
Abstract
To combat the antimicrobial and anticancer drug resistance by pathogens and cancerous cells, efforts has been made to study the pharmacological activities of newly synthesized N-(4-(4-bromophenyl)thiazol-2-yl)-2-chloroacetamide derivatives. The molecular structures of the synthesized derivatives were confirmed by their physicochemical properties and spectroanalytical data (NMR, IR and elemental). The synthesized compounds were evaluated for their in vitro antimicrobial activity against bacterial (Gram positive and Gram negative) and fungal species using turbidimetric method and anticancer activity against oestrogen receptor positive human breast adenocarcinoma cancer cell line (MCF7) by Sulforhodamine B (SRB) assay. Molecular docking studies were carried out to study the binding mode of active compounds with receptor using Schrodinger v11.5. The antimicrobial activity results revealed that compounds d1, d2 and d3 have promising antimicrobial activity. Anticancer screening results indicated that compounds d6 and d7 were found to be the most active ones against breast cancer cell line. Furthermore, the molecular docking study demonstrated that compounds d1, d2, d3, d6 and d7 displayed good docking score within binding pocket of the selected PDB ID (1JIJ, 4WMZ and 3ERT) and has the potential to be used as lead compounds for rational drug designing.

Similar content being viewed by others
Introduction
The indiscriminate use of antimicrobial agents has resulted in microbial resistance which has reached on alarming level [1]. This necessitates the need for discovery and development of new molecules from the natural or synthetic sources with novel mode of action to treat microbial infections [2, 3]. Cancer, one of the most dreadful diseases of the current era, is characterized by uncontrolled growth of cells. Its incidence is rising in developed as well as in undeveloped countries. Malignancy is caused by abnormalities in cells, which may be due to inherited genes or caused by outside exposure of the body to chemicals, radiations etc. [4, 5]. To date, there is no ideal cure of cancer. Multidisciplinary scientific approaches are being employed to overcome the various challenges of cancer treatment [6].
The heterocyclic thiazole nucleus reported to have various medicinal properties like anti-inflammatory [7, 8], antibacterial [9, 10], antifungal [11, 12], antitubercular [13] and antitumor [2, 14] activities etc. Thiazole nucleus exhibited its antimicrobial activity by blocking the biosynthesis of certain bacterial lipids and/or by additional mechanisms against various bacterial species [2]. Based on literature reports, it was found that thiazole ring is essential for the antimicrobial [1] and anticancer [15, 16] activities. The presence of electron withdrawing group as para-aromatic substituent on 1,3-thiazole nucleus and amide linkage within the molecule can be considered as a useful template for antitumor activity [14, 17]. Prajapti et al. and Singh et al. reported that amino thiazole derivatives with amide linkage exhibit good antibacterial and antifungal activity [12, 18]. Literature based design of the proposed thiazole molecules with antimicrobial and anticancer potential is shown in Fig. 1.

Design of proposed thiazole molecules for antimicrobial and anticancer activity based on literature
Molecular docking, a cost effective and time saving tool has become an effective and competent approach for rational drug designing. Molecular docking software’s that are available in drug research industry includes AutoDock/Vina, GOLD, FlexX, FRED, DOCK, GLIDE. Docking is an in silico screening technique for the search of suitable ligand–protein complex that fits both energetically and geometrically in the protein’s active site [19]. In light of above facts in continuation of our efforts in search for novel antimicrobial and anticancer agents [20, 21], the present study was undertaken to design, synthesize, molecularly dock and evaluate the biological potentials of N-(4-(4-bromophenyl)thiazol-2-yl)-2-chloroacetamide derivatives.
Experimental part
Materials and methods
Preliminary material required to carry out the research work was obtained from the commercial sources [Loba Chemie, Pvt Ltd. Mumbai, India Central Drug House (CDH) Pvt. Ltd., New Delhi, India] and used without further purification. Reaction progress was observed by thin layer chromatography making use of commercial silica gel plates (Merck), Silica gel F254 on aluminum sheets, using TLC mobile phase {Chloroform: Toluene (7:3)}. Melting points were determined in open capillary tubes on a Sonar melting point apparatus. Infrared (IR, KBr cm−1) spectra were recorded on a Bruker FTIR spectrometer. 1H-NMR at 600 MHz and 13C-NMR at150 MHz was recorded on Bruker Avance III 600 NMR spectrometer, using appropriate deuterated solvents (DMSO-d6). The results are conveyed in parts per million (δ, ppm) downfield from tetramethyl silane (internal standard). Proton NMR data are given as multiplicity (s, singlet; d, doublet; t, triplet; m, multiplet) and number of protons. The mass spectral data were recorded on Waters Q-TOF micromass (ESI–MS). Elemental analysis was performed by Perkin-Elmer 2400 C, H and N analyzer.
General procedure for the synthetic scheme-1
Step a: Synthesis of 4-(4-bromophenyl)thiazol-2-amine (Int.-I)
A mixture of p-bromoacetophenone (0.1 mol), thiourea (0.2 mol) and iodine (0.1 mol) was taken in a round bottom flask and refluxed for 12 h. The reaction mixture was cooled and washed with diethyl ether to remove the unreacted acetophenone and iodine. The reaction mixture was allowed to cool at room temperature, poured in the solution of ammonium hydroxide and the precipitated crude product was filtered [22].
Step b: Synthesis of N-(4-(4-bromophenyl)-thiazol-2-yl)-2-chloroacetamide (Int.-II)
Chloro acetyl chloride (0.05 mol) and few drops of triethylacetic acid were added in ethanol (30 ml) and the mixture was stirred on water bath for 10 min. The 4-(4-bromo-phenyl)thiazol-2-amine (prepared in step a) (0.05 mol) was added drop wise to above reaction mixture and refluxed for 2–3 h. The reaction mixture was then cooled and poured into ice cold water and the resultant precipitate was filtered [23].
Step c: Synthesis of N-(4-(4-bromophenyl)-thiazol-2-yl)-2-(substituted phenylamino) acetamide derivatives (d1-d9)
A mixture of N-(4-(4-bromophenyl)thiazol-2-yl)-2-chloracetamide (0.01 mol) and corresponding substituted aniline (0.02 mol) was refluxed for 7–8 h. Then the mixture was cooled, poured into ice cold water and separated product was filtered, washed with water and dried [24].
Spectral data
4-(4-Bromophenyl)thiazol-2-amine (Int-I)
1H-NMR δ: 7.19–7.56 (m, 4H, ArH), 6.90 (s, 1H, CH of thiazole), 4.12 (s, 2H, –NH2); 13C-NMR δ: 128.4, 127.4, 131.7, 124.3, 122.5, 130.2 (6C of phenyl nucleus), 167.1, 146.4, 98.6 (3C of thiazole); IR: 3113 (C–H str., phenyl nucleus), 1586 (C=C skeletal str., phenyl), 1196 (C–C skeletal str., phenyl), 666 (C–Br str., C6H5Br), 725 (C–S str., thiazole), 1632 (C=N skeletal str., thiazole), 817 (N–H str., NH2), 1265 (C–N str., Ar–NH2); Mass: m/z 256 [M++1]; CHN: C9H7BrN2S: Theoretical: C, 42.37; H, 2.77; N, 10.98; Found: C, 42.29; H, 2.57; N, 10.78.
N-(4-(4-Bromophenyl)thiazol-2-yl)-2-chloroacetamide (Int-II)
1H-NMR δ: 7.54–7.65 (m, 4H, Ar CH), 8.34 (s, 1H, NH), 7.34 (s, 1H, CH of thiazole), 3.18 (s, 2H, CH2–Cl), 7.62 (s, 1H, –CONH); 13C-NMR δ: 127.6, 130.1, 122.5, 124.2, 127.3, 132.1 (6C of phenyl nucleus), 161.4, 102.5, 151.2 (3C of thiazole); IR: 3444 (C–H str., phenyl nucleus), 1572 (C=C skeletal str., phenyl), 1199 (C–C skeletal str., phenyl), 629 (C–Br str., C6H5Br), 829 (C–S str., thiazole), 1633 (C=N skeletal str., thiazole), 817 (C–N str., Ar–NH2), 1604 (C=O str.), 746 (C–Cl str., CH2Cl); Mass: m/z 332 [M++1]; CHN: C9H7BrN2S: Theoretical: C, 39.84; H, 2.43; N, 8.45; Found: C, 39.26; H, 2.32; N, 8.41.
N-(4-(4-Bromophenyl)thiazol-2-yl)-2-((4-chloro-3-nitrophenyl)amino)acetamide (d1)
1H-NMR δ: 7.32–7.52 (m, 7H, ArH), 6.84 (s, 1H, CH of thiazole), 7.64 (s, 1H, –CONH) 3.50 (s, 2H, CH2), 3.18 (s, 1H, Ar–NH); 13C-NMR δ: 128.4, 131.1, 121.6, 127.2, 129.3, 131.2, 119.2, 129.5, 113.4, 146.0, 105.4, 143.3 (12C of aromatic nucleus), 163.2, 104.4, 149.5 (3C of thiazole), 164.7 (1C of amide); IR: 3117 (C–H str., phenyl nucleus), 1587 (C=C skeletal str., phenyl), 1671 (C=O str., CONH), 706 (C–Br str., C6H5Br), 832 (C–S–C str.), 1156 (C–N str., CONH), 1597 (N–H in plane bending, CONH), 1567 (N–O str., NO2), 757 (C–Cl str., Ar–Cl); Mass: m/z 469 [M++1]; CHN: C17H12BrClN4O3S: Theoretical: C, 43.65; H, 2.59; N, 11.98; Found: C, 42.35; H, 2.36; N, 11.67.
N-(4-(4-Bromophenyl)thiazol-2-yl)-2-((2-chloro-4-nitrophenyl)amino)acetamide (d2)
1H-NMR δ: 6.70–7.38 (m, 8H, ArH), 7.90 (s, 1H, CONH), 3.46 (s, 2H, CH2), 4.10 (s, 1H, Ar–NH); 13C-NMR δ: 128.3, 131.0, 122.7, 128.2, 129.3, 132.4, 121.1, 112.8, 148.2, 120.0, 124.3, 134.2 (12C, aromatic nucleus), 164.1, 103.4, 151.2 (3C of thiazole), 166.1 (1C of amide); IR: 3094 (C–H str., phenyl nucleus), 1588 (C=C skeletal str., phenyl), 1633 (C=O str., CONH), 647 (C–Br str., C6H5Br), 746 (C–S–C str.), 1505 (N–H in plane bending, CONH), 1561 (N–O, NO2 str.), 746 (C–Cl str., Ar–Cl); Mass: m/z 468 [M++1]; CHN: C17H12BrClN4O3S: Theoretical: C, 43.65; H, 2.59; N, 11.98; Found: C, 41.74; H, 2.17; N, 10.98.
2-((4-Bromophenyl)amino)-N-(4-(4-bromophenyl)thiazol-2-yl)acetamide (d3)
1H-NMR δ: 6.69–7.70 (m, 8H, Ar–NH), 6.61 (s, 1H, C–H of thiazole), 7.90 (s, 1H, CONH), 3.51 (s, 1H, Ar–NH), 3.33 (s, 2H, CH2); 13C-NMR δ: 128.3, 131.04, 122.7, 128.2, 129.3, 132.4, 131.2, 112.7, 130.2, 142.0, 145.1, 115.0 (12C of aromatic nucleus), 164.1, 103.4, 151.2 (3C of thiazole), 168.2 (1C of amide); IR: 3220 (C–H str., phenyl nucleus), 1585 (C=C skeletal str., phenyl), 1627 (C=O str., CONH), 631 (C–Br str., C6H5Br), 807 (C–S–C str.), 1505 (N–H in plane bending, CONH), 751 (C–Cl str., Ar–Cl); Mass: m/z 469 [M++1]; CHN: C17H13Br2N3OS: Theoretical: C, 43.71; H, 2.80; N, 8.99; Found: C, 42.74; H, 2.69; N, 8.76.
N-(4-(4-Bromophenyl)thiazol-2-yl)-2-((2-nitrophenyl)amino)acetamide (d4)
1H-NMR δ: 6.63–7.39 (m, 8H, Ar–C–H), 6.63 (s, 1H, C–H of thiazole), 7.40 (s, 1H, CONH), 4.10 (s, 1H, Ar–NH), 3.18 (s, 2H, CH2); 13C-NMR δ: 135.3, 118.1, 124.4, 129.7, 143.3, 110.4, 117.2, 122.5, 129.2, 143.1, 111.4, 131.7 (12C of phenyl nucleus), 163.2, 148.4, 104.5 (3C of thiazole), 166.3 (1C of amide); IR: 3339 (C–H str., phenyl nucleus), 1603 (C=C skeletal str., phenyl), 1622 (C=O str., CONH), 631 (C–Br str., C6H5Br), 744 (C–S–C str.), 1500 (N–H in plane bending, CONH), 1586 (N–O str. NO2); Mass: m/z 434 [M++1]; CHN: C17H13BrN4O3S: Theoretical: C, 47.12; H, 3.02; N, 12.93; Found: C, 47.10; H, 3.01; N, 12.89.
N-(4-(4-Bromophenyl)thiazol-2-yl)-2-((4-chlorophenyl)amino)acetamide (d5)
1H-NMR δ: 7.30–7.70 (m, 8H, Ar–C–H), 6.8 (s, 1H, C–H of thiazole), 7.90 (s, 1H, CONH), 3.90 (s, 1H, Ar–NH), 3.85 (s, 2H, CH2); 13C-NMR δ: 130.1, 122.4, 131.2, 128.6, 131.3, 127.4, 145.2, 113.2, 128.6, 125.7, 131.2, 113.9 (12C of phenyl nucleus), 163.2, 150.1, 104.8 (3C of thiazole), 167.7 (1C of amide); IR: 3116 (C–H str., phenyl nucleus), 1622 (C=C skeletal str., phenyl), 1671 (C=O, CONH), 662 (C–Br str., C6H5Br), 812 (C–S–C str.), 1567 (N–H in plane bending, CONH), 755 (C–Cl str., Ar–Cl); Mass: m/z 434 [M++1]; CHN: C17H12BrClN2OS: Theoretical: C, 47.12; H, 3.02; N, 12.93; Found: C, 47.10; H, 3.01; N,12.89.
N-(4-(4-Bromophenyl)thiazol-2-yl)-2-((2-methyl-5-nitrophenyl)amino)acetamide (d6)
1H-NMR δ: 7.15–7.47 (m, 7H, Ar–H), 7.18 (s, 1H, C–H of thiazole), 7.59 (s, 1H, CONH), 3.17 (s, 1H, CH2), 4.00 (s, 1H, Ar–NH), 2.36 (s, 3H, CH3); 13C-NMR δ: 131.2, 121.3, 132.0, 126.8, 129.1, 126.3, 143.1, 127.2, 129.4, 121.7, 141.3, 104.4 (12C of phenyl nucleus), 162.2, 147.1, 103.2 (3C of thiazole), 165.8 (1C of amide); IR: 3126 (C–H str., phenyl nucleus), 1596 (C=C skeletal str., phenyl), 1630 (C=O str., CONH), 647 (C–Br str., C6H5Br), 813 (C–S–C str.), 1543 (N–H in plane bending, CONH), 1454 (N–O str., NO2); Mass: m/z 448 [M++1]; CHN: C18H15BrN4O3S: Theoretical: C, 48.33; H, 3.38; N, 12.53; Found: C, 48.21; H, 3.30; N, 12.48.
N-(4-(4-Bromophenyl)thiazol-2-yl)-2-((3-nitrophenyl)amino)acetamide (d7)
1H-NMR δ: 6.96–7.46 (m, 8H, ArH), 5.79 (s, 1H, C–H of thiazole), 7.48 (s, 1H, CONH), 3.51 (s, 1H, CH2), 4.14 (s, 1H, Ar–NH), 3.51 (s, 3H, CH3); 13C-NMR δ: 122.3, 130.2, 126.2, 129.1, 132.0, 125.4, 147.1, 128.2, 105.4, 117.9, 147.3, 111.4 (12C of phenyl nucleus), 163.4, 146.5, 104.0 (3C of thiazole), 165.8 (1C of amide); IR: 3070 (C–H str., phenyl nucleus), 1471 (C=C skeletal str., phenyl), 1614 (C=O, CONH), 1590 (N–H in plane bending, CONH), 678 (C–Br str., C6H5Br), 752 (C–S–C str.), 1340 (C–N str., CONH), 1570 (N–O, NO2 str.); Mass: m/z 419 [M++1]; CHN: C17H12BrN3O3S: Theoretical: C, 47.12; H, 3.02; N, 12.93; Calculated: C, 47.06; H, 3.01; N, 12.78.
N-(4-(4-Bromophenyl)thiazol-2-yl)-2-((4-chloro-2-nitrophenyl)amino)acetamide (d8)
1H-NMR δ: 7.07–7.44 (m, 7H, ArH), 7.05 (s, 1H, C–H of thiazole), 7.54 (s, 1H, CONH), 3.18 (s, 2H, CH2), 3.38 (s, 1H, Ar–NH); 13C-NMR δ: 122.4, 131.9, 130.3, 127.4, 130.1, 124.2, 121.1, 125.4, 128.4, 141.6, 114.2, 132.5 (12C of phenyl nucleus), 161.5, 148.2, 102.1 (3C of thiazole), 167.8 (1C of amide); IR: 3095 (C–H str., phenyl nucleus), 1454 (C=C skeletal str., phenyl), 1633 (C=O str., CONH), 647 (C–Br str., C6H5Br), 763 (C–S–C str.), 1602 (N–H in plane bending, CONH), 1560 (N–O str., NO2), 722 (C–Cl str., Ar–Cl); Mass: m/z 453 [M++1]; CHN: C17H11BrClN3O3S: Theoretical: C, 43.65; H, 2.59; N, 11.98; Found: C, 43.54; H, 2.61; N, 11.76.
N-(4-(4-Bromophenyl)thiazol-2-yl)-2-((2-methyl-4-nitrophenyl)amino)acetamide (d9)
1H-NMR δ: 6.64–7.64 (m, 8H, Ar–H), 6.66 (s, 1H, CH of thiazole), 7.85 (s, 1H, CONH), 3.43 (s, 2H, CH2), 4.15 (s, 1H, Ar–NH), 2.52 (s, 3H, CH3); 13C-NMR δ: 129.3, 122.8, 130.1, 123.7, 130.1, 128.4, 135.4, 123.2, 150.7, 112.1, 118.3, 126.3 (12C of aromatic nucleus), 161.4, 145.2, 106.7 (3C of thiazole), 162.8 (1C of amide); IR: 3125 (C–H str., phenyl nucleus), 1500 (C=C skeletal str., phenyl), 1672 (C=O str., CONH), 685 (C–Br str., C6H5Br), 722 (C–S–C str.), 1599 (N–H in plane bending, CONH), 1561 (N–O str., NO2 str.); Mass: m/z 453 [M++1]; CHN: C18H14BrN3O3S: Theoretical: C, 48.33; H, 3.38; N, 12.53; Found: C, 48.17; H, 3.32; N, 12.51.
Antimicrobial evaluation (in vitro)
The antimicrobial activity of the synthesized compounds of N-(4-(4-bromophenyl) thiazol-2-yl)-2-chloroacetamide was evaluated against Gram positive bacteria [Staphylococcus aureus (MTCC3160) and Bacillus subtilis (MTCC441)], Gram negative bacterium Escherichia coli (MTCC443) and fungal strains- Aspergillus niger (MTCC281); Candida albicans (MTCC227) by tube dilution method. The stock solution was prepared for the test compounds (d1–d9) and for the standard drugs (norfloxacin and fluconazole) in acetone to get a concentration of 100 μg/ml and this stock solution was further serially tube diluted [25]. Dilution of test and standard compounds were prepared with double strength nutrient broth-I.P (antibacterial) and sabouraud dextrose broth-I.P (antifungal). The samples were incubated at 37 ± 1 °C for 24 h (bacteria), 25 ± 1 °C for 7 days (A. niger and C. albicans), respectively and results were recorded in terms of MIC.
Anticancer evaluation (in vitro)
The anticancer screening of synthesized N-(4-(4-bromophenyl)-thiazol-2-yl)-2-chloroacetamide derivatives was conducted against the oestrogen receptor positive human breast adenocarcinoma, MCF7, in comparison to a standard drug (5-fluorouracil) using the SRB assay [26]. Briefly, MCF7 cell was exposed to the compounds for 72 h. Treated cell was being fixed with trichloroacetic acid and then stained with 0.4% (w/v) SRB in 1% acetic acid. Unbound dye was removed by five washes with 1% acetic acid solution. Protein-bound dye was solubilized with 10 mM Tris base prior to reading of optical density using a computer-interfaced, 96-well microtiter plate reader. The anticancer activity results were expressed as mean IC50 value of at least triplicates.
Molecular docking study
The selected target proteins (PDB ID: 3ERT, 4WMZ and 1JIJ) required for molecular docking studies were obtained from the RCSB Protein data bank (http://www.rcsb.org/pdb/home/home.do). The selected PDB file was prepared for the molecular docking study using Protein Preparation Wizard (preprocessed, optimized and minimized). A grid is generated around the co crystallized ligand so that it can be excluded and new compounds can be attached to the same active site to study their interactions with receptor. The molecular structures of compounds that are to be docked must be in good representations of as they would appear in a protein–ligand complex. LigPrep module of Schrodinger v11.5 was used to prepare the ligand (compounds) for docking in Maestro format. The prepared ligand and receptor are docked using extra precision (XP). XP module docked the compounds with better precision and accuracy. The XP parameters like docking score, glide energy and glide emodel value were calculated within the Schrodinger v11.5 [27,28,29,30].
Results and discussion
Chemistry
Syntheses of the intermediate and target derivatives (d1–d9) were carried out as stated in Scheme 1. Initially, p-bromoacetophenone and thiourea were reacted in the presence of catalyst iodine to afford the intermediate 4-(4-bromophenyl) thiazol-2-amine (Step a, Int-I). The Int-I was further reacted with chloroacetyl chloride to generate (Int-II). The Int-II was then reacted with corresponding substituted aniline to yield the target compounds (d1–d9). The molecular structures of the synthesized compounds were confirmed by physicochemical properties (Table 1) and spectral characteristics (IR, NMR, elemental analysis and Mass spectra). The characteristic IR band at 817 cm−1 and 666 cm−1 indicated the presence of N–H str. of NH2 and C–Br str. of C6H5Br, respectively in Int-I and II. The IR stretch present at 725 cm−1 and 1632 cm−1 showed the C–S and C=N linkage, respectively. Therefore, these linkages indicated the existence of thiazole nucleus within the structure of the Int-I and II. The presence of CONH linkage in int-II was confirmed by the presence of C=O group within the range of 1680–1630 cm−1 and N–H in plane bending lie within the range of 1500–1640 cm−1. The occurrence of band at 3070–3444 cm−1 and 1505–1596 cm−1 indicated the presence of C–H skeletal and C=C skeletal structure, respectively within the phenyl nucleus. The stretch band present at 1454–1587 cm−1 showed the presence of nitro group within the compounds (d1, d2, d4 and d6–d9). The molecular structures of synthesized compounds were further confirmed by 1H NMR spectral data. The 1H-NMR spectrum of int-I showed singlet at 6.9 δ ppm, indicating the presence of NH2 group. The 1H-NMR spectra of synthesized derivatives displayed multiplet at 6.939–7.52 δ ppm, indicating the aromatic C–H linkage. The presence of singlet at 7.4–7.902 δ ppm indicated the CONH connectivity, confirming of the presence of amide linkage within the synthesized derivatives. All compounds showed singlet at 6.9–7.80 δ ppm which were due to the existence of C–H in thiazole ring. 13C-NMR spectra of the thiazole derivatives was recorded in DMSO-d6 and displayed the fine conformity of their proposed molecular structure i.e. the carbon atoms of phenyl nucleus appeared at 119.2, 127.6, 128.4, 131.2 143.32 δ ppm, carbon atoms of thiazole appearance and carbon of CONH group appeared at 167.6 δ ppm. Elemental analysis of the thiazole derivatives was within the limits of ± 0.5% of the theoretical results.

For the synthesis of N-(4-(4-bromophenyl)thiazol-2-yl)-2-chloroacetamide derivatives
Antimicrobial activity
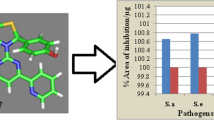
Antimicrobial screening results indicated that compound d3 (MICsa = 13.4 µM) was exhibited promising activity against S. aureus. Compound d1, on the other hand, was found to be most active against E. coli (MICec = 26.7 µM). Compound d2 was found to be the active one against E. coli (MICec = 26.7 µM) and B. subtilis (MICbs = 26.7 µM), respectively. Antifungal activity results demonstrated that compound d2 (MICca = 13.4 µM) and compound d3 (MICan = 13.4 µM) were found to be most active against C. albicans and A. niger, respectively. The results of antibacterial and antifungal evaluation were expressed as minimum inhibitory concentration (MIC) (Table 2, Figs. 2 and 3).

Graphical representation of antibacterial activity of synthesized compounds

Graphical representation of antifungal activity of synthesized compounds
Anticancer activity
Anticancer activity results demonstrated that compounds d6 (IC50 = 38.0 µM) and d7 (IC50 = 40.6 µM) were the two most active compounds against MCF7 cancer cell line (Table 2 and Fig. 4). However, the standard anticancer drug, 5-fluorouracil (IC50 = 5.2 µM), was more potent when compared to compounds d6 and d7. Compounds d6 and d7 may be used as lead molecule for further development of novel anticancer agents.

Graphical representation of anticancer activity of synthesized compounds
Molecular docking
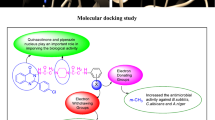
Molecular docking was performed to study the binding mode of the synthesized N-(4-(4-bromophenyl)thiazol-2-yl)-2-chloroacetamide derivatives with their respective receptors. The PDB files required was identified through literature survey. Docking studies of the most active compounds were carried out using GLIDE module of docking software Schrodinger v11.5. Docking score values were used to rank the conformations of these ligand-receptor complexes. Molecular docking study of the most active antibacterial compounds, d1, d2 and d3 and standard drug (norfloxacin) was done in the active sites of topoisomerse II (PDB ID:1JIJ) obtained from the protein data bank (Table 3). The ligand interaction diagram (2D) and pictorial presentation (3D) of docked compounds and standard drug are as shown in Fig. 5. The 2D ligand interaction diagrammatic view depicted that nitrogen and oxygen atoms of amide nucleus of compounds, d2 and d3 and oxygen atom of d1 formed H-bond with Asp40 amino acid residue. In compound d2 an additional H-bond was generated with the oxygen atom of nitro group with Gln190 amino acid residue. Molecular docking study of the most active antifungal compounds, d2 and d3 and standard drug (fluconazole) was done in the active sites of lantosterol alpha demethylase (PDB:4WMZ) obtained from the protein data bank (Table 4). The ligand interaction diagram (2D) and pictorial presentation (3D) of docked compounds and standard drug are as shown in Fig. 6. The nitrogen atom of compound d2 showed H-bond interaction with Tyr140 amino acid residue. The nitrogen and oxygen atoms of compound d3 showed H-bond interaction with the amino acid His468 and Arg385, respectively.

Pictorial presentation (3D) and Ligand interaction diagram (2D) of compounds (d1 to d3) and norfloxacin

Pictorial presentation (3D) and Ligand interaction diagram (2D) of antifungal compounds (d2 and d3) and fluconazole
The two most active anticancer compounds, d6 and d7 were docked in the binding pocket of ER-alpha of MCF7 (PDB ID-3ERT) co-crystallized with tamoxifen ligand. The results were examined based on docking score and glide energy values obtained by molecular docking study (Table 5, Fig. 7). The docking scores and glide energy values was illustrated in the negative terms. The more negative the docking score value, the better the binding affinity of ligand with the receptor. The ligand interaction diagram interpretated that nitro group of compound d6 showed hydrogen bonding with the Glu353 and Arg394 amino acid residues and the nitrogen atom of the amide group formed hydrogen bonding with the Thr347 residue. The ligand interaction diagram interpretated that nitrogen of the amide group and nitrogen atom from aniline of compound d7 formed hydrogen bonding with the Asp351 amino acid residue.

Pictorial presentation (3D) and Ligand interaction diagram (2D) of anticancer compounds (d6 and d7) and 5-fluorouracil
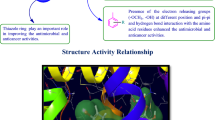
Structure activity relationship study
The in vitro antimicrobial, antiproliferative and molecular docking results demonstrated the following structure activity relationship for N-(4-(4-bromophenyl)thiazol-2-yl)-2-chloroacetamide derivatives (Fig. 8):

Structural activity relationship study of synthesized derivatives
-
Electron withdrawing group (–Br) present at para-position of ring A improved the antimicrobial and anticancer activities of N-(4-(4-bromophenyl)thiazol-2-yl)-2-chloroacetamide derivatives
-
The presence of electron withdrawing groups (–Br, –Cl, –NO2) at ortho, meta and para-position of ring B, improved antimicrobial activity against E. coli, S. aureus,
-
B. subtilis, C. albicans, A. niger and anticancer activity against cancer cell line (MCF7).
-
The presence of amide linkage with the synthesized compounds helps in H-bonding formation with the amino acid residues that leads to better fitting of compounds within the receptor and exhibited good antimicrobial and anticancer activities.
Conclusion
In conclusion, synthesis, molecular docking and pharmacological potentials of new synthesized thiazole derivatives are presented in this paper. The synthesized compounds were evaluated for in vitro antimicrobial and antiproliferative activities against using microorganisms (bacterial and fungal) and cancer cell line (human breast adenocarcinoma). Among the synthesized derivatives, compounds d1, d2 and d3 showed promising antimicrobial activity and compounds d6 and d7 displayed better anticancer activity against human breast adenocarcinoma cancer cell line. Further, the molecular docking study indicated that compounds d1–d3, d6 and d7 showed the good docking score within binding pocket and comparable to the standard drugs. The docking results are consistent with the antimicrobial and cytotoxicity assays. Docking data remain in good correlation with antimicrobial and cytotoxic activity results and these active compounds may be used as a lead for rational drug designing.
References
Khan KM, Ambreen N, Karim A, Saied S, Amyn A, Ahmed A, Perveen S (2012) Synthesis of Schiff bases of thiazole as antibacterial and antifungal agents. J Pharm Res 5:651–656
Desai NC, Somani H, Trivedi A (2013) Synthesis, antimicrobial and cytotoxic activities of some novel thiazole clubbed 1,3,4-oxadiazoles. Eur J Med Chem 67:54–59
Zhang HJ, Qin X, Liu K, Zhu DD, Wang XM, Zhu HL (2011) Synthesis, antibacterial activities and molecular docking studies of Schiff bases derived from N-(2/4-benzaldehyde-amino)phenyl-N′-phenyl-thiourea. Biorg Med Chem 19:5708–5715
Hejamdi M (2010). Introduction to cancer biology, 2nd edn, p 7
Al-omary FAM, Hassan GS, EI-Messery SM, EI-Subbagh HI (2012) Substituted thiazoles V. Synthesis and antitumor activity of novel thiazolo[2,3-b]quinazoline and pyrido[4,3-d]thiazolo[3,2-a]pyrimidine analogues. Eur J Med Chem 47:65–72
Sanghani HV, Ganatra SH, Pande R (2012) Molecular—docking studies of potent anticancer agent. J Comput Sci Syst Biol 5(1):12–15
Shamsudeen J (2014) Synthesis and evaluation of anti inflammatory activity of novel derivatives of 2 aminothiazole and oxadiazole. Int J Pharm Sci Invent 3(5):1–3
Sharma RN, Xavier FP, Vasu KK, Chaturvedi SC, Pancholi SS (2009) Synthesis of 4-benzyl-1,3-thiazole derivatives as potential anti-inflammatory agents: an analogue-based drug design approach. J Enzyme Inhib Med Chem 24(3):890–897
Cheng K, Xue JY, Zhu HL (2013) Design, synthesis and antibacterial activity studies of thiazole derivatives as potent ecKAS III inhibitors. Bioorg Med Chem Lett 23:4235–4238
Lv K, Wu Wang J, Liu M, Wei Z, Cao J, Sun Y, Guo H (2013) Synthesis and in vitro antibacterial activity of quinolone/naphthyridone derivatives containing 3-alkoxyimino-4-(methyl)aminopiperidine scaffolds. Bioorg Med Chem Lett 23:1754–1759
Liaras K, Geronikaki A, Glamocˇlija J, Ciric A, Sokovic M (2011) Thiazole-based chalcones as potent antimicrobial agents: synthesis and biological evaluation. Bioorg Med Chem Lett 19:3135–3140
Prajapti AK, Modi VP (2010) Synthesis and biological evaluation of some substituted amino thiazole derivatives. J Chil Chem Soc 55:240–243
Pattan SR, Bukitagar AA, Pattan JS, Kapadnis PD, Jadhav SJ (2009) Synthesis and evaluation of some new substituted phenylthiazole derivatives and their antitubercular activity. Indian J Chem 48B:1033–1037
Hassan GS, EI-Messery SM, Al-omary FAM, EI-Subbagh HI (2012) Substituted thiazoles VII synthesis and antitumor activity of certain 2-(substituted amino)-4-phenyl-1,3-thiazole analogs. Bioorg Med Chem Lett 22:6318–6323
Romagnoli R, Baraldi PG, Salvador MK, Camacho ME, Preti D, Tabrizi MA, Bassetto M, Brancale A, Hamel E, Bortolozzi R, Basso G, Viola G (2012) Synthesis and biological evaluation of 2-substituted-4-(3′,4′,5′-trimethoxyphenyl)-5-aryl thiazoles as anticancer agents. Biorg Med Chem 20:7083–7094
Braga SFP, Fonseca NC, Ramos JP, Souza- Fagundes EM, Oliveira RB (2016) Synthesis and cytotoxicity evaluation of thiosemicarbazones and their thiazole derivatives. Braz J Pharm Sci 52:299–307
Yurttas L, Ozkay Y, Çiftçi GK, Yıldırım SU (2013) Synthesis and anticancer activity evaluation of N-[4-(2-methylthiazol-4-yl)phenyl]acetamide derivatives containing (benz)azole moiety. J Enzyme Inhib Med Chem 29(2):175–184
Singh N, Sharma US, Sutar N, Kumar S, Sharma UK (2010) Synthesis and antimicrobial activity of some novel 2-amino thiazole derivatives. J Chem Pharm Res 2(3):691–698
Azam SS, Abbasi SW (2013) Molecular docking studies for the identification of novel melatoninergic inhibitors for acetylserotonin-O-methyltransferase using different docking routines. Theor Biol Med Model 10(63):1–16
Kumar S, Lim SM, Ramasamy K, Mani V, Shah SAA, Narasimhan B (2018) Design, synthesis, antimicrobial and cytotoxicity study on human colorectal carcinoma cell line of new 4,4′-(1,4-phenylene)bis(pyrimidin-2-amine) derivatives. Chem Cent J 12(73):1–13
Kumar S, Lim SM, Ramasamy K, Vasudevan M, Shah SAA, Selvaraj M, Narasimhan B (2017) Synthesis, molecular docking and biological evaluation of bis-pyrimidine Schiff base derivatives. Chem Cent J 11(89):1–16
Shruthy VS, Yusuf S (2014) In silico, design, docking, synthesis and evaluation of thiazole Schiff bases. Int J Pharm Pharm Sci 6(3):271–275
Naik TA, Chikhalia KH (2007) Studies on synthesis of pyrimidine derivatives and their pharmacological evaluation. E-J Chem 4(1):60–66
Kumar S, Lim SM, Ramasamy K, Vasudevan M, Shah SAA, Narasimhan B (2017) Bis-pyrimidine acetamides: design, synthesis and biological evaluation. Chem Cent J 11(80):1–14
Cappucino JC, Sherman N (1999) Microbiology-a laboratory manual. Addison Wesley, California, p 263
Skehan P, Storeng R, Scudiiero D, Monks A, Mcmohan J, Vistica D, Wareen JT, Bokesch H, Kenney S, Boyd MR (1990) New colorimetric cytotoxicity assay for anticancer drug screening. J Natl Cancer Inst 82:1107–1112
Gullapelli K, Brahmeshwari G, Ravichander M, Kusuma U (2017) Synthesis, antibacterial and molecular docking studies of new benzimidazole derivatives. Egypt J Basic Appl Sci 4:303–309
Stana A, Vodnar DC, Tamaian R, Au AP, Vlase L, Ionut L, Oniga O, Tiperciuc BS (2017) Design, synthesis and antifungal activity evaluation of new thiazolin-4-ones as potential Lanosterol 14 α-demethylase inhibitors. Int J Mol Sci 18(177):1–25
Muchtaridi M, Dermawan D, Yusuf M (2018) Molecular docking, 3D structure-based pharmacophore modeling and ADME prediction of alpha mangostin and its derivatives against estrogen receptor alpha. J Young Pharm 10(3):252–259
Kumar S, Singh J, Narasimhan B, Shah SAA, Lim SM, Ramasamy K, Mani V (2018) Reverse pharmacophore mapping and molecular docking studies for discovery of GTPase HRas as promising drug target for bis-pyrimidine derivatives. Chem Cent J 12(106):1–11
Authors’ contributions
Authors BN, DS—performed synthesis, biological evaluation and docking study of anticancer active compounds; SK- performed docking study of antimicrobial active compounds and KR, SML, SAAS and VM—performed spectral characterization and antiproliferative study of synthesized thiazole compounds. All authors read and approved the final manuscript.
Acknowledgements
The authors are thankful to Head, Department of Pharmaceutical Sciences, Maharshi Dayanand University, Rohtak, for providing necessary facilities to carry out this research work
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Not applicable.
Funding
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Sharma, D., Kumar, S., Narasimhan, B. et al. Synthesis, molecular modelling and biological significance of N-(4-(4-bromophenyl) thiazol-2-yl)-2-chloroacetamide derivatives as prospective antimicrobial and antiproliferative agents. BMC Chemistry 13, 46 (2019). https://doi.org/10.1186/s13065-019-0564-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-019-0564-0