
Abstract
Axons are the slender, cable-like, up to meter-long projections of neurons that electrically wire our brains and bodies. In spite of their challenging morphology, they usually need to be maintained for an organism's lifetime. This makes them key lesion sites in pathological processes of ageing, injury and neurodegeneration. The morphology and physiology of axons crucially depends on the parallel bundles of microtubules (MTs), running all along to serve as their structural backbones and highways for life-sustaining cargo transport and organelle dynamics. Understanding how these bundles are formed and then maintained will provide important explanations for axon biology and pathology. Currently, much is known about MTs and the proteins that bind and regulate them, but very little about how these factors functionally integrate to regulate axon biology. As an attempt to bridge between molecular mechanisms and their cellular relevance, we explain here the model of local axon homeostasis, based on our own experiments in Drosophila and published data primarily from vertebrates/mammals as well as C. elegans. The model proposes that (1) the physical forces imposed by motor protein-driven transport and dynamics in the confined axonal space, are a life-sustaining necessity, but pose a strong bias for MT bundles to become disorganised. (2) To counterbalance this risk, MT-binding and -regulating proteins of different classes work together to maintain and protect MT bundles as necessary transport highways. Loss of balance between these two fundamental processes can explain the development of axonopathies, in particular those linking to MT-regulating proteins, motors and transport defects. With this perspective in mind, we hope that more researchers incorporate MTs into their work, thus enhancing our chances of deciphering the complex regulatory networks that underpin axon biology and pathology.
Similar content being viewed by others
Introduction
Axons are the slender, cable-like extensions of nerve cells which form the nerves and nerve tracts that wire our brain and body, sending neuronal messages in highly regulated manners. With diameters of only 0.1-15μm [1], they extend over distances of up to a meter in humans. To adopt such a unique morphology and physiology, axons display many specialised features (Fig. 1).
Axons are indispensable for nervous system function, as illustrated by paralysis in spinal cord injury caused by the interruption of ascending and descending axon tracts [2, 3]. Axons are key lesion sites in injury-induced trauma and coma [4,5,6,7], and axon decay is believed to be an important trigger for neuronal decay in ageing and many neurodegenerative disorders [8, 9]. Notably, most neurons cannot be replaced, and compensation of lost axons through collateral branching of intact neighbouring axons has obvious limitations [9, 10].
This means that most axons have to be maintained for an organism's life time, i.e. up to a century in humans; unsurprisingly, mammals tend to lose almost half their axon mass towards high age [11, 12]. This trend is severely enhanced in neurodegenerative disorders, as illustrated by gradually increasing paralysis in spastic paraplegia or motorneuron disease [13, 14].
Research into neurodegenerative disorders typically approaches the problem by describing observed phenotypes and unravelling the molecular mechanisms performed by proteins linked to the disease. However, this approach rarely leads to satisfactory explanations of the pathology [15]. We believe that more profound understanding will arise when widening the scope from molecular to cellular mechanisms, by studying how proteins work within regulatory networks to underpin observable processes of axon biology - thus performing investigations at the same level of complexity at which pathology becomes manifest. Here we will illustrate this approach by focussing on the axonal cytoskeleton.
The importance of microtubule bundles for axon biology
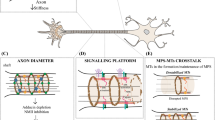
As illustrated in Fig. 1, the cytoskeleton of the axon shaft consists of straight parallel bundles of MTs, which are interspersed with intermediate filaments (not shown [16]) and longitudinal actin fibres called 'actin trails' [17, 18] - all running through a sleeve of cortical actin [19] which is now known to consist of evenly spaced periodic rings; these rings have been proposed to consist either of short and adducin-capped actin filaments [20, 21] or of two long intertwined actin filaments [22]. Significant deviations from this organisation, not to be considered in this review, exist at axon initial segments (not shown in Fig. 1), growth cones and synapses [23,24,25,26].

Specific properties of axons. Axons during the growth cone stage are shown in (a) and after synaptic maturation in (b), differing primarily in certain stage-specific specialisations including growth cones, synapses, electrical properties and glial interactions (here myelination [389, 392]). The core machinery in the axon shaft can be expected to be similar at both stages: parallel continuous bundles of extended but discontinuous MTs run all along axons serving as a structural backbone (see Fig. 2), a transport highway for axonal trafficking (driven by motor proteins), and a source for 'off-track' MTs contributing to morphogenetic processes including branch formation, directed axon growth and synapse formation/plasticity (green, orange, blue curved arrows); MT bundles are interspersed with longitudinal actin trails [18, 24], continuous networks of (smooth) endoplasmic reticulum [44, 393], and other membranous organelles including mitochondria [45]; axonal membranes display regularly spaced periodic rings of cortical actin [20, 21], a high number of ion-specific channel proteins and transporters to conduct nerve impulses [394], as well as adhesions with external structures including fasciculating parallel axons (not shown), glial processes [395] and synaptic partner cells [396]; a degree of independence from cell-body derived proteins is provided by local translation machinery [397,398,399] or supply from surrounding glia cells (not shown; [400,401,402,403]). Note that the axon diameter in the region between glia cells in B (referred to as Node of Ranvier) usually has a much smaller diameter than the rest of the axon [1]
Of the three cytoskeleton classes, intermediate filaments were suggested by anatomical, developmental and genetic studies to regulate axon diameters, and their axonal aggregation is a hallmark of many neurodegenerative diseases [1, 27,28,29,30]. However, intermediate filament aggregations are not necessarily the cause, but can be the consequence of axon decay [30,31,32]. Notably, Neurofilament-H-lacZ mutant mice or Quiver mutant quail completely lack axonal intermediate filaments, but develop and breed fairly normally [33, 34]. Furthermore, various arthropods form axons of defined diameters in the absence of any axonal intermediate filaments [35,36,37]. In contrast to the moderate roles of intermediate filaments, actin and microtubules (MT) are essential for all stages of neuronal development and maintenance [37,38,39]. This review will be dedicated to the role and regulation of MTs.
Axons contain bundles of MTs that run along the entire length of their shafts (Fig. 1); these bundles are essential for axon biology in at least three ways (details in Table 1): as structural backbones (Fig. 2), as highways for axonal transport and organelle dynamics, and as source for splaying MTs that can contribute to axon morphogenesis or physiology. Maintaining MT bundles is therefore crucial for axon longevity. Accordingly, there are prominent and numerous genetic links from MT regulators to hereditary neurodegenerative disorders (Suppl. Mat. in [46]), and axon decay is a frequent side effect of MT-targeting chemotherapies [53,54,55,56].

Axonal response to mechanical challenges. Continuous bundles of discontinuous MTs which are flexibly cross-linked (likely involving slip-bonds) are thought to provide a structural element that can respond to different forms of mechanical impact (as indicated in blue)
Of particular interest for this review are reports of pathological axon swellings where MT bundles have disintegrated into loops or waves (bottom of Fig. 3), as observed in ageing, after injury and in certain in vivo models of axonopathies [7, 34, 57,58,59,60,61,62,63,64,65,66]. Notably, one study suggests that MT aberration upon ageing could cause swellings that trap and damage mitochondria, thus triggering axon degeneration [67]. However, MTs are surprisingly often ignored or side-lined in existing studies of axon pathology and there are simply not enough data to deduce meaningful correlations between axon degeneration and MT bundle decay. But even if this were to reveal a close correlation, this still would not exclude that, depending on the pathological condition, MT bundle deterioration may be a mere consequence rather than cause of axon decay (details in Fig. 4). Ultimate clarification will only arise from developing a better understanding of MT bundle-forming and -maintaining machinery. Here we propose a conceptual framework that may facilitate such developments.

An interactome of MT-regulating and -associated mechanisms expected to contribute within the model of local axon homeostasis. Developing and mature neurons are shown at the bottom indicating that the close-up (magenta frame) might apply in both contexts. 1-16) Potential mechanisms that can 'tame' MTs into bundled conformation: MT polymerisation (blue stippled arrows) is driven by molecular machinery centred on Eb1 (blue balls), further influenced by the tubulin supply machinery (not shown) and shaft-binding proteins (7); polymerisation generates new MTs required for bundle formation (8) and turn-over (14); to integrate into bundles, extending MTs require guidance via actin-Eb1 cross-linkage along the axonal surface (5; Shot) or along pre-existing MTs through MT-MT cross-linkers (9; brown L). The same or other cross-linkers provide the structural glue that holds MT bundles together (12; brown L); some of them can also bind to actin (2), they protect from (or recruit) MT severing activity (15), and influence motor protein dynamics (a). MTs which have escaped any cross-linkage are inhibited by cortical collapse factors when approaching the axonal surface (4; Efa6) or by MT-severing factors at MT-MT cross-points (6). The bundled MTs are discontinuous; their free minus ends are stabilised by CAMSAP/Patronin (Ptrn) together with katanin (black scissors; 13), whereas non-polymerising MT plus ends are stabilised by other factors (not shown; e.g. CLASP or the Dynactin subunit p150/Glued [404, 405]). The dynein/Dynactin complex is believed to link cortical actin to MT bundles and drive them anterogradely (10), whereas Ptrn at minus ends may anchor MTs via spectraplakins to the axon cortex (1); spectraplakins may also link MTs directly to cortical actin (2) or to transmembrane receptors (3), and they are expected to perform further, still unexplored actin-independent bundle-promoting roles through their PRR domains (11). Tear-and-wear damages MTs (dashed green line), potentially affecting interaction with MT-binding proteins (16; red X); MT severing proteins might selectively eliminate such MTs (16; scissors), or MTs undergo repair (not shown). Nucleation of MTs (17) is mediated by ɣTuRC directionally anchored to MT lattices via the augmin/HAUS complex (AUG). A-E) Mechanisms closely 'associated' with MT bundles: MT-associated motor proteins ('motor', solid orange arrows) drive axonal transport of (protein-loaded) vesicles (A), cytoplasmic factors including proteins, translational machinery (ribosomes) or RNAs (B), move other MTs (B, sliding), and position/rearrange organelles including mitochondria (C, mitos), endoplasmic reticulum, peroxisomes and endosome (D) - and this likely includes mitochondrial fission and fusion (E). a-e) The motor-associated functions all act downstream of MT bundles because they require them to walk on; but they also act upstream: for example, the forces they generate (stippled orange arrows) are the potential cause for MT disorganisation (buckling shown in d); transport delivers important regulators and building blocks for bundle-maintaining processes (b); the proper regulation of organelles/endocytic compartments provides systemic factors that can orchestrate MT bundle-taming mechanisms, including intracellular free calcium or reactive oxygen species (Ca2+, ROS; yellow cloud [202, 203]) as well as ATP required for many processes including actin dynamics, MT severing and MT motor activity (red stippled arrows; note that vesicular transport uses glycolysis to generate its own ATP; yellow star); vice versa, the MT severer spastin also regulates the ER through ATP-independent mechanisms (e), and MT-associated proteins (APC) regulate local translation events (c)

MT bundle defects as cause or consequence of axon decay. 1) Disease-inducing mutations/conditions can affect a MT bundle regulator (e.g. dystonin [90]), thus causing MT bundle defects first which, in turn, can trigger axon decay. 2) Disease-inducing mutations/conditions can affect systemic factors which, in turn cause MT bundle defects as an intermediate causative step in the cascade leading to axon decay (e.g. axonal transport fails, leading to MT bundle defects which then contribute to axon decay, as is the case in Alzheimer's disease or ALS [302, 406, 407]); this may occur even if MT regulators are affected, but these regulators mainly act in the cell body (e.g. dysregulation of the Golgi [408]). 3) MT bundle deterioration may be a mere consequence of axon decay, although this case will be difficult to disentangle from option 2, since MT bundle disintegration and axonal disassembly may occur in parallel, as observed in developmental or injury-induced axon degeneration [409,410,411]). All MT-related phenotypes in this graph are indicated with a frame
From work in Drosophila to the integrated model of local axon homeostasis
The foundations for this conceptual framework were laid when we took the decision to use the fruit fly Drosophila melanogaster as a means to study how cytoskeletal regulators collaborate in orchestrating the morphogenetic changes that drive axon growth [68]. Drosophila is certainly not a miniature human, but it has many advantages and provides powerful means to uncover the regulatory concepts behind the roles and regulations of axonal MTs, which then often apply to higher organisms (Table 2; [15, 69,70,71]). Through using Drosophila neurons as a consistent, standardised cell system, our group alone performed functional analyses of over 50 actin- and/or MT-binding or -associating regulators ([46]; unpublished data); these studies form an unprecedented pool of data on the basis of which to develop novel concepts [21, 72,73,74,75,76].
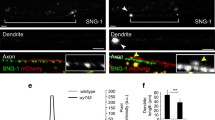
For example, loss-of-function conditions of 24 MT-binding or -associating (2nd order) proteins that we analysed in cultured primary neurons, revealed MT disorganisation in more than half the cases. Interestingly, the MT disorganisation found in these various conditions appears to display certain common characteristics: axons display areas in which their bundles are dissolved into chaotic, intertwined, crisscrossing arrangements of curled MTs (see examples in Fig. 5). These phenotypes were surprising when considering that MTs usually behave like rigid rods [84,85,86]. Notably, when using some of the same genetic conditions in vivo, comparable phenotypes were observed in the fly brain [74]. Such in vivo phenotypes in the fly are reminiscent of the curled MT conformations in pathological axon swellings of mammalian models mentioned in the previous section. Potential evolutionary conservation of this phenomenon is further supported by the occurrence of similar MT curling and disorganisation in mouse and rat primary neurons [87, 88] - and we are certain that more reports will emerge once researchers consider MT disorganisation to be a phenotype worth quantifying.

Disorganisation of axonal MTs upon loss of different MT regulators in Drosophila primary neurons. a Normal neuron (wild-type, wt) with soma (asterisk), axon shaft (curved arrow) and growth cone (tip of most distal MT indicated by arrow head). b Eb15 mutant neuron where the area of MT disorganisation is framed by a red stippled box and shown as close-up on the right. c-e Similar close-ups shown for Efa6GX6[w-], Khc27 and shot3 mutant neurons. Note that the four mutated factors perform fundamentally different molecular functions, with Eb1 being a MT plus-end binder ('8' in Fig. 3), Efa6 a cortical collapse factor ('4' in Fig. 3), Khc a kinesin-1 motor protein ('A-E' in Fig. 3) and Shot a multi-functional cross-linker ('1-3, 5, 11' in Fig. 3). All neurons were derived from wild-type or homozygous mutant embryos, mechanically and chemically dissociated, kept for 7 days in pre-culture in a centrifuge tube to deplete any maternal gene product, mechanically and chemically dissociated again, cultured on concanavalin A-coated glass coverslips for 1day at 21°C, fixed and stained with anti-α-tubulin (DM1A, Sigma; procedures detailed elsewhere: [78]); images were taken by A.V. using STED (stimulated emission depletion) microscopy. Scale bar in A represents 10 μm for the two neurons and 4 μm in close-ups
As an attempt to explain the occurrence of this MT phenotype across mutant conditions and animal groups, we developed the model of 'local axon homeostasis' [37, 89], based on two fundamental elements:
(1) The model proposes that MTs in axons show a strong bias to become disorganised and curl up. As detailed further below, this is most likely induced by the force-generating motor proteins that drive transport of large cargoes in the narrow axonal environment crowded with physical obstacles posed by organelles and protein complexes (Fig. 1, 'A-E' in Fig. 3). Once MT disorganisation occurs, e.g. through off-track polymerisation or buckling ('4' and 'd' in Fig. 3), it can form a seed that gradually develops into pathological axon swellings.
(2) The model further proposes that this risk is contained through the actions of different classes of MT-associating and -regulating proteins, which co-operate and complement each other to form robust machinery that 'tames' MTs into bundles ('1-17' in Fig. 3).
In this model, each axon segment uses locally acting MT regulators to maintain its MT bundles (hence 'local axon homeostasis'). Hereditary or acquired loss of single regulators would be expected to weaken this machinery and increase the statistical risk of MT disorganisation. Such heightened probability might explain why many axonopathies affect primarily long axons [54], and why certain disorders linked to MT regulators display late onset of axon decay [90].
In the next two sections, we discuss potential causes explaining the bias of axonal MTs to become disorganised. We will then summarise experimentally demonstrated MT bundle-maintaining mechanisms, and speculate about further mechanisms based on existing knowledge of known classes of axonal MT-regulating proteins.
Understanding the unusual curling behaviours of MTs in axons
Although curvature is a key driver of MT plus end dynamics during de-/polymerisation [91, 92], MT lattices in vitro usually behave as rigid rods with a persistence length of 1-10 mm (as compared to ∼12 μm measured for actin filaments [84,85,86]). MTs are polar polymers composed of α/ß-tubulin heterodimers which are arranged in a head-to-tail fashion into linear protofilaments; usually 13 of these protofilaments are laterally aligned forming a straight tube of roughly 25 nm outer diameter (Fig. 6a, c). But MTs can deviate from this norm, and this may be one factor introducing an intrinsic bias towards disorder: for example, axonal MTs were reported to contain 13 protofilaments in frog olfactory or goldfish brain axons, but 11 or 15 in C. elegans, and 12 in certain neurons of Drosophila, crayfish and lobster [66, 93,94,95]. Deviation from the straight 13 protofilament conformation appears to equip MTs with functionally relevant physical properties [96, 97]. But it also introduces a skew into the MT structure, which causes a supertwist of the tubule (Fig. 6d [98,99,100]); this supertwist forces motor proteins to rotate around MTs [101] and is a potential explanation for supercoil of entire axons observed under destabilising conditions [83, 102].

A molecular perspective of microtubule properties. a Cross-section of a MT with 14 protofilaments (PF) and lateral view of a 13 PF MT, both in B-lattice configuration, where α-tubulins make lateral bonds with α-tubulins and ß with ß, except at the seam (magenta line: seam; dashed red line: PF). b Close-up of an α/ß-tubulin heterodimer showing the various post-translational modification sites as indicated; note that the GTP of ß-tubulin in lattices is usually hydrolysed (GDP). c A 13 PF MT (top), cut open at the seam and rolled out (bottom); the yellow line shows the diameter, the white line follows the helical rise of laterally bonded tubulins; in 13 PF MTs, tubulins are precisely aligned at the seam (yellow arrow head) but shifted by three positions (3-start helix). d When deviating from the 13 PF prototype, tubulins are misaligned at the seam (orange arrow head); when forced into alignment, the PFs skew (deviation of the magenta line from the white stippled line), causing a super-twist of the MT as described by the 'lattice accommodation model' [98, 412]; for certain PF numbers, MTs can form two alternative alignments, of which usually the version with the lower helix start value (left) has a left-handed super-twist, whereas the higher value is right-handed [98]. e MTs behave like rigid rods with a persistence length of up to 10 mm, but can be bent down to diameters of curvature of ~1μm before they break; it has been reported that their cross-sectional profile may flatten above a certain threshold (black arrow head), thus softening the tube. f Lattices of GDP-tubulin are 1-3% shorter than MTs that were polymerised with the non-hydrolysable GTP analogue GMPCPP, or stabilised with taxol (orange structure binding α-tubulin in a 1:1 ratio, according to [413]); binding of kinesin-1 causes similar lengthening of tubulin (and additional compactions in the tubulin structure: yellow stars) which may cause cooperative binding of further kinesins and induce curvature if occurring only on one side of the MT; in extended taxol-bound MTs, bending forces were suggested to change tubulins on the concave side into their short conformation as an energetically favoured condition. For further references see main text
Furthermore, MTs are structurally active: their physical properties can change when proteins bind to them (e.g. kinesins, see below) or when the 'tubulin code' is altered. The tubulin code is determined by the incorporation of different existing isotypes of α- and ß-tubulin into the MT lattice, and the addition of a range of distinct post-translational modifications (Fig. 6b [103,104,105,106]). Some modifications influence the interaction with MT-binding proteins (e.g. poly-glutamylation attracts spastin and tau [107, 108]). Others are believed to structurally protect MTs from damage or depolymerisation: for example, poly-aminations on various residues stabilises against depolymerisation [109], or acetylation of luminal lysine 40 makes MTs more flexible and break-resistant (Fig. 6b; [110,111,112,113,114]). Notably, site-directed mutation of lysine 40 in Drosophila α1-tubulin was able to demonstrate that intraluminal MT acetylation is physiologically relevant [115, 116]. In addition, the MT lumen may contain MIPs (MT inner proteins) that likely also modify MT stability [117].
These intrinsic or acquired physical properties are likely to determine how MTs respond to external forces - and we can expect such forces to be highly enriched in axons (see next section). Some ideas about how forces may impact on axonal MTs can be derived from in vitro experiments. For example, MTs in flow chambers that are anchored at one end, will bend when applying flow and rapidly return to straight confirmation when flow is stopped; if certain shaft-binding proteins (e.g. doublecortin or non-motile kinesin-1) are added, MTs become locked in bent conformation and fail to re-straighten [118,119,120].
Another example is provided by so-called in vitro gliding assays, where MTs are moved around on carpets of active motor proteins. On carpets of (axonemal) dynein, MTs move plus-end-first; they undergo collisions at high frequency, but seem to stay fairly straight and form vortices at the millimetre scale [121]. In contrast, if similarly prepared MTs are on kinesin carpets, they move minus-end-first and undergo fewer collisions because they can pass over one another, likely owed to the adaptable length and dynamic MT binding properties of kinesins [121,122,123,124]. However, if they collide or become pinned to the substrate (e.g. by dead kinesins) they can undergo dramatic shape changes including fishtailing and arc or loop formation at the micrometre scale [125,126,127]. The smallest diameters of curvature observed are similar to those of curled MTs in axons with values as low as 1-3 μm (Table 3, Fig. 5; [87, 88]) - and below 1μm MTs are believed to break [128, 129].
If MTs on kinesin carpets are reversibly cross-linked with biotin-streptavidin, they coalesce into bundles containing dozens of MTs which frequently curl up into spools with inner diameters similar to those of loops (details in Table 3). Spools can take on similar appearances as looped MT bundles observed in growth cones of fly or mammalian neurons [77, 130, 131]. Furthermore, single MTs can escape from spools which may trigger spool disassembly [130, 132, 133], bearing some resemblance with off-track MTs leaving axonal bundles ('4' in Fig. 3).
Parameters known to influence loops and spools in vitro might provide mechanistic insights into similar behaviours of MTs in axons (details in Table 3). Firstly, loop formation is favoured by high density of MTs and/or kinesins [134, 135], i.e. conditions that are clearly given in axons. Potential explanations are offered by reports that kinesins directly impact on the structural properties of MTs (see below), but they can also cause pinning events in gliding assays, which could be seen as a potential proxy for the abundant obstacles faced by extending or sliding MTs in the narrow axons. Secondly, higher MT rigidity results in larger diameter curls and spools in gliding assays [136]. Thirdly, right- versus left-handed supertwist of the MTs involved in gliding assays determines whether curls and spools have a clockwise or counter-clockwise directionality (Fig. 6d; [132, 137]). Furthermore, exposure to non-polar interfaces (e.g. n-heptane or air bubbles) induces strong curling [138], and this may be relevant in axons: changes in physical and chemical parameters of neurons upon ageing or in degenerative diseases promote liquid-liquid phase separation [139]; liquid compartments likely are of low polarity [140] and might therefore influence the curling bias of MTs. All these parameters observed in vitro can be expected to apply also in axons and might contribute to the observed curling behaviours (Fig. 5).
MT loops in gliding assays can be surprisingly stable (frequently >5 mins, as reported in [134]). To explain this, it has been proposed that tubulin-heterodimers on the concave side of the tube take on a shorter conformation than those on the convex side, and that this asymmetric distribution can be maintained as an energetically favoured state (Fig. 6f, bottom right [141]). Conformational length variations underlying this model were observed in non-hydrolysed GMPCPP-MTs where tubulins are 1-3% longer than hydrolysed GDP-tubulin; taxol added after (but not during) polymerisation achieves a similar elongation (Fig. 6f; [142,143,144,145,146]). Notably, this conformational length change seems physiologically relevant, as its suppression by the T238A mutation in yeast ß-tubulin stabilises MTs in vivo and causes mitotic defects [147, 148].
Such intrinsic properties of MTs may contribute to MT curling in axons, further influenced by MT lattice-associating proteins, such as tau, doublecortin or kinesin-1 which were reported to bind differently to curved versus straight MTs [118,119,120, 149,150,151]. In particular kinesins-1 was shown to extend MT lattices to similar degrees as taxol [120] through mechanisms that involve local compaction of tubulin different from taxol- or GMPCPP-induced effects [152, 153]. Since kinesin-1 has a preference for convex MT surfaces and was reported to undergo cooperative binding, this may lead to a curvature-enhancing and -stabilising snowball effect with an estimated diameter of curvature of 3.2 μm [120, 154, 155]. In this way, kinesin carpets in gliding assays might induce stable yet reversible curling, as has been suggested by mathematical modelling (top right in Fig. 6f, [156, 157]).
Naturally, current models are in their infancy and further findings need to be incorporated. For example, MTs behave as elastic cylinders (comparable to a garden hose) and can undergo softening through cross-sectional flattening when strongly bent (Fig. 6e [158, 159]). In this same vein, conformational changes of MTs upon kinesin-1 binding were reported to soften MTs locally [160]. If confirmed, this would have important implications for any existing models; together with the kinesin-induced tubulin compaction (yellow asterisks in Fig. 6f), it might be a mechanism to absorb energy and reduce the shear force load on MTs. Notably, softening of MTs is also observed upon taxol application (usually used in gliding assays; Table 3 [144]) or MT acetylation (abundant in axons [111, 114]), and might be a common prerequisite for curling behaviours.
To conclude, loop and spool formation in gliding assays are considered processes of 'active self-organisation' [125]. We strongly feel that this term might apply also to the formation of MT disorganisation in axons, and that potential mechanisms underlying MT curling in axons can be learned from in vitro assays. Notably, motor proteins, in particular kinesins, are being highlighted as key factors in both gliding and flow chamber assays. In the next section we will therefore summarise roles of kinesins during axon pathology.
The intricate relationship between MTs and their associated motor proteins
Several kinesins display direct roles in MT regulation [161]: they may promote MT polymerisation (kinesin-2, -5 [162,163,164]), drive MT depolymerisation (kinesin-8, -13 [165]), stabilise MT-minus ends (kinesin-14 [166]), cross-link MTs (kinesin-5, -6, -12; see section on bundling), and regulate MT orientation as a feature of neuronal polarity [167,168,169].
However, in axons most attention is usually given to cargo and organelle transport/dynamics (Fig. 3a-e; see section on axonal cytoskeleton) driven retrogradely by the minus end-directed dynein/Dynactin complex, and anterogradely by plus-end directed kinesins (primarily kinesin-1, -2, and -3 [40, 170]). The forces imposed by these dynamics and/or the size of cargoes moved in the constrained environment of axons rich in physical obstacles, poses an obvious challenge to MT bundles [171] and might be an important factor leading to MT disorganisation.
Clearly, there is an intricate mutual regulatory relationship and finely tuned balance between the amount of transport and the structural properties of MT bundles as the transport highways [23, 171]. Thus, MT bundle properties influence transport: firstly, MT density is higher in small calibre axons than in large axons, with MT numbers and densities ranging to enormous degrees (4-130 MTs per axon, ~4-150 MTs/μm2); correlative studies and mathematical modelling suggest that higher MT numbers promote axonal transport ([172,173,174] and references therein). How MT numbers are so precisely controlled is an important but entirely unresolved issue that likely involves the nucleation machinery (see section on nucleation/polymerisation). Secondly, MT length correlates with transport rates [81]. Thirdly, the tubulin isotype composition of MTs, their posttranslational modifications, and the physical presence of certain MT-binding proteins influence motor protein dynamics ('a' in Fig. 3; [149, 175,176,177,178,179,180]).
Vice versa, transport affects MT bundles: for example, binding of kinesin changes the physical properties of MTs (see previous section), and binding and buckling through motor proteins cause damage to the MTs they walk on, triggering maintenance responses including MT repair or potentially even replacement ('14' in Fig. 3; [120, 181,182,183,184,185]). Close links between MT organisation and transport are also illustrated by charge-changing mutations in the H12 helix of C. elegans α-tubulin, reported to impact on axon transport whilst causing MT bundle aberrations [66].
Tipping the balance in this mutual relationship can easily be imagined to cause reciprocal deficiencies in transport rate and MT bundle organisation. For example, disorganisation or partial breakage of MTs has been reported to cause pathological transport deficits (option '1' in Fig. 4; [7, 67]). Furthermore, the space required for large cargo movements is likely generated through dynamic rearrangements of local MT-MT crosslinking networks (see section on cross-linkage); in this scenario, deviating from the right amount of cross-linkers may be a path to bundle aberration. Vice versa, demyelination upon immunological lesioning [186, 187], was reported to initially cause transport defects, which were then followed by MT disorganisation ('2' in Fig. 4; [60]). Analogously, we observe that loss of certain transport kinesins (Kinesin heavy chain/Khc/Kif5A or B, Unc-104/Kif1A) causes severe MT disorganisation in Drosophila primary neurons (Y.T.L. and A.V., unpublished data; Khc shown in Fig. 5e).
How loss of these kinesins may cause MT disorganisation can currently only be hypothesised. There are potential biomechanical and/or biochemical explanations. For example, it has been reported for dendrites that kinesin-1 migrates on acetylated and kinesin-3 on tyrosinated MTs [167]. Provided the same is true in axons, the loss of kinesin-1 would relieve acetylated MTs, but tyrosinated MTs would still bear their full transport load - and vice versa. Such imbalances in transport distribution within MT bundles could lead to shear forces that buckle MTs and seed MT disorganisation. Similar mechanisms may explain why MT disorganisation was observed at the axon initial segment upon directional changes in motor traffic caused by deficiency of the dynein regulator NDEL1 [188].
Loss of kinesins could have impact on MTs also through biochemical routes. For example, the bundle-maintaining machinery may simply suffer from aberrant supply of cargoes including (a) tubulin heterodimers as building blocks, (b) the MT-binding proteins that execute MT bundle maintenance work ('b' in Fig. 3), or (c) organelles which can be expected to play major roles in MT bundle maintenance (see Table 4 for details).
Functional interdependencies between transport and MT organisation provide potential explanations for a number of observations. For example, they may explain why axonal swellings induced by senile plaques in the Tg-swAPPPrp mouse (overexpressing an amyloid precursor protein carrying a familial Alzheimer's disease-linked mutation [189]) were strongly enhanced when removing one copy of the KLC1 gene (kinesin light chain; a linker required for kinesin-1 mediated veciscular transport) - and this effect is conserved in Drosophila [190]. They may explain why different types of Charcot-Marie-Tooth disease or hereditary spastic paraplegias can be caused through motor proteins as well as regulators of membranous compartments [191, 192]. They may also explain why MT-stabilising drugs can be beneficial in animal models of neurodegeneration as diverse as SPG4 (Table 4) and Alzheimer's disease [193, 194].
Naturally, the argumentative framework presented here is highly speculative, given the enormous complexity of the relationships between MT bundle organisation, motor protein activity and organelle-dependent systemic factors. But we hope that these reflections will motivate experimenters to have a closer look at MTs in future studies of axon biology and pathology, and include statements in their reports as to whether MTs are affected. More data are urgently needed, which does often not require more than analysing neuronal morphology with antisera against tubulin (rather than restricting to intermediate filaments), or having a closer look at MTs in ultrastructural studies by increasing the resolution. In the following sections we will explore the mechanisms that are potentially used to form and maintain MT bundles against the odds of motor-induced aberration or damage.
MT nucleation and polymerisation as fundamental requirements for bundle maintenance
The de novo formation of MT bundles during developmental, plastic or regenerative axon growth ('8' in Fig. 3) requires MT nucleation and polymerisation. Also in axons of mature and fully grown neurons, MTs undergo nucleation and polymerisation [37, 218], for example to drive MT repair and/or turn-over in order to maintain a steady state and prevent MT senescence ('14' in Fig. 1; [183, 184]). A well-regulated machinery of MT nucleation/polymerisation and disassembly is therefore needed to keep the numbers of healthy axonal MTs in balance with the transport load (see previous section; [172]).
Mechanisms of MT nucleation have long been known to be independent of centrosomes [219, 220] and should therefore involve cytoplasmic assembly or non-centrosomal MTOCs (MT organising centres; [221, 222]). For example, tau was reported to form condensations on MTs in vitro [179, 180], and such condensed phases of tau could theoretically have nucleation capacity [223, 224]. Furthermore, new MTs could arise from MT fragments (see Section on severing proteins below), potentially anchored via CAMSAP (calmodulin-regulated spectrin-associated protein)/Patronin to polymerise in the accurate direction towards the axon tip [113, 225]. Best demonstrated so far are mechanisms dependent on ɣTuRC (ɣ-tubulin ring complexes) and their anchorage via augmin/HAUS complexes to MTs ('17' in Fig. 3): depletion of either ɣ-tubulin or different HAUS proteins causes severe axon shortening and reduction in MT density; in addition, HAUS depletion causes polarity defects reflected in frequent MT polymerisation events towards the soma [226,227,228], suggesting that regulated nucleation is doubly important for axonal MT bundle maintenance. As the underlying mechanism it has been proposed that the augmin/HAUS complex anchors ɣTuRC to other MTs and points them distally ('17' in Fig. 3), and such a view is consistent with live imaging in Drosophila S2 cells [229].
The machinery of MT de-/polymerisation (blue stippled arrows in Fig. 3) requires at least three sub-machineries [37]: (1) dynamic protein complexes at the MT plus end that directly regulate polymerisation (blue balls, 'Eb1' in Fig. 3); (2) a complex regulatory network that supplies mature α/β-tubulin heterodimers as building blocks and that is closely co-regulated with MT dynamics ('B' in Fig. 3; [230,231,232]); (3) proteins binding or post-translationally modifying MT lattices that have impact on plus end dynamics, for example by stabilising MTs against depolymerisation or by promoting rescue ('7' in 'Fig. 3).
The fine-tuning of the net rates of MT nucleation and polymerisation appears to depend on complex regulation. For example, we recently found that loss of cortical actin rings in the axon shaft of Drosophila primary neurons (Fig. 1) caused a reduction in MT polymerisation speed, eventually affecting MT bundle integrity; simultaneous genetic or pharmacological destabilisation of MTs exacerbated these effects, frequently even eliminating entire axons [21]. Similar dependencies of MT polymerisation on actin networks are suggested by other reports: (1) parallel loss of spectrin and tau causes a reduction in axonal MT numbers in C. elegans [83]; (2) axon-shortening induced by the MT-stabiliser taxol can be ameliorated through co-application of actin-destabilising drugs (in both chick and Drosophila neurons [77, 233]); (3) application of actin-destabilising drugs changes the tubulin-to-microtubule ratio in PC12 cells [234] and causes axon retraction in chick dorsal root ganglia neurons ([235]; see also Table 5). Explanations for the mechanistic links from actin networks to net MT polymerisation remain speculative: they might involve biochemical pathways since cortical actin rings have recently been reported to act as signalling hubs [236], or might work through biomechanical mechanisms (see Table 5).
Maintaining MT bundles through cortical guidance and elimination of polymerising MTs
Whilst MT nucleation and polymerisation are essential for axon formation and maintenance, they also pose a risk: for example, extending MTs may be obstructed by the abundant organelles or protein complexes in axons, thus causing accidental 'off-track' MTs that project out of the bundle towards the cortex ('4' in Fig. 3). Apart from MT buckling, off-track MTs may therefore be a second cause for axonal MT disorganisation.
A key factor preventing this from happening is Eb1 (end binding protein 1; Figs. 3 and 5b; [75]). Eb1 directly binds at extending MT plus ends where it promotes polymerisation [237] and serves as a scaffold for many other proteins [238]. Upon absence of Eb1 in Drosophila primary neurons, MTs are severely disorganised, indicating important roles in MT bundle maintenance (Fig. 5B [75]). One underlying mechanism is the guidance of polymerising MTs through binding of Eb1 to Short stop (Shot); Shot is a well-conserved spectraplakin, able to cross-link cortical actin, MTs and Eb1 ('5' in Fig. 3), thus guiding polymerising MTs in parallel to the axonal surface and laying them out into parallel bundles [75]. Accordingly, also loss of Shot causes severe MT disorganisation in fly axons (Fig. 5e) - and the same is true for functional loss of its two mammalian homologues ACF7 and dystonin in culture and in vivo [57, 59, 87, 90]. Of these, dystonin mutations link to the axonopathy HSAN6 (type 6 hereditary sensory and autonomic neuropathy [239]).
Cortical guidance is complemented by at least one quality control mechanism [74]: MTs that have (accidentally) left their bundled arrangements and extend towards the cortex in Drosophila neurons, get inhibited by Efa6 (exchange factor for ARF6; '4' in Fig. 3). Efa6 is a cortical collapse factor that associates with the axonal membrane via its C-terminal plekstrin homology domain and blocks MT polymerisation via its N-terminal MTED (MT elimination domain); when Efa6 is absent, off-track MTs outside axonal MT bundles persist for longer and are higher in number. Consistent with the known roles of off-track MTs in axon growth, branching and MT disorganisation (see Table 1 and above), fly neurons in culture and in vivo lacking Efa6 display longer axons, more branches and prominent MT disorganisation (Fig. 5d [74]).
Our model would predict that mutant phenotypes caused by loss of Shot and Efa6 should enhance each other because they are caused through complementary mechanisms of MT bundle regulation. Accordingly, we found enhanced MT disorganisation when removing both Shot and Efa6, and over-expression of Shot could rescue Efa6 mutant phenotypes [74]. We propose therefore that Shot and Eb1 keep MTs away from the membrane, whereas Efa6 acts as a quality control factor inhibiting occasional accidental off-track MTs. This elimination seems to occur in moderate, well-balanced amounts so that 'intended' off-track MTs required for axon growth and branching can persist and perform their function.
Interestingly, the cortical collapse function of fly Efa6 is not conserved in vertebrates [74]. Nevertheless, the concepts derived from Efa6 studies appear relevant, because loss of the unrelated neuronal cortical collapse factor KIF21A (kinesin family member 21A; a type 4 kinesin) causes analogous phenotypes in mammalian neurons. Thus, KIF21A mutations linked to the neurodevelopmental disorder CFEOM1 (type 1 congenital fibrosis of the extraocular muscles) affect axon growth and axonal branching just like Efa6 [74, 263] - and might as well cause MT disorganisation, but no data are currently available.
However, guidance along cortical actin seems not the only mechanism through which Eb1 and Shot keep MTs on track. This is illustrated by the simple fact that MT disorganisation observed upon loss of Shot or Eb1 in primary fly neurons (Fig. 5b, e) does not occur when removing actin from axon shafts [21, 75, 77]. This suggests that both factors perform additional, actin-independent functions or interactions to promote MT bundles.
For example, the unusual Shot-PH isoform, which is highly enriched in the nervous system and harbours a plakin repeat region (PRR; conserved in mammalian spectraplakins), is a likely candidate for such roles that still await investigation ('11' in Fig. 3; [79, 90]). Apart from spectraplakins, Eb proteins have a long list of further interactors [238], and some of them might associate with MTs and guide extending plus ends along pre-existing bundles ('9' in Fig. 3); for example, APC (adenomatous polyposis coli) or GAS2-LIKE family members are good candidates, known to bind both MTs and Eb1 in mammals and Drosophila [264,265,266]. In this context, Eb1-APC-kinesin complexes were already suggested to contribute to MT guidance [169, 267]. Furthermore, MT guidance through septins might offer new explanations for axonal growth defects observed upon septin deficiencies [268,269,270].
Potential roles of severing proteins and MT-destabilising kinesins in MT bundle maintenance
Apart from cortical MT elimination, also MT severing and/or depolymerisation in the cytoplasm may play important roles in maintaining axonal MT bundles. This is supported by axonal MT disorganisation observed upon the losses of Drosophila katanin (Y.T.L., unpublished results) or mammalian spastin [62,63,64,65].
As explained in the previous section, MTs leaving the bundled conformation can drive axonal growth, branching and MT disorganisation, and cortical collapse factors negatively regulate all three processes. In line with this argumentation, also Kif2A (a MT-depolymerising kinesin-13 family member; [271]) and MT severing proteins (spastin, katanin and fidgetin) were reported to inhibit neurite growth and/or branching [272,273,274]. However, other studies of spastin, katanin and fidgetin led to contradictory findings, describing them as promoters rather than inhibitors of neurite growth and branching [63, 275,276,277,278,279,280,281,282]. Such stark, potentially context-dependent deviations reflect the complex regulation of these proteins.
Spastin, katanin and fidgetin are all members of the superfamily of AAA proteins (ATPases associated with diverse cellular activities; [201, 283, 284]), but their severing activity is differentially regulated through their individual responses to (a) posttranslational MT modifications (in particular acetylation and poly-glutamylation; [107, 274, 285,286,287,288]), (b) competition with other MT shaft-binding proteins such as tau ('15' in Fig. 3; [275, 289, 290]), or (c) spatial recruitment through specifically localised proteins such as CAMSAP ('13' in Fig. 3 [291]). Furthermore, katanin has the ability to depolymerise MTs in an ATP-independent manner [292].
Through this context-dependent spatiotemporal regulation of their activities, severing proteins can have two diametrically opposed outcomes: they either eliminate MTs, or they break them up into stable fragments that may serve as seeds for MT amplification [113, 201, 293]. In the following, we will briefly speculate how either of these outcomes could be used to prevent MT disorganisation:
First, MT severing proteins could complement roles of cortical collapse factors ('4' in Fig. 3) by serving as quality control factors that eliminate disorganised MTs within the cytoplasm ('6' in Fig. 3). For example, katanin in plant cells was reported to localise and sever preferentially at MT cross-points, which can be used to take out non-aligned MTs [201].
Second, MT shortening functions of katanin are required at MT minus ends. Thus, in both mammals and Drosophila, the minus-end capper CAMSAP/Patronin protects against MT disassembly, and recruits katanin to counterbalance against uncontrolled minus-end polymerisation ('13' in Fig. 3; [225, 291, 294]); uncontrolled minus end extension upon katanin deficiency may cause MTs to go off-track or potentially buckle through extra forces produced. Note that CAMSAP3 also plays roles in maintaining non-acetylated MTs, thus further complicating interpretations [295].
Third, MT elimination functions could prevent MT bundle senescence. For example, MTs suffer from damage through tear-and-wear [120, 181,182,183, 185], which might cause bundle aberration by abrogating interactions with MT-binding proteins (red cross at '16' in Fig. 3). MT fractures or holes can be repaired through mechanisms involving katanin or spastin [183, 232, 293, 296, 297]. However, more subtle features of senescence (e.g. irreversible modifications, loss of tubulin C-tails) might require selective elimination of ageing MTs through severing factors (as similarly suggested for kinesin-8 or -13 [298]) which could then trigger compensatory polymerisation ('14' in Fig. 3). For example, spastin deficiency in the SpΔ mouse model caused a drop in MT polymerisation (potentially reflecting reduced turn-over) accompanied by a rise in MT disorganisation (potentially caused by precocious MT senescence; [65]).
However, the MT phenotypes observed in the SpΔ mouse model could likewise be explained through the opposite role of spastin in promoting MT multiplication through generating nucleation seeds: in the absence of such a function, MT numbers might gradually decline and cause transport interruptions and eventually axonal pathology (see section on motor proteins; [214, 299]). Curiously, axon swellings in the SpΔ mouse model were reduced with low doses of MT-stabilising or -destabilising drugs [65], thus failing to provide any clues as to whether spastin works through MT turn-over or amplification in this context.
Understanding spastin is important because it is by far the most prominent factor linking to spastic paraplegias worldwide [300, 301], and axonal swellings are a hallmark of the disease [192, 302]. Most SPG4-linked mutations lie within the AAA-ATPase domain [303], suggesting that MT severing is key to the disease pathology. However, point mutations might generate versions of spastin, which either act as dominant negative alleles (forming dysfunctional complexes that titrate out other spastin-interacting factors), or acquire gain-of-function qualities by diffusing away to perform very different roles. One such MT-independent role of spastin is the isoform-specific regulation of the endoplasmic reticulum ('e' in Fig. 3), including its shape, its interaction with the endosome and its production of lipid droplets [304,305,306,307]. It is therefore difficult to exclude that at least part of those SPG4-linked mutations triggers axon decay through other routes than the direct induction of MT aberrations ('2' or '3' versus '1' in Fig. 4).
Potential roles of MT-MT cross-linkage in MT bundle maintenance
MT-MT cross-linkage appears an obvious means of suppressing MT disorganisation ('12' in Fig. 3) and is likely the oldest mechanistic concept put forward by neurobiologists to explain MT bundles [16, 308, 309]. Mathematical models support MT-MT cross-linkage as an important structural feature of axons (e.g. [310,311,312,313]), and physical cross-linking strands of varying length between axonal MTs were observed decades ago [16, 314]. Such MT-MT cross-linkers would be expected to detach upon super-threshold pull or compression, and re-attach thereafter (Fig. 2; slip-bonds), giving axons properties approximating those of active fluids (K. Miller, personal communication). However, the molecular players mediating MT-MT cross-linkage in axons remain surprisingly controversial to this day [315], as briefly explained in the following.
First, showing that a neuronal linker expressed in non-neuronal cells induces MT bundling, is insufficient proof: MT bundling can even be achieved through expression of isolated MT-binding domains, or the application of the MT-stabilising drug taxol; intriguingly, taxol-induced bundles display ultrastructural cross-bridges that are indistinguishable from those induced by tau or MAP 2 [308, 316,317,318,319]. As a further example, dynamin is linked to Charcot-Marie-Tooth disease and has been shown to bundle MTs in vitro; however, the physiological relevance of this is questionable, because dynamin in vivo seems to bind primarily membranes [320,321,322].
Second, neurofilaments were reported to fill spaces between MTs especially in larger diameter axons and to form lateral extension that link to MTs around them [323]. However, as mentioned in the second section, lack of neurofilaments is not a lethal condition, suggesting that cross-linking roles of intermediate filaments are not crucial in axons.
Third, MAP 1B appears an ideal cross-linker at first sight, because it possesses an N- and a C-terminal MT-binding domain, but it is not a convincing bundler when expressed in non-neuronal cells [324] - although the Drosophila homologue Futsch was reported to promote MT spools at synaptic terminals [325]. We are aware of only two reports mentioning axonal bundle defects upon loss of MAP 1B or Futsch [326, 327] and another where loss of Futsch reduces the spacing between axonal MTs [328]. Rather than MT bundle structure, most insights into MAP 1B/Futsch functions concern axon development, which likely reflects its major role [324, 329,330,331]. Another candidate with N- and C-terminal MT-binding domains is MTCL1 (microtubule cross-linking factor 1) which displays prominent bundling activity when expressed in non-neuronal cells and is prominently expressed at the AIS (axon initial segment) of cerebellar Purkinje cells [317, 332, 333]. Ultrastructural analyses of a range of neuron types have revealed that MTs at AISs are not distributed throughout the axonal lumen as observed along axon shafts, but are grouped up into parallel sheet-like arrangements cross-linked by ~25nm long spacers ('Axon' chapter in [334]). In Purkinje cells, these parallel arrangements are affected upon loss of MTCL1, as is in agreement with its proposed role as cross-linker [332]. However, due to its limited expression in the cerebellum, MTCL1 cannot explain similar sheet-like arrangements in the AISs of other neuron types. A more likely candidate is TRIM-46 (tripartite motif-containing protein 46) which is expressed in AISs in many nervous tissues, contains only one central MT-binding domain, can induce sheet-like arrangements when expressed in non-neuronal cells, localises to the cross-bridges, and its knock-down in cultured neurons causes reduced cross-linkage [335, 336].
Fifth, the conserved linker candidate tau, has one central MT-binding region and seems to achieve physical MT-MT linkage through N-terminal dimerisation [337,338,339]. However, its dwell time on MTs is very short [150, 340]. Similar to MAP 1B/Futsch, we are aware of only rare reports of fairly mild bundle aberration upon loss of tau [83, 341], and most tau-deficient phenotypes concern neuro-developmental defects instead [331].
Pinpointing roles of tau or MAP 1B/Futsch in axonal MT-MT cross-linkage is enormously complicated by the fact that both proteins seem to perform a whole array of further molecular functions relevant for MT dynamics. For example, tau can protect MTs from severing by katanin [289], bind tubulin hetero-dimers [342], switch between bundled and single MT states [343], cross-link MTs with actin or the cortex [344,345,346,347], stabilise MTs [315, 348], maintain labile domains along MT shafts [290, 349], regulate end-binding proteins [350], compete with kinesins or regulate their traffic [179, 180, 351], and promote MT nucleation and polymerisation [331, 352]. A similarly broad functional pleiotropy has been reported for MAP 1B [324].
Gaining experimental proof for MT-MT cross-linking activities in axons is also complicated by functional redundancies. For example, enhanced phenotypes are observed when mutations of MAP1B and tau or of spectraplakins and tau are combined in the same neurons, or when Futsch and Tau are co-expressed. Such functional redundancies likely extend to further potential cross-linkers. For example, kinesin-5 (KIF11), kinesin-6 (KIF23, Pavarotti in Drosophila) and kinesin-12 (KIF15) slide anti-parallel MTs in the mitotic spindle [354]; since axonal MTs are arranged in parallel, these kinesins seem therefore to inhibit sliding in this cellular compartment [355,356,357,358,359,360], suggesting that they cross-link MTs. In support of this idea, we observe that loss of Pavarotti causes axonal MT disorganisation in Drosophila primary neurons which might reflect potential linker function (Y.T.L., unpublished data).
In conclusion, MT-MT cross-linkage is a long proposed concept, but pinpointing the responsible molecular factors in axons remains a key challenge. We seem to have come a step closer at the AIS, but are far from understanding the situation in the axon shaft. We even cannot fully exclude a model where MT bundles are held together by the corset of contractile cortical actin rings (Fig. 1), and cross-linkers merely separate MTs to generate space for transport [241, 309]. It is therefore pivotal to decipher the true molecular nature of MT-MT spacing/cross-linkage in axons; overcoming functional redundancies between different classes of linker candidates might be an important strategy to this end.
Does MT bundle maintenance involve their anchorage to the axonal surface?
Apart from cross-linking MTs within axonal bundles, they might also be anchored to the axon wall, as a further means to prevent MT buckling and bundle deformation caused by the enormous forces imposed by axonal cargo transport. For example, axolinin in squid giant axon has been discussed as a potential MT-cortex linker [361]. Certainly, large ankyrin isoforms can anchor MTs to the cortex at the AIS of mammalian neurons [25, 362, 363] and along axon shafts in Drosophila [328]. In the case of Drosophila it was proposed that MT-cortex linkage through ankyrin combined with spacer function of the MAP 1B homologue Futsch (which contains an unusual central domain of 60 repeats with neurofilament homology [329]) form networks that sustain a large axon diameter, thus replacing roles of neurofilaments in mammals (see above).
Relevant in this context is the phenomenon of slow (ca. 0.5-5mm/day) somatofugal flow of MTs in developing axons, first observed in 1940 using axonal constrictions [364] and later confirmed in vertebrates and flies alike [365,366,367,368]. Forces contributing to this process could be derived from an increase in MT volume through polymerisation along the axon shaft [77], pulling forces in the rear of growth cones [246], thermal motion of MT-MT cross-linkers [369], kinesins actively sliding MTs along other MTs ('B' in Fig. 3; [370]), or dyneins sliding MTs along cortical F-actin ('10' in Fig. 3; [88, 258, 371, 372]).
Potential MT sliding along cortical actin would represent one form of tethering MT bundles to the axonal surface. Such anchorage is also suggested by observed co-drift of the axolemma with the axon core [248, 251, 373]. But anchorage would not have to be static; for example, it might involve an interface of slip-bonds, as similarly suggested for actin networks that flow across, whilst dynamically anchoring to, stable focal adhesion sites [374]. MTs could anchor to cortical actin (Fig. 1; '2' in Fig. 3; [20]) or to membrane-associated or transmembrane proteins including ion channels, ion transporters or adhesion factors (Fig. 1; '3' in Fig. 3). Links to transmembrane proteins could be used as mechano-sensing modules [375] that measure local shear forces generated between MT bundles and the axonal environment (Fig. 1). Such mechano-sensing properties could explain local regulation phenomena: for example, net rates of mitochondrial movement along the axon are fairly constant, but the slow transport component (driven by MT bundle flow) is low in proximal and high in distal axon segments; this gradual increase in the amount of slow transport is compensated for by inverse proportional amounts of fast transport (high proximal, low distal [366]). This well-balanced regional amount of fast mitochondrial transport could potentially be regulated by mechano-sensing, i.e. measuring the local MT drift rate relative to the outer axonal environment in each axon segment.
Apart from dynein (see above), other potential anchoring mechanisms can be deduced from the literature. For example, spectraplakins are good candidates, as suggested by distal shift of axonal MTs in fly neurons lacking the Drosophila spectraplakin Shot and treated with the MT-stabilising drug taxol [90]. Three distinct mechanisms could account for spectraplakin-mediated MT anchorage: Firstly, spectraplakins could directly cross-link actin and MTs ('2' and '5' in Fig. 3). Secondly, they could link to membrane-associated proteins; for example, the mammalian spectraplakin dystonin can link to ß4-integrin and transmembrane collagen XVII ('3' in Fig. 3; [90]), and Drosophila Shot is able to regulate the axonal localisation of the cell adhesion molecule Fasciclin 2, potentially cross-linking Fasciclin 2 to MT bundles [376, 377]. Thirdly, spectraplakins were shown, in non-neuronal cells of fly and mammals, to anchor MT minus ends to the cortex ('1' in Fig. 3; [225, 378, 379]); this mechanism requires interaction with the MT minus end-stabilising factor CAMSAP/Patronin, a factor that is known to be relevant for neuronal morphology [380].
Also other MT-binding proteins, such as tau, MAP1B, APC and dynamin, might be involved in anchorage since they were also reported to bind to actin or to the cortex ('2' in Fig. 3; [324, 344, 346, 347, 381,382,383,384]). Potential MT-actin cross-linkage in the axon may not only occur at the cortex, but as well at central longitudinal actin trails (Fig. 1; [17, 24]), thus further contributing to the intricate cross-linking networks expected to stabilise MT bundles. Deciphering MT bundle cross-linkage, internally or with the axonal surface, stays a major challenge for future research, but its understanding will teach us important lessons about axon biology and pathology.
Conclusions and future perspectives
Here we have presented a conceptual view by describing our vision of a functional interactome of cross-regulatory networks acting at the local level in axons. This model sits right at the interface of research into molecular mechanisms and modern bioinformatics approaches of functional network analysis. We propose that there has to be a fine balance between damaging effects inflicted by life-sustaining motor movements ('associated', 'A-E' in Fig. 3) and those factors that maintain the highways required for this movement (MT-'taming' mechanisms; '1-17' in Fig. 3); both are fine-tuned through a number of cross-regulatory mechanisms ('a-e' in Fig. 3).
Our model integrates a broad range of findings from the literature. But its original foundations are derived from our own work in Drosophila neurons, as a consistent cellular system in which it is feasible to study a wide range of genetic factors in parallel and in combination, as a means to understand redundancies, hierarchies and cooperation [46, 70, 72]. This offers unique possibilities to tackle the daunting task of disentangling the enormous complexity of axonal MT bundle regulation. For this, the model of local axon homeostasis could provide a useful basis, helping to develop testable working hypotheses; a good starting point might be to break down the local axon homeostasis machinery into classifiable sub-machineries, like those discussed in the different sections of this review.
This approach also means that we need to recognise the value of incremental long-term approaches that gradually assemble known and newly discovered molecular mechanisms into an integrated understanding of how axon biology is orchestrated at the cellular level, i.e. the organisation level at which axonopathies become manifest. As B.A. Cohen put it: "Research that results in models that reliably and quantitatively predict the outcomes of genetic, biochemical, or pharmacological perturbations should be valued highly, and rewarded, regardless of whether such models invoke novel phenomena" [385].
For the studies of MTs in neurons, we need to take into consideration that knowledge derived from non-neuronal cells might only partly apply [72]. Furthermore, the interactome shown in Fig. 3 makes clear that we will need quantitative approaches: we know increasingly well how factors bind to MTs and partly understand how they might compete with each other. But how crowded can a single MT be, how many molecules are there in its surrounding at any time point, and how much dynamic exchange is taking place? Computational modelling will be an unavoidable means to make sense of existing data and make reasonable predictions to inform experimentation [386, 387].
Integrated understanding of axon biology will also improve our knowledge of the next higher level of complexity, i.e. the signalling networks and systemic factors (e.g. second messengers, ATP, ROS, the 'tubulin code' [104, 113, 212, 388]) that govern axon homeostasis and that maintain balance even during phases of change (e.g. when switching from growth to differentiation, or during stress, injury, regeneration) - or that tip the balance and induce degeneration in disease or ageing. Also the roles of glial cells, likely acting as important external influencers of such systemic processes [389], will become clearer.
Finally, MTs have been recognised as promising therapeutic targets [302, 390, 391], and urgently needed advance on this translational path will be facilitated by a better understanding of the axonal MT homeostasis system. A larger focus of the research community on MTs and, in turn, an improved availability of MT-related data that can be incorporated into our understanding, would be a key prerequisite to make such progress.
Availability of data and materials
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
Abbreviations
- AAA:
-
ATPases associated with diverse cellular activities
- ACF7:
-
Actin cross-linking factor 7
- APC:
-
Adenomatous polyposis coli
- CAMSAP:
-
Calmodulin-regulated spectrin-associated protein
- CFEOM1:
-
type 1 congenital fibrosis of the extraocular muscles
- Eb1:
-
End binding protein 1
- Efa6:
-
Exchange factor for ARF6
- ɣTuRC:
-
ɣ-tubulin ring complex
- HSAN6:
-
type 6 hereditary sensory and autonomic neuropathy
- iPSC:
-
induced pluripotent stem cell
- Khc:
-
Kinesin heavy chain
- KIF:
-
Kinesin family member
- KLC:
-
Kinesin light chain
- MIP:
-
Microtubule inner proteins
- MT:
-
Microtubule
- MTED:
-
Microtubule elimination domain
- MTOC:
-
MT organising centres
- PF:
-
Protofilament
- PRR:
-
Plakin repeat region
- ROS:
-
Reactive oxygen species
- Shot:
-
Short stop
- SPG4:
-
Spastin-linked spastic paraplegia
- STED:
-
Stimulated emission depletion
References
Hoffman PN. Review : The Synthesis, Axonal Transport, and Phosphorylation of Neurofilaments Determine Axonal Caliber in Myelinated Nerve Fibers. Neuroscientist. 1995;1:76–83. https://doi.org/10.1177/107385849500100204.
Bichenback J. International perspectives on spinal cord injury. WHO, ISCOS: Switzerland; 2013.
Tedeschi A, Bradke F. Spatial and temporal arrangement of neuronal intrinsic and extrinsic mechanisms controlling axon regeneration. Curr Opin Neurobiol. 2016;42:118–27 http://www.ncbi.nlm.nih.gov/pubmed/28039763.
Medana IM, Esiri MM. Axonal damage: a key predictor of outcome in human CNS diseases. Brain. 2003;126:515–30 http://www.ncbi.nlm.nih.gov/pubmed/12566274.
Gaetz M. The neurophysiology of brain injury. Clin Neurophysiol. 2004;115:4–18 http://www.ncbi.nlm.nih.gov/pubmed/14706464.
Smith DH, Nonaka M, Miller R, Leoni M, Chen XH, Alsop D, Meaney DF. Immediate coma following inertial brain injury dependent on axonal damage in the brainstem. J Neurosurg. 2000;93:315–22 http://www.ncbi.nlm.nih.gov/pubmed/10930019.
Tang-Schomer MD, Johnson VE, Baas PW, Stewart W, Smith DH. Partial interruption of axonal transport due to microtubule breakage accounts for the formation of periodic varicosities after traumatic axonal injury. Exp Neurol. 2012;233:364–72 http://www.ncbi.nlm.nih.gov/pubmed/22079153.
Salvadores N, Sanhueza M, Manque P, Court FA. Axonal degeneration during aging and its functional role in neurodegenerative disorders. Front Neurosci. 2017;11:451 https://www.frontiersin.org/article/10.3389/fnins.2017.00451.
Adalbert R, Coleman MP. Axon pathology in age-related neurodegenerative disorders. Neuropathol Appl Neurobiol. 2012;39:90–108 http://www.ncbi.nlm.nih.gov/pubmed/23046254.
Sturrock RR. Age-related changes in the number of myelinated axons and glial cells in the anterior and posterior limbs of the mouse anterior commissure. J Anat. 1987;150:111–27 https://www.ncbi.nlm.nih.gov/pubmed/3654327.
Marner L, Nyengaard JR, Tang Y, Pakkenberg B. Marked loss of myelinated nerve fibers in the human brain with age. J Comp Neurol. 2003;462:144–52 http://www.ncbi.nlm.nih.gov/pubmed/12794739.
Calkins DJ: Age-Related Changes in the Visual Pathways: Blame It on the AxonAge-Related Changes in the Visual Pathways. Invest Ophthalmol Vis Sci 2013, 54:ORSF 37-41. https://doi.org/10.1167/iovs.13-12784
Blackstone C, O'Kane CJ, Reid E. Hereditary spastic paraplegias: membrane traffic and the motor pathway. Nat Rev Neurosci. 2011;12:31–42 http://www.ncbi.nlm.nih.gov/pubmed/21139634.
Riancho J, Gonzalo I, Ruiz-Soto M, Berciano J. Why do motor neurons degenerate? Actualization in the pathogenesis of amyotrophic lateral sclerosis. Neurología. 2019;34:27–37 http://www.elsevier.es/en-revista-neurologia-english-edition-495-articulo-why-do-motor-neurons-degenerate-S2173580817301633.
Aguzzi A: ‘Forward genetics’ and the causes of ALS. Nature Reviews Molecular Cell Biology 2019, 20:67-67. https://doi.org/10.1038/s41580-018-0062-6
Hirokawa N: Molecular architecture and dynamics of the neuronal cytoskeleton. In The neuronal cytoskeleton. Edited by Burgoyne R. New York: Wiley-Liss; 1991: 5-74
Fath KR, Lasek RJ. Two classes of actin microfilaments are associated with the inner cytoskeleton of axons. J Cell Biol. 1988;107:613–21 http://www.ncbi.nlm.nih.gov/pubmed/3417765.
Ganguly A, Tang Y, Wang L, Ladt K, Loi J, Dargent B, Leterrier C, Roy S. A dynamic formin-dependent deep F-actin network in axons. J Cell Biol. 2015;210:401–17 http://jcb.rupress.org/content/early/2015/07/21/jcb.201506110.abstract.
Hirokawa N. Cross-linker system between neurofilaments, microtubules, and membranous organelles in frog axons revealed by the quick-freeze, deep-etching method. J Cell Biol. 1982;94:129–42 http://www.ncbi.nlm.nih.gov/pubmed/6181077.
Xu K, Zhong G, Zhuang X. Actin, Spectrin, and associated proteins form a periodic cytoskeletal structure in axons. Science. 2013;339:452–6 http://www.ncbi.nlm.nih.gov/pubmed/23239625.
Qu Y, Hahn I, Webb SED, Pearce SP, Prokop A. Periodic actin structures in neuronal axons are required to maintain microtubules. Mol Biol Cell. 2017;28:296–308 http://www.molbiolcell.org/content/early/2016/11/21/mbc.E16-10-0727.
Vassilopoulos S, Gibaud S, Jimenez A, Caillol G, Leterrier C: Ultrastructure of the axonal periodic scaffold reveals a braid-like organization of actin rings. bioRxiv 2019:636217. https://www.biorxiv.org/content/10.1101/636217v2
Prokop A. The intricate relationship between microtubules and their associated motor proteins during axon growth and maintenance. Neur Dev. 2013;8:17 http://www.neuraldevelopment.com/content/8/1/17.
Leterrier C, Dubey P, Roy S. The nano-architecture of the axonal cytoskeleton. Nature Reviews Neuroscience. 2017;18:713–26. https://doi.org/10.1038/nrn.2017.129.
Leterrier C. The axon initial segment: an updated viewpoint. J Neurosci. 2018;38:2135–45 http://www.jneurosci.org/content/jneuro/38/9/2135.full.pdf.
Dent EW, Gupton SL, Gertler FB. The growth cone cytoskeleton in axon outgrowth and guidance. Cold Spring Harb Perspect Biol. 2011;3:a001800 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=21106647.
Friede RL, Samorajski T. Axon caliber related to neurofilaments and microtubules in sciatic nerve fibers of rats and mice. Anat Rec. 1970;167:379–87 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=5454590.
Sakaguchi T, Okada M, Kitamura T, Kawasaki K. Reduced diameter and conduction velocity of myelinated fibers in the sciatic nerve of a neurofilament-deficient mutant quail. Neurosci Lett. 1993;153:65–8 http://www.ncbi.nlm.nih.gov/pubmed/8510825.
Rao MV, Campbell J, Yuan A, Kumar A, Gotow T, Uchiyama Y, Nixon RA. The neurofilament middle molecular mass subunit carboxyl-terminal tail domains is essential for the radial growth and cytoskeletal architecture of axons but not for regulating neurofilament transport rate. J Cell Biol. 2003;163:1021–31 http://www.ncbi.nlm.nih.gov/pubmed/14662746.
Perrot R, Berges R, Bocquet A, Eyer J. Review of the multiple aspects of neurofilament functions, and their possible contribution to neurodegeneration. Mol Neurobiol. 2008;38:27–65 http://www.ncbi.nlm.nih.gov/pubmed/18649148.
Eyer J, Cleveland DW, Wong PC, Peterson AC. Pathogenesis of two axonopathies does not require axonal neurofilaments. Nature. 1998;391:584–7 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=9468135.
Nguyen MD, Lariviere RC, Julien JP. Reduction of axonal caliber does not alleviate motor neuron disease caused by mutant superoxide dismutase 1. Proc Natl Acad Sci U S A. 2000;97:12306–11 http://www.ncbi.nlm.nih.gov/pubmed/11050249.
Eyer J, Peterson A. Neurofilament-deficient axons and perikaryal aggregates in viable transgenic mice expressing a neurofilament-beta-galactosidase fusion protein. Neuron. 1994;12:389–405 http://www.ncbi.nlm.nih.gov/pubmed/8110465.
Yamasaki H, Itakura C, Mizutani M. Hereditary hypotrophic axonopathy with neurofilament deficiency in a mutant strain of the Japanese quail. Acta Neuropathol. 1991;82:427–34 http://www.ncbi.nlm.nih.gov/pubmed/1785256.
Allen MJ, Godenschwege TA, Tanouye MA, Phelan P. Making an escape: development and function of the Drosophila giant fibre system. Semin Cell Dev Biol. 2006;17:31–41 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16378740.
Hirokawa N. 270K microtubule-associated protein cross-reacting with anti-MAP 2 IgG in the crayfish peripheral nerve axon. J Cell Biol. 1986;103:33–9 http://www.ncbi.nlm.nih.gov/pubmed/3722268.
Voelzmann A, Hahn I, Pearce S, Sánchez-Soriano NP, Prokop A. A conceptual view at microtubule plus end dynamics in neuronal axons. Brain Res Bulletin. 2016;126:226–37 http://www.sciencedirect.com/science/article/pii/S0361923016301885.
Sakakibara A, Ando R, Sapir T, Tanaka T. Microtubule dynamics in neuronal morphogenesis. Open Biology. 2013;3:130061 https://royalsocietypublishing.org/doi/abs/10.1098/rsob.130061.
Tas RP, Kapitein LC. Exploring cytoskeletal diversity in neurons. Science. 2018;361:231–2 https://science.sciencemag.org/content/sci/361/6399/231.full.
Hirokawa N, Niwa S, Tanaka Y. Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron. 2010;68:610–38 http://www.ncbi.nlm.nih.gov/pubmed/21092854.
Goldstein AY, Wang X, Schwarz TL. Axonal transport and the delivery of pre-synaptic components. Curr Opin Neurobiol. 2008;18:495–503 http://www.ncbi.nlm.nih.gov/pubmed/18950710.
Pfenninger KH. Plasma membrane expansion: a neuron's Herculean task. Nat Rev Neurosci. 2009;10:251–61 http://www.ncbi.nlm.nih.gov/pubmed/19259102.
Gondre-Lewis MC, Park JJ, Loh YP. Cellular mechanisms for the biogenesis and transport of synaptic and dense-core vesicles. Int Rev Cell Mol Biol. 2012;299:27–115 http://www.ncbi.nlm.nih.gov/pubmed/22959301.
Gonzalez C, Couve A. The axonal endoplasmic reticulum and protein trafficking: Cellular bootlegging south of the soma. Semin Cell Dev Biol. 2014;27:23–31 http://www.ncbi.nlm.nih.gov/pubmed/24361785.
Saxton WM, Hollenbeck PJ. The axonal transport of mitochondria. J Cell Sci. 2012;125:2095–104 http://www.ncbi.nlm.nih.gov/pubmed/22619228.
Prokop A, Beaven R, Qu Y, Sánchez-Soriano N. Using fly genetics to dissect the cytoskeletal machinery of neurons during axonal growth and maintenance. J Cell Sci. 2013;126:2331–41. https://doi.org/10.1242/jcs.126912.
Miller KE, Suter DM. An integrated cytoskeletal model of neurite outgrowth. Front Cell Neurosci. 2018;12:477 https://www.frontiersin.org/article/10.3389/fncel.2018.00447.
Acebes A, Ferrus A. Cellular and molecular features of axon collaterals and dendrites. Trends Neurosci. 2000;23:557–65 http://www.ncbi.nlm.nih.gov/pubmed/11074265.
Kalil K, Dent EW. Branch management: mechanisms of axon branching in the developing vertebrate CNS. Nat Rev Neurosci. 2014;15:7–18 http://www.ncbi.nlm.nih.gov/pubmed/24356070.
Tymanskyj SR, Yang B, Falnikar A, Lepore AC, Ma L. MAP 7 regulates axon collateral branch development in dorsal root ganglion neurons. J Neurosci. 2017;37:1648–61 http://www.ncbi.nlm.nih.gov/pubmed/28069923.
Tint I, Jean D, Baas PW, Black MM. Doublecortin associates with microtubules preferentially in regions of the axon displaying actin-rich protrusive structures. J Neurosci. 2009;29:10995–1010 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=19726658.
Bodaleo FJ, Gonzalez-Billault C. The presynaptic microtubule cytoskeleton in physiological and pathological conditions: lessons from Drosophila Fragile X Syndrome and Hereditary Spastic Paraplegias. Front Mol Neurosci. 2016;9:60 https://www.ncbi.nlm.nih.gov/pubmed/27504085.
Wozniak KM, Vornov JJ, Wu Y, Liu Y, Carozzi VA, Rodriguez-Menendez V, Ballarini E, Alberti P, Pozzi E, Semperboni S, et al. Peripheral Neuropathy Induced by Microtubule-Targeted Chemotherapies: Insights into Acute Injury and Long-term Recovery. Cancer Res. 2018;78:817–29 http://www.ncbi.nlm.nih.gov/pubmed/29191802.
Prior R, Van Helleputte L, Benoy V, Van Den Bosch L. Defective axonal transport: A common pathological mechanism in inherited and acquired peripheral neuropathies. Neurobiol Dis. 2017;105:300–20 http://www.ncbi.nlm.nih.gov/pubmed/28238949.
Wu Y, Li J, Zhou J, Feng Y. Dynamic long-term microstructural and ultrastructural alterations in sensory nerves of rats of paclitaxel-induced neuropathic pain. Chin Med J (Engl). 2014;127:2945–52 http://www.ncbi.nlm.nih.gov/pubmed/25131233.
Malacrida A, Meregalli C, Rodriguez-Menendez V, Nicolini G. Chemotherapy-induced peripheral neuropathy and changes in cytoskeleton. Int J Mol Sci. 2019;20:2287 https://www.mdpi.com/1422-0067/20/9/2287.
Bernier G, Kothary R. Prenatal onset of axonopathy in Dystonia musculorum mice. Dev Genet. 1998;22:160–8 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=9581287.
Adalbert R, Nogradi A, Babetto E, Janeckova L, Walker SA, Kerschensteiner M, Misgeld T, Coleman MP. Severely dystrophic axons at amyloid plaques remain continuous and connected to viable cell bodies. Brain. 2009;132:402–16 http://www.ncbi.nlm.nih.gov/pubmed/19059977.
Dalpe G, Leclerc N, Vallee A, Messer A, Mathieu M, De Repentigny Y, Kothary R. Dystonin is essential for maintaining neuronal cytoskeleton organization. Mol Cell Neurosci. 1998;10:243–57 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=9604204.
Sorbara CD, Wagner NE, Ladwig A, Nikic I, Merkler D, Kleele T, Marinkovic P, Naumann R, Godinho L, Bareyre FM, et al. Pervasive axonal transport deficits in multiple sclerosis models. Neuron. 2014;84:1183–90 http://www.ncbi.nlm.nih.gov/pubmed/25433639.
Yin X, Kidd GJ, Ohno N, Perkins GA, Ellisman MH, Bastian C, Brunet S, Baltan S, Trapp BD. Proteolipid protein-deficient myelin promotes axonal mitochondrial dysfunction via altered metabolic coupling. J Cell Biol. 2016;215:531–42 http://www.ncbi.nlm.nih.gov/pubmed/27872255.
Denton KR, Lei L, Grenier J, Rodionov V, Blackstone C, Li XJ. Loss of spastin function results in disease-specific axonal defects in human pluripotent stem cell-based models of hereditary spastic paraplegia. Stem Cells. 2014;32:414–23 http://www.ncbi.nlm.nih.gov/pubmed/24123785.
Havlicek S, Kohl Z, Mishra HK, Prots I, Eberhardt E, Denguir N, Wend H, Plotz S, Boyer L, Marchetto MC, et al. Gene dosage-dependent rescue of HSP neurite defects in SPG4 patients' neurons. Hum Mol Genet. 2014;23:2527–41 http://www.ncbi.nlm.nih.gov/pubmed/24381312.
Tarrade A, Fassier C, Courageot S, Charvin D, Vitte J, Peris L, Thorel A, Mouisel E, Fonknechten N, Roblot N, et al. A mutation of spastin is responsible for swellings and impairment of transport in a region of axon characterized by changes in microtubule composition. Hum Mol Genet. 2006;15:3544–58 http://www.ncbi.nlm.nih.gov/pubmed/17101632.
Fassier C, Tarrade A, Peris L, Courageot S, Mailly P, Dalard C, Delga S, Roblot N, Lefevre J, Job D, et al. Microtubule-targeting drugs rescue axonal swellings in cortical neurons from spastin knockout mice. Dis Model Mech. 2013;6:72–83 http://www.ncbi.nlm.nih.gov/pubmed/22773755.
Hsu J-M, Chen C-H, Chen Y-C, McDonald KL, Gurling M, Lee A, Garriga G, Pan C-L. Genetic analysis of a novel tubulin mutation that redirects synaptic vesicle targeting and causes neurite degeneration in C. elegans. PLoS Genet. 2014;10:e1004715. https://doi.org/10.1371/journal.pgen.1004715.
Fiala JC, Feinberg M, Peters A, Barbas H. Mitochondrial degeneration in dystrophic neurites of senile plaques may lead to extracellular deposition of fine filaments. Brain Struct Funct. 2007;212:195–207 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17717688.
Sánchez-Soriano N, Tear G, Whitington P, Prokop A. Drosophila as a genetic and cellular model for studies on axonal growth. Neural Develop. 2007;2:9 https://tinyurl.com/y3yjg8u4.
Elden AC, Kim H-J, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, Armakola M, Geser F, Greene R, Lu MM, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466:1069. https://doi.org/10.1038/nature09320.
Prokop A. Why funding fruit fly research is important for the biomedical sciences. Open Access Govern. 2018;20:198–201 https://tinyurl.com/y7b25jpm.
Bellen HJ, Tong C, Tsuda H. 100 years of Drosophila research and its impact on vertebrate neuroscience: a history lesson for the future. Nat Rev Neurosci. 2010;11:514–22 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=20383202.
Beaven R, Dzhindzhev NS, Qu Y, Hahn I, Dajas-Bailador F, Ohkura H, Prokop A. Drosophila CLIP-190 and mammalian CLIP-170 display reduced microtubule plus end association in the nervous system. Mol Biol Cell. 2015;26:1491–508 http://www.molbiolcell.org/content/26/8/1491.abstract.
Gonçalves-Pimentel C, Gombos R, Mihály J, Sánchez-Soriano N, Prokop A. Dissecting regulatory networks of filopodia formation in a Drosophila growth cone model. PLoS One. 2011;6:e18340 http://www.plosone.org/article/info%3Adoi%2F10.1371%2Fjournal.pone.0018340.
Qu Y, Hahn I, Lees M, Parkin J, Voelzmann A, Dorey K, Rathbone A, Friel C, Allan V, Okenve Ramos P, et al: Efa6 protects axons and regulates their growth and branching by inhibiting microtubule polymerisation at the cortex. bioRxiv 2018, 10.1101/385658https://www.biorxiv.org/content/10.1101/385658v4
Alves-Silva J, Sánchez-Soriano N, Beaven R, Klein M, Parkin J, Millard T, Bellen H, Venken KJT, Ballestrem C, Kammerer RA, Prokop A. Spectraplakins promote microtubule-mediated axonal growth by functioning as structural microtubule-associated proteins and EB1-dependent +TIPs (Tip Interacting Proteins). J Neurosci. 2012;32:9143–58 http://www.jneurosci.org/content/32/27/9143.full.
Voelzmann A, Okenve-Ramos P, Qu Y, Chojnowska-Monga M, del Caño-Espinel M, Prokop A, Sánchez-Soriano N. Tau and spectraplakins promote synapse formation and maintenance through Jun kinase and neuronal trafficking. eLife. 2016;5:e14694 https://elifesciences.org/content/5/e14694.
Sánchez-Soriano N, Gonçalves-Pimentel C, Beaven R, Haessler U, Ofner L, Ballestrem C, Prokop A. Drosophila growth cones: a genetically tractable platform for the analysis of axonal growth dynamics. Dev Neurobiol. 2010;70:58–71 https://onlinelibrary.wiley.com/doi/abs/10.1002/dneu.20762.
Prokop A, Küppers-Munther B, Sánchez-Soriano N: Using primary neuron cultures of Drosophila to analyse neuronal circuit formation and function. In The making and un-making of neuronal circuits in Drosophila. Volume 69. Edited by Hassan BA. New York: Humana Press; 2012: 225-247.[Walz W (Series Editor): Neuromethods]. http://www.springerlink.com/content/t07618161235u475/#section=1102403&page=1
Hahn I, Ronshaugen M, Sánchez-Soriano N, Prokop A. Functional and genetic analysis of spectraplakins in Drosophila. Methods Enzymol. 2016;569:373–405 https://tinyurl.com/y4vhld56.
Roote J, Prokop A. How to design a genetic mating scheme: a basic training package for Drosophila genetics. G3 (Bethesda). 2013;3:353–8 http://www.g3journal.org/content/3/2/353.full.
Yogev S, Cooper R, Fetter R, Horowitz M, Shen K. Microtubule organization determines axonal transport dynamics. Neuron. 2016;92:449–60 http://www.sciencedirect.com/science/article/pii/S0896627316306377.
Yogev S, Maeder CI, Cooper R, Horowitz M, Hendricks AG, Shen K. Local inhibition of microtubule dynamics by dynein is required for neuronal cargo distribution. Nat Commun. 2017;8:15063 http://www.ncbi.nlm.nih.gov/pubmed/28406181.
Krieg M, Stühmer J, Cueva JG, Fetter R, Spilker K, Cremers D, Shen K, Dunn AR, Goodman MB: Genetic defects in β-spectrin and tau sensitize C. elegans axons to movement-induced damage via torque-tension coupling. Elife 2017, 6:e20172. https://doi.org/10.7554/eLife.20172
Fletcher DA, Mullins RD. Cell mechanics and the cytoskeleton. Nature. 2010;463:485–92 http://www.ncbi.nlm.nih.gov/pubmed/20110992.
Howard J. Mechanics of motorproteins and the cytoskeleton. Sunderland: Sinauer Assoc.; 2001.
Hawkins T, Mirigian M, Selcuk Yasar M, Ross JL. Mechanics of microtubules. J Biomech. 2010;43:23–30 http://www.ncbi.nlm.nih.gov/pubmed/19815217.
Sánchez-Soriano N, Travis M, Dajas-Bailador F, Goncalves-Pimentel C, Whitmarsh AJ, Prokop A. Mouse ACF7 and Drosophila Short stop modulate filopodia formation and microtubule organisation during neuronal growth. Journal of cell science. 2009;122:2534–42 http://jcs.biologists.org/content/122/14/2534.long.
Ahmad FJ, He Y, Myers KA, Hasaka TP, Francis F, Black MM, Baas PW. Effects of dynactin disruption and dynein depletion on axonal microtubules. Traffic. 2006;7:524–37 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16643276.
Prokop A. Fruit flies in biological research. Biological Sciences Review. 2016;28:10–4 https://tinyurl.com/ybvpoqmw.
Voelzmann A, Liew Y-T, Qu Y, Hahn I, Melero C, Sánchez-Soriano N, Prokop A. Drosophila Short stop as a paradigm for the role and regulation of spectraplakins. Sem Cell Dev Biol. 2017;69:40–57 http://www.sciencedirect.com/science/article/pii/S1084952117302124.
Brouhard GJ, Rice LM. Microtubule dynamics: an interplay of biochemistry and mechanics. Nat Rev Mol Cell Biol. 2018;19:451–63. https://doi.org/10.1038/s41580-018-0009-y.
van Haren J, Wittmann T. Microtubule plus end dynamics − do we know how microtubules grow? BioEssays. 2019;41:1800194. https://doi.org/10.1002/bies.201800194.
Burton PR, Hinkley RE, Pierson GB. Tannic acid-stained microtubules with 12, 13, and 15 protofilaments. J Cell Biol. 1975;65:227–33 http://www.ncbi.nlm.nih.gov/pubmed/47861.
Benshalom G, Reese TS. Ultrastructural observations on the cytoarchitecture of axons processed by rapid-freezing and freeze-substitution. J Neurocytol. 1985;14:943–60 http://www.ncbi.nlm.nih.gov/pubmed/2420942.
Savage C, Hamelin M, Culotti JG, Coulson A, Albertson DG. Chalfie M: mec-7 is a beta-tubulin gene required for the production of 15-protofilament microtubules in Caenorhabditis elegans. Genes Dev. 1989;3:870–81 http://www.ncbi.nlm.nih.gov/pubmed/2744465.
Chalfie M, Thomson JN. Structural and functional diversity in the neuronal microtubules of Caenorhabditis elegans. J Cell Biol. 1982;93:15–23 http://www.ncbi.nlm.nih.gov/pubmed/7068753.
Chaaban S, Brouhard GJ. A microtubule bestiary: structural diversity in tubulin polymers. Mol Biol Cell. 2017;28:2924–31 http://www.molbiolcell.org/content/28/22/2924.abstract.
Chrétien D, Fuller SD. Microtubules switch occasionally into unfavorable configurations during elongation. J Mol Biol. 2000;298:663–76 http://www.ncbi.nlm.nih.gov/pubmed/10788328.
Chrétien D, Wade RH. New data on the microtubule surface lattice. Biol Cell. 1991;71:161–74 http://www.ncbi.nlm.nih.gov/pubmed/1912942.
Chrétien D, Kenney JM, Fuller SD, Wade RH. Determination of microtubule polarity by cryo-electron microscopy. Structure. 1996;4:1031–40 http://www.ncbi.nlm.nih.gov/pubmed/8805589.
Ray S, Meyhofer E, Milligan RA, Howard J. Kinesin follows the microtubule's protofilament axis. J Cell Biol. 1993;121:1083–93 http://www.ncbi.nlm.nih.gov/pubmed/8099076.
Shaw G, Bray D. Movement and extension of isolated growth cones. Exp Cell Res. 1977;104:55–62 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=556695.
Vemu A, Atherton J, Spector JO, Moores CA, Roll-Mecak A. Tubulin isoform composition tunes microtubule dynamics. Mol Biol Cell. 2017;28:3564–72 http://www.ncbi.nlm.nih.gov/pubmed/29021343.
Park JH, Roll-Mecak A. The tubulin code in neuronal polarity. Curr Opin Neurobiol. 2018;51:95–102 http://www.ncbi.nlm.nih.gov/pubmed/29554585.
Ti S-C, Alushin GM, Kapoor TM. Human β-tubulin isotypes can regulate microtubule protofilament number and stability. Developmental Cell. 2018;47:175–190.e175 http://www.sciencedirect.com/science/article/pii/S1534580718306841.
Janke C, Kneussel M. Tubulin post-translational modifications: encoding functions on the neuronal microtubule cytoskeleton. Trends Neurosci. 2010;33:362–72 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=20541813.
Valenstein ML, Roll-Mecak A. Graded control of microtubule severing by tubulin glutamylation. Cell. 2016;164:911–21 https://www.sciencedirect.com/science/article/pii/S0092867416000593?via%3Dihub.
Boucher D, Larcher JC, Gros F, Denoulet P. Polyglutamylation of tubulin as a progressive regulator of in vitro interactions between the microtubule-associated protein Tau and tubulin. Biochemistry. 1994;33:12471–7 http://www.ncbi.nlm.nih.gov/pubmed/7522559.
Song Y, Kirkpatrick LL, Schilling AB, Helseth DL, Chabot N, Keillor JW, Johnson GV, Brady ST. Transglutaminase and polyamination of tubulin: posttranslational modification for stabilizing axonal microtubules. Neuron. 2013;78:109–23 http://www.ncbi.nlm.nih.gov/pubmed/23583110.
Soppina V, Herbstman JF, Skiniotis G, Verhey KJ. Luminal localization of alpha-tubulin K40 acetylation by cryo-EM analysis of fab-labeled microtubules. PLoS One. 2012;7:e48204 http://www.ncbi.nlm.nih.gov/pubmed/23110214.
Xu Z, Schaedel L, Portran D, Aguilar A, Gaillard J, Marinkovich MP, Théry M, Nachury MV. Microtubules acquire resistance from mechanical breakage through intralumenal acetylation. Science. 2017;356:328–32 http://science.sciencemag.org/content/sci/356/6335/328.full.
Howes SC, Alushin GM, Shida T, Nachury MV, Nogales E. Effects of tubulin acetylation and tubulin acetyltransferase binding on microtubule structure. Mol Biol Cell. 2014;25:257–66 http://www.ncbi.nlm.nih.gov/pubmed/24227885.
Baas PW, Rao AN, Matamoros AJ, Leo L. Stability properties of neuronal microtubules. Cytoskeleton (Hoboken). 2016;73:442–60 http://www.ncbi.nlm.nih.gov/pubmed/26887570.
Portran D, Schaedel L, Xu Z, Thery M, Nachury MV. Tubulin acetylation protects long-lived microtubules against mechanical ageing. Nat Cell Biol. 2017;19:391–8 https://doi.org/10.1038/ncb3481.
Yan C, Wang F, Peng Y, Williams CR, Jenkins B, Wildonger J, Kim HJ, Perr JB, Vaughan JC, Kern ME, et al: Microtubule acetylation is required for mechanosensation in Drosophila. Cell Rep 2018, 25:1051-1065 e1056. http://www.ncbi.nlm.nih.gov/pubmed/30355484
Jenkins BV, Saunders HAJ, Record HL, Johnson-Schlitz DM, Wildonger J. Effects of mutating alpha-tubulin lysine 40 on sensory dendrite development. J Cell Sci. 2017;130:4120–31 http://www.ncbi.nlm.nih.gov/pubmed/29122984.
Ichikawa M, Bui KH. Microtubule inner proteins: a meshwork of luminal proteins stabilizing the doublet microtubule. BioEssays. 2018;40:1700209. https://doi.org/10.1002/bies.201700209.
Bechstedt S, Lu K, Brouhard Gary J. Doublecortin recognizes the longitudinal curvature of the microtubule end and lattice. Curr Biol. 2014;24:2366–75 http://www.sciencedirect.com/science/article/pii/S0960982214010525.
Ettinger A, van Haren J, Ribeiro SA, Wittmann T. Doublecortin is excluded from growing microtubule ends and recognizes the GDP-microtubule lattice. Curr Biol. 2016;26:1549–55 http://www.ncbi.nlm.nih.gov/pubmed/27238282.
Peet DR, Burroughs NJ, Cross RA. Kinesin expands and stabilizes the GDP-microtubule lattice. Nat Nanotechnol. 2018;13:386–91 http://www.ncbi.nlm.nih.gov/pubmed/29531331.
Sumino Y, Nagai KH, Shitaka Y, Tanaka D, Yoshikawa K, Chate H, Oiwa K. Large-scale vortex lattice emerging from collectively moving microtubules. Nature. 2012;483:448–52 http://www.ncbi.nlm.nih.gov/pubmed/22437613.
Kerssemakers J, Howard J, Hess H, Diez S. The distance that kinesin-1 holds its cargo from the microtubule surface measured by fluorescence interference contrast microscopy. Proc Natl Acad Sci U S A. 2006;103:15812–7 http://www.ncbi.nlm.nih.gov/pubmed/17035506.
Palacci H, Idan O, Armstrong MJ, Agarwal A, Nitta T, Hess H. Velocity fluctuations in kinesin-1 gliding motility assays originate in motor attachment geometry variations. Langmuir. 2016;32:7943–50 http://www.ncbi.nlm.nih.gov/pubmed/27414063.
VanDelinder V, Imam ZI, Bachand G. Kinesin motor density and dynamics in gliding microtubule motility. Scientific Reports. 2019;9:7206. https://doi.org/10.1038/s41598-019-43749-8.
Lam AT, VanDelinder V, Kabir AMR, Hess H, Bachand GD, Kakugo A. Cytoskeletal motor-driven active self-assembly in in vitro systems. Soft Matter. 2016;12:988–97. https://doi.org/10.1039/C5SM02042E.
Amos L, Amos W. The bending of sliding microtubules imaged by confocal light microscopy and negative stain electron microscopy. J Cell Sci. 1991;1991:95–101 http://jcs.biologists.org/content/joces/1991/Supplement_14/95.full.pdf.
Weiss D, Langford G, Seitz-Tutter D, Maile W. Analysis of the gliding, fishtailing and circling motions of native microtubules. Acta Histochem Suppl. 1991;41:81–105.
Waterman-Storer CM, Salmon ED. Actomyosin-based retrograde flow of microtubules in the lamella of migrating epithelial cells influences microtubule dynamic instability and turnover and is associated with microtubule breakage and treadmilling. J Cell Biol. 1997;139:417–34 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=9334345.
Odde DJ, Ma L, Briggs AH, DeMarco A, Kirschner MW. Microtubule bending and breaking in living fibroblast cells. Journal of cell science. 1999;112:3283–8 http://jcs.biologists.org/content/joces/112/19/3283.full.pdf.
Hess H, Clemmens J, Brunner C, Doot R, Luna S, Ernst KH, Vogel V. Molecular self-assembly of "nanowires"and "nanospools" using active transport. Nano Lett. 2005;5:629–33 http://www.ncbi.nlm.nih.gov/pubmed/15826099.
Dent EW, Kalil K. Axon branching requires interactions between dynamic microtubules and actin filaments. J Neurosci. 2001;21:9757–69 http://www.jneurosci.org.manchester.idm.oclc.org/content/21/24/9757.long.
Liu H, Spoerke ED, Bachand M, Koch SJ, Bunker BC, Bachand GD. Biomolecular Motor-Powered Self-Assembly of Dissipative Nanocomposite Rings. Advanced Materials. 2008;20:4476–81. https://doi.org/10.1002/adma.200801291.
VanDelinder V, Brener S, Bachand GD. Mechanisms underlying the active self-assembly of microtubule rings and spools. Biomacromolecules. 2016;17:1048–56 http://www.ncbi.nlm.nih.gov/pubmed/26842978.
Liu L, Tuzel E, Ross JL. Loop formation of microtubules during gliding at high density. J Phys Condens Matter. 2011;23:374104 http://www.ncbi.nlm.nih.gov/pubmed/21862840.
Lam AT, Curschellas C, Krovvidi D, Hess H. Controlling self-assembly of microtubule spools via kinesin motor density. Soft Matter. 2014;10:8731–6. https://doi.org/10.1039/C4SM01518E.
Wada S, Rashedul Kabir AM, Ito M, Inoue D, Sada K, Kakugo A. Effect of length and rigidity of microtubules on the size of ring-shaped assemblies obtained through active self-organization. Soft Matter. 2015;11:1151–7. https://doi.org/10.1039/C4SM02292K.
Kawamura R, Kakugo A, Shikinaka K, Osada Y, Gong JP. Ring-Shaped Assembly of Microtubules Shows Preferential Counterclockwise Motion. Biomacromolecules. 2008;9:2277–82. https://doi.org/10.1021/bm800639w.
Rashedul Kabir AM, Wada S, Inoue D, Tamura Y, Kajihara T, Mayama H, Sada K, Kakugo A, Gong JP. Formation of ring-shaped assembly of microtubules with a narrow size distribution at an air-buffer interface. Soft Matter. 2012;8:10863–7 https://doi.org/10.1039/C2SM26441B.
Alberti S, Hyman AA. Are aberrant phase transitions a driver of cellular aging? BioEssays. 2016;38:959–68 https://www.ncbi.nlm.nih.gov/pubmed/27554449.
Nakashima KK, Vibhute MA, Spruijt E. Biomolecular Chemistry in Liquid Phase Separated Compartments. Frontiers in Molecular Biosciences. 2019;6:21 https://www.frontiersin.org/article/10.3389/fmolb.2019.00021.
Ziebert F, Mohrbach H, Kulić IM. Why Microtubules Run in Circles: Mechanical Hysteresis of the Tubulin Lattice. Physical Review Letters. 2015;114:148101. https://doi.org/10.1103/PhysRevLett.114.148101.
Arnal I, Wade RH. How does taxol stabilize microtubules? Curr Biol. 1995;5:900–8 http://www.ncbi.nlm.nih.gov/pubmed/7583148.
Alushin GM, Lander GC, Kellogg EH, Zhang R, Baker D, Nogales E. High-resolution microtubule structures reveal the structural transitions in alphabeta-tubulin upon GTP hydrolysis. Cell. 2014;157:1117–29 http://www.ncbi.nlm.nih.gov/pubmed/24855948.
Castle BT, McCubbin S, Prahl LS, Bernens JN, Sept D, Odde DJ. Mechanisms of kinetic stabilization by the drugs paclitaxel and vinblastine. Mol Biol Cell. 2017;28:1238–57. https://doi.org/10.1091/mbc.e16-08-0567.
Hyman AA, Chretien D, Arnal I, Wade RH. Structural changes accompanying GTP hydrolysis in microtubules: information from a slowly hydrolyzable analogue guanylyl-(alpha,beta)-methylene-diphosphonate. J Cell Biol. 1995;128:117–25 http://www.ncbi.nlm.nih.gov/pubmed/7822409.
Amos LA, Löwe J. How taxol stabilises microtubule structure. Chem Biol. 1999;6:R65–9 http://www.ncbi.nlm.nih.gov/pubmed/10074470.
Geyer EA, Burns A, Lalonde BA, Ye X, Piedra F-A, Huffaker TC, Rice LM. A mutation uncouples the tubulin conformational and GTPase cycles, revealing allosteric control of microtubule dynamics. Elife. 2015;4:e10113. https://doi.org/10.7554/eLife.10113.
Machin NA, Lee JM, Barnes G. Microtubule stability in budding yeast: characterization and dosage suppression of a benomyl-dependent tubulin mutant. Mol Biol Cell. 1995;6:1241–59. https://doi.org/10.1091/mbc.6.9.1241.
Balabanian L, Berger CL, Hendricks AG. Acetylated microtubules are preferentially bundled leading to enhanced kinesin-1 motility. Biophys J. 2017;113:1551–60 http://www.sciencedirect.com/science/article/pii/S0006349517308664.
Samsonov A, Yu JZ, Rasenick M, Popov SV. Tau interaction with microtubules in vivo. J Cell Sci. 2004;117:6129–41 http://www.ncbi.nlm.nih.gov/pubmed/15564376.
Fourniol FJ, Sindelar CV, Amigues B, Clare DK, Thomas G, Perderiset M, Francis F, Houdusse A, Moores CA. Template-free 13-protofilament microtubule–MAP assembly visualized at 8 Å resolution. J Cell Biol. 2010;191:463–70 http://jcb.rupress.org/content/jcb/191/3/463.full.pdf.
Krebs A, Goldie KN, Hoenger A. Complex formation with kinesin motor domains affects the structure of microtubules. J Mol Biol. 2004;335:139–53 http://www.ncbi.nlm.nih.gov/pubmed/14659746.
Morikawa M, Yajima H, Nitta R, Inoue S, Ogura T, Sato C, Hirokawa N. X-ray and Cryo-EM structures reveal mutual conformational changes of Kinesin and GTP-state microtubules upon binding. EMBO J. 2015;34:1270–86 http://www.ncbi.nlm.nih.gov/pubmed/25777528.
Muto E, Sakai H, Kaseda K. Long-range cooperative binding of kinesin to a microtubule in the presence of ATP. J Cell Biol. 2005;168:691–6 http://www.ncbi.nlm.nih.gov/pubmed/15738263.
Cross RA. Microtubule lattice plasticity. Curr Opin Cell Biol. 2019;56:88–93 http://www.sciencedirect.com/science/article/pii/S0955067418301418.
Pearce SP, Heil M, Jensen OE, Jones GW, Prokop A. Curvature-sensitive kinesin binding can explain microtubule ring formation and reveals chaotic dynamics in a mathematical model. Bull Math Biol. 2018;80:3002–22 https://tinyurl.com/yd43ncb9.
Man Y, Kanso E. Morphological transitions of axially-driven microfilaments. Soft Matter. 2019;15:5163–73. https://doi.org/10.1039/C8SM02397B.
Memet E, Hilitski F, Morris MA, Schwenger WJ, Dogic Z, Mahadevan L. Microtubules soften due to cross-sectional flattening. Elife. 2018;7:e34695 http://www.ncbi.nlm.nih.gov/pubmed/29856317.
Kononova O, Kholodov Y, Theisen KE, Marx KA, Dima RI, Ataullakhanov FI, Grishchuk EL, Barsegov V. Tubulin bond energies and microtubule biomechanics determined from nanoindentation in silico. J Am Chem Soc. 2014;136:17036–45 https://doi.org/10.1021/ja506385p.
Kabir AM, Inoue D, Hamano Y, Mayama H, Sada K, Kakugo A. Biomolecular motor modulates mechanical property of microtubule. Biomacromolecules. 2014;15:1797–805 http://www.ncbi.nlm.nih.gov/pubmed/24697688.
Sturgill EG, Ohi R. Microtubule-regulating kinesins. Current Biology. 2013;23:R946–8 http://www.sciencedirect.com/science/article/pii/S0960982213009883.
Guzik-Lendrum S, Rayment I, Gilbert SP. Homodimeric kinesin-2 KIF3CC promotes microtubule dynamics. Biophys J. 2017;113:1845–57 http://www.ncbi.nlm.nih.gov/pubmed/29045878.
Chen Y, Hancock WO. Kinesin-5 is a microtubule polymerase. Nat Commun. 2015;6:8160 http://www.ncbi.nlm.nih.gov/pubmed/26437877.
Gumy LF, Chew DJ, Tortosa E, Katrukha EA, Kapitein LC, Tolkovsky AM, Hoogenraad CC, Fawcett JW. The kinesin-2 family member KIF3C regulates microtubule dynamics and is required for axon growth and regeneration. J Neurosci. 2013;33:11329–45 http://www.jneurosci.org/content/jneuro/33/28/11329.full.pdf.
Walczak CE, Gayek S, Ohi R. Microtubule-depolymerizing kinesins. Annu Rev Cell Dev Biol. 2013;29:417–41 http://www.ncbi.nlm.nih.gov/pubmed/23875646.
Strothman C, Farmer V, Arpağ G, Rodgers N, Podolski M, Norris S, Ohi R, Zanic M: Microtubule minus-end stability is dictated by the tubulin off-rate. J Cell Biol 2019:jcb.201905019. http://jcb.rupress.org/content/jcb/early/2019/08/14/jcb.201905019.full.pdf
Tas RP, Chazeau A, Cloin BMC, Lambers MLA, Hoogenraad CC, Kapitein LC. Differentiation between oppositely oriented microtubules controls polarized neuronal transport. Neuron. 2017;96:1264–71 https://www.sciencedirect.com/science/article/pii/S0896627317310711.
Zheng Y, Wildonger J, Ye B, Zhang Y, Kita A, Younger SH, Zimmerman S, Jan LY, Jan YN. Dynein is required for polarized dendritic transport and uniform microtubule orientation in axons. Nat Cell Biol. 2008;10:1172–80 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=18758451.
Chen Y, Rolls MM, Hancock WO. An EB1-kinesin complex is sufficient to steer microtubule growth in vitro. Curr Biol. 2014;24:316–21 http://www.ncbi.nlm.nih.gov/pubmed/24462004.
Allan VJ. Cytoplasmic dynein. Biochem Soc Trans. 2011;39:1169–78 http://www.ncbi.nlm.nih.gov/pubmed/21936784.
Appert-Rolland C, Ebbinghaus M, Santen L. Intracellular transport driven by cytoskeletal motors: General mechanisms and defects. Physics Reports. 2015;593:1–59 http://www.sciencedirect.com/science/article/pii/S037015731500335X.
Wortman JC, Shrestha UM, Barry DM, Garcia ML, Gross SP, Yu CC. Axonal transport: how high microtubule density can compensate for boundary effects in small-caliber axons. Biophys J. 2014;106:813–23 https://www.cell.com/biophysj/fulltext/S0006-3495(14)00075-7.
Pannese E, Ledda M, Arcidiacono G, Rigamonti L, Procacci P. A comparison of the density of microtubules in the central and peripheral axonal branches of the pseudounipolar neurons of lizard spinal ganglia. Anat Rec. 1984;208:595–605 http://www.ncbi.nlm.nih.gov/pubmed/6731865.
Fadić R, Vergara J, Alvarez J. Microtubules and caliber of central and peripheral processes of sensory axons. J Comp Neurol. 1985;236:258–64. https://doi.org/10.1002/cne.902360209.
Sirajuddin M, Rice LM, Vale RD. Regulation of microtubule motors by tubulin isotypes and post-translational modifications. Nat Cell Biol. 2014;16:335–44. https://doi.org/10.1038/ncb2920.
Monroy BY, Sawyer DL, Ackermann BE, Borden MM, Tan TC, Ori-McKenney KM. Competition between microtubule-associated proteins directs motor transport. Nat Commun. 2018;9:1487. https://doi.org/10.1038/s41467-018-03909-2.
Subramaniyan Parimalam S, Tarhan MC, Karsten SL, Fujita H, Shintaku H, Kotera H, Yokokawa R. On-chip microtubule gliding assay for parallel measurement of tau protein species. Lab on a Chip. 2016;16:1691–7. https://doi.org/10.1039/C5LC01486G.
Nakata T, Niwa S, Okada Y, Perez F, Hirokawa N. Preferential binding of a kinesin-1 motor to GTP-tubulin-rich microtubules underlies polarized vesicle transport. J Cell Biol. 2011;194:245–55 http://www.ncbi.nlm.nih.gov/pubmed/21768290.
Siahaan V, Krattenmacher J, Hernandez-Vega A, Hyman AA, Diez S, Lansky Z, Braun M. Kinetically distinct phases of tau on microtubules regulate kinesin motors and severing enzymes. Nat Cell Biol. 2019;21:1086–92. https://doi.org/10.1038/s41556-019-0374-6.
Tan R, Lam AJ, Tan T, Han J, Nowakowski DW, Vershinin M, Simo S, Ori-McKenney KM, McKenney RJ. Microtubules gate tau condensation to spatially regulate microtubule functions. Nat Cell Biol. 2019;21:1078–85. https://doi.org/10.1038/s41556-019-0375-5.
Dumont E. L. P., Do C, Hess H: Molecular wear of microtubules propelled by surface-adhered kinesins. Nat Nano. 2015;10:166–9. https://doi.org/10.1038/nnano.2014.334.
VanDelinder V, Adams PG, Bachand GD. Mechanical splitting of microtubules into protofilament bundles by surface-bound kinesin-1. Sci Rep. 2016;6:39408 http://www.ncbi.nlm.nih.gov/pubmed/28000714.
Triclin S, Inoue D, Gaillard J, Htet ZM, De Santis M, Portran D, Derivery E, Aumeier C, Schaedel L, John K, et al: Self-repair protects microtubules from their destruction by molecular motors. bioRxiv 2018:499020. https://www.biorxiv.org/content/biorxiv/early/2018/12/17/499020.full
Akhmanova A. Strengthening Microtubules by Cuts that Heal. Dev Cell. 2018;47:400–1 http://www.sciencedirect.com/science/article/pii/S1534580718309201.
Schaedel L, John K, Gaillard J, Nachury MV, Blanchoin L, Thery M. Microtubules self-repair in response to mechanical stress. Nat Mater. 2015;14:1156–63 http://www.ncbi.nlm.nih.gov/pubmed/26343914.
Baker D, O'Neill JK, Gschmeissner SE, Wilcox CE, Butter C, Turk JL. Induction of chronic relapsing experimental allergic encephalomyelitis in Biozzi mice. J Neuroimmunol. 1990;28:261–70 http://www.ncbi.nlm.nih.gov/pubmed/2373763.
Abdul-Majid KB, Jirholt J, Stadelmann C, Stefferl A, Kjellen P, Wallstrom E, Holmdahl R, Lassmann H, Olsson T, Harris RA. Screening of several H-2 congenic mouse strains identified H-2(q) mice as highly susceptible to MOG-induced EAE with minimal adjuvant requirement. J Neuroimmunol. 2000;111:23–33 http://www.ncbi.nlm.nih.gov/pubmed/11063818.
Kuijpers M, van de Willige D, Freal A, Chazeau A, Franker Mariella A, Hofenk J, Rodrigues Ricardo JC, Kapitein Lukas C, Akhmanova A, Jaarsma D, Hoogenraad Casper C. Dynein regulator NDEL1 controls polarized cargo transport at the axon initial segment. Neuron. 2016;89:461–71 http://www.sciencedirect.com/science/article/pii/S0896627316000477.
Stokin GB, Almenar-Queralt A, Gunawardena S, Rodrigues EM, Falzone T, Kim J, Lillo C, Mount SL, Roberts EA, McGowan E, et al. Amyloid precursor protein-induced axonopathies are independent of amyloid-beta peptides. Hum Mol Genet. 2008;17:3474–86 http://www.ncbi.nlm.nih.gov/pubmed/18694898.
Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, Goldstein LS. Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science. 2005;307:1282–8 http://www.ncbi.nlm.nih.gov/pubmed/15731448.
Bucci C, Bakke O, Progida C. Charcot-Marie-Tooth disease and intracellular traffic. Prog Neurobiol. 2012;99:191–225 http://www.ncbi.nlm.nih.gov/pubmed/22465036.
Blackstone C. Hereditary spastic paraplegia. Handb Clin Neurol. 2018;148:633–52 http://www.ncbi.nlm.nih.gov/pubmed/29478605.
Brunden KR, Trojanowski JQ, Smith AB 3rd, Lee VM, Ballatore C. Microtubule-stabilizing agents as potential therapeutics for neurodegenerative disease. Bioorg Med Chem. 2014;22:5040–9 http://www.ncbi.nlm.nih.gov/pubmed/24433963.
Zhang B, Carroll J, Trojanowski JQ, Yao Y, Iba M, Potuzak JS, Hogan AM, Xie SX, Ballatore C, Smith AB 3rd, et al. The microtubule-stabilizing agent, epothilone D, reduces axonal dysfunction, neurotoxicity, cognitive deficits, and Alzheimer-like pathology in an interventional study with aged tau transgenic mice. J Neurosci. 2012;32:3601–11 http://www.ncbi.nlm.nih.gov/pubmed/22423084.
Sheng ZH. The interplay of axonal energy homeostasis and mitochondrial trafficking and anchoring. Trends Cell Biol. 2017;27:403–16 http://www.ncbi.nlm.nih.gov/pubmed/28228333.
Skruber K, Read T-A, Vitriol EA: Reconsidering an active role for G-actin in cytoskeletal regulation. J Cell Sci 2018, 131:10.1242/jcs.203760. http://jcs.biologists.org/content/joces/131/1/jcs203760.full
Krendel M, Mooseker MS. Myosins: tails (and heads) of functional diversity. Physiology (Bethesda). 2005;20:239–51 http://www.ncbi.nlm.nih.gov/pubmed/16024512.
Bogoyevitch MA, Fairlie DP. A new paradigm for protein kinase inhibition: blocking phosphorylation without directly targeting ATP binding. Drug Discov Today. 2007;12:622–33 http://www.ncbi.nlm.nih.gov/pubmed/17706543.
Berg JM, Tymoczko JL, Stryer L: Biochemistry (5th edition). New York: W H Freeman; 2002.
Hall A, Lalli G. Rho and Ras GTPases in axon growth, guidance, and branching. Cold Spring Harb Perspect Biol. 2010;2:a001818 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=20182621.
McNally FJ, Roll-Mecak A. Microtubule-severing enzymes: From cellular functions to molecular mechanism. J Cell Biol. 2018;217:4057–69 http://jcb.rupress.org/content/early/2018/10/31/jcb.201612104.
Zala D, Hinckelmann M-V, Yu H, da Cunha L, Menezes M, Liot G, Cordelières Fabrice P, Marco S, Saudou F. Vesicular glycolysis provides on-board energy for fast axonal transport. Cell. 2013;152:479–91 http://www.sciencedirect.com/science/article/pii/S0092867412015516.
Hinckelmann MV, Virlogeux A, Niehage C, Poujol C, Choquet D, Hoflack B, Zala D, Saudou F. Self-propelling vesicles define glycolysis as the minimal energy machinery for neuronal transport. Nat Commun. 2016;7:13233 http://www.ncbi.nlm.nih.gov/pubmed/27775035.
McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Curr Biol. 2006;16:R551–60 http://www.ncbi.nlm.nih.gov/pubmed/16860735.
Wu Y, Whiteus C, Xu CS, Hayworth KJ, Weinberg RJ, Hess HF, De Camilli P. Contacts between the endoplasmic reticulum and other membranes in neurons. Proc Natl Acad Sci U S A. 2017;114:E4859–e4867 https://www.pnas.org/content/114/24/E4859.short.
Rieusset J. Endoplasmic reticulum-mitochondria calcium signaling in hepatic metabolic diseases. Biochim Biophys Acta Mol Cell Res. 1864;2017:865–76 http://www.ncbi.nlm.nih.gov/pubmed/28064001.
Kapur M, Wang W, Maloney MT, Millan I, Lundin VF, Tran TA, Yang Y. Calcium tips the balance: a microtubule plus end to lattice binding switch operates in the carboxyl terminus of BPAG1n4. EMBO Rep. 2012;13:1021–9 http://www.ncbi.nlm.nih.gov/pubmed/22995871.
McVicker DP, Millette MM, Dent EW. Signaling to the microtubule cytoskeleton: an unconventional role for CaMKII. Dev Neurobiol. 2015;75:423–34 http://www.ncbi.nlm.nih.gov/pubmed/25156276.
O'Brien ET, Salmon ED, Erickson HP. How calcium causes microtubule depolymerization. Cell Motility. 1997;36:125–35 https://onlinelibrary.wiley.com/doi/abs/10.1002/%28SICI%291097-0169%281997%2936%3A2%3C125%3A%3AAID-CM3%3E3.0.CO%3B2-8.
Fransen M, Lismont C, Walton P. The Peroxisome-Mitochondria Connection: How and Why? Int J Mol Sci. 2017;18:E1126 http://www.ncbi.nlm.nih.gov/pubmed/28538669.
Pascual-Ahuir A, Manzanares-Estreder S, Proft M. Pro- and antioxidant functions of the peroxisome-mitochondria connection and its impact on aging and disease. Oxid Med Cell Longev. 2017;2017:9860841 http://www.ncbi.nlm.nih.gov/pubmed/28811869.
Wilson C, Gonzalez-Billault C. Regulation of cytoskeletal dynamics by redox signaling and oxidative stress: implications for neuronal development and trafficking. Front Cell Neurosci. 2015;9:381 http://www.ncbi.nlm.nih.gov/pubmed/26483635.
Liu Z, Zhou T, Ziegler AC, Dimitrion P, Zuo L. Oxidative Stress in Neurodegenerative Diseases: From Molecular Mechanisms to Clinical Applications. Oxid Med Cell Longev. 2017;2017:2525967 http://www.ncbi.nlm.nih.gov/pubmed/28785371.
Wali G, Sutharsan R, Fan Y, Stewart R, Tello Velasquez J, Sue CM, Crane DI, Mackay-Sim A. Mechanism of impaired microtubule-dependent peroxisome trafficking and oxidative stress in SPAST-mutated cells from patients with Hereditary Spastic Paraplegia. Sci Rep. 2016;6:27004. https://doi.org/10.1038/srep27004.
Winckler B, Faundez V, Maday S, Cai Q, Guimas Almeida C, Zhang H. The endolysosomal system and proteostasis: from development to degeneration. J Neurosci. 2018;38:9364–74 http://www.ncbi.nlm.nih.gov/pubmed/30381428.
Poruchynsky MS, Sackett DL, Robey RW, Ward Y, Annunziata C, Fojo T. Proteasome inhibitors increase tubulin polymerization and stabilization in tissue culture cells: A possible mechanism contributing to peripheral neuropathy and cellular toxicity following proteasome inhibition. Cell Cycle. 2008;7:940–9. https://doi.org/10.4161/cc.7.7.5625.
Staff NP, Podratz JL, Grassner L, Bader M, Paz J, Knight AM, Loprinzi CL, Trushina E, Windebank AJ. Bortezomib alters microtubule polymerization and axonal transport in rat dorsal root ganglion neurons. Neurotoxicology. 2013;39:124–31 http://www.ncbi.nlm.nih.gov/pubmed/24035926.
Kleele T, Marinković P, Williams PR, Stern S, Weigand EE, Engerer P, Naumann R, Hartmann J, Karl RM, Bradke F, et al. An assay to image neuronal microtubule dynamics in mice. Nat Commun. 2014;5:4827. https://doi.org/10.1038/ncomms5827.
Stiess M, Maghelli N, Kapitein LC, Gomis-Ruth S, Wilsch-Brauninger M, Hoogenraad CC, Tolic-Norrelykke IM, Bradke F. Axon extension occurs independently of centrosomal microtubule nucleation. Science. 2010;327:704–7 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=20056854.
Basto R, Lau J, Vinogradova T, Gardiol A, Woods CG, Khodjakov A, Raff JW. Flies without centrioles. Cell. 2006;125:1375–86 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16814722.
Sulimenko V, Hájková Z, Klebanovych A, Dráber P. Regulation of microtubule nucleation mediated by γ-tubulin complexes. Protoplasma. 2017;254:1187–99 https://doi.org/10.1007/s00709-016-1070-z.
Tann J, Moore A: MTOC organization and competition during neuron differentiation. In The Golgi apparatus and centriole Results and problems in cell differentiation. Volume 67. Edited by M K: Springer, Cham; 2019: 337-357. https://doi.org/10.1007/978-3-030-23173-6_14
Thawani A, Kadzik RS, Petry S. XMAP215 is a microtubule nucleation factor that functions synergistically with the γ-tubulin ring complex. Nature Cell Biology. 2018;20:575–85. https://doi.org/10.1038/s41556-018-0091-6.
Brandt R, Lee G. The balance between tau protein's microtubule growth and nucleation activities: implications for the formation of axonal microtubules. J Neurochem. 1993;61:997–1005 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=8360696.
Nashchekin D, Fernandes Artur R, St Johnston D. Patronin/Shot cortical foci assemble the noncentrosomal microtubule array that specifies the Drosophila anterior-posterior axis. Dev Cell. 2016;38:61–72. https://doi.org/10.1016/j.devcel.2016.06.010.
Cunha-Ferreira I, Chazeau A, Buijs RR, Stucchi R, Will L, Pan X, Adolfs Y, van der Meer C, Wolthuis JC, Kahn OI, et al. The HAUS complex is a key regulator of non-centrosomal microtubule organization during neuronal development. Cell Rep. 2018;24:791–800. https://doi.org/10.1016/j.celrep.2018.06.093.
Sanchez-Huertas C, Freixo F, Viais R, Lacasa C, Soriano E, Lüders J. Non-centrosomal nucleation mediated by augmin organizes microtubules in post-mitotic neurons and controls axonal microtubule polarity. Nat Commun. 2016;7:12187. https://doi.org/10.1038/ncomms12187.
Chen L, Stone MC, Tao J, Rolls MM. Axon injury and stress trigger a microtubule-based neuroprotective pathway. Proc Natl Acad Sci U S A. 2012;109:11842–7 https://www.pnas.org/content/pnas/109/29/11842.full.pdf.
Verma V, Maresca TJ: Direct observation of branching MT nucleation in living animal cells. J Cell Biol 2019:jcb.201904114. http://jcb.rupress.org/content/jcb/early/2019/07/23/jcb.201904114.full.pdf
Al-Bassam J. Revisiting the tubulin cofactors and Arl2 in the regulation of soluble αβ-tubulin pools and their effect on microtubule dynamics. Mol Biol Cell. 2017;28:359–63 http://www.molbiolcell.org/content/28/3/359.abstract.
Preitner N, Quan J, Nowakowski Dan W, Hancock Melissa L, Shi J, Tcherkezian J, Young-Pearse Tracy L, Flanagan John G. APC is an RNA-binding protein, and its interactome provides a link to neural development and microtubule assembly. Cell. 2014;158:368–82 http://www.sciencedirect.com/science/article/pii/S0092867414007478.
Gasic I, Mitchison TJ. Autoregulation and repair in microtubule homeostasis. Curr Opin Cell Biol. 2018;56:80–7 http://www.ncbi.nlm.nih.gov/pubmed/30415186.
Letourneau PC, Shattuck TA, Ressler AH. "Pull" and "push" in neurite elongation: observations on the effects of different concentrations of cytochalasin B and taxol. Cell Motil Cytoskeleton. 1987;8:193–209 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=2891448.
Dennerll TJ, Joshi HC, Steel VL, Buxbaum RE, Heidemann SR. Tension and compression in the cytoskeleton of PC-12 neurites. II: Quantitative measurements. J Cell Biol. 1988;107:665–74 http://www.ncbi.nlm.nih.gov/pubmed/3417767.
Datar A, Ameeramja J, Bhat A, Srivastava R, Bernal R, Prost J, Callan-Jones A, Pullarkat PA: The roles of microtubules and membrane tension in axonal beading, retraction, and atrophy. bioRxiv 2019:10.1101/575258. https://www.biorxiv.org/content/biorxiv/early/2019/03/12/575258.full
Zhou R, Han B, Xia C, Zhuang X. Membrane-associated periodic skeleton is a signaling platform for RTK transactivation in neurons. Science. 2019;365:929–34 https://science.sciencemag.org/content/sci/365/6456/929.full.pdf.
Zanic M, Widlund PO, Hyman AA, Howard J. Synergy between XMAP215 and EB1 increases microtubule growth rates to physiological levels. Nat Cell Biol. 2013;15:688–93. https://doi.org/10.1038/ncb2744.
Gupta KK, Alberico EO, Nathke IS, Goodson HV. Promoting microtubule assembly: A hypothesis for the functional significance of the +TIP network. Bioessays. 2014;36:818–26 http://www.ncbi.nlm.nih.gov/pubmed/24943963.
Edvardson S, Cinnamon Y, Jalas C, Shaag A, Maayan C, Axelrod FB, Elpeleg O. Hereditary sensory autonomic neuropathy caused by a mutation in dystonin. Ann Neurol. 2012;71:569–72 http://www.ncbi.nlm.nih.gov/pubmed/22522446.
de Rooij R, Kuhl E, Miller KE. Modeling the axon as an active partner with the growth cone in axonal elongation. Biophys J. 2018;115:1783–95 http://www.ncbi.nlm.nih.gov/pubmed/30309611.
Fan A, Tofangchi A, Kandel M, Popescu G, Saif T. Coupled circumferential and axial tension driven by actin and myosin influences in vivo axon diameter. Sci Rep. 2017;7:14188 http://www.ncbi.nlm.nih.gov/pubmed/29079766.
Heidemann SR, Buxbaum RE. Tension as a regulator and integrator of axonal growth. Cell Motil Cytoskeleton. 1990;17:6–10 http://www.ncbi.nlm.nih.gov/pubmed/2225090.
Koch D, Rosoff WJ, Jiang J, Geller HM, Urbach JS. Strength in the periphery: growth cone biomechanics and substrate rigidity response in peripheral and central nervous system neurons. Biophys J. 2012;102:452–60 http://www.ncbi.nlm.nih.gov/pubmed/22325267.
Chan CE, Odde DJ. Traction dynamics of filopodia on compliant substrates. Science. 2008;322:1687–91 http://www.ncbi.nlm.nih.gov/pubmed/19074349.
Lamoureux P, Buxbaum RE, Heidemann SR. Direct evidence that growth cones pull. Nature. 1989;340:159–62 https://www.nature.com/articles/340159a0.
O'Toole M, Lamoureux P, Miller Kyle E. Measurement of subcellular force generation in neurons. Biophys J. 2015;108:1027–37 http://www.sciencedirect.com/science/article/pii/S0006349515001150.
Buxbaum RE, Heidemann SR. An absolute rate theory model for tension control of axonal elongation. J Theor Biol. 1992;155:409–26 http://www.ncbi.nlm.nih.gov/pubmed/1619959.
Lamoureux P, Heidemann SR, Martzke NR, Miller KE. Growth and elongation within and along the axon. Dev Neurobiol. 2010;70:135–49 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=19950193.
Bray D. Axonal growth in response to experimentally applied mechanical tension. Dev Biol. 1984;102:379–89. https://doi.org/10.1016/0012-1606(84)90202-1.
Pfister BJ, Iwata A, Meaney DF, Smith DH. Extreme stretch growth of integrated axons. J Neurosci. 2004;24:7978–83 http://www.ncbi.nlm.nih.gov/pubmed/15356212.
Zheng J, Lamoureux P, Santiago V, Dennerll T, Buxbaum RE, Heidemann SR. Tensile regulation of axonal elongation and initiation. J Neurosci. 1991;11:1117–25 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=2010807.
Ahmad FJ, Hughey J, Wittmann T, Hyman A, Greaser M, Baas PW. Motor proteins regulate force interactions between microtubules and microfilaments in the axon. Nat Cell Biol. 2000;2:276–80 http://www.ncbi.nlm.nih.gov/pubmed/10806478.
Ketschek AR, Jones SL, Gallo G. Axon extension in the fast and slow lanes: substratum-dependent engagement of myosin II functions. Dev Neurobiol. 2007;67:1305–20 http://www.ncbi.nlm.nih.gov/pubmed/17638383.
Turney SG, Ahmed M, Chandrasekar I, Wysolmerski RB, Goeckeler ZM, Rioux RM, Whitesides GM, Bridgman PC. Nerve growth factor stimulates axon outgrowth through negative regulation of growth cone actomyosin restraint of microtubule advance. Mol Biol Cell. 2016;27:500–17 http://www.molbiolcell.org/content/early/2015/11/29/mbc.E15-09-0636.abstract.
Wylie SR, Chantler PD. Myosin IIC: a third molecular motor driving neuronal dynamics. Mol Biol Cell. 2008;19:3956–68 http://www.ncbi.nlm.nih.gov/pubmed/18614800.
Kaverina I, Krylyshkina O, Beningo K, Anderson K, Wang YL, Small JV. Tensile stress stimulates microtubule outgrowth in living cells. Journal of cell science. 2002;115:2283–91 http://www.ncbi.nlm.nih.gov/pubmed/12006613.
Winding M, Kelliher MT, Lu W, Wildonger J, Gelfand VI. Role of kinesin-1-based microtubule sliding in Drosophila nervous system development. Proc Natl Acad Sci U S A. 2016;113:E4985–94 http://www.ncbi.nlm.nih.gov/pubmed/27512046.
Roossien DH, Lamoureux P, Miller KE. Cytoplasmic dynein pushes the cytoskeletal meshwork forward during axonal elongation. J Cell Sci. 2014;127:3593–602 http://www.ncbi.nlm.nih.gov/pubmed/24951117.
Hamant O, Inoue D, Bouchez D, Dumais J, Mjolsness E. Are microtubules tension sensors? Nat Commun. 2019;10:2360. https://doi.org/10.1038/s41467-019-10207-y.
Franze K, Gerdelmann J, Weick M, Betz T, Pawlizak S, Lakadamyali M, Bayer J, Rillich K, Gogler M, Lu YB, et al. Neurite branch retraction is caused by a threshold-dependent mechanical impact. Biophys J. 2009;97:1883–90 http://www.ncbi.nlm.nih.gov/pubmed/19804718.
Song Y, Li D, Farrelly O, Miles L, Li F, Kim SE, Lo TY, Wang F, Li T, Thompson-Peer KL, et al: The mechanosensitive ion channel Piezo inhibits axon regeneration. Neuron 2019, 102:373-389 e376. http://www.ncbi.nlm.nih.gov/pubmed/30819546
Koser DE, Thompson AJ, Foster SK, Dwivedy A, Pillai EK, Sheridan GK, Svoboda H, Viana M, Costa LD, Guck J, et al. Mechanosensing is critical for axon growth in the developing brain. Nat Neurosci. 2016;19:1592–8 http://www.ncbi.nlm.nih.gov/pubmed/27643431.
van der Vaart B, van Riel WE, Doodhi H, Kevenaar Josta T, Katrukha Eugene A, Gumy L, Bouchet Benjamin P, Grigoriev I, Spangler Samantha A, Yu Ka L, et al. CFEOM1-associated kinesin KIF21A is a cortical microtubule growth inhibitor. Developmental Cell. 2013;27:145–60. https://doi.org/10.1016/j.devcel.2013.09.010.
Pines MK, Housden BE, Bernard F, Bray SJ, Roper K. The cytolinker Pigs is a direct target and a negative regulator of Notch signalling. Development. 2010;137:913–22 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=20150280.
Stroud MJ, Nazgiewicz A, McKenzie EA, Wang Y, Kammerer RA, Ballestrem C. GAS2-like proteins mediate communication between microtubules and actin through interactions with end-binding proteins. J Cell Sci. 2014;127:2672–82 http://www.ncbi.nlm.nih.gov/pubmed/24706950.
van de Willige D, Hummel JJ, Alkemade C, Kahn OI, Au FK, Qi RZ, Dogterom M, Koenderink GH, Hoogenraad CC, Akhmanova A: Cytolinker Gas2L1 regulates axon morphology through microtubule-modulated actin stabilization. EMBO Reports 2019, 0:e47732. https://www.embopress.org/doi/abs/10.15252/embr.201947732
Mattie FJ, Stackpole MM, Stone MC, Clippard JR, Rudnick DA, Qiu Y, Tao J, Allender DL, Parmar M, Rolls MM. Directed microtubule growth, +TIPs, and kinesin-2 are required for uniform microtubule polarity in dendrites. Curr Biol. 2010;20:2169–77 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=21145742.
Ageta-Ishihara N, Miyata T, Ohshima C, Watanabe M, Sato Y, Hamamura Y, Higashiyama T, Mazitschek R, Bito H, Kinoshita M. Septins promote dendrite and axon development by negatively regulating microtubule stability via HDAC6-mediated deacetylation. Nat Commun. 2013;4:2532. https://doi.org/10.1038/ncomms3532.
Bowen JR, Hwang D, Bai X, Roy D, Spiliotis ET. Septin GTPases spatially guide microtubule organization and plus end dynamics in polarizing epithelia. J Cell Biol. 2011;194:187–97 www.jcb.org/cgi/doi/10.1083/jcb.201102076.
Hu J, Bai X, Bowen JR, Dolat L, Korobova F, Yu W, Baas PW, Svitkina T, Gallo G, Spiliotis ET. Septin-driven coordination of actin and microtubule remodeling regulates the collateral branching of axons. Curr Biol. 2012;22:1109–15 http://www.ncbi.nlm.nih.gov/pubmed/22608511.
Homma N, Takei Y, Tanaka Y, Nakata T, Terada S, Kikkawa M, Noda Y, Hirokawa N. Kinesin superfamily protein 2A (KIF2A) functions in suppression of collateral branch extension. Cell. 2003;114:229–39 http://www.ncbi.nlm.nih.gov/pubmed/12887924.
Mao C-X, Xiong Y, Xiong Z, Wang Q, Zhang YQ, Jin S. Microtubule-severing protein Katanin regulates neuromuscular junction development and dendritic elaboration in Drosophila. Development. 2014;141:1064–74 http://dev.biologists.org/content/141/5/1064.abstract.
Tao J, Feng C, Rolls MM. The microtubule-severing protein fidgetin acts after dendrite injury to promote their degeneration. J Cell Sci. 2016;129:3274–81 http://www.ncbi.nlm.nih.gov/pubmed/27411367.
Leo L, Yu W, D'Rozario M, Waddell EA, Marenda DR, Baird MA, Davidson MW, Zhou B, Wu B, Baker L, et al. Vertebrate fidgetin restrains axonal growth by severing labile domains of microtubules. Cell Rep. 2015;12:1723–30. https://doi.org/10.1016/j.celrep.2015.08.017.
Yu W, Qiang L, Solowska JM, Karabay A, Korulu S, Baas PW. The microtubule-severing proteins spastin and katanin participate differently in the formation of axonal branches. Mol Biol Cell. 2008;19:1485–98 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=18234839.
Stewart A, Tsubouchi A, Rolls MM, Tracey WD, Sherwood NT. Katanin p60-like1 promotes microtubule growth and terminal dendrite stability in the larval class IV sensory neurons of Drosophila. J Neurosc. 2012;32:11631–42 http://www.jneurosci.org/content/jneuro/32/34/11631.full.
Wood JD, Landers JA, Bingley M, McDermott CJ, Thomas-McArthur V, Gleadall LJ, Shaw PJ, Cunliffe VT. The microtubule-severing protein Spastin is essential for axon outgrowth in the zebrafish embryo. Hum Mol Genet. 2006;15:2763–71 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16893913.
Stone MC, Rao K, Gheres KW, Kim S, Tao J, La Rochelle C, Folker CT, Sherwood NT, Rolls MM. Normal spastin gene dosage is specifically required for axon regeneration. Cell Rep. 2012;2:1340–50 http://www.ncbi.nlm.nih.gov/pubmed/23122959.
Karabay A, Yu W, Solowska JM, Baird DH, Baas PW. Axonal growth is sensitive to the levels of katanin, a protein that severs microtubules. J Neurosci. 2004;24:5778–88 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15215300.
Ahmad FJ, Yu W, McNally FJ, Baas PW. An essential role for katanin in severing microtubules in the neuron. J Cell Biol. 1999;145:305–15 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=10209026.
Butler R, Wood JD, Landers JA, Cunliffe VT. Genetic and chemical modulation of spastin-dependent axon outgrowth in zebrafish embryos indicates a role for impaired microtubule dynamics in hereditary spastic paraplegia. Dis Model Mech. 2010;3:743–51 http://dmm.biologists.org/content/3/11-12/743.long.
Riano E, Martignoni M, Mancuso G, Cartelli D, Crippa F, Toldo I, Siciliano G, Di Bella D, Taroni F, Bassi MT, et al. Pleiotropic effects of spastin on neurite growth depending on expression levels. J Neurochem. 2009;108:1277–88. https://doi.org/10.1111/j.1471-4159.2009.05875.x.
Zhang D, Rogers GC, Buster DW, Sharp DJ. Three microtubule severing enzymes contribute to the "Pacman-flux" machinery that moves chromosomes. J Cell Biol. 2007;177:231–42 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17452528.
Sharp DJ, Ross JL. Microtubule-severing enzymes at the cutting edge. J Cell Sci. 2012;125:2561–9 http://www.ncbi.nlm.nih.gov/pubmed/22595526.
Shin SC, Im S-K, Jang E-H, Jin KS, Hur E-M, Kim EE: Structural and molecular basis for katanin-mediated severing of glutamylated microtubules. Cell Reports 2019, 26:1357-1367.e1355. https://doi.org/10.1016/j.celrep.2019.01.020
Bailey ME, Sackett DL, Ross JL. Katanin Severing and Binding Microtubules Are Inhibited by Tubulin Carboxy Tails. Biophys J. 2015;109:2546–61 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4699919/?otool=igbumllib.
Lacroix B, van Dijk J, Gold ND, Guizetti J, Aldrian-Herrada G, Rogowski K, Gerlich DW, Janke C. Tubulin polyglutamylation stimulates spastin-mediated microtubule severing. J Cell Biol. 2010;189:945–54 http://www.ncbi.nlm.nih.gov/pubmed/20530212.
Sudo H, Baas PW. Acetylation of microtubules influences their sensitivity to severing by katanin in neurons and fibroblasts. J Neurosci. 2010;30:7215–26 http://www.ncbi.nlm.nih.gov/pubmed/20505088.
Qiang L, Yu W, Andreadis A, Luo M, Baas PW. Tau protects microtubules in the axon from severing by katanin. J Neurosci. 2006;26:3120–9 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16554463.
Qiang L, Sun X, Austin TO, Muralidharan H, Jean DC, Liu M, Yu W, Baas PW: Tau does not stabilize axonal microtubules but rather enables them to have long labile domains. Curr Biol 2018, 28:2181-2189 e2184. http://www.ncbi.nlm.nih.gov/pubmed/30008334
Jiang K, Faltova L, Hua S, Capitani G, Prota AE, Landgraf C, Volkmer R, Kammerer RA, Steinmetz MO, Akhmanova A: Structural basis of formation of the microtubule minus-end-regulating CAMSAP-katanin complex. Structure 2018, 26:375-382 e374. http://www.ncbi.nlm.nih.gov/pubmed/29395789
Belonogov L, Bailey ME, Tyler MA, Kazemi A, Ross JL. Katanin catalyzes microtubule depolymerization independently of tubulin C- terminal tails. Cytoskeleton (Hoboken). 2019;76:254–68 http://www.ncbi.nlm.nih.gov/pubmed/30980604.
Vemu A, Szczesna E, Zehr EA, Spector JO, Grigorieff N, Deaconescu AM, Roll-Mecak A: Severing enzymes amplify microtubule arrays through lattice GTP-tubulin incorporation. Science 2018, 361:eaau1504. http://www.ncbi.nlm.nih.gov/pubmed/30139843
Goodwin SS, Vale RD. Patronin regulates the microtubule network by protecting microtubule minus ends. Cell. 2010;143:263–74 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=20946984.
Pongrakhananon V, Saito H, Hiver S, Abe T, Shioi G, Meng W, Takeichi M. CAMSAP3 maintains neuronal polarity through regulation of microtubule stability. Proc Natl Acad Sci U S A. 2018;115:9750–5 https://www.pnas.org/content/pnas/115/39/9750.full.pdf.
Diaz-Valencia JD, Morelli MM, Bailey M, Zhang D, Sharp DJ, Ross JL. Drosophila katanin-60 depolymerizes and severs at microtubule defects. Biophys J. 2011;100:2440–9 http://www.ncbi.nlm.nih.gov/pubmed/21575578.
Davis LJ, Odde DJ, Block SM, Gross SP. The importance of lattice defects in katanin-mediated microtubule mevering in vitro. Biophysical Journal. 2002;82:2916–27. https://doi.org/10.1016/S0006-3495(02)75632-4.
Gardner MK, Zanic M, Gell C, Bormuth V, Howard J. Depolymerizing kinesins Kip3 and MCAK shape cellular microtubule architecture by differential control of catastrophe. Cell. 2011;147:1092–103 http://www.ncbi.nlm.nih.gov/pubmed/22118464.
Wali G, Sue CM, Mackay-Sim A. Patient-Derived Stem Cell Models in SPAST HSP: Disease Modelling and Drug Discovery. Brain Sci. 2018;8:E142 http://www.ncbi.nlm.nih.gov/pubmed/30065201.
Koh K, Ishiura H, Tsuji S, Takiyama Y. JASPAC: Japan Spastic Paraplegia Research Consortium. Brain Sci. 2018;8:E153 http://www.ncbi.nlm.nih.gov/pubmed/30104498.
Schüle R, Wiethoff S, Martus P, Karle KN, Otto S, Klebe S, Klimpe S, Gallenmüller C, Kurzwelly D, Henkel D, et al. Hereditary spastic paraplegia: Clinicogenetic lessons from 608 patients. Ann Neurol. 2016;79:646–58. https://doi.org/10.1002/ana.24611.
Zempel H, Mandelkow EM. Tau missorting and spastin-induced microtubule disruption in neurodegeneration: Alzheimer Disease and Hereditary Spastic Paraplegia. Mol Neurodegener. 2015;10:68 http://www.ncbi.nlm.nih.gov/pubmed/26691836.
Shoukier M, Neesen J, Sauter SM, Argyriou L, Doerwald N, Pantakani DV, Mannan AU. Expansion of mutation spectrum, determination of mutation cluster regions and predictive structural classification of SPAST mutations in hereditary spastic paraplegia. Eur J Hum Genet. 2009;17:187–94 http://www.ncbi.nlm.nih.gov/pubmed/18701882.
Park SH, Zhu PP, Parker RL, Blackstone C. Hereditary spastic paraplegia proteins REEP1, spastin, and atlastin-1 coordinate microtubule interactions with the tubular ER network. J Clin Invest. 2010;120:1097–110 https://www.jci.org/articles/view/40979.
Allison R, Edgar JR, Pearson G, Rizo T, Newton T, Gunther S, Berner F, Hague J, Connell JW, Winkler J, et al. Defects in ER-endosome contacts impact lysosome function in hereditary spastic paraplegia. J Cell Biol. 2017;216:1337–55 http://www.ncbi.nlm.nih.gov/pubmed/28389476.
Papadopoulos C, Orso G, Mancuso G, Herholz M, Gumeni S, Tadepalle N, Jungst C, Tzschichholz A, Schauss A, Honing S, et al. Spastin binds to lipid droplets and affects lipid metabolism. PLoS Genet. 2015;11:e1005149 http://www.ncbi.nlm.nih.gov/pubmed/25875445.
Solowska JM, Baas PW. Hereditary spastic paraplegia SPG4: what is known and not known about the disease. Brain. 2015;138:2471–84 http://www.ncbi.nlm.nih.gov/pubmed/26094131.
Lee G, Brandt R. Microtubule bundling studies revisited: is there a role for MAPs? Trends in Cell Biology. 1992;2:286–9 http://www.sciencedirect.com/science/article/pii/096289249290106W.
Matus A. Microtubule-associated proteins and neuronal morphogenesis. J Cell Sci Suppl. 1991;15:61–7 http://www.ncbi.nlm.nih.gov/pubmed/1824108.
Peter SJ, Mofrad MR. Computational modeling of axonal microtubule bundles under tension. Biophys J. 2012;102:749–57 https://www.ncbi.nlm.nih.gov/pubmed/22385845.
de Rooij R, Kuhl E. Microtubule polymerization and cross-link dynamics explain axonal stiffness and damage. Biophys J. 2018;114:201–12 https://www.sciencedirect.com/science/article/pii/S0006349517312390.
Lazarus C, Soheilypour M, Mofrad Mohammad RK. Torsional behavior of axonal microtubule bundles. Biophys J. 2015;109:231–9 http://www.sciencedirect.com/science/article/pii/S0006349515006128.
Li S, Wang C, Nithiarasu P. Effects of the cross-linkers on the buckling of microtubules in cells. J Biomech. 2018;72:167–72 http://www.sciencedirect.com/science/article/pii/S0021929018301544.
Chen J, Kanai Y, Cowan NJ, Hirokawa N. Projection domains of MAP 2 and tau determine spacings between microtubules in dendrites and axons. Nature. 1992;360:674–7 http://www.ncbi.nlm.nih.gov/pubmed/1465130.
Bodakuntla S, Jijumon AS, Villablanca C, Gonzalez-Billault C, Janke C. Microtubule-Associated Proteins: Structuring the Cytoskeleton. Trends Cell Biol. 2019. https://doi.org/10.1016/j.tcb.2019.1007.1004 http://www.sciencedirect.com/science/article/pii/S0962892419301205.
Chapin SJ, Bulinski JC, Gundersen GG. Microtubule bundling in cells. Nature. 1991;349:24 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=1670738.
Kader MA, Satake T, Yoshida M, Hayashi I, Suzuki A. Molecular basis of the microtubule-regulating activity of microtubule crosslinking factor 1. PLoS One. 2017;12:e0182641 https://doi.org/10.1371/journal.pone.0182641.
DeBonis S, Neumann E, Skoufias DA. Self protein-protein interactions are involved in TPPP/p25 mediated microtubule bundling. Sci Rep. 2015;5:13242 http://www.ncbi.nlm.nih.gov/pubmed/26289831.
Goriounov D, Leung CL, Liem RK. Protein products of human Gas2-related genes on chromosomes 17 and 22 (hGAR17 and hGAR22) associate with both microfilaments and microtubules. Journal of cell science. 2003;116:1045–58 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=12584248.
Scaife R, Margolis RL. Biochemical and immunochemical analysis of rat brain dynamin interaction with microtubules and organelles in vivo and in vitro. J Cell Biol. 1990;111:3023–33 http://www.ncbi.nlm.nih.gov/pubmed/2148566.
Shpetner HS, Vallee RB. Identification of dynamin, a novel mechanochemical enzyme that mediates interactions between microtubules. Cell. 1989;59:421–32 http://www.ncbi.nlm.nih.gov/pubmed/2529977.
Züchner S, Noureddine M, Kennerson M, Verhoeven K, Claeys K, De Jonghe P, Merory J, Oliveira SA, Speer MC, Stenger JE, et al. Mutations in the pleckstrin homology domain of dynamin 2 cause dominant intermediate Charcot-Marie-Tooth disease. Nat Genet. 2005;37:289–94 http://www.ncbi.nlm.nih.gov/pubmed/15731758.
Pant HC. Veeranna: Neurofilament phosphorylation. Biochem Cell Biol. 1995;73:575–92 http://www.ncbi.nlm.nih.gov/pubmed/8714676.
Villarroel-Campos D, Gonzalez-Billault C. The MAP 1B case: an old MAP that is new again. Dev Neurobiol. 2014;74:953–71 http://www.ncbi.nlm.nih.gov/pubmed/24700609.
Roos J, Hummel T, Ng N, Klämbt C, Davis GW. Drosophila Futsch regulates synaptic microtubule organisation and is necessary for synaptic growth. Neuron. 2000;26:371–82 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=10839356.
Feltrin D, Fusco L, Witte H, Moretti F, Martin K, Letzelter M, Fluri E, Scheiffele P, Pertz O. Growth cone MKK7 mRNA targeting regulates MAP 1b-dependent microtubule bundling to control neurite elongation. PLoS Biol. 2012;10:e1001439 http://www.ncbi.nlm.nih.gov/pubmed/23226105.
Bettencourt da Cruz A, Schwarzel M, Schulze S, Niyyati M, Heisenberg M, Kretzschmar D. Disruption of the MAP 1B-related protein FUTSCH leads to changes in the neuronal cytoskeleton, axonal transport defects, and progressive neurodegeneration in Drosophila. Mol Biol Cell. 2005;16:2433–42 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15772149.
Stephan R, Goellner B, Moreno E, Frank CA, Hugenschmidt T, Genoud C, Aberle H, Pielage J. Hierarchical microtubule organization controls axon caliber and transport and determines synaptic structure and stability. Dev Cell. 2015;33:1–17 http://www.sciencedirect.com/science/article/pii/S1534580715001057.
Hummel T, Krukkert K, Roos J, Davis G, Klämbt C. Drosophila Futsch/22C10 is a MAP 1B-like protein required for dendritic and axonal development. Neuron. 2000;26:357–70 https://www.cell.com/neuron/fulltext/S0896-6273(00)81169-1.
Migh E, Götz T, Földi I, Szikora S, Gombos R, Darula Z, Medzihradszky KF, Maléth J, Hegyi P, Sigrist S, Mihály J: Microtubule organization in presynaptic boutons relies on the formin DAAM. Development 2018:dev.158519. http://dev.biologists.org/content/develop/early/2018/02/21/dev.158519.full
Penazzi L, Bakota L, Brandt R. Microtubule dynamics in neuronal development, plasticity, and neurodegeneration. Int Rev Cell Mol Biol. 2016;321:89–169 http://www.ncbi.nlm.nih.gov/pubmed/26811287.
Satake T, Yamashita K, Hayashi K, Miyatake S, Tamura-Nakano M, Doi H, Furuta Y, Shioi G, Miura E, Takeo YH, et al. MTCL1 plays an essential role in maintaining Purkinje neuron axon initial segment. EMBO J. 2017;36:1227–42 http://emboj.embopress.org/content/embojnl/36/9/1227.full.
Sato Y, Akitsu M, Amano Y, Yamashita K, Ide M, Shimada K, Yamashita A, Hirano H, Arakawa N, Maki T, et al. The novel PAR-1-binding protein MTCL1 has crucial roles in organizing microtubules in polarizing epithelial cells. Journal of cell science. 2013;126:4671–83 http://www.ncbi.nlm.nih.gov/pubmed/23902687.
Peters A, Palay SL. Webster Hd: The fine structure of the nervous system: neurons and their supporting cells. 3rd ed. New York, Oxford: Oxford University Press; 1991. https://books.google.co.uk/books/about/The_Fine_Structure_of_the_Nervous_System.html?id=eapqAAAAMAAJ&source=kp_cover&redir_esc=y
Harterink M, Vocking K, Pan X, Soriano Jerez EM, Slenders L, Freal A, Tas RP, van de Wetering WJ, Timmer K, Motshagen J, et al. TRIM46 organizes microtubule fasciculation in the axon initial segment. J Neurosci. 2019;39:4864–73 http://www.ncbi.nlm.nih.gov/pubmed/30967428.
van Beuningen Sam FB, Will L, Harterink M, Chazeau A, van Battum EY, Frias Cátia P, Franker Mariella AM, Katrukha Eugene A, Stucchi R, Vocking K, et al. TRIM46 controls neuronal polarity and axon specification by driving the formation of parallel microtubule arrays. Neuron. 2015;88:1208–26 https://doi.org/10.1016/j.neuron.2015.11.012.
Chung PJ, Song C, Deek J, Miller HP, Li Y, Choi MC, Wilson L, Feinstein SC, Safinya CR. Tau mediates microtubule bundle architectures mimicking fascicles of microtubules found in the axon initial segment. Nat Commun. 2016;7:12278 https://doi.org/10.1038/ncomms12278.
Rosenberg KJ, Ross JL, Feinstein HE, Feinstein SC, Israelachvili J. Complementary dimerization of microtubule-associated tau protein: Implications for microtubule bundling and tau-mediated pathogenesis. Proc Natl Acad Sci U S A. 2008;105:7445–50 http://www.ncbi.nlm.nih.gov/pubmed/18495933.
Méphon-Gaspard A, Boca M, Pioche-Durieu C, Desforges B, Burgo A, Hamon L, Piétrement O, Pastré D. Role of tau in the spatial organization of axonal microtubules: keeping parallel microtubules evenly distributed despite macromolecular crowding. Cell Mol Life Sci. 2016;73:3745–60 https://www.ncbi.nlm.nih.gov/pubmed/27076215.
Janning D, Igaev M, Sundermann F, Bruhmann J, Beutel O, Heinisch JJ, Bakota L, Piehler J, Junge W, Brandt R. Single-molecule tracking of tau reveals fast kiss-and-hop interaction with microtubules in living neurons. Mol Biol Cell. 2014;25:3541–51 http://www.ncbi.nlm.nih.gov/pubmed/25165145.
Harada A, Oguchi K, Okabe S, Kuno J, Terada S, Ohshima T, Sato-Yoshitake R, Takei Y, Noda T, Hirokawa N. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature. 1994;369:488–91 http://www.ncbi.nlm.nih.gov/pubmed/8202139.
Shin S, Lim S, Jeong H, Kwan L, Kim Y. Visualization of tau–tubulin interaction in a living cell using bifluorescence complementation technique. International Journal of Molecular Sciences. 2018;19:2978 http://www.mdpi.com/1422-0067/19/10/2978.
Prezel E, Elie A, Delaroche J, Stoppin-Mellet V, Bosc C, Serre L, Fourest-Lieuvin A, Andrieux A, Vantard M, Arnal I, Zhu X. Tau can switch microtubule network organizations: from random networks to dynamic and stable bundles. Molecular Biology of the Cell. 2018;29:154–65 https://www.molbiolcell.org/doi/abs/10.1091/mbc.E17-06-0429.
Biswas S, Kalil K. The microtubule-associated protein tau mediates the organization of microtubules and their dynamic exploration of actin-rich lamellipodia and filopodia of cortical growth cones. J Neurosci. 2018;38:291–307 http://www.ncbi.nlm.nih.gov/pubmed/29167405.
Cabrales Fontela Y, Kadavath H, Biernat J, Riedel D, Mandelkow E, Zweckstetter M. Multivalent cross-linking of actin filaments and microtubules through the microtubule-associated protein Tau. Nature Communications. 2017;8:1981 https://doi.org/10.1038/s41467-017-02230-8.
Maas T, Eidenmuller J, Brandt R. Interaction of tau with the neural membrane cortex is regulated by phosphorylation at sites that are modified in paired helical filaments. J Biol Chem. 2000;275:15733–40 http://www.jbc.org.manchester.idm.oclc.org/content/275/21/15733.
Elie A, Prezel E, Guerin C, Denarier E, Ramirez-Rios S, Serre L, Andrieux A, Fourest-Lieuvin A, Blanchoin L, Arnal I. Tau co-organizes dynamic microtubule and actin networks. Sci Rep. 2015;5:9964 http://www.ncbi.nlm.nih.gov/pubmed/25944224.
Brandt R. Cytoskeletal mechanisms of axon outgrowth and pathfinding. Cell Tissue Res. 1998;292:181–9 https://link-springer-com.manchester.idm.oclc.org/article/10.1007%2Fs004410051049.
Baas PW, Qiang L. Tau: it’s not what you yhink. Trends Cell Biol. 2019;29:452–61 http://www.sciencedirect.com/science/article/pii/S0962892419300388.
Sayas CL, Tortosa E, Bollati F, Ramirez-Rios S, Arnal I, Avila J. Tau regulates the localization and function of End-binding proteins 1 and 3 in developing neuronal cells. J Neurochem. 2015;133:653–67 http://www.ncbi.nlm.nih.gov/pubmed/25761518.
Trinczek B, Ebneth A, Mandelkow EM, Mandelkow E. Tau regulates the attachment/detachment but not the speed of motors in microtubule-dependent transport of single vesicles and organelles. Journal of cell science. 1999;112:2355–67 http://jcs.biologists.org/content/joces/112/14/2355.full.
Hernández-Vega A, Braun M, Scharrel L, Jahnel M, Wegmann S, Hyman BT, Alberti S, Diez S, Hyman AA. Local nucleation of microtubule bundles through tubulin concentration into a condensed tau phase. Cell Rep. 2017;20:2304–12 https://doi.org/10.1016/j.celrep.2017.08.042.
Takei Y, Teng J, Harada A, Hirokawa N. Defects in axonal elongation and neuronal migration in mice with disrupted tau and map 1b genes. J Cell Biol. 2000;150:989–1000 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=10973990.
Baas PW. Microtubules and neuronal polarity: lessons from mitosis. Neuron. 1999;22:23–31 https://www.ncbi.nlm.nih.gov/pubmed/10027286.
Liu M, Nadar VC, Kozielski F, Kozlowska M, Yu W, Baas PW. Kinesin-12, a mitotic microtubule-associated motor protein, impacts axonal growth, navigation, and branching. J Neurosci. 2010;30:14896–906 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=21048148.
Lin S, Liu M, Mozgova OI, Yu W, Baas PW. Mitotic motors coregulate microtubule patterns in axons and dendrites. J Neurosci. 2012;32:14033–49 http://www.ncbi.nlm.nih.gov/pubmed/23035110.
Nadar VC, Lin S, Baas PW. Microtubule redistribution in growth cones elicited by focal inactivation of kinesin-5. J Neurosci. 2012;32:5783–94 http://www.ncbi.nlm.nih.gov/pubmed/22539840.
Myers KA, Baas PW. Kinesin-5 regulates the growth of the axon by acting as a brake on its microtubule array. J Cell Biol. 2007;178:1081–91 http://www.ncbi.nlm.nih.gov/pubmed/17846176.
Lu W, Fox P, Lakonishok M, Davidson Michael W, Gelfand Vladimir I. Initial neurite outgrowth in Drosophila neurons is driven by Kinesin-powered microtubule sliding. Current Biology. 2013;23:1018–23 http://www.sciencedirect.com/science/article/pii/S0960982213004910.
Dong Z, Wu S, Zhu C, Wang X, Li Y, Chen X, Liu D, Qiang L, Baas PW, Liu M. Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9-mediated kif15 mutations accelerate axonal outgrowth during neuronal development and regeneration in zebrafish. Traffic. 2019;20:71–81 http://www.ncbi.nlm.nih.gov/pubmed/30411440.
Baines AJ: Neuronal plasma membrane associated cytoskeleton. In The neuronal cytoskeleton. Edited by Burgoyne R. New York: Wiley-Liss; 1991: 161-183
Huang CY-M, Rasband MN. Axon initial segments: structure, function, and disease. Annals of the New York Academy of Sciences. 2018;1420:46–61 https://nyaspubs.onlinelibrary.wiley.com/doi/abs/10.1111/nyas.13718.
Jones SL, Svitkina TM. Axon initial segment cytoskeleton: architecture, development, and role in neuron polarity. Neural Plasticity. 2016;2016:19 https://doi.org/10.1155/2016/6808293.
Weiss P, Hiscoe HB. Experiments on the mechanism of nerve growth. J Exp Zool. 1948;107:315–95 http://www.ncbi.nlm.nih.gov/pubmed/18915618.
Reinsch SS, Mitchison TJ, Kirschner M. Microtubule polymer assembly and transport during axonal elongation. J Cell Biol. 1991;115:365–79 http://www.ncbi.nlm.nih.gov/pubmed/1717484.
Miller KE, Sheetz MP. Direct evidence for coherent low velocity axonal transport of mitochondria. J Cell Biol. 2006;173:373–81 http://www.ncbi.nlm.nih.gov/pubmed/16682527.
Roossien DH, Lamoureux P, Van Vactor D, Miller KE. Drosophila growth cones advance by forward translocation of the neuronal cytoskeletal meshwork in vivo. PLoS One. 2013;8:e80136 http://www.ncbi.nlm.nih.gov/pubmed/24244629.
O'Toole M, Lamoureux P, Miller KE. A physical model of axonal elongation: force, viscosity, and adhesions govern the mode of outgrowth. Biophys J. 2008;94:2610–20 http://www.ncbi.nlm.nih.gov/pubmed/18178646.
Lansky Z, Braun M, Lüdecke A, Schlierf M, ten Wolde PR, Janson Marcel E, Diez S. Diffusible crosslinkers generate directed forces in microtubule networks. Cell. 2015;160:1159–68 http://www.cell.com/cell/abstract/S0092-8674(15)00129-4.
Lu W, Gelfand VI: Moonlighting Motors: Kinesin, Dynein, and Cell Polarity. Trends Cell Biol 2017http://www.ncbi.nlm.nih.gov/pubmed/28284467
He Y, Francis F, Myers KA, Yu W, Black MM, Baas PW. Role of cytoplasmic dynein in the axonal transport of microtubules and neurofilaments. J Cell Biol. 2005;168:697–703 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15728192.
Myers KA, He Y, Hasaka TP, Baas PW. Microtubule transport in the axon: Re-thinking a potential role for the actin cytoskeleton. Neuroscientist. 2006;12:107–18 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16514008.
Popov S, Brown A, Poo MM. Forward plasma membrane flow in growing nerve processes. Science. 1993;259:244–6 http://www.ncbi.nlm.nih.gov/pubmed/7678471.
Case LB, Waterman CM. Integration of actin dynamics and cell adhesion by a three-dimensional, mechanosensitive molecular clutch. Nat Cell Biol. 2015;17:955–63 http://www.ncbi.nlm.nih.gov/pubmed/26121555.
Yap AS, Duszyc K, Viasnoff V. Mechanosensing and Mechanotransduction at Cell-Cell Junctions. Cold Spring Harb Perspect Biol. 2018;10:a028761 http://www.ncbi.nlm.nih.gov/pubmed/28778874.
Bottenberg W, Sánchez-Soriano N, Alves-Silva J, Hahn I, Mende M, Prokop A. Context-specific requirements of functional domains of the Spectraplakin Short stop in vivo. Mech Dev. 2009;126:489–502 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=19409984.
Prokop A, Uhler J, Roote J, Bate MC. The kakapo mutation affects terminal arborisation and central dendritic sprouting of Drosophila motorneurons. J Cell Biol. 1998;143:1283–94 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=9832556.
Noordstra I, Liu Q, Nijenhuis W, Hua S, Jiang K, Baars M, Remmelzwaal S, Martin M, Kapitein LC, Akhmanova A. Control of apico-basal epithelial polarity by the microtubule minus-end-binding protein CAMSAP3 and spectraplakin ACF7. J Cell Sci. 2016;129:4278–88 http://www.ncbi.nlm.nih.gov/pubmed/27802168.
Ning W, Yu Y, Xu H, Liu X, Wang D, Wang J, Wang Y, Meng W. The CAMSAP3-ACF7 complex couples noncentrosomal microtubules with actin filaments to coordinate their dynamics. Dev Cell. 2016;39:61–74 http://www.sciencedirect.com/science/article/pii/S1534580716305998.
Yau KW, van Beuningen SF, Cunha-Ferreira I, Cloin BM, van Battum EY, Will L, Schatzle P, Tas RP, van Krugten J, Katrukha EA, et al. Microtubule minus-end binding protein CAMSAP2 controls axon specification and dendrite development. Neuron. 2014;82:1058–73 http://www.ncbi.nlm.nih.gov/pubmed/24908486.
Blanchoin L, Michelot A. Actin Cytoskeleton: A Team Effort during Actin Assembly. Curr Biol. 2012;22:R643–5 http://www.ncbi.nlm.nih.gov/pubmed/22917514.
Gu C, Yaddanapudi S, Weins A, Osborn T, Reiser J, Pollak M, Hartwig J, Sever S. Direct dynamin–actin interactions regulate the actin cytoskeleton. The EMBO Journal. 2010;29:3593–606 http://emboj.embopress.org/content/embojnl/29/21/3593.full.pdf.
Mohan R, John A. Microtubule-associated proteins as direct crosslinkers of actin filaments and microtubules. IUBMB Life. 2015;67:395–403. https://doi.org/10.1002/iub.1384.
Brandt R, Leger J, Lee G. Interaction of tau with the neural plasma membrane mediated by tau's amino-terminal projection domain. J Cell Biol. 1995;131:1327–40 http://www.ncbi.nlm.nih.gov/pubmed/8522593.
Cohen BA. How should novelty be valued in science? Elife. 2017;6:e28699. https://doi.org/10.7554/eLife.28699.
Gunawardena J. Models in biology: 'accurate descriptions of our pathetic thinking'. BMC Biology. 2014;12:29 http://www.biomedcentral.com/1741-7007/12/29.
Cohen JE. Mathematics is biology's next microscope, only better; biology is mathematics' next physics, only better. PLoS Biol. 2004;2:e439 http://www.ncbi.nlm.nih.gov/pubmed/15597117.
Schelski M, Bradke F. Neuronal polarization: From spatiotemporal signaling to cytoskeletal dynamics. Mol Cell Neurosci. 2017;84:11–28 http://www.ncbi.nlm.nih.gov/pubmed/28363876.
Pan S, Chan JR. Regulation and dysregulation of axon infrastructure by myelinating glia. Journal Cell Biol. 2017;216:3903–16 http://jcb.rupress.org/content/jcb/216/12/3903.full.pdf.
Eira J, Silva CS, Sousa MM, Liz MA. The cytoskeleton as a novel therapeutic target for old neurodegenerative disorders. Progr Neurobiol. 2016;141:61–82 http://www.sciencedirect.com/science/article/pii/S0301008215300800.
Baas PW, Ahmad FJ. Beyond taxol: microtubule-based treatment of disease and injury of the nervous system. Brain. 2013;136:2937–51 http://www.ncbi.nlm.nih.gov/pubmed/23811322.
Meyer K, Kaspar BK. Glia-neuron interactions in neurological diseases: Testing non-cell autonomy in a dish. Brain Res. 1656;2017:27–39 https://www.ncbi.nlm.nih.gov/pubmed/26778174.
Tsukita S, Ishikawa H. Three-dimensional distribution of smooth endoplasmic reticulum in myelinated axons. J Electron Microsc (Tokyo). 1976;25:141–9 http://www.ncbi.nlm.nih.gov/pubmed/1025229.
Kandel ER, Schwartz JH, Jessell TM, Siegelbaum S, Hudspeth AJ: Principles of neural science (5th edition). 5th edn: McGraw-Hill Publishing; 2012. https://ebookcentral.proquest.com/lib/manchester/detail.action?docID=4959346#
Pronker MF, Lemstra S, Snijder J, Heck AJR, Thies-Weesie DME, Pasterkamp RJ, Janssen BJC. Structural basis of myelin-associated glycoprotein adhesion and signalling. Nat Comm. 2016;7:13584. https://doi.org/10.1038/ncomms13584.
Koper A, Schenck A, Prokop A. Analysis of adhesion molecules and basement membrane contributions to synaptic adhesion at the Drosophila embryonic NMJ. PLoS One. 2012;7:e36339 http://www.ncbi.nlm.nih.gov/pubmed/22558441.
Giuditta A, Kaplan BB, van Minnen J, Alvarez J, Koenig E. Axonal and presynaptic protein synthesis: new insights into the biology of the neuron. Trends Neurosci. 2002;25:400–4 http://www.ncbi.nlm.nih.gov/pubmed/12127756.
Cioni JM, Koppers M, Holt CE. Molecular control of local translation in axon development and maintenance. Curr Opin Neurobiol. 2018;51:86–94 http://www.ncbi.nlm.nih.gov/pubmed/29549711.
Shigeoka T, Koppers M, Wong HH-W, Lin JQ, Dwivedy A, Nascimento JdF, Cagnetta R, van Tartwijk F, Strohl F, Cioni J-M, et al: On-site ribosome remodeling by locally synthesized ribosomal proteins in axons. bioRxiv 2018:500033. https://www.biorxiv.org/content/biorxiv/early/2018/12/19/500033.full
Frühbeis C, Fröhlich D, Kuo WP, Amphornrat J, Thilemann S, Saab AS, Kirchhoff F, Möbius W, Goebbels S, Nave KA, et al. Neurotransmitter-triggered transfer of exosomes mediates oligodendrocyte-neuron communication. PLoS Biol. 2013;11:e1001604 http://www.ncbi.nlm.nih.gov/pubmed/23874151.
Rajendran L, Bali J, Barr MM, Court FA, Kramer-Albers EM, Picou F, Raposo G, van der Vos KE, van Niel G, Wang J, Breakefield XO. Emerging roles of extracellular vesicles in the nervous system. J Neurosci. 2014;34:15482–9 http://www.ncbi.nlm.nih.gov/pubmed/25392515.
Court FA, Midha R, Cisterna BA, Grochmal J, Shakhbazau A, Hendriks WT, Van Minnen J. Morphological evidence for a transport of ribosomes from Schwann cells to regenerating axons. Glia. 2011;59:1529–39. https://doi.org/10.1002/glia.21196.
Giuditta A, Eyman M, Kaplan BB. Gene expression in the squid giant axon: neurotransmitter modulation of RNA transfer from periaxonal glia to the axon. Biol Bull. 2002;203:189–90. https://doi.org/10.2307/1543389.
Lazarus JE, Moughamian AJ, Tokito MK, Holzbaur EL. Dynactin subunit p150(Glued) is a neuron-specific anti-catastrophe factor. PLoS Biol. 2013;11:e1001611 http://www.ncbi.nlm.nih.gov/pubmed/23874158.
Hur EM. Saijilafu, Lee BD, Kim SJ, Xu WL, Zhou FQ: GSK3 controls axon growth via CLASP-mediated regulation of growth cone microtubules. Genes Dev. 2011;25:1968–81 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=21937714.
Brandt R, Bakota L. Microtubule dynamics and the neurodegenerative triad of Alzheimer's disease: The hidden connection. J Neurochem. 2017;173:409–17. https://doi.org/10.1111/jnc.14011.
Farah CA, Nguyen MD, Julien JP, Leclerc N. Altered levels and distribution of microtubule-associated proteins before disease onset in a mouse model of amyotrophic lateral sclerosis. J Neurochem. 2003;84:77–86 http://www.ncbi.nlm.nih.gov/pubmed/12485403.
Ferrier A, Boyer JG, Kothary R. Cellular and molecular biology of neuronal Dystonin. Int Rev Cell Mol Biol. 2013;300:85–120 http://www.ncbi.nlm.nih.gov/pubmed/23273860.
Wang JT, Medress ZA, Barres BA. Axon degeneration: molecular mechanisms of a self-destruction pathway. J Cell Biol. 2012;196:7–18 http://www.ncbi.nlm.nih.gov/pubmed/22232700.
Yaron A, Schuldiner O. Common and divergent mechanisms in developmental neuronal remodeling and dying back neurodegeneration. Curr Biol. 2016;26:R628–39 http://www.ncbi.nlm.nih.gov/pubmed/27404258.
Bradke F, Fawcett JW, Spira ME. Assembly of a new growth cone after axotomy: the precursor to axon regeneration. Nat Rev Neurosci. 2012;13:183–93 http://www.ncbi.nlm.nih.gov/pubmed/22334213.
Langford GM. Arrangement of subunits in microtubules with 14 profilaments. J Cell Biol. 1980;87:521–6 http://www.ncbi.nlm.nih.gov/pubmed/7430256.
Nogales E, Wolf SG, Khan IA, Luduena RF, Downing KH. Structure of tubulin at 6.5 A and location of the taxol-binding site. Nature. 1995;375:424–7 https://www.nature.com/articles/375424a0.
Luria I, Crenshaw J, Downs M, Agarwal A, Seshadri SB, Gonzales J, Idan O, Kamcev J, Katira P, Pandey S, et al. Microtubule nanospool formation by active self-assembly is not initiated by thermal activation. Soft Matter. 2011;7:3108–15. https://doi.org/10.1039/C0SM00802H.
Liu H, Bachand GD. Effects of Confinement on Molecular Motor-Driven Self-Assembly of Ring Structures. Cellular and Molecular Bioengineering. 2013;6:98–108. https://doi.org/10.1007/s12195-012-0256-5.
Crenshaw JD, Liang T, Hess H, Phillpot SR. A cellular automation approach to the simulation of active self-assembly of kinesin-powered molecular shuttles. J Comp Theoret Nanosci. 2011;8:1999–2005. https://doi.org/10.1166/jctn.2011.1916.
Gosselin P, Mohrbach H, Kulić IM, Ziebert F. On complex, curved trajectories in microtubule gliding. Physica D: Nonlinear Phenomena. 2016;318–319:105–11 http://www.sciencedirect.com/science/article/pii/S0167278915002183.
Acknowledgements
Work underpinning this article was made possible through support by the BBSRC to A. P (BB/I002448/1, BB/P020151/1, BB/L000717/1, BB/M007553/1), by parents to Y.-T.L., by the Leverhulme Trust to I.H. (ECF-2017-247). The Manchester Bioimaging Facility microscopes used in this study were purchased with grants from the BBSRC, The Wellcome Trust and The University of Manchester Strategic Fund. The Fly Facility has been supported by funds from The University of Manchester and the Wellcome Trust (087742/Z/08/Z). We thank Anthony Brown for introducing A.P. to the arguments that support a role of intermediate filaments in axon diameter, Anna Akhmanova as well as Casper Hoogenraad and his group for constructive and helpful feedback that has hopefully been well addressed in our last version, and Kyle Miller for major contributions to the contents of Table 5.
Funding
All sources of funding for the research reported are declared in the acknowledgement section.
Author information
Authors and Affiliations
Contributions
AP has done most of the writing and designed all figures, IH and BCG have contributed substantially to the writing, AV and YTL have contributed unpublished data shown in Fig. 4. All authors read and approved the final manuscript.
Authors' information
No additional information
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hahn, I., Voelzmann, A., Liew, YT. et al. The model of local axon homeostasis - explaining the role and regulation of microtubule bundles in axon maintenance and pathology. Neural Dev 14, 11 (2019). https://doi.org/10.1186/s13064-019-0134-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13064-019-0134-0