
Abstract
Over the past few decades, RNA sequencing has significantly progressed, becoming a paramount approach for transcriptome profiling. The revolution from bulk RNA sequencing to single-molecular, single-cell and spatial transcriptome approaches has enabled increasingly accurate, individual cell resolution incorporated with spatial information. Cancer, a major malignant and heterogeneous lethal disease, remains an enormous challenge in medical research and clinical treatment. As a vital tool, RNA sequencing has been utilized in many aspects of cancer research and therapy, including biomarker discovery and characterization of cancer heterogeneity and evolution, drug resistance, cancer immune microenvironment and immunotherapy, cancer neoantigens and so on. In this review, the latest studies on RNA sequencing technology and their applications in cancer are summarized, and future challenges and opportunities for RNA sequencing technology in cancer applications are discussed.
Similar content being viewed by others
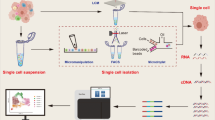

Background
Cancer remains one of the major malignant diseases that endangers human life and health and comprises complex biological systems that require accurate and comprehensive analysis. Since the first appearance of high-throughput sequencing in 2005 [1], it has become possible to understand life activities at the molecular level and to conduct detailed research to elucidate the genome and transcriptome. As an essential part of high-throughput sequencing, RNA sequencing (RNA-seq), especially single-cell RNA sequencing (scRNA-seq), provides biological information on a single tumor cell, analyzes the determinants of intratumor expression heterogeneity and identifies the molecular basis of formation of many oncological diseases [2, 3]. Thus, RNA sequencing offers invaluable insights for cancer research and treatment. With the advent of the era of precision medicine, RNA sequencing will be widely used for research on many different types of cancer. This review summarizes the history of the development of RNA sequencing and focuses on the latest studies of RNA sequencing technology in cancer applications, especially single-cell RNA sequencing and spatial transcriptome sequencing. In addition, we provide a general introduction to the current bioinformatics analysis tools used for RNA sequencing and discuss future challenges and opportunities for RNA sequencing technology in cancer applications.
The development of RNA sequencing technologies
It was not until 1953 when Watson and Crick proposed the double-helix structure did people truly realize at the molecular level that the essence of life is the result of gene interactions [4]. The continuous development of RNA sequencing has ushered transcriptome analysis into a new era, with higher efficiency and lower cost. The timeline of RNA sequencing technologies is shown in Fig. 1.

The development timeline of RNA sequencing technologies
The first-generation sequencing technology is also called Sanger sequencing. The chain termination method was initiated by Sanger in 1977, followed by the chemical degradation method developed by Maxam and Gilbert [5, 6]. The same year, Sanger determined the 5368 bp genome of phage φX174, which is the first DNA genome sequenced [7]. The DNA microarray has aided significant progress in many fields since it was first introduced. However, microarrays require prior knowledge of gene sequences and are unable to identify novel gene expression [8]. After the first high-throughput sequencing platform appeared in 2005 [1], multiple next-generation sequencing platforms followed (Table 1, Figs. 2, 3). The accuracy and reproducibility among different platforms depended on several factors, including the inherent features of the platform and the corresponding analysis pipelines [9, 10]. Pyrosequencing that was no longer supported after 2016, developed by 454 Life Sciences, used a “sequencing by synthesis” method [1, 11,12,13]. The ion torrent sequencing platform is also based on the “sequencing by synthesis” method, which outperforms pyrosequencing with respect to sensitivity. SOLiD (Sequencing by Oligonucleotide Ligation and Detection) exhibits high accuracy, as each base is sequenced twice, but the read length is short [11,12,13]. DNBS (DNA nanoball sequencing) enables large collection of DNA nanoballs for simultaneous sequencing. Illumina-based sequencing technology represents a “reversible terminator sequencing” method. High-throughput sequencing has the advantage of fast speed, low sequencing cost and high accuracy, otherwise known as next-generation sequencing (NGS). Compared to microarray, it can detect unknown gene expression sequences but is time intensive [14].

RNA extraction and template preparation before RNA-sequencing. RNA was extracted from tissues, and after fragmentation, fragmented DNA molecules were converted into cDNA by reverse transcription then amplified by emulsion PCR or bridge PCR to prepare sequencing library

Three kinds of sequencing methods. These methods contain sequencing by synthesis, sequencing by reversible terminator and sequencing by ligation. And their different mechanisms are shown in detail
In addition to NGS, there is third-generation sequencing, which allows for long-read sequencing of individual RNA molecules [15]. Single-molecule RNA sequencing enables the generation of full-length cDNA transcripts without clonal amplification or transcript assembly. Thus, third-generation sequencing is free from the shortcomings generated by PCR amplification and read mapping. It can greatly reduce the false positive rate of splice sites and capture the diversity of transcript isoforms [15]. Single-molecule sequencing platforms comprise Pacific Biosciences (PacBio) single-molecule real-time (SMRT) sequencing [16], Helicos single-molecule fluorescent sequencing [17] and Oxford Nanopore Technologies (ONT) nanopore sequencing [18]. Furthermore, RNA-seq recently evolved from bulk sequencing to single-cell sequencing. Single-cell RNA sequencing was first published in 2009 to profile the transcriptome at single-cell resolution [19]. Drop-Seq and InDrop were initially reported in 2015 by analyzing mouse retina cell and embryonic stem cell transcriptomes, identifying novel cell types. Sci-RNA-seq, single-cell combinatorial indexing RNA sequencing, was developed in 2017, and SPLiT-seq (split-pool ligation-based transcriptome sequencing) was first reported in 2018. Both approaches use a combinatorial indexing strategy in which attached RNAs are labeled with barcodes that indicate their cellular origin [20, 21].
Though single-cell data enable single-cell transcriptomics, it may lose spatial information during single-cell isolation. To solve this problem, spatial transcriptomics has emerged. Spatial transcriptomics employs unique positional barcodes to visualize RNA distributions in RNA sequencing of tissue sections and was first published in 2016 [22]. Slide-seq, reported in 2019, uses DNA barcode beads with specific positional information [23]. Geo-seq was introduced in 2017 and integrated scRNA-seq with laser capture microdissection (LCM), which can isolate individual cells [24]. In situ sequencing refers to targeted sequencing of RNA fragments in morphologically preserved tissues or cells without RNA extraction, including in situ cDNA synthesis by padlock probes or stably cross-linked cDNA amplicons in fluorescent in situ RNA sequencing (FISSEQ) and in situ amplification by rolling-circle amplification (RCA) [25, 26]. Furthermore, various new technologies based on RNA-seq have been developed for specific applications. For example, a type of targeted RNA sequencing, CaptureSeq, employs biotinylated oligonucleotide probes and results in the enrichment of certain transcripts to identify gene fusion [27, 28].
Computational analysis of RNA sequencing data
Computational analysis tools for RNA sequencing have dramatically increased during the past decade. The choice of a particular tool should be based on the purpose and accuracy of application [29,30,31]. A general RNA sequencing data analysis process involves the quality control of raw data, read alignment and transcript assembly, expression quantification and differential expression analysis (Fig. 4).

Bioinformatics tools commonly used in RNA-seq data analysis. These tools are primarily used in the four main processes of RNA-seq data analysis, including quality control, read alignment and transcript assembly, expression quantification and differential expression analysis
The first step of data analysis is to assess and clean the raw sequencing data, which is usually provided in the form of FASTQ files [32]. Quality control visually reflects the quality of the sequencing and purposefully discards low-quality reads, eliminates poor-quality bases and trims adaptor sequences [31]. Common tools include FASTQ [33], NGSQC [34], RNA-SeQC [35], Trimmomatic [36], PRINSEQ [37] and Soapnuke [38].
The next step is to map the clean reads to either a genome or a transcriptome. There are some mapping tools available, including Tophat2 [39], HISAT2 [40], STAR [41], BWA [42] and Bowtie [43]. After alignment, another type of software, such as Cufflinks [44], StringTie [45], Trinity [46], SOAPdenovoTrans [47] and Trans-AByS [48] can be used to assemble transcripts from short-reads. When the transcript model is established, its expression can be quantified at the gene, transcript and exon levels. Commonly used software for gene-level quantification includes FeatureCount [49] and HTSeq-count [50]. Transcript level quantitative software includes Cufflinks [44], eXpress [51] and RSEM [52]. DEXSeq is a software for exon level quantification [53]. In addition, there are some alignment-free quantification tools such as Kallisto [54], Sailfish [55] and Salmon [56], which have the advantage of marked computational resource saving. After normalizing, an expression matrix is generated, and statistical methods can be used to identify differentially expressed genes. DESeq2 [57] and edgeR [58] are commonly used to perform this task.
Applications of RNA-sequencing in cancer research
Genomic data, such as RNA-seq, have become widely available due to the popularity of high-throughput sequencing technology [59]. As an important part of next-generation sequencing, RNA sequencing has made great contributions in various fields, especially cancer research, including studies on differential gene expression analysis and cancer biomarkers, cancer heterogeneity and evolution, cancer drug resistance, the cancer microenvironment and immunotherapy, neoantigens, etc. (Fig. 5).

Applications of RNA-seq in differential expression analysis and cancer biomarkers, cancer heterogeneity and drug resistance, cancer immune microenvironment, immunotherapy and neoantigen. a Differential expression analysis by RNA sequencing can identify potential biomarkers, including fusion transcript, lncRNA, miRNA and circRNA. b The heterogeneity and drug resistance of cancer cells identified by RNA-seq. c Novel molecular signature, regulatory protein and unknown subtypes in cancer infiltrating immune cells and potential resistance effector in immunotherapy can be identified by RNA-seq; d Neoantigen profiling by RNA-seq and TCR modification targeted neoantigens
Differential gene expression analysis and cancer biomarkers
Differential gene expression analysis is one of the most common applications of RNA sequencing [60]. Samples from different backgrounds (different species, tissues and periods) can be used for RNA sequencing to identify differentially expressed genes, revealing their function and potential molecular mechanisms [61]. More importantly, differential gene expression analysis facilitates the discovery of potential cancer biomarkers [62]. Many studies have shown that gene fusions are closely related to oncogenesis and are appreciated as both ideal cancer biomarkers and therapeutic targets [63]. Gene fusions in clinical samples are primarily detected by RNA-CaptureSeq. Compared to whole transcriptome sequencing, RNA-CaptureSeq has significantly higher sequencing depth [27, 64, 65]. It has been reported that the NUP98-PHF23 fusion gene is likely to be a novel therapeutic target in acute myeloid leukemia (AML) [66]. Recently, a variety of recurrent gene fusions, including ESR1-CCDC170, SEC16A-NOTCH1, SEC22B-NOTCH2 and ESR1-YAP1, have been identified in breast cancer, indicating that recurrent gene fusion is one of the key drivers for cancer [67]. Several novel configurations of BRAF, NTRK3 and RET gene fusions have been identified in colorectal cancer [68]. These fusions may promote the development of malignancy and provide new targets for personalized treatment [68]. In addition, some special genomic factors have been discovered as biomarkers by RNA sequencing, including miRNA, lncRNA and circRNA, which are widely present in various types of cancer [69,70,71]. A recent example is circRNA_0001178 and circRNA_0000826, which are biomarkers of colorectal cancer metastasis to the liver [72]. By applying both RNA sequencing and small RNA sequencing, a study on pancreatic cancer identified differential expression of simple repetitive sequences (SSRs) and demonstrated that the frequency of SSR motifs changed dramatically, which is expected to become a tumor biomarker [73]. In addition to nucleic acid biomarkers, RNA-seq combined with immunohistochemistry and western blot has also identified certain proteins as cancer biomarkers, such as nuclear COX2 (cyclooxygenase2) in combination with HER2 (human epidermal growth factor receptor type 2), which may serve as potential biomarkers for the diagnosis and prognosis of colorectal cancer [74]. Similar examples identified using RNA-seq profiling analysis include ISG15 (Interferon-stimulated gene 15) in nasopharyngeal carcinoma [75] and DMGDH (dimethylglycine dehydrogenase) in hepatocellular carcinoma [76]. Data-mining analysis of RNA sequencing data and other clinical data has identified that the isoforms of peroxiredoxins also can be expected the prognostic biomarkers for predicting overall survival and relapse-free survival in breast cancer [77]. Increasing differentially expressed genes are being identified by RNA sequencing, and new potential cancer biomarkers are being continuously discovered (Table 2). However, sufficient clinical practice is needed to confirm the diagnostic and predictive applications of these biomarkers in cancer.
RNA-seq could detect early mutations as well as high molecular risk mutations, thus can discover novel cancer biomarkers and potential therapeutic targets, monitoring of diseases and guiding targeted therapy during early treatment decisions. Tumor mutation burden (TMB) is considered as a potential biomarker for immune checkpoint therapy and prognosis [78, 79]. RNA-seq can be used to explore the application value of TMB in diffuse glioma [78]. Through the RNA-seq, MET exon 14 mutation and isocitrate dehydrogenase 1 (IDH1) mutation were identified as new potential therapeutic targets in lung adenocarcinoma and chondrosarcoma patients, respectively [80, 81]. Several studies have shown that RNA sequencing can effectively improve the detection rate on the basis of DNA sequencing, provide more comprehensive detection results and achieve a better curative effect for targeted therapy [82]. In addition, it has been proved that IDH mutation is a good prognostic marker for glioma by RNA-seq [83]. Targeted therapy is also considered to enhance or replace cytotoxic chemotherapy regimen in cancer including AML [84,85,86].
ScRNA-seq also has some new discoveries in diagnosis. For example, scRNA-seq data can be used to infer copy number variations (CNV) and to distinguish malignant from non-malignant cells. The infer CNV algorithm, which was used in the study of glioblastoma, uses averaging relative expression levels over large genomic regions to infer chromosome copy number variation [87]. Similar examples include head and neck cancer [88] and human oligodendroglioma [89]. It is reported that RNA sequence of tumor-educated blood platelets (TEPs) can also become a blood-based cancer diagnosis method [90]. It should be noted that the lack of detailed functional implications of the identified RNAs in platelets in the field of platelet RNA research is also an urgent problem to be solved [91].
Cancer heterogeneity and evolution
Heterogeneity has always existed during the transformation of normal cells to cancer cells. The continuous accumulation of heterogeneity may reflect the evolution of cancer [109]. Early RNA sequencing detected all RNA transcripts in a given tissue or cell group, ignoring differences in individual cells. Transcriptome profiling of single-cell RNA sequencing solves this problem by providing single-cell resolution of the transcriptome [3]. In melanoma, single-cell RNA-seq was used to analyze 4645 tumor cells from 19 patients, including cancer cells, immune cells, mesenchymal cells and endothelial cells. Transcriptomic data from different single cells revealed that heterogeneity of cells within the same cancer is associated with cell cycle, spatial background and drug resistance [110]. A recent single-cell RNA-seq study of 49 samples of metastatic lung cancer revealed changes in plasticity induced by non-small cell lung cancer treatment, providing new directions for clinical treatment [111]. Single-cell RNA sequencing also integrates a variety of information in a single cancer cell, deciphering the secrets of cancer heterogeneity and evolution [112]. Compared with scRNA-seq, another emerging technology spatial transcriptome sequencing incorporates information on the spatial location of cells. In prostate cancer, using spatial transcriptomics technology, the transcriptome of nearly 6750 tissue regions was analyzed, revealing the whole-tissue gene expression heterogeneity of the entire multifocal prostate cancer and accurately describing the range of cancer foci [113]. In a study of breast cancer tissues, the results of spatial transcriptome sequencing revealed that gene expression among different regions was surprisingly highly heterogeneous [22]. In recent years, single-nucleus RNA sequencing (snRNA-seq) has also received extensive attention due to its solving the problem that single-cell RNA sequencing cannot be applied to frozen specimens and cannot obtain all cell types in a given tissue [114, 115]. The emerging technology of RNA sequencing will contribute to research on cancer heterogeneity and evolution.
Cancer drug resistance
Drug resistance is a main reason leading to cancer treatment failure. However, the molecular mechanisms underlying drug resistance are still poorly understood [116]. RNA sequencing became a vital tool for revealing the mechanisms of cancer drug resistance. In breast cancer, single-cell RNA sequencing identified a tumor-infiltrating immunosuppressive immature myeloid cell that leads to drug resistance [117]. Another study identified a new COX7B gene related to platinum resistance and a surrogate marker CD63 in cancer cells by single-cell RNA-seq [118]. RNA sequencing has also demonstrated that cancer cells that wake up from a dormant state produce large amounts of BORIS (brother of the regulator of imprinted sites), which can regulate the expression of survival genes in drug-resistant neuroblastoma cells [119]. Identifying special molecules that mediate these processes could help us understand the occurrence of drug resistance. Single-cell transcriptomics can be used to study different modes of chemoresistance in tumor cells and has shown that pre-existing drug-resistant cells can be selected through higher phenotypic intratumoral heterogeneity, while phenotypic homogeneous cells use other mechanisms to trans-differentiate under drug-selection [120]. In one study of pancreatic ductal adenocarcinoma, human pancreatic cancer (PANC-1) cells and gemcitabine-resistant PANC-1 cell lines were compared by RNA sequencing, and two circRNAs were identified as both novel biomarkers and potential therapeutic targets for gemcitabine resistant patients [121]. RNA-seq has also conducted in-depth research on the drug resistance of hematological malignancies. Through RNA-seq, it has been found that non-coding RNAs and fusion genes play an important role in mediating the drug resistance of hematological malignancies [122]. A good example is to compare the circRNA expression profile of the drug-resistant acute myeloid leukemia cell with its parent cell, and determine the circRNAs involved in drug resistance [123]. Similarly, the novel MEF2D-BCL9 fusion transcript identified by RNA-seq was found to increase HDAC9 (histone deacetylase 9) expression and to enhance the resistance to dexamethasone in acute lymphocytic leukemia (ALL) [124]. Leukemia stem cells (LSCs), a rare cell population assumed to be responsible for relapse, is crucial to improve the prognosis of patients [125, 126]. RNA-seq analysis showed that LSCs have a unique lncRNA signature with functional relevance and therapeutic potential, providing an explanation for chemotherapy resistance and disease recurrence [127].
The cancer microenvironment and immunotherapy
The immune system plays a critical role in the cancer microenvironment, affecting several stages of cancer development, including tumorigenesis, progression and metastasis, through tumor-infiltrating lymphocytes (TILs) [128]. TILs and their interactions with malignant cells and stromal cells make up the cancer immune microenvironment. Due to the heterogeneity of cancer, it is difficult to define the exact pro- or anti-cancer function of certain immune cells. Cancer heterogeneity also causes the varied clinical efficacy observed in patients treated with immunotherapies due to different responses of different subclones [129]. Transcriptomic profiling by RNA-seq, in particular scRNA-seq, provides comprehensive information on cellular activity and interactions among cells in the tumor microenvironment (TME). ScRNA-seq enables genomic and molecular profiling of high quantity and quality individual immune cells and assessment of cellular heterogeneity to depict the immune system spectrum in the cancer microenvironment [130,131,132]. ScRNA-seq data demonstrated that compared to normal tissues, cancer tissues exhibited significantly higher heterogeneity in the immune microenvironment, and a continuity in T cell activation resulting from polyclonal T cells and heterogeneous antigen-presenting cells has been identified [133].
ScRNA-seq of tumor-infiltrating T cells in metastatic melanoma identified transcription factor NFATC1 (nuclear factor of activated T cells 1) as a potential molecular signature of T cell exhaustion programs and revealed the depletion of low-exhaustion T cells in expanded clones of T cells [110]. Combining scRNA-seq with assembled T cell receptor (TCR) sequences, 11 T cell subsets, such as CD8 + T cells and CD8 + FOXP3 + regulatory-like cells, and their genomic signatures, were identified in hepatocellular carcinoma (HCC), providing valuable insights for understanding the immune landscape of infiltrating T cells in HCC [134]. Cancer infiltrating T cells also play an anti-tumor role through impairment of an autophagy protein, LC3 (microtubule-associated protein 1A/1B-light chain 3, often short for LC3)-associated phagocytosis (LAP), demonstrating the role of autophagy in oncogenesis and suppression revealed by scRNA-seq [135]. In addition to solid tumors, scRNA-seq of acute myeloid leukemia patients detected diverse immunomodulatory genes that suppress T cell function [136]. By CSOmap, a computational tool for scRNA-seq, the CCL4-CCR8 directed interaction between Tregs and Texs, as well as reduced proliferation of Texs, was characterized [137]. Notably, findings also revealed that tumor-infiltrating T cells exhibited more interactions among themselves than with T cells from peripheral blood and different interactions between tumors and T cells, indicating a varied response to immunotherapy and a potential trend for immune escape [137].
In the cancer immune microenvironment, neutrophils, in addition to T cells, are also key components of cancer progression and cancer drug resistance [138,139,140,141]. Through scRNA-seq of murine sarcomas and certain human cancers, neutrophils with CSF3R (colony stimulating factor 3 receptor) expression were found to be a part of type 1 antitumor immunity associated with unconventional CD4−CD8−αβ T cells (UTCαβ) in anti-cancer immunity, indicating better prognosis [142]. ScRNA-seq of metastatic breast cancer and CD45 cells from primary cancer identified neutrophils as pro- and anti-tumorigenic or metastatic, in which pro-tumorigenic and metastatic neutrophils are induced by IL11 expressing cancer subclones, resulting in polyclonal metastasis [143]. This observation also provides new insight into anti-cancer immunotherapy by targeting neutrophils [143]. With scRNA-seq of CD4 and CD8 T cells, several crucial pathways with anti-cancer function were revealed [144].
The balance between immune reaction and immune tolerance is the basis of immune homeostasis, which is also involved in anti-cancer immunity and oncogenesis. scRNA-seq of monocytes and dendritic cells (DCs) separated from a single lymph node melanoma metastasis revealed a conserved homeostatic module regulated by suppressor-of-cytokine-2 (SOCS2) protein and IFNγ [145]. SOCS2 serves an essential regulatory role in anti-tumor immunity and T cell priming through DCs. This highly conserved homeostatic program establishes a connection between autoimmune prevention and immune surveillance in cancer [145].
Immunotherapies, especially immune checkpoint blockade (ICB), has opened a new chapter for anti-cancer therapy with remarkable responses from targeting programmed death 1 (PD1), programmed death-ligand 1 (PD-L1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA4) [146,147,148]. However, only a few patients benefit from ICB, and severe side effects were observed [149, 150]. Obviously, various unknown determinants are correlated with the outcome of immunotherapies in addition to well-known factors such as PD1/PD-L1/CTLA-4 expression and mismatch repair deficiency [151,152,153,154,155]. Therefore, it is paramount to identify potential effectors for ICB efficacy. By analyzing RNA-seq data from melanoma patients who underwent anti-PD1 and anti-CTLA4 treatment, a potential ICB resistance effector SERPINB9 (a member of the serine protease inhibitor (serpin) family) and the connection between cytotoxic T lymphocytes (CTL) infiltration level and ICB response were characterized [156]. A sub-population cells with immunotherapy persistence have been identified by scRNA-seq and were found to have stem cell-like states with the expression of stem cell antigen-1 (Sca-1) and Snai1 [157].
Another immunotherapy, myeloid-targeted immunotherapy, is based on the complexity of tumor-infiltrating myeloid cells, including DCs and tumor-associated macrophages (TAMs) revealed by scRNA-seq [158]. Through scRNA-seq of immune cells from colorectal cancer patients, C1QC + and SPP1 + TAMs, two subsets of TAMs, were identified, and the mechanism of myeloid-targeted immunotherapy, such as anti-CSF1R (colony stimulating factor 1 receptor) and CD40 agonist, was revealed [159]. Intracellular staining and sequencing (INs-seq), a novel technology integrating scRNA-seq and intracellular protein activity measurements, revealed novel Arg1 + Trem2 + regulatory myeloid (Mreg) cells and demonstrated that depletion of Trem2 led to deduction of exhausted CD8 T cells with increased NK and cytotoxic T cells and cancer suppression by reducing accumulation of intratumoral Mreg cells [160].
Cancer neoantigens
Neoantigens, human leukocyte antigen (HLA)-bound peptides derived from cancer-specific somatic mutations or gene fusions during tumor growth, are another crucial regulator of the clinical response to immunotherapy [161]. Higher intratumor neoantigen heterogeneity and clonal neoantigen burden increases sensitivity to ICB and contributes to better clinical outcome in patients with melanoma and advanced non-small cell lung cancer [162]. This kind of antigen is an optimal target for anti-cancer immunotherapy, enhancing neoantigen-specific T cell activity, and a vaccine targeting personal neoantigens for melanoma patients has been developed [163, 164]. Given all these promising features of personalized medicine targeting neoantigens in tumors, massive parallel profiling of tumor neoantigen burden is necessary for improving clinical efficacy and a deeper understanding of the neoantigen landscape. An RNA-seq-based transcriptomic approach is an efficient tool for neoantigen profiling in many studies. It was revealed that homology of neoantigen and somatic-mutation induced pathogens are important in response prediction in anti-CTLA4 treated melanoma [165]. In addition to melanoma, other studies found reduced neoantigen load in triple-negative and HER2 breast cancers [166], diverse neoantigen abundance in non-small-cell lung cancer patients with different treatment strategies [167], a decreased ratio of neoantigen expression to predicted neoantigens in recurrent glioma due to immune selection pressure [168], a negative correlation between neoantigen abundance and clinical outcome in selected solid tumors [169] and different neoantigen landscapes in immune filtration and T cell dysfunction based on histology in salivary gland carcinoma (SGC) patients [170].
A neoantigen prediction program, Neopepsee, based on RNA-seq data and somatic mutation, can be utilized to detect potential neoantigens for personal vaccine development with reduced false-positive rate compared to binding affinity prediction [171]. ScanNeo is another prediction computational pipeline based on RNA-seq that aims to identify insertion and deletion derived neoantigens, which was validated in prostate cancer [172], and ASNEO, which identifies personal-specific alternative splicing derived neoantigens [173]. Several neoantigens have been identified to be related to cancer prognosis and might be potential targets of immunotherapies, such as the TP53 neoantigen for HCC patients [174]. For the anti-neoantigen immunotherapy in cancer, a new strategy involving neoantigen-specific TCRs modification has been proposed, and scRNA-seq has been applied to isolate neoantigen-specific TCRs for further clinical application [175].
Conclusions and perspectives
High-throughput RNA-seq technology has been a major tool to explore the transcriptome. The rapid development of RNA-seq technology not only saves time and cost but also sheds light on many new research fields. However, there are still limitations of RNA-seq technology that need to be improved.
For short-read length RNA-seq technologies, bias and imperfections are primarily generated in sequencing library preparation and short read assembly. It is difficult for these methods to correctly identify multiple isoforms from a certain gene. To overcome the disadvantage of short read length, improved read coverage and sequencing depth is required. Long-read length RNA-seq technologies avoid shortcomings in template amplification, reduce the false positive rate in splice junction detection and enable the identification of unannotated longer transcripts, overcoming the common limitations of short-read sequencing [176, 177]. However, this method suffers from the drawback of reduced throughput, higher cost and higher sequencing error rate, especially insertion-deletion errors. To reduce random errors, PacBio circular consensus-sequencing (CCS) was developed to increase sequencing depth by rereading molecules several times. However, it also reduces the identification rate of unique isoforms. In addition, the sensitivity of long-read sequencing for identification of differentially expressed genes is lower compared to short-read sequencing [178,179,180]. Thus, hybridization of long-read and short-read sequencing has been reported to yield a more comprehensive and accurate analysis [181].
Improvements in the throughput of RNA sequencing technology have resulted in billions of sequencing reads, bringing great challenges to the computational process, such as data storage, transmission, quality control and data analysis, including read mapping, transcript assembly and read normalization. Therefore, it is important for bioinformatics to keep pace with the continuous developments of RNA-seq technologies. Notably, bias could be produced due to differences in read data handling, necessitating the improvement of current bioinformatics pipelines.
RNA-seq measures gene expression by the read counts, which always containing missing values, thus results in information loss of specific gene and negative impact on downstream analysis. To overcome this problem, missing data need to be imputed and analyzed by several methods, such as optimal clustering with missing values [182]. For scRNA-seq, the proportion of genes with zero or low expression varies across cells due to biological or technical bias. For example, batch effects can come from cells captured and sequenced in different conditions [183]. Imputation methods, such as SAVER, MAGIC and kNN-smoothing, are recommended for scRNA-seq [184]. Another method named batch effects correction with unknown subtypes for scRNA-seq data (BUSseq) utilizes Bayesian hierarchical model and can also be used to correct batch effects and missing data [185].
Combination of data from multi-omics sequencing can undoubtedly expand the application of RNA-seq. For example, Assay for Transposase-Accessible Chromatin using sequencing (ATAC-seq) was developed by utilizing hyperactive Tn5 transposase to identify open chromatin region and transcriptional factor (TF) binding sites [186]. The integration of ATAC-seq and RNA-seq enables the reveal of TF-targeted genes and their transcripts [187, 188]. Chromatin conformation capture analysis (3C) technology and its several derivatives including circular chromosome conformation capture (4C), carbon copy chromosome conformation capture (5C), ChIP-Loop, Hi-C and capture Hi-C were developed and improved to detect chromatin structure as well as unknown interacting regions [189,190,191]. It has been reported that combined analysis of RNA-seq and chromatin structure can detect structure variation-related differentially expressed genes [192,193,194].
Epitranscriptomics is a crucial part of gene expression, and methylation of adenosine at the N6 position (m6A) is the most abundant [195]. Traditional RNA-seq needs reverse transcription before sequencing and thus easily loses the information of transcriptome complexity. This shortcoming can be overcome by directly sequencing native RNA molecules using methods such as nanopore sequencing. Transcript modifications could be inferred from the current signal as the modified RNA molecules passing nanopore cause a characteristic temporary current blockade, which enables the detection of diverse modifications such as m6A or 5-methylcytosine (m5C) [196,197,198].
ScRNA-seq is a powerful technology to facilitate further exploration in cancer research and also has been employed in the detection of cancer stem cell subpopulation, metabolic switch in cancer-draining lymph nodes and therapy-induced adaption of cancer cells [111, 199, 200]. Combined with cell sorting or ligand-receptor interaction, scRNA-seq was utilized in cellular interaction, cell spatial organization as well as molecular crosstalk characterization [137, 201, 202]. Coupling of parallel CRISPR (clustered regularly interspaced short palindromic repeats)-pooled screen, scRNA-seq enables the simultaneous analysis of genomic perturbation and transcriptional activity to detect heterogeneous cell type as well as crucial factors of complexity regulatory mechanism [203,204,205]. ScNT-seq, single-cell metabolically labeled new RNA tagging sequencing, brings RNA-seq into time resolution by identifying RNAs transcribed at different stage [206]. Utilizing SNP-based demultiplexing of scRNA-seq data, MIX-Seq was developed to study cancer cell reaction to pharmacologic treatment [207]. Another technology, snRNA-seq, is invaluable for detecting cellular heterogeneity of cancer and has been employed to identification of a sub-population of adipocytes regulating cancer genesis [208].
Taken together, RNA-seq has been applied in an impressively wide range of cancer research. All applications in cancer research rely on the boost of advanced RNA-seq technologies, especially the combination of scRNA-seq and spatial transcriptomics as well as data from multi-omics, which will bring RNA-seq technologies into single-cell resolution and tissue-level transcriptomics, providing new insight into cancer diagnosis, treatment and prevention.
Availability of data and materials
Not applicable.
Abbreviations
- RNA-seq:
-
RNA sequencing
- ScRNA-seq:
-
Single-cell RNA sequencing
- SOLiD:
-
Sequencing by Oligonucleotide Ligation and Detection
- DNBS:
-
DNA nanoball sequencing
- NGS:
-
Next-generation sequencing
- PacBio SMRT:
-
Pacific Biosciences single-molecule real-time
- ONT:
-
Oxford Nanopore Technologies
- Sci-RNA-seq:
-
Single-cell combinatorial indexing RNA sequencing
- SPLiT-seq:
-
Split-pool ligation-based transcriptome sequencing
- LCM:
-
Laser capture microdissection
- FISSEQ:
-
Fluorescent in situ RNA sequencing
- RCA:
-
Rolling-circle amplification
- AML:
-
Acute myeloid leukemia
- SSRs:
-
Simple repetitive sequences
- COX2:
-
Cyclooxygenase2
- HER2:
-
Human epidermal growth factor receptor type 2
- ISG15:
-
Interferon-stimulated gene 15
- DMGDH:
-
Dimethylglycine dehydrogenase
- TMB:
-
Tumor mutation burden
- IDH:
-
Isocitrate dehydrogenase
- CNV:
-
Copy number variations
- TEPs:
-
Tumor-educated blood platelets
- SnRNA-seq:
-
Single-nucleus RNA-sequencing
- BORIS:
-
Brother of the regulator of imprinted sites
- ALL:
-
Acute lymphocytic leukemia
- LSCs:
-
Leukemia stem cells
- TILs:
-
Tumor-infiltrating lymphocytes
- TME:
-
Tumor microenvironment
- NFATC1:
-
Nuclear factor of activated T cells 1
- TCR:
-
T cell receptor
- HCC:
-
Hepatocellular carcinoma
- LAP:
-
Microtubule-associated protein 1A/1B-light chain 3-associated phagocytosis
- CSF3R:
-
Colony stimulating factor 3 receptor
- UTCαβ:
-
Unconventional CD4−CD8− αβ T cells
- DCs:
-
Dendritic cells
- SOCS2:
-
Suppressor-of-cytokine-2 protein
- ICB:
-
Immune checkpoint blockade
- PD1:
-
Targeting programmed death 1
- PD-L1:
-
Programmed death-ligand 1
- CTLA4:
-
Cytotoxic T-lymphocyte-associated protein 4
- CTL:
-
Cytotoxic T lymphocytes
- Sca-1:
-
Stem cell antigen-1
- TAMs:
-
Tumor-associated macrophages
- CSF1R:
-
Colony stimulating factor 1 receptor
- INs-seq:
-
Intracellular staining and sequencing
- Mreg:
-
Regulatory myeloid cells
- HLA:
-
Human leukocyte antigen
- SGC:
-
Salivary gland carcinoma
- CCS:
-
Circular consensus-sequencing
- ATAC-seq:
-
Assay for Transposase-Accessible Chromatin using sequencing
- TF:
-
Transcriptional factor
- 3C:
-
Chromatin conformation capture analysis
- 4C:
-
Circular chromosome conformation capture
- 5C:
-
Carbon copy chromosome conformation capture
- m6A:
-
N6 position
- m5C:
-
5-Methylcytosine
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
References
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–80.
Cieślik M, Chinnaiyan AM. Cancer transcriptome profiling at the juncture of clinical translation. Nat Rev Genet. 2018;19:93–109.
Suvà ML, Tirosh I. Single-cell RNA sequencing in cancer: lessons learned and emerging challenges. Mol Cell. 2019;75:7–12.
Watson JD, Crick FH. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature. 1953;171:737–8.
Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–7.
Maxam AM, Gilbert W. A new method for sequencing DNA. Proc Natl Acad Sci USA. 1977;74:560–4.
Sanger F, Air GM, Barrell BG, Brown NL, Coulson AR, Fiddes CA, et al. Nucleotide sequence of bacteriophage phi X174 DNA. Nature. 1977;265:687–95.
Russo G, Zegar C, Giordano A. Advantages and limitations of microarray technology in human cancer. Oncogene. 2003;22:6497–507.
Li S, Tighe S, Nicolet C, Grove D, Levy S, Farmerie W, et al. Multi-platform assessment of transcriptome profiling using RNA-seq in the ABRF next-generation sequencing study. Nat Biotechnol. 2014;32:915–25.
A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the Sequencing Quality Control Consortium. Nat Biotechnol. 2014;32:903–14.
Mardis E. Next-generation DNA sequencing methods. Annu Rev Genomics Hum Genet. 2008;9:387–402.
Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26:1135–45.
Mardis E. Next-generation sequencing platforms. Annu Rev Anal Chem (Palo Alto Calif). 2013;6:287–303.
Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008;18:1509–17.
Schadt EE, Turner S, Kasarskis A. A window into third-generation sequencing. Hum Mol Genet. 2010;19:R227–40.
Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323:133–8.
Hart C, Lipson D, Ozsolak F, Raz T, Steinmann K, Thompson J, et al. Single-molecule sequencing: sequence methods to enable accurate quantitation. Methods Enzymol. 2010;472:407–30.
Bayley H. Nanopore sequencing: from imagination to reality. Clin Chem. 2015;61:25–31.
Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods. 2009;6:377–82.
Cao J, Packer J, Ramani V, Cusanovich D, Huynh C, Daza R, et al. Comprehensive single-cell transcriptional profiling of a multicellular organism. Science. 2017;357:661–7.
Rosenberg A, Roco C, Muscat R, Kuchina A, Sample P, Yao Z, et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science. 2018;360:176–82.
Ståhl P, Salmén F, Vickovic S, Lundmark A, Navarro J, Magnusson J, et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science. 2016;353:78–82.
Rodriques SG, Stickels RR, Goeva A, Martin CA, Murray E, Vanderburg CR, et al. Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science. 2019;363:1463–7.
Chen J, Suo S, Tam PP, Han JJ, Peng G, Jing N. Spatial transcriptomic analysis of cryosectioned tissue samples with geo-seq. Nat Protoc. 2017;12:566–80.
Ke R, Mignardi M, Pacureanu A, Svedlund J, Botling J, Wählby C, et al. In situ sequencing for RNA analysis in preserved tissue and cells. Nat Methods. 2013;10:857–60.
Lee JH, Daugharthy ER, Scheiman J, Kalhor R, Yang JL, Ferrante TC, et al. Highly multiplexed subcellular RNA sequencing in situ. Science. 2014;343:1360–3.
Mercer TR, Gerhardt DJ, Dinger ME, Crawford J, Trapnell C, Jeddeloh JA, et al. Targeted RNA sequencing reveals the deep complexity of the human transcriptome. Nat Biotechnol. 2011;30:99–104.
Heyer EE, Deveson IW, Wooi D, Selinger CI, Lyons RJ, Hayes VM, et al. Diagnosis of fusion genes using targeted RNA sequencing. Nat Commun. 2019;10:1388.
Soverini S, Abruzzese E, Bocchia M, Bonifacio M, Galimberti S, Gozzini A, et al. Next-generation sequencing for BCR-ABL1 kinase domain mutation testing in patients with chronic myeloid leukemia: a position paper. J Hematol Oncol. 2019;12:131.
Chatterjee A, Ahn A, Rodger EJ, Stockwell PA, Eccles MR. A guide for designing and analyzing RNA-seq data. Methods Mol Biol. 2018;1783:35–80.
Conesa A, Madrigal P, Tarazona S, Gomez-Cabrero D, Cervera A, McPherson A, et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016;17:13.
Cock PJ, Fields CJ, Goto N, Heuer ML, Rice PM. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2010;38:1767–71.
Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–90.
Patel RK, Jain M. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS ONE. 2012;7:e30619.
DeLuca DS, Levin JZ, Sivachenko A, Fennell T, Nazaire MD, Williams C, et al. RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics. 2012;28:1530–2.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27:863–4.
Chen Y, Chen Y, Shi C, Huang Z, Zhang Y, Li S, et al. SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience. 2018;7:1–6.
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36.
Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–60.
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21.
Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–60.
Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25.
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–5.
Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33:290–5.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–52.
Hurgobin B. Short read alignment using SOAP2. Methods Mol Biol. 2016;1374:241–52.
Robertson G, Schein J, Chiu R, Corbett R, Field M, Jackman SD, et al. De novo assembly and analysis of RNA-seq data. Nat Methods. 2010;7:909–12.
Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–30.
Anders S, Pyl PT, Huber W. HTSeq: a python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–9.
Roberts A, Pachter L. Streaming fragment assignment for real-time analysis of sequencing experiments. Nat Methods. 2013;10:71–3.
Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011;12:323.
Anders S, Reyes A, Huber W. Detecting differential usage of exons from RNA-seq data. Genome Res. 2012;22:2008–17.
Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 2016;34:525–7.
Patro R, Mount SM, Kingsford C. Sailfish enables alignment-free isoform quantification from RNA-seq reads using lightweight algorithms. Nat Biotechnol. 2014;32:462–4.
Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 2017;14:417–9.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40.
Moon M, Nakai K. Stable feature selection based on the ensemble L (1)-norm support vector machine for biomarker discovery. BMC Genomics. 2016;17:1026.
Zubovic L, Piazza S, Tebaldi T, Cozzuto L, Palazzo G, Sidarovich V, et al. The altered transcriptome of pediatric myelodysplastic syndrome revealed by RNA sequencing. J Hematol Oncol. 2020;13:135.
Oshlack A, Robinson MD, Young MD. From RNA-seq reads to differential expression results. Genome Biol. 2010;11:220.
Govindarajan M, Wohlmuth C, Waas M, Bernardini MQ, Kislinger T. High-throughput approaches for precision medicine in high-grade serous ovarian cancer. J Hematol Oncol. 2020;13:134.
Wu H, Li X, Li H. Gene fusions and chimeric RNAs, and their implications in cancer. Genes Dis. 2019;6:385–90.
Reeser JW, Martin D, Miya J, Kautto EA, Lyon E, Zhu E, et al. Validation of a targeted RNA sequencing assay for kinase fusion detection in solid tumors. J Mol Diagn. 2017;19:682–96.
Mercer TR, Clark MB, Crawford J, Brunck ME, Gerhardt DJ, Taft RJ, et al. Targeted sequencing for gene discovery and quantification using RNA CaptureSeq. Nat Protoc. 2014;9:989–1009.
Togni M, Masetti R, Pigazzi M, Astolfi A, Zama D, Indio V, et al. Identification of the NUP98-PHF23 fusion gene in pediatric cytogenetically normal acute myeloid leukemia by whole-transcriptome sequencing. J Hematol Oncol. 2015;8:69.
Veeraraghavan J, Ma J, Hu Y, Wang XS. Recurrent and pathological gene fusions in breast cancer: current advances in genomic discovery and clinical implications. Breast Cancer Res Treat. 2016;158:219–32.
Kloosterman WP, Coebergh van den Braak RRJ, Pieterse M, van Roosmalen MJ, Sieuwerts AM, Stangl C, et al. A systematic analysis of oncogenic gene fusions in primary colon cancer. Cancer Res. 2017;77:3814–22.
Sun YM, Chen YQ. Principles and innovative technologies for decrypting noncoding RNAs: from discovery and functional prediction to clinical application. J Hematol Oncol. 2020;13:109.
Zhou X, Zhan L, Huang K, Wang X. The functions and clinical significance of circRNAs in hematological malignancies. J Hematol Oncol. 2020;13:138.
Liu Y, Cheng Z, Pang Y, Cui L, Qian T, Quan L, et al. Role of microRNAs, circRNAs and long noncoding RNAs in acute myeloid leukemia. J Hematol Oncol. 2019;12:51.
Xu H, Wang C, Song H, Xu Y, Ji G. RNA-Seq profiling of circular RNAs in human colorectal Cancer liver metastasis and the potential biomarkers. Mol Cancer. 2019;18:8.
Alisoltani A, Fallahi H, Shiran B, Alisoltani A, Ebrahimie E. RNA-seq SSRs and small RNA-seq SSRs: new approaches in cancer biomarker discovery. Gene. 2015;560:34–43.
Zhou FF, Huang R, Jiang J, Zeng XH, Zou SQ. Correlated non-nuclear COX2 and low HER2 expression confers a good prognosis in colorectal cancer. Saudi J Gastroenterol. 2018;24:301–6.
Chen RH, Du Y, Han P, Wang HB, Liang FY, Feng GK, et al. ISG15 predicts poor prognosis and promotes cancer stem cell phenotype in nasopharyngeal carcinoma. Oncotarget. 2016;7:16910–22.
Liu G, Hou G, Li L, Li Y, Zhou W, Liu L. Potential diagnostic and prognostic marker dimethylglycine dehydrogenase (DMGDH) suppresses hepatocellular carcinoma metastasis in vitro and in vivo. Oncotarget. 2016;7:32607–16.
Mei J, Hao L, Liu X, Sun G, Xu R, Wang H, et al. Comprehensive analysis of peroxiredoxins expression profiles and prognostic values in breast cancer. Biomark Res. 2019;7:16.
Wang L, Ge J, Lan Y, Shi Y, Luo Y, Tan Y, et al. Tumor mutational burden is associated with poor outcomes in diffuse glioma. BMC Cancer. 2020;20:213.
Jiang T, Shi J, Dong Z, Hou L, Zhao C, Li X, et al. Genomic landscape and its correlations with tumor mutational burden, PD-L1 expression, and immune cells infiltration in Chinese lung squamous cell carcinoma. J Hematol Oncol. 2019;12:75.
Seo JS, Ju YS, Lee WC, Shin JY, Lee JK, Bleazard T, et al. The transcriptional landscape and mutational profile of lung adenocarcinoma. Genome Res. 2012;22:2109–19.
Nakagawa M, Nakatani F, Matsunaga H, Seki T, Endo M, Ogawara Y, et al. Selective inhibition of mutant IDH1 by DS-1001b ameliorates aberrant histone modifications and impairs tumor activity in chondrosarcoma. Oncogene. 2019;38:6835–49.
Davies KD, Lomboy A, Lawrence CA, Yourshaw M, Bocsi GT, Camidge DR, et al. DNA-based versus RNA-based detection of MET Exon 14 skipping events in lung cancer. J Thorac Oncol. 2019;14:737–41.
Unruh D, Zewde M, Buss A, Drumm MR, Tran AN, Scholtens DM, et al. Methylation and transcription patterns are distinct in IDH mutant gliomas compared to other IDH mutant cancers. Sci Rep. 2019;9:8946.
Yu J, Jiang PYZ, Sun H, Zhang X, Jiang Z, Li Y, et al. Advances in targeted therapy for acute myeloid leukemia. Biomark Res. 2020;8:17.
Yang X, Wang J. Precision therapy for acute myeloid leukemia. J Hematol Oncol. 2018;11:3.
Gu R, Yang X, Wei H. Molecular landscape and targeted therapy of acute myeloid leukemia. Biomark Res. 2018;6:32.
Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–401.
Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, et al. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell. 2017;171(1611–24):e24.
Tirosh I, Venteicher AS, Hebert C, Escalante LE, Patel AP, Yizhak K, et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature. 2016;539:309–13.
Best MG, Sol N, Kooi I, Tannous J, Westerman BA, Rustenburg F, et al. RNA-seq of tumor-educated platelets enables blood-based pan-cancer, multiclass, and molecular pathway cancer diagnostics. Cancer Cell. 2015;28:666–76.
Best MG, Vancura A, Wurdinger T. Platelet RNA as a circulating biomarker trove for cancer diagnostics. J Thromb Haemost. 2017;15:1295–306.
Zhu L, Li J, Gong Y, Wu Q, Tan S, Sun D, et al. Exosomal tRNA-derived small RNA as a promising biomarker for cancer diagnosis. Mol Cancer. 2019;18:74.
Nie Y, Jiao Y, Li Y, Li W. Investigation of the clinical significance and prognostic value of the lncRNA ACVR2B-As1 in liver cancer. Biomed Res Int. 2019;2019:4602371.
Gong W, Yang L, Wang Y, Xian J, Qiu F, Liu L, et al. Analysis of survival-related lncRNA landscape identifies a role for LINC01537 in energy metabolism and lung cancer progression. Int J Mol Sci. 2019;20.
Hang D, Zhou J, Qin N, Zhou W, Ma H, Jin G, et al. A novel plasma circular RNA circFARSA is a potential biomarker for non-small cell lung cancer. Cancer Med. 2018;7:2783–91.
Hua Q, Jin M, Mi B, Xu F, Li T, Zhao L, et al. LINC01123, a c-Myc-activated long non-coding RNA, promotes proliferation and aerobic glycolysis of non-small cell lung cancer through miR-199a-5p/c-Myc axis. J Hematol Oncol. 2019;12:91.
Wang Z, Qin B. Prognostic and clinicopathological significance of long noncoding RNA CTD-2510F5.4 in gastric cancer. Gastric Cancer. 2019;22:692–704.
Luo T, Zhao J, Lu Z, Bi J, Pang T, Cui H, et al. Characterization of long non-coding RNAs and MEF2C-AS1 identified as a novel biomarker in diffuse gastric cancer. Transl Oncol. 2018;11:1080–9.
Wang D, Wan X, Zhang Y, Kong Z, Lu Y, Sun X, et al. A novel androgen-reduced prostate-specific lncRNA, PSLNR, inhibits prostate-cancer progression in part by regulating the p53-dependent pathway. Prostate. 2019;79:1362–77.
Silva-Fisher JM, Dang HX, White NM, Strand MS, Krasnick BA, Rozycki EB, et al. Long non-coding RNA RAMS11 promotes metastatic colorectal cancer progression. Nat Commun. 2020;11:2156.
Yamada A, Yu P, Lin W, Okugawa Y, Boland CR, Goel A. A RNA-Sequencing approach for the identification of novel long non-coding RNA biomarkers in colorectal cancer. Sci Rep. 2018;8:575.
Bo H, Fan L, Li J, Liu Z, Zhang S, Shi L, et al. High Expression of lncRNA AFAP1-AS1 Promotes the Progression of Colon Cancer and Predicts Poor Prognosis. J Cancer. 2018;9:4677–83.
Chen Q, Hu L, Chen K. Construction of a nomogram based on a hypoxia-related lncRNA signature to improve the prediction of gastric cancer prognosis. Front Genet. 2020;11:570325.
Guo YZ, Sun HH, Wang XT, Wang MT. Transcriptomic analysis reveals key lncRNAs associated with ribosomal biogenesis and epidermis differentiation in head and neck squamous cell carcinoma. J Zhejiang Univ Sci B. 2018;19:674–88.
Yao Y, Chen X, Lu S, Zhou C, Xu G, Yan Z, et al. Circulating long noncoding RNAs as biomarkers for predicting head and neck squamous cell carcinoma. Cell Physiol Biochem. 2018;50:1429–40.
Gong X, Siprashvili Z, Eminaga O, Shen Z, Sato Y, Kume H, et al. Novel lincRNA SLINKY is a prognostic biomarker in kidney cancer. Oncotarget. 2017;8:18657–69.
James AR, Schroeder MP, Neumann M, Bastian L, Eckert C, Gökbuget N, et al. Long non-coding RNAs defining major subtypes of B cell precursor acute lymphoblastic leukemia. J Hematol Oncol. 2019;12:8.
Li S, Ma Y, Tan Y, Ma X, Zhao M, Chen B, et al. Profiling and functional analysis of circular RNAs in acute promyelocytic leukemia and their dynamic regulation during all-trans retinoic acid treatment. Cell Death Dis. 2018;9:651.
Guo M, Peng Y, Gao A, Du C, Herman JG. Epigenetic heterogeneity in cancer. Biomark Res. 2019;7:23.
Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–96.
Maynard A, McCoach CE, Rotow JK, Harris L, Haderk F, Kerr DL, et al. Therapy-induced evolution of human lung cancer revealed by single-cell RNA sequencing. Cell. 2020;182(1232–51):e22.
Nam AS, Chaligne R, Landau DA. Integrating genetic and non-genetic determinants of cancer evolution by single-cell multi-omics. Nat Rev Genet. 2020.
Berglund E, Maaskola J, Schultz N, Friedrich S, Marklund M, Bergenstråhle J, et al. Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity. Nat Commun. 2018;9:2419.
Bakken TE, Hodge RD, Miller JA, Yao Z, Nguyen TN, Aevermann B, et al. Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLoS ONE. 2018;13:e0209648.
Selewa A, Dohn R, Eckart H, Lozano S, Xie B, Gauchat E, et al. Systematic comparison of high-throughput single-cell and single-nucleus transcriptomes during cardiomyocyte differentiation. Sci Rep. 2020;10:1535.
Lim ZF, Ma PC. Emerging insights of tumor heterogeneity and drug resistance mechanisms in lung cancer targeted therapy. J Hematol Oncol. 2019;12:134.
Wang Q, Guldner IH, Golomb SM, Sun L, Harris JA, Lu X, et al. Single-cell profiling guided combinatorial immunotherapy for fast-evolving CDK4/6 inhibitor-resistant HER2-positive breast cancer. Nat Commun. 2019;10:3817.
Tanaka N, Katayama S, Reddy A, Nishimura K, Niwa N, Hongo H, et al. Single-cell RNA-seq analysis reveals the platinum resistance gene COX7B and the surrogate marker CD63. Cancer Med. 2018;7:6193–204.
Debruyne DN, Dries R, Sengupta S, Seruggia D, Gao Y, Sharma B, et al. BORIS promotes chromatin regulatory interactions in treatment-resistant cancer cells. Nature. 2019;572:676–80.
Sharma A, Cao EY, Kumar V, Zhang X, Leong HS, Wong AML, et al. Longitudinal single-cell RNA sequencing of patient-derived primary cells reveals drug-induced infidelity in stem cell hierarchy. Nat Commun. 2018;9:4931.
Shao F, Huang M, Meng F, Huang Q. Circular RNA SIgnature predicts gemcitabine resistance of pancreatic ductal adenocarcinoma. Front Pharmacol. 2018;9:584.
Wang WT, Han C, Sun YM, Chen TQ, Chen YQ. Noncoding RNAs in cancer therapy resistance and targeted drug development. J Hematol Oncol. 2019;12:55.
Shang J, Chen WM, Liu S, Wang ZH, Wei TN, Chen ZZ, et al. CircPAN3 contributes to drug resistance in acute myeloid leukemia through regulation of autophagy. Leuk Res. 2019;85:106198.
Suzuki K, Okuno Y, Kawashima N, Muramatsu H, Okuno T, Wang X, et al. MEF2D-BCL9 fusion gene is associated with high-risk acute B-cell precursor lymphoblastic leukemia in adolescents. J Clin Oncol. 2016;34:3451–9.
Pallarès V, Unzueta U, Falgàs A, Sánchez-García L, Serna N, Gallardo A, et al. An Auristatin nanoconjugate targeting CXCR4+ leukemic cells blocks acute myeloid leukemia dissemination. J Hematol Oncol. 2020;13:36.
Ding Y, Gao H, Zhang Y, Li Y, Vasdev N, Gao Y, et al. Alantolactone selectively ablates acute myeloid leukemia stem and progenitor cells. J Hematol Oncol. 2016;9:93.
Bill M, Papaioannou D, Karunasiri M, Kohlschmidt J, Pepe F, Walker CJ, et al. Expression and functional relevance of long non-coding RNAs in acute myeloid leukemia stem cells. Leukemia. 2019;33:2169–82.
Fan X, Rudensky AY. Hallmarks of tissue-resident lymphocytes. Cell. 2016;164:1198–211.
Burrell RA, Swanton C. Re-evaluating clonal dominance in cancer evolution. Trends Cancer. 2016;2:263–76.
Shalek AK, Satija R, Adiconis X, Gertner RS, Gaublomme JT, Raychowdhury R, et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature. 2013;498:236–40.
Pan Y, Lu F, Fei Q, Yu X, Xiong P, Yu X, et al. Single-cell RNA sequencing reveals compartmental remodeling of tumor-infiltrating immune cells induced by anti-CD47 targeting in pancreatic cancer. J Hematol Oncol. 2019;12:124.
Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161:1202–14.
Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, et al. Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell. 2018;174:1293–308.
Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, Guo X, et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell. 2017;169:1342–56.
Cunha LD, Yang M, Carter R, Guy C, Harris L, Crawford JC, et al. LC3-associated phagocytosis in myeloid cells promotes tumor immune tolerance. Cell. 2018;175:429–41.
van Galen P, Hovestadt V, Wadsworth Ii MH, Hughes TK, Griffin GK, Battaglia S, et al. Single-cell RNA-seq reveals AML hierarchies relevant to disease progression and immunity. Cell. 2019;176:1265–81.
Ren X, Zhong G, Zhang Q, Zhang L, Sun Y, Zhang Z. Reconstruction of cell spatial organization from single-cell RNA sequencing data based on ligand-receptor mediated self-assembly. Cell Res. 2020;30:763–78.
Wculek SK, Malanchi I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature. 2015;528:413–7.
Coffelt SB, Wellenstein MD, de Visser KE. Neutrophils in cancer: neutral no more. Nat Rev Cancer. 2016;16:431–46.
Singhal S, Bhojnagarwala PS, O’Brien S, Moon EK, Garfall AL, Rao AS, et al. Origin and role of a subset of tumor-associated neutrophils with antigen-presenting cell features in early-stage human lung cancer. Cancer Cell. 2016;30:120–35.
Massara M, Bonavita O, Savino B, Caronni N, Mollica Poeta V, Sironi M, et al. ACKR2 in hematopoietic precursors as a checkpoint of neutrophil release and anti-metastatic activity. Nat Commun. 2018;9:676.
Ponzetta A, Carriero R, Carnevale S, Barbagallo M, Molgora M, Perucchini C, et al. Neutrophils driving unconventional T cells mediate resistance against murine sarcomas and selected human tumors. Cell. 2019;178(346–60):e24.
Janiszewska M, Tabassum DP, Castaño Z, Cristea S, Yamamoto KN, Kingston NL, et al. Subclonal cooperation drives metastasis by modulating local and systemic immune microenvironments. Nat Cell Biol. 2019;21:879–88.
Rath J, Bajwa G, Carreres B, Hoyer E, Gruber I, Martínez-Paniagua M, et al. Single-cell transcriptomics identifies multiple pathways underlying antitumor function of TCR- and CD8αβ-engineered human CD4 T cells. Sci Adv. 2020;6:eaaz7809.
Nirschl CJ, Suárez-Fariñas M, Izar B, Prakadan S, Dannenfelser R, Tirosh I, et al. IFNγ-dependent tissue-immune homeostasis is co-opted in the tumor microenvironment. Cell. 2017;170(127–41):e15.
Wang D, Lin J, Yang X, Long J, Bai Y, Yang X, et al. Combination regimens with PD-1/PD-L1 immune checkpoint inhibitors for gastrointestinal malignancies. J Hematol Oncol. 2019;12:42.
Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27:450–61.
Zhao Z, Zheng L, Chen W, Weng W, Song J, Ji J. Delivery strategies of cancer immunotherapy: recent advances and future perspectives. J Hematol Oncol. 2019;12:126.
Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168:707–23.
Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov. 2015;14:561–84.
Yi M, Yu S, Qin S, Liu Q, Xu H, Zhao W, et al. Gut microbiome modulates efficacy of immune checkpoint inhibitors. J Hematol Oncol. 2018;11:47.
Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371:2189–99.
Li Z, Song W, Rubinstein M, Liu D. Recent updates in cancer immunotherapy: a comprehensive review and perspective of the 2018 China Cancer Immunotherapy Workshop in Beijing. J Hematol Oncol. 2018;11:142.
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–20.
Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16:275–87.
Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med. 2018;24:1550–8.
Sehgal K, Portell A, Ivanova E, Lizotte P, Mahadevan N, Greene J, et al. Dynamic single-cell RNA sequencing identifies immunotherapy persister cells following PD-1 blockade. J Clin Invest. 2020. https://doi.org/10.1172/JCI135038.
Zilionis R, Engblom C, Pfirschke C, Savova V, Zemmour D, Saatcioglu HD, et al. Single-cell transcriptomics of human and mouse lung cancers reveals conserved myeloid populations across individuals and species. Immunity. 2019;50:1317–34.
Zhang L, Li Z, Skrzypczynska KM, Fang Q, Zhang W, O’Brien SA, et al. Single-cell analyses inform mechanisms of myeloid-targeted therapies in colon cancer. Cell. 2020;181:442–59.
Katzenelenbogen Y, Sheban F, Yalin A, Yofe I, Svetlichnyy D, Jaitin DA, et al. Coupled scRNA-Seq and intracellular protein activity reveal an immunosuppressive role of TREM2 in cancer. Cell. 2020;182:872–85.
Jiang T, Shi T, Zhang H, Hu J, Song Y, Wei J, et al. Tumor neoantigens: from basic research to clinical applications. J Hematol Oncol. 2019;12:93.
McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463–9.
Hacohen N, Fritsch EF, Carter TA, Lander ES, Wu CJ. Getting personal with neoantigen-based therapeutic cancer vaccines. Cancer Immunol Res. 2013;1:11–5.
Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547:217–21.
Nathanson T, Ahuja A, Rubinsteyn A, Aksoy BA, Hellmann MD, Miao D, et al. Somatic mutations and neoepitope homology in melanomas treated with CTLA-4 blockade. Cancer Immunol Res. 2017;5:84–91.
Safonov A, Jiang T, Bianchini G, Győrffy B, Karn T, Hatzis C, et al. Immune gene expression is associated with genomic aberrations in breast cancer. Cancer Res. 2017;77:3317–24.
Karasaki T, Nagayama K, Kuwano H, Nitadori JI, Sato M, Anraku M, et al. Prediction and prioritization of neoantigens: integration of RNA sequencing data with whole-exome sequencing. Cancer Sci. 2017;108:170–7.
Nejo T, Matsushita H, Karasaki T, Nomura M, Saito K, Tanaka S, et al. Reduced neoantigen expression revealed by longitudinal multiomics as a possible immune evasion mechanism in glioma. Cancer Immunol Res. 2019;7:1148–61.
Lv JW, Zheng ZQ, Wang ZX, Zhou GQ, Chen L, Mao YP, et al. Pan-cancer genomic analyses reveal prognostic and immunogenic features of the tumor melatonergic microenvironment across 14 solid cancer types. J Pineal Res. 2019;66:e12557.
Linxweiler M, Kuo F, Katabi N, Lee M, Nadeem Z, Dalin MG, et al. The immune microenvironment and neoantigen landscape of aggressive salivary gland carcinomas differ by subtype. Clin Cancer Res. 2020;26:2859–70.
Kim S, Kim HS, Kim E, Lee MG, Shin EC, Paik S, et al. Neopepsee: accurate genome-level prediction of neoantigens by harnessing sequence and amino acid immunogenicity information. Ann Oncol. 2018;29:1030–6.
Wang TY, Wang L, Alam SK, Hoeppner LH, Yang R. ScanNeo: identifying indel-derived neoantigens using RNA-Seq data. Bioinformatics. 2019;35:4159–61.
Zhang Z, Zhou C, Tang L, Gong Y, Wei Z, Zhang G, et al. ASNEO: Identification of personalized alternative splicing based neoantigens with RNA-seq. Aging (Albany NY). 2020;12:14633–48.
Yang H, Sun L, Guan A, Yin H, Liu M, Mao X, et al. Unique TP53 neoantigen and the immune microenvironment in long-term survivors of Hepatocellular carcinoma. Cancer Immunol Immunother. 2020. https://doi.org/10.1007/s00262-020-02711-8.
Lu YC, Zheng Z, Robbins PF, Tran E, Prickett TD, Gartner JJ, et al. An efficient single-cell RNA-seq approach to identify neoantigen-specific T cell receptors. Mol Ther. 2018;26:379–89.
Engström P, Steijger T, Sipos B, Grant G, Kahles A, Rätsch G, et al. Systematic evaluation of spliced alignment programs for RNA-seq data. Nat Methods. 2013;10:1185–91.
Sharon D, Tilgner H, Grubert F, Snyder M. A single-molecule long-read survey of the human transcriptome. Nat Biotechnol. 2013;31:1009–14.
Quail MA, Smith M, Coupland P, Otto TD, Harris SR, Connor TR, et al. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics. 2012;13:341.
Tilgner H, Grubert F, Sharon D, Snyder MP. Defining a personal, allele-specific, and single-molecule long-read transcriptome. Proc Natl Acad Sci USA. 2014;111:9869–74.
Stark R, Grzelak M, Hadfield J. RNA sequencing: the teenage years. Nat Rev Genet. 2019;20:631–56.
Antipov D, Korobeynikov A, McLean J, Pevzner P. hybridSPAdes: an algorithm for hybrid assembly of short and long reads. Bioinformatics. 2016;32:1009–15.
Boluki S, Zamani Dadaneh S, Qian X, Dougherty E. Optimal clustering with missing values. BMC Bioinform. 2019;20:321.
Hicks S, Townes F, Teng M, Irizarry R. Missing data and technical variability in single-cell RNA-sequencing experiments. Biostatistics. 2018;19:562–78.
Hou W, Ji Z, Ji H, Hicks S. A systematic evaluation of single-cell RNA-sequencing imputation methods. Genome biol. 2020;21:218.
Song F, Chan G, Wei Y. Flexible experimental designs for valid single-cell RNA-sequencing experiments allowing batch effects correction. Nat Commun. 2020;11:3274.
Buenrostro J, Giresi P, Zaba L, Chang H, Greenleaf W. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10:1213–8.
Yang C, Ma L, Xiao D, Ying Z, Jiang X, Lin Y. Sparassis latifolia Integration of ATAC-Seq and RNA-Seq Identifies Key Genes in Light-Induced Primordia Formation of. Int J Mol Sci. 2019;21.
Wu X, Yang Y, Zhong C, Guo Y, Wei T, Li S, et al. Epinephelus coioides Integration of ATAC-seq and RNA-seq Unravels Chromatin Accessibility during Sex Reversal in Orange-Spotted Grouper (Epinephelus coioides). Int J Mol Sci. 2020;21.
Simonis M, Kooren J, de Laat W. An evaluation of 3C-based methods to capture DNA interactions. Nat Methods. 2007;4:895–901.
Lieberman-Aiden E, van Berkum N, Williams L, Imakaev M, Ragoczy T, Telling A, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–93.
Mifsud B, Tavares-Cadete F, Young A, Sugar R, Schoenfelder S, Ferreira L, et al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat Genet. 2015;47:598–606.
Crane E, Bian Q, McCord R, Lajoie B, Wheeler B, Ralston E, et al. Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature. 2015;523:240–4.
Chen H, Seaman L, Liu S, Ried T, Rajapakse I. Chromosome conformation and gene expression patterns differ profoundly in human fibroblasts grown in spheroids versus monolayers. Nucleus. 2017;8:383–91.
Vara C, Paytuví-Gallart A, Cuartero Y, Le Dily F, Garcia F, Salvà-Castro J, et al. Three-dimensional genomic structure and cohesin occupancy correlate with transcriptional activity during spermatogenesis. Cell Rep. 2019;28(352–67):e9.
Zeng C, Huang W, Li Y, Weng H. Roles of METTL3 in cancer: mechanisms and therapeutic targeting. J Hematol Oncol. 2020;13:117.
Jenjaroenpun P, Wongsurawat T, Wadley T, Wassenaar T, Liu J, Dai Q, et al. Decoding the epitranscriptional landscape from native RNA sequences. Nucleic Acids Res. 2020. https://doi.org/10.1093/nar/gkaa620.
Parker M, Knop K, Sherwood A, Schurch N, Mackinnon K, Gould P, et al. Nanopore direct RNA sequencing maps the complexity of Arabidopsis mRNA processing and mA modification. eLife. 2020;9.
Zhang S, Li R, Zhang L, Chen S, Xie M, Yang L, et al. New insights into Arabidopsis transcriptome complexity revealed by direct sequencing of native RNAs. Nucleic Acids Res. 2020;48:7700–11.
Pan X, Zhang H, Xu D, Chen J, Chen W, Gan S, et al. Identification of a novel cancer stem cell subpopulation that promotes progression of human fatal renal cell carcinoma by single-cell RNA-seq analysis. Int J Biol Sci. 2020;16:3149–62.
Li Y, Chen C, Chen J, Lai Y, Wang S, Jiang S, et al. Single-cell analysis reveals immune modulation and metabolic switch in tumor-draining lymph nodes. Oncoimmunology. 2020;9:1830513.
Giladi A, Cohen M, Medaglia C, Baran Y, Li B, Zada M, et al. Dissecting cellular crosstalk by sequencing physically interacting cells. Nat Biotechnol. 2020;38:629–37.
Caruso F, Garofano L, D'Angelo F, Yu K, Tang F, Yuan J, et al. A map of tumor-host interactions in glioma at single-cell resolution. GigaScience. 2020;9.
Jaitin D, Weiner A, Yofe I, Lara-Astiaso D, Keren-Shaul H, David E, et al. Dissecting immune circuits by linking CRISPR-pooled screens with single-cell RNA-seq. Cell. 2016;167:1883–96.
Datlinger P, Rendeiro A, Schmidl C, Krausgruber T, Traxler P, Klughammer J, et al. Pooled CRISPR screening with single-cell transcriptome readout. Nat Methods. 2017;14:297–301.
Genga R, Kernfeld E, Parsi K, Parsons T, Ziller M, Maehr R. Single-cell RNA-sequencing-based CRISPRi screening resolves molecular drivers of early human endoderm development. Cell Rep. 2019;27:708–18.
Qiu Q, Hu P, Qiu X, Govek K, Cámara P, Wu H. Massively parallel and time-resolved RNA sequencing in single cells with scNT-seq. Nat Methods. 2020;17:991–1001.
McFarland J, Paolella B, Warren A, Geiger-Schuller K, Shibue T, Rothberg M, et al. Multiplexed single-cell transcriptional response profiling to define cancer vulnerabilities and therapeutic mechanism of action. Nat commun. 2020;11:4296.
Sun W, Dong H, Balaz M, Slyper M, Drokhlyansky E, Colleluori G, et al. snRNA-seq reveals a subpopulation of adipocytes that regulates thermogenesis. Nature. 2020;587:98–102.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (81300398 and 81974436); Key-Area Research and Development Program of Guangdong Province (2020B1111030005); The Program for Guangdong Introducing Innovative and Entrepreneurial Teams (2019ZT08Y485); Key Program of Marine Economy Development (Six Marine Industries) Special Foundation of Department of Natural Resources of Guangdong Province (GDNRC [2020]070); The Fundamental Research Funds for the Central Universities (19ykzd06) and the Opening Fund of Guangdong Key Laboratory of Marine Materia Medica (LMM2020-4).
Author information
Authors and Affiliations
Contributions
M.H. and S.T. wrote and edited this manuscript and created figures and tables. L.Z., L-T. D., X-M. H., S-H. H., S-J. X., Z-D. X and H.Z. reviewed and revised the manuscript. Z-D. X and H.Z. provided direction and guidance throughout the preparation of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hong, M., Tao, S., Zhang, L. et al. RNA sequencing: new technologies and applications in cancer research. J Hematol Oncol 13, 166 (2020). https://doi.org/10.1186/s13045-020-01005-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-020-01005-x