Jin H, Serrano GE, Anantharam V, Kanthasamy A, et al. Blinded RT-QuIC analysis of α-synuclein biomarker in skin tissue from Parkinson’s disease patients. Mov Disord. 2020;35(12):2230–9.
Bargar C, Wang W, Gunzler SA, LeFevre A, Wang Z, Lerner AJ, et al. Streamlined alpha-synuclein RT-QuIC assay for various biospecimens in Parkinson’s disease and dementia with Lewy bodies. Acta Neuropathol Commun. 2021;9(1):62.
Bongianni M, Catalan M, Perra D, Fontana E, Janes F, Bertolotti C, et al. Olfactory swab sampling optimization for α-synuclein aggregate detection in patients with Parkinson’s disease. Translational Neurodegeneration. 2022;11(1):37.
Manne S, Kondru N, Jin H, Anantharam V, Huang X, Kanthasamy A, et al. α-Synuclein real-time quaking-induced conversion in the submandibular glands of Parkinson’s disease patients. Mov Disord. 2020;35(2):268–78.
Hall S, Orrù CD, Serrano GE, Galasko D, Hughson AG, Groveman BR, et al. Performance of αSynuclein RT-QuIC in relation to neuropathological staging of Lewy body disease. Acta Neuropathol Commun. 2022;10(1):90.
Arnold MR, Coughlin DG, Brumbach BH, Smirnov DS, Concha-Marambio L, Farris CM, et al. α-Synuclein seed amplification in CSF and brain from patients with different brain distributions of pathological α-synuclein in the context of co-pathology and non-LBD diagnoses. Ann Neurol. 2022;92(4):650–62.
Laske C, Fallgatter AJ, Stransky E, Hagen K, Berg D, Maetzler W. Decreased α-synuclein serum levels in patients with Lewy body dementia compared to Alzheimer’s disease patients and control subjects. Dement Geriatr Cogn Disord. 2011;31(6):413–6.
Tsai CL, Liang CS, Yang CP, Lee JT, Ho TH, Su MW, et al. Indicators of rapid cognitive decline in amnestic mild cognitive impairment: the role of plasma biomarkers using magnetically labeled immunoassays. J Psychiatr Res. 2020;129:66–72.
Lv S, Zhou X, Li Y, Zhang S, Wang Y, Jia S, et al. The association between plasma α-synuclein (α-syn) protein, urinary Alzheimer-associated neuronal thread protein (AD7c-NTP), and apolipoprotein epsilon 4 (ApoE ε4) alleles and cognitive decline in 60 patients with Alzheimer’s disease compared with 28 age-matched normal individuals. Med Sci Monit. 2021;27: e932998.
Shim KH, Kim SC, Youn YC, Sung Y-H, An SSA. Decreased plasma α-synuclein in idiopathic Parkinson’s disease patients after adjusting hemolysis factor. Mol Cell Toxicol. 2020;16(4):477–84.
Li X-Y, Yang W, Li X, Li X-R, Li W, Song Q, et al. Phosphorylated alpha-synuclein in red blood cells as a potential diagnostic biomarker for multiple system atrophy: a pilot study. Parkinson’s Disease. 2020;2020:8740419.
Li X-Y, Li W, Li X, Li X-R, Sun L, Yang W, et al. Alterations of erythrocytic phosphorylated alpha-synuclein in different subtypes and stages of Parkinson’s disease. Frontiers in Aging Neuroscience. 2021;13:623977.
Chen W-R, Chen J-C, Chang S-Y, Chao C-T, Wu Y-R, Chen C-M, et al. Phosphorylated α-synuclein in diluted human serum as a biomarker for Parkinson’s disease. Biomed J. 2022;45(6):914–22.
Li X-Y, Yang W, Li X, Li X, Chen Z, Wang C, et al. editors. Levels of oligomeric alpha-synuclein in red blood cells are elevated in patients with Parkinson’s disease and affected by brain alpha-synuclein expression Int J Clin Exp Med. 2019;12:10470–7.
Acknowledgements
The authors wish to thank the anonymous reviewers for the initial draft of this manuscript.
Funding
This work was supported by a National Research Foundation of Korea grant funded by the Ministry of Education (No. 2020R1F1A1075666 and 2021R1A2C1093218).
Author information
Authors and Affiliations
Contributions
Conceptualization of the manuscript: KHS, MJK, YCY, SSAA, and SYK; investigation: KHS and MJK; writing—original draft preparation: KHS and MJK; writing—review and editing: YCY, SSAA, and SYK; funding acquisition: KHS and MJK; and supervision: SSAA and SYK. The authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Shim, K.H., Kang, M.J., Youn, Y.C. et al. Alpha-synuclein: a pathological factor with Aβ and tau and biomarker in Alzheimer’s disease. Alz Res Therapy 14, 201 (2022). https://doi.org/10.1186/s13195-022-01150-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13195-022-01150-0