
Abstract
In the present study, a series of 2-(4-(2-chloroacetyl)piperazin-1-yl)-N-(2-(4-chlorophenyl)-4-oxoquinazolin-3(4H)-yl)acetamide derivatives was synthesized and its chemical structures were confirmed by physicochemical and spectral characteristics. The synthesized compounds were evaluated for their in vitro antimicrobial (tube dilution technique) and anticancer (MTT assay) activities along with molecular docking study by Schrodinger 2018-1, maestro v11.5. The antimicrobial results indicated that compounds 3, 8, 11 and 12 displayed the significant antimicrobial activity and comparable to the standards drugs (ciprofloxacin and fluconazole). The anticancer activity results indicated that compound 5 have good anticancer activity among the synthesized compounds but lower active than the standard drugs (5-fluorouracil and tomudex). Molecular docking study demonstrated that compounds 5 and 7 displayed the good docking score with better anticancer potency within the binding pocket and these compounds may be used as a lead for rational drug designing for the anticancer molecules.

Similar content being viewed by others

Introduction
The ever-increasing microbial antibiotic resistance leads to ongoing testing of new biologically efficient compounds of either natural or synthetic origin for infectious diseases [1]. Subsequently, compounds carrying heterocyclic nuclei gained much attention in the growth of novel antimicrobial agents due to their chemotherapeutic significance. Quinazoline nucleus is an exciting molecule with two nitrogen atoms in its structure among the most significant classes of aromatic bicyclic compounds. Quinazoline is one of the most widespread scaffolds among natural and synthetic bioactive compounds. Quinazoline heterocyclic compound resembles both the purine nucleus and the pteridine one and exhibited wide spectrum medicinal values i.e. antihypertensive, antitumor, antiplasmodial, antiviral and anti-inflammatory activities [2, 3].
Interest in quinazolinones as anticancer agents has further increased since the discovery of raltitrexed (Tomudex®) (Fig. 1) as an antimetabolite drug used in cancer chemotherapy. Quinazolinone derivatives have been reported to have potent anticancer activities viz. aurora kinase inhibitors [4], α-folate receptor inhibitors [5], CDK-inhibitors [6], activin-like kinase (ALK) inhibitors [7], EGFR inhibitors [8], topoisomerase inhibitors [9], pin1 (Protein interaction with NIMA1) inhibitors [10], T cell proliferation inhibitors on peripheral blood mononuclear cells (PBMC) and jurkat cells [11] and VEGFR inhibitors [12].

Structure of Raltitrexed (Tomudex)
Molecular docking analyses provide the most comprehensive illustration of the interaction between drug receptors and produced a modern rational approach to drug design [13]. The RAW 264.7 cells are monocyte/macrophage like cells from the BALB/c mouse modified cell line from Abelson leukemia virus. These cells are defined as a suitable macrophage model. Pinocytosis and phagocytosis can be performed. RAW 264.7 cells raise the production of nitric oxide (NO) at LPS stimulation and improve phagocytosis. In addition, these cells can destroy target cells by cytotoxicity dependent on antibodies [14]. Macrophages are immune cells found in many distinct tissues, performing a broad variety of biological activities. They are highly plastic in their pattern of protein expression and can be activated by a variety of cytokines and pathogen-associated molecules like lipopolysaccharide. The quinazoline and other heterocyclic compounds reported have a mass variety of less than 500 ppm or more [15,16,17,18].
Recently Kiruthiga et al. [19] reported that quinazolinone moiety is (I) essential for antimicrobial activity. Rajveer et al. [20] found that acetamide nucleus attached at N-1 position to quinazoline moiety (II) possessed good antimicrobial activity. Rajasekaran et al. [1] proposed that acetamide group of quinazolinone moiety (III) attached with heterocyclic compound showed better antimicrobial activity. Kumar et al. [21] found that halogenated phenyl moiety (chlorine substitution at the 2nd position) (IV) attached at 2nd position on the quinazolinone nucleus possesses a good antimicrobial activity. Desai et al. [3] showed that quinazolinones bearing piperazine moiety (V) having asserted antimicrobial activity.
Hour et al. [22] identified that the presence of phenyl ring at 2nd position of quinazolinone nucleus (VI) improved the anticancer activity. Xia et al. [23] revealed that substitution with halogens at 4th position on phenyl ring attached at 2nd position of quinazolinone moiety (VII) increased anticancer activity. Raghavendra et al. [24] reported that acetamide group at N-1 position of quinazolinone nucleus (VIII) enhanced the anticancer activity. Tobe et al. [25] reported that quinazolinone nucleus having a piperazine ring substitution (IX) exerted an anticancer activity by suppression of T cell proliferation (Fig. 2).

Design of proposed quinazolinone molecules based on literature
Rational behind the selection of cyclin dependent kinase (CDK8)
Quinazolin derivatives are protein kinase inhibitors. Protein kinases are the most important class of human enzymes that regulate the sequence of events such as cell cycle progression, cell division and cell proliferation [26]. Developing new quinazoline derivatives as an anticancer agent is considered a promising area and researchers around the world are continuously exploring this region to generate new drug candidates [26]. CDK activity is controlled by association with CDK-inhibiting regulatory subunits (cyclines) and proteins, their phosphorylation status and ubiquitin-mediated proteolysis. Since the loss of cell cycle control leading to deregulated cell proliferation is one of cancer’s hallmarks, it is anticipated that the inhibition of CDKs will provide an effective tumor growth control strategy and thus impact cancer therapy. Many organisations researched CDK inhibition and used a range of structural templates with different degrees of selectivity and activity [27, 28].
In the light of above facts and in continuation of our effort to develop novel anticancer and antimicrobial agents [29,30,31], the present study is aimed to design, molecular docking and synthesize of 2-(4-(2-chloroacetyl)piperazin-1-yl)-N-(2-(4-chlorophenyl)-4-oxoquinazolin-3(4H)-yl)acetamides as prospective anticancer and antimicrobial agents.
Experimental
Materials and methods
Starting materials were obtained from commercial sources and used without further purification. The microbial strains for the antimicrobial evaluation were obtained from the Microbial Type Culture Collection and Gene Bank MTCC, Chandigarh. Thin layer chromatography (TLC) using commercial silica gel plates (Merck), Silica gel F254 on aluminum sheets, has noted reaction improvements. Infrared (KBr pellets, cm−1) spectra were recorded on an Agilent Resolutions Pro FT-IR spectrometer. Melting points were determined in open capillary tubes. Mass spectra were recorded using Waters Micromass Q-Tof micro instrument. 1H-NMR (DMSO) and 13C–NMR (DMSO) were recorded at 600 MHz and 150 MHz, respectively on Bruker Avance III 600 NMR spectrometer. Perkin–Elmer 2400 C, H and N analyzer used for elemental analysis.
Synthetic procedure for the synthesis of quinazolinone derivatives (1–17)
2-(4-Chlorophenyl)-4H-benzo[e] [ 1, 3 ] oxazin-4-one (I)
2-Aminobenzoic acid (0.01 mol) was stirred for 3 h at room temperature with 4-chloro benzoyl chloride (0.01 mol) in the presence of pyridine. The resultant mixture was treated with 5% sodium bicarbonate solution to get I, which was filtered, dried and recrystallized with ethanol.
3-Amino-2-(4-chlorophenyl)quinazolin-4(3H)-one (II)
2-(4-Chlorophenyl)-4H-benzo[e] [1, 3] oxazin-4-one (I) (0.01 mol) was reacted with hydrazine hydrate (0.02 mol) in the presence of ethanol and refluxed for 3 h (30 °C) to obtain II. The reaction mixture was cooled and the resultant precipitate was filtered off and recrystallized with ethanol [32].
2-Chloro-N-(2-(4-chlorophenyl)-4-oxoquinazolin-3(4H)-yl)acetamide (III)
A mixture of 3-amino-2-(4-chlorophenyl)quinazolin-4(3H)-one (II) (0.01 mol) and chloroacetyl chloride (0.01 mol) with a few drops of glacial acetic acid in absolute ethanol (20 mL) was refluxed for 8 h (30 °C). The reaction mixture was then cooled in ice cold water and resultant precipitate of III was filtered, washed with water, dried and recrystallized with ethanol.
N-(2-(4-Chlorophenyl)-4-oxoquinazolin-3(4H)-yl)-2-(piperazin-1-yl)acetamide (IV)
A mixture of 2-chloro-N-(2-(4-chlorophenyl)-4-oxoquinazolin-3(4H)-yl)acetamide (III) (0.01 mol) and piperazine (0.01 mol) with a few drops of glacial acetic acid in absolute ethanol (20 mL) was refluxed for 10 h (40 °C). The reaction mixture was cooled in ice cold water and resultant precipitate of IV was filtered, washed with water, dried and recrystallized with ethanol.
2-(4-(2-Chloroacetyl)piperazin-1-yl)-N-(2-(4-chlorophenyl)-4-oxoquinazolin-3(4H)-l) acetamide (V)
N-(2-(4-Chlorophenyl)-4-oxoquinazolin-3(4H)-yl)-2-(piperazin-1-yl)acetamide (IV) (0.01 mol) was further treated with chloroacetyl chloride (0.01 mol) in the presence of few drops of glacial acetic acid in absolute ethanol (20 mL) and refluxed for 8 h (30 °C) yielded of V. The reaction mixture was then cooled in ice cold water and resultant precipitate was filtered, washed with water, dried and recrystallized with ethanol. 2-(4-(2-Chloroacetyl)piperazin-1-yl)-N-(2-(4-chlorophenyl)-4-oxoquinazolin-3(4H)-yl)acetamide (V) (0.01 mol) was reacted with different corresponding aniline (0.01 mol) with a few drops of glacial acetic acid in absolute ethanol was refluxed for 10 h (40 °C) to synthesize the title compounds (1–17). The reaction mixture was then cooled in ice cold water and the resultant precipitate was filtered, washed with water, dried and recrystallized with ethanol.
Antimicrobial evaluation (in vitro)
The synthesized derivatives of quinazolinone were tested using ciprofloxacin (antibacterial) and fluconazole (antifungal) as reference drugs for their in vitro antimicrobial potential. The antimicrobial activity towards Gram-positive bacteria: S. aureus MTCC3160, B. subtilis MTCC441, Gram-negative bacterium: E. coli MTCC443 and fungal species: C. albicans MTCC227 and A. niger MTCC281 was determined by tube dilution method [33]. To give a concentration of 100 µg/ml, the standard and test samples were dissolved in DMF. Dilutions of test and standard compounds were prepared in double strength nutrient broth I.P. (bacteria) or Sabouraud dextrose broth I.P. (fungi) [34].
Anticancer evaluation (in vitro)
It has been determined the activity of synthesized compounds and control drugs against human colon (HCT116) and cancer cell lines of the mouse monocyte macrophage leukaemic (RAW 264.7). In RPMI 1640 (Sigma) the selected cancer cell strains were developed, supplemented by 10% heat-inactivated bovine fetal serum (FBS) (PAA Laboratories) and 1% penicillin/streptomycin (PAA Laboratories). Cultures were kept at 37 °C in a humidified incubator in an environment of 5% CO2. Anticancer activity of synthesized compounds was assessed at distinct concentrations using MTT assay, as Mosmann (1983) explained but with minor alteration after 72 h of incubation. Using a spectrophotometer at 520 nm, assay sheets were read. Data produced were used to determine a dose–response curve from which the concentration of test compounds needed to kill 50% of the cell population (IC50) was determined [35].
Molecular docking
Target identification
Kinase inhibitors are extremely efficient in the therapy of cancer, specifically targeting specific mutations that mainly drive tumorigenesis. They are categorized according to their capacity to catalyze ATP terminal phosphate transfer to substrates that usually contain serine, threonine or tyrosine residues [36]. Cycline-dependent kinases (CDKs) are a family of significant regulatory proteins that regulate different cell activity and are primarily engaged in the cell cycle and transcription. It is not surprising that many diseases, especially cancer, are common in their aberrant activities, given the fundamental biological functions played by CDKs. Different cell cycle proteins such as CDKs and cyclines induce development of the cell cycle as they are the main regulators of the cell cycle. Previous trials have shown that quinazoline derivatives therapy arrests cancer cells in the G2/M stage CDK activity enables the orderly transition between cell cycle stages. Cell cycle progression inhibition and apoptosis are the most prevalent causes of inhibition of cell growth [37]. The macrophage-like cell line RAW264.7 promotes replication of murine noroviruses as opposed to most other mouse-derived cell cultures and is commonly used for this purpose. In addition, RAW264.7 was discovered to be unique in research of a mouse model of serious respiratory disease for the propagation of the causative agent, pneumonia virus of mice and for the measurement of pro-inflammatory mediators associated with infection [38, 39].
Docking
To investigate the binding mode of synthesized quinazolinone compounds and standard drugs with selected PDB ID for cancer cell lines, molecular docking research was implemented i.e. human colorectal carcinoma and mouse leukaemic monocyte macrophage, was retrieved from protein data bank. GLIDE (Schrodinger 2018-1, maestro v11.5) acquired the docking score and targeted the binding site and formed the grid. The active site grid covered the major amino acids that interact with the receptor. Using their specific PDB ID: 5FGK for human colorectal carcinoma and PDB ID: 5JVY for mouse monocyte macrophage (Additional file 1), the protein’s 3-D structure was acquired from the protein database. The protein structure has been prepared using the protein preparation wizard in the Schrodinger maestro v11.5. A set of quinazolinone compounds have been selected as ligands for the docking studies and their structures have been drawn using the workspace of the maestro and converted into 3D form [40, 41].
Results and discussion
Chemistry
A new series of quinazoline derivatives was synthesized using synthetic Scheme 1 (1–17) (Additional file 2). The physicochemical and spectral characteristics of the synthesized derivatives are presented in Tables 1 and 2, respectively. The molecular structures of the quinazoline compounds were confirmed by FT-IR, 1H-NMR, 13C–NMR, MS and elemental analysis. The IR spectrum of compound showed around 2928–3077 cm−1 and 1561–1592 cm−1 indicated the presence of C–H and C=C groups, respectively. The presence of C=O group in compounds displayed in the scale of 1665–1727 cm−1. The presence of an arylalkyl ether group (Ar-OCH3) in synthesized compounds is established by the existence of an IR absorption band around 1195 cm−1. The appearance of IR stretching 1591–1636 cm−1 in the spectral data of compounds (1–17) specified the existence of N=CH group. The appearance of IR stretching 1250–1253 cm−1 in the spectral data of synthesized compounds specified the existence of C–N group. The proton NMR displayed the multiplet signals between 6.03 and 8.05 δ ppm in the aromatic ring of the synthesized compounds. The compounds exhibited singlet at 8.91 δ ppm due to the existence of N–NH group. Compounds displayed singlet at 2.36–2.38 δ ppm due to the existence of –CH3 group. The compounds displayed singlet at 3.71δ ppm due to the existence of OCH3 of Ar-OCH3. The 13C–NMR spectra of aromatic ring exhibited the carbon atoms in the range of 170.9, 164.9, 164.6, 161.3, 151.9, 138.4, 135.8, 135.5, 133.5, 132.3, 129.5, 129.4, 128.0, 127.9, 127.6, 127.4, 126.9, 122.4, 121.7, 120.6, 118.3, 114.6, 58.7, 52.5, 52.4, 52.3, 46.8, 46.7 of the synthesized compounds. The elemental analysis was found within ± 0.4% of the theoretical results of compounds.

Synthesis of 2-(4-(2-chloroacetyl)piperazin-1-yl)-N-(2-(4-chlorophenyl)-4-oxoquinazolin-3(4H)-yl)acetamide derivatives 1–17
Antimicrobial screening results
The synthesized compounds were tested by tube dilution technique against Gram positive and Gram negative bacterial and fungal strains for their in vitro antimicrobial activity and the MIC values of control drugs and synthetic compounds are shown in Table 3. Antimicrobial screening results indicated that compound 3 (MICbs = 4.81 µM) emerged as most effective antibacterial agent toward B. subtilis. Compounds 11 and 12 (MICsa = 2.56 µM) displayed the promising activity toward S. aureus. Compound 8 (MICec = 5.54 µM) emerged as most active candidate against Gram-negative bacterium E. coli and comparable to standard drug ciprofloxacin. In the case of antifungal activity, compound 3 (MICca = 4.81 and MICan = 2.40 µM) was found to be the most potent antifungal agent against C. albicans and A. niger and had better antifungal activity than standard drug fluconazole (MICca,an = 5.09 µM). Results of antimicrobial activity indicated that the synthesized compounds possessed a higher antifungal activity than antibacterial activity. As far as antibacterial activity is concerned, the synthesized compounds were more active against Gram negative bacterium (E. coli) than Gram positive bacteria (S. aureus and B. subtilis).
Anticancer screening results
The synthesized compounds were evaluated for their in vitro anticancer activity against human colorectal cancer (HCT116) and mouse monocyte macrophage (RAW 264.7) cancer cell lines using MTT assay. Compound 5 (IC50 = 0.82 and 12.39 µM against RAW 264.7 and HCT116, respectively) was found to be the most active one and compared to the 5-fluorouracil and Tomudex (used as standard drugs). The anticancer screening (IC50 = µM) results are shown in Table 4.
Molecular docking results
The synthesized derivatives of quinazolinone showed excellent docking performance and the chosen proteins were discovered to communicate with significant amino acids. Molecular docking was performed to analyze the binding mode of active compounds 5 and 7 for the cancer cell lines of human colorectal carcinoma (HCT116) and mouse leukaemic monocyte macrophage (RAW 264.7). The compound 5 and standard drugs (5-fluorouracil and tomudex) were docked in the active site of the cycline-dependent kinase cdk8 (PDB ID: 5FGK) co-crystallized acuity 5XG ligand and in the active site of the S121P murine COX-2 mutant (PDB ID: 5JVY) co-crystallized acuity ligand COH were also docked. The binding mode of native ligand 5XG have docked score (− 8.72) and COH ligand have docked score (− 8.93) showed good interaction with crucial amino acids residues with their respective proteins (Fig. 3). The binding mode of compound 5 (Fig. 4) (using PDB ID: 5FGK for HCT116), scored docked score (− 8.011) with moderate anticancer potency (12.39 μM) and formed H-bond with crucial amino acids and compared to the docked score of 5-fluorouracil (Fig. 5) have lowest docked score (− 5.753) with better anticancer activity (4.6 μM) and tomudex (− 10.86) have good docked score with better anticancer potency (9.05 μM) (Fig. 6a). The binding mode of active compound 5 (using PDB ID: 5JVY for mouse leukaemic monocyte macrophage) have docked score (− 11.054) with better anticancer activity (0.82 μM) which also developed H-bond with crucial amino acid (Fig. 7) and compound 7 have docked score (− 11.284) with better anticancer potency (1.20 μM) (Fig. 8) and comparable to 5-fluorouracil have lowest docked score (− 4.122) with better anticancer potency (0.60 μM) (Fig. 9) and also compared to the tomudex (− 10.83) have better docked score with better anticancer potency (2.81 μM) (Fig. 6b). The docking results of synthesized compounds (5 and 7), native ligands and standard drug (tomudex) which showed good to better docking score with their respective proteins. Docking results and interacting residues are shown in Tables 5 and 6.

Binding surface and ligand interaction diagram of native ligand 5XG (a) and COH (b) with their respective protein

Binding surface and ligand interaction diagram of compound 5

Binding surface and ligand interaction diagram of 5-fluorouracil

Binding surface and ligand interaction diagram of Raltitrexed (Tomudex) with 5FGK (a) and 5JVY (b) proteins

Binding surface and ligand interaction diagram of compound 5

Binding surface and ligand interaction diagram of compound 7

Binding surface and ligand interaction diagram of 5-fluorouracil
The docking findings therefore indicate that the synthesized compounds may be of excellent importance in effective chemotherapy. The selected database of protein i.e. (PDB ID: 5FGK) for human colorectal carcinoma and (PDB ID: 5JVY) mouse monocyte macrophage may be the target protein of derivatives of quinazolinone for their anticancer activity (Additional file 3).
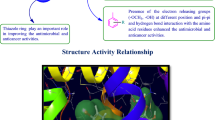
Structure activity relationship studies (SAR)
The SAR of synthesized compounds can be summarized (Fig. 10) as follow:

Structure activity relationship study of the synthesized compounds
-
a.
Results of antimicrobial activity indicated that substitution of phenyl amino ring attached to the chlorophenyl quinazolinone piperazine-acetamide nucleus with electron donating methyl group enhanced the antimicrobial activity against B. subtilis, C. albicans and A. niger.
-
b.
In the case of antibacterial activity towards S. aureus and E. coli, electron withdrawing groups i.e. chloro (Cl) and chloro with nitro (NO2) substitution on the phenylamino ring attached to the chlorophenyl quinazolinone piperazine-acetamide nucleus increased the antibacterial activity.
-
c.
Results of anticancer activity revealed that the presence of electron withdrawing groups i.e. dichloro on the phenylamino ring attached to the chlorophenyl quinazolinone piperazine acetamide nucleus increased the anticancer activity against both cancer cell lines (RAW 264.7 and HCT116).
Conclusion
In the present study, the synthesized quinazolinone derivatives i.e. compound 3 showed promising antimicrobial activity due to the presence of electron releasing group at meta–position of the substituted benzylidene nucleus and comparable to the control drugs. In case of anticancer activity indicated that compound 5 (meta/para-Cl) and compound 7 (meta-NO2) displayed moderate anticancer activity towards human colorectal carcinoma and mouse leukaemic monocyte macrophage cancer cell lines due to the presence of EWG on the substituted benzylidene nucleus. Molecular docking analysis demonstrated that compounds 5 and 7 showed the better docked score with better potency and comparable to the standard drug and native ligands of the proteins. The findings of the docking are compatible with the assays of anticancer. Docking information stay in excellent correlation with the outcomes of anticancer activity and these molecules may be used as a lead in the design of new anticancer agents.
Abbreviations
- ALK:
-
activin-like kinase
- EGFR:
-
epidermal growth factor receptor
- PBMC:
-
peripheral blood mononuclear cells
- VEGFR:
-
vascular endothelial growth factor
- ATP:
-
adinosine triphosphate
- CDK:
-
cyclin-dependent kinase
- NO:
-
nitric oxide
- RNA:
-
ribonucleic acid
- NMR:
-
nuclear magnetic resonance
- IR:
-
infrared
- MS:
-
mass spectrum
- CHN:
-
carbon hydrogen nitrogen
- TLC:
-
thin-layer chromatography
- Str:
-
starching
- DMSO:
-
dimethyl sulfoxide
- rt:
-
room temperature
- FBS:
-
fetal bovine serum
- MIC:
-
minimum inhibitory concentration
- MTCC:
-
Microbial Type Culture Collection
- E. coli :
-
Escherichia coli
- C. albicans :
-
Candida albicans
- S. aureus :
-
Staphylococcus aureus
- B. subtilis :
-
Bacillus subtilis
- A. niger :
-
Aspergillus niger
- MTT:
-
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- HCT116:
-
human colorectal carcinoma116
- RAW 264.7:
-
murine macrophaga 264.7
- 5-Fu:
-
5-fluorouracil
- PDB:
-
protein data bank
- XP:
-
extra precision
- 3D:
-
3 dimensional
References
Rajasekaran S, Rao G (2012) Synthesis, antibacterial and antioxidant activity of some 2,3-susbtituted quinazolin-4(3H)-ones. Der Pharm Lett 4(1):349–353
Marzaro G, Guiotto A, Chilin A (2012) Quinazoline derivatives as potential anticancer agents: a patent review (2007–2010). Expert Opin Ther Patents 2012:223–252
Desai NC, Dodiya A, Bhatt N, Kumar M (2012) Dimeric 2-(2-chlorophenyl)- quinazolin-4-ones as potential antimicrobial agents. Med Chem Res 21:1127–1135
Foote K, Mortlock A, Heron N, Jung F, Hill G, Pasquet G, Brady M, Green S, Heaton S, Kearney S, Keen N, Odedra R, Wedge S, Wilkinson R (2008) Synthesis and SAR of 1-acetanilide-4-aminopyrazole substituted quinazolines: selective inhibitors of Aurora B kinase with potent antitumor activity. Bioorg Med Chem Lett 18:1904–1909
Henderson E, Bavetsias V, Theti D, Wilson S, Clauss R, Jackman A (2006) Targeting the alpha-folate receptor with cyclopenta[g]quinazoline-based inhibitors of thymidylate synthase. Bioorg Med Chem 14:5020–5042
Loge C, Testard A, Thiery V, Lozach O, Blairvacq M, Meijer L, Besson T (2008) Novel 9-oxo-thiazolo[5,4-f]quinazoline-2-carbonitrile derivatives as dual cyclin-dependent kinase 1(CDK1)/glycogen synthase kinase-3 (GSK-3) inhibitors: synthesis, biological evaluation and molecular modeling studies. Eur J Med Chem 43:1469–1477
Gellibert F, Fouchet M, Nguyen V, Wang R, Krysa G, De Gouville A, Huet S, Dodic N (2009) Design of novel quinazoline derivatives and related analogues as potent and selective ALK5 inhibitors. Bioorg Med Chem Lett 19:2277–2281
Noolvi M, Patel H (2013) Synthesis, method optimization, anticancer activity of 2,3,7- trisubstituted quinazoline derivatives and targeting EGFR-tyrosine kinase by rational approach:1st Cancer Update. Arab J Chem 6(1):35–48
Park HJ, Kim Y, Kim J, Lee E, Yi Y, Hwang H, Suh M, Ryu C, Lee S (2004) 6-Arylamino-7-chloro-quinazoline-5,8-diones as novel cytotoxic and DNA topoisomerase inhibitory agents. Bioorg Med Chem Lett 14:3385–3388
Zhu L, Jin J, Liu C, Zhang C, Sun Y, Guo Y, Fu D, Chen X, Xu B (2011) Synthesis and biological evaluation of novel quinazoline-derived human Pin1 inhibitors. Bioorg Med Chem 19:2797–2807
Sagiv-Barfi I, Weiss E, Levitzki A (2010) Design, synthesis, and evaluation of quinazoline T cell proliferation inhibitors. Bioorg Med Chem 18:6404–6413
Ple AP, Jung F, Ashton S, Hennequin L, Laine R, Morgentin R, Pasquet G, Taylor S (2012) Discovery of AZD2932, a new quinazoline ether inhibitor with high affinity for VEGFR-2 and PDGFR tyrosine kinases. Bioorg Med Chem Lett 22:262–266
Kakkar S, Kumar S, Lim SM, Ramasamy K, Mani V, Shah SAA, Narasimhan B (2018) Design, synthesis and biological evaluation of 3-(2-aminooxazol-5-yl)-2H-chromen-2-one derivatives. Chem Cent J 12(130):1–13
Taciak B, Biaøasek M, Braniewska A, Sas Z, Sawicka P, Kiraga L, Rygiel T, Krol M (2018) Evaluation of phenotypic and functional stability of raw 2647 cell line through serial passages. PLoS ONE 13(6):1–13
Guo M, Hartlova A, Dill BD, Prescott AR, Gierlinski M, Trost M (2015) High-resolution quantitative proteome analysis reveals substantial differences between phagosomes of RAW 264.7 and bone marrow derived macrophages. Proteomics 15:3169–3174
Rajasekaran S, Rao G, Pai PNS (2010) Synthesis, antitubercular, antibacterial and antioxidant activity of some 2-phenyl-3-substituted quinazolin-4(3H)-ones. Der Pharma Chem 2(5):153–163
Mohamed MS, Kamel MM, Kassem EMM, Abotaleb N, Khedr M, Ahmed MF (2011) Synthesis, biological evaluation and molecular docking of quinazoline-4(1H)-one derivatives as anti-inflammatory and analgesic agents. Acta Pol Pharm Drug Res 68(5):665–675
Vashi RT, Patel SB, Patel H (2013) Synthesis, characterization and antifungal activity of 2-[(8-hydroxyquinolinyl)-5-aminomethyl]-3-(4-bromophenyl)-6-bromo-3(H)-quinazolin-4-one and its metal chelates. Int J Pharm Bio Sci 4(1):172–178
Kiruthiga B, Ilango K, Valentina P, Umarani N, Patel R (2009) Synthesis of some new 2-substituted quinazolin-4-one derivatives and their biological activities. Int J Pharm Tech Res 1(4):1503–1506
Rajveer C, Kumaraswamy D, Sudharshini S, Rathinaraj BS (2010) Synthesis of some 6-bromo quinazolinone derivatives for their pharmacological activities. Int J Pharm Bio Sci 1(3):1–10
Kumar S, Mishra G, Singh P, Jha KK, Khosa RL, Gupta SK (2011) Quinazolin-4-one: a highly important heterocyclic with diverse biological activities: A review. Der Chemica Sinica 2(4):36–58
Hour M, Yang J, Lien J, Kuo S, Huang L (2007) Synthesis and cytotoxicity of 6- pyrrolidinyl-2-(2-substitutedphenyl)-4-quinazolinones. J Chin Chem Soc 54:785–790
Xia Y, Yang Z, Hour M, Kuo S, Xia P, Bastow KF, Nakanishi Y, Nampoothiri P, Hackl T, Hamel E, Lee K (2001) Antitumor agents. part 204: 1 synthesis and biological evaluation of substituted 2-aryl quinazolinones. Bioorg Med Chem Lett 11:1193–1196
Raghavendra NM, Gurubasavarajaswamy PM, Nagaranavile SK, Parameshwaran T (2009) Antitumor actions of imidazolyl-(4-oxoquinazolin-3(4H)-yl)-acetamides against Ehrlich ascites carcinoma. Arch Pharm Res 32(3):431–436
Tobe M, Isobe Y, Tomizawa H, Matsumoto M, Obara F, Nagasaki T, Hayashi H (2001) Structure activity relationship of quinazoline derivatives: dual-acting compounds with inhibitory activities toward both TNF-α production and T cell proliferation. Bioorg Med Chem Lett 11:545–548
Shagufta Ahmad I (2017) An insight into the therapeutic potential of quinazoline derivatives as anticancer agents. Med Chem Commun 2017(8):871–885
Chu X-J, DePinto W, Bartkovitz D, So S-S, Vu BT, Packman K, Lukacs C, Ding Q, Jiang N, Wang K, Goelzer P, Yin X, Smith MA, Higgins BX, Chen Y, Xiang Q, Moliterni J, Kaplan G, Graves B, Lovey A, Fotouhi N (2006) Discovery of [4-amino-2-(1-methanesulfonyl-piperidin-4-ylamino)pyrimidin-5-yl](2,3-difluoro-6-methoxyphenyl) methanone (R547), A potent and selective cyclin-dependent kinase inhibitor with significant in vivo antitumor activity. J Med Chem 49:6549–6560
Koehler MFT, Bergeron P, Blackwood EM, Bowman K, Clark KR, Firestein R, Kiefer JR, Maskos K, McCleland ML, Orren L, Salphati L, Schmidt S, Schneider FV, Wu J, Beresini MH (2016) Development of a potent, specific CDK8 kinase inhibitor which phenocopies CDK8/19 knockout cells. ACS Med Chem Lett 7:223–228
Sigroha S, Narasimhan B, Kumar P, Khatkar A, Ramasamy K, Mani V, Mishra RK, Majeed ABA (2012) Design, synthesis, antimicrobial, anticancer evaluation, and QSAR studies of 4-(substituted benzylidene-amino)-1,5-dimethyl-2-phenyl-1,2-dihydropyrazol-3-ones. Med Chem Res 21:3863–3875
Kumar H, Kumar P, Narasimhan B, Kalavathy R, Mani V, Mishra RK, Majeed ABA (2012) Synthesis, in vitro antimicrobial, antiproliferative and QSAR studies of N-(substituted phenyl)-2/4-(1H-indol-3-ylazo)-benzamides. Med Chem Res 22:1957–1971
Kumar R, Kumar P, Kumar M, Narasimhan B (2012) Synthesis, antimicrobial evaluation and QSAR studies of 4-amino-3-hydroxy-naphthalene-1-sulfonic acid derivatives. Med Chem Res 21:4301–4310
Mariappan G, Saha BP, Dutta S, Majumdar A, Saha S (2010) Synthesis and biological evaluation of quinazoline fused Schiff bases. Ind J Pharm Edu Res 45:78–81
Cappuccino JC, Sherman N (1999) Microbiology — A Laboratory Manual. Addison Wesley, California, p 263
Pharmacopoeia of India (2007) Controller of Publications, Ministry of Health Department, Government of India, New Delhi, 1:37
Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65:55–63
Bhullar KS, Lagarón NO, McGowan EM, Parmar I, Jha A, Hubbard BP, Rupasinghe HPV (2018) Kinase-targeted cancer therapies: progress, challenges and future directions. Mol Cancer 17(48):1–20
Shaik TB, Malik SM, Seddigid ZS, Routhu SR, Kamala A (2017) Evaluation of anticancer and anti-mitotic properties of quinazoline and quinazolino-benzothiadiazine derivatives. bioRxiv. https://doi.org/10.1101/108654
Hartley JW, Evans LH, Green KY, Naghashfar Z, Macias AR, Zerfas PM, Ward JM (2008) Expression of infectious murine leukemia viruses by RAW264.7 cells, a potential complication for studies with a widely used mouse macrophage cell line. Retrovirology 5(1):1–6
Hernández-Santoyo A, Tenorio-Barajas AY, Altuzar V, Vivanco-Cid H, Mendoza-Barrera C (2013) Protein-protein and protein-ligand docking, protein engineering-technology and application, tomohisa ogawa. IntechOpen. https://doi.org/10.5772/56376. https://www.intechopen.com/books/protein-engineering-technology andapplication/protein-protein-and-protein-ligand-docking
Singh J, Kumar M, Mansuri R, Sahoo GC, Deep A (2016) Inhibitor designing, virtual screening and docking studies for methyltrans-ferase: a potential target against dengue virus. J Pharm Bioallied Sci 8(3):188–194
Kumar S, Singh J, Narasimhan B, Shah SAA, Lim SM, Ramasamy K, Mani V (2018) Reverse pharmacophore mapping and molecular docking studies for discovery of GTPase HRas as promising drug target for bis-pyrimidine derivatives. Chem Cent J 12(106):1–11
Acknowledgements
The authors are thankful to Head, Department of Pharmaceutical Sciences, Maharshi Dayanand University, Rohtak, for providing necessary facilities to carry out this research work.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
Authors BN, RKM, SM- Performed synthesis and antimicrobial activity and SK-performed molecular docking study of the most active anticancer compounds; KR, SAAS, SML and VM-Performed characterization and antiproliferative study of synthesized compounds. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
Web link for PDB ID: 5FGK and 5JVY proteins.
Additional file 2.
Synthetic scheme with chemical structures.
Additional file 3.
Docking results of active compounds.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Mehta, S., Kumar, S., Marwaha, R.K. et al. Synthesis, molecular docking and biological potentials of new 2-(4-(2-chloroacetyl) piperazin-1-yl)-N-(2-(4-chlorophenyl)-4-oxoquinazolin-3(4H)-yl)acetamide derivatives. BMC Chemistry 13, 113 (2019). https://doi.org/10.1186/s13065-019-0629-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-019-0629-0