
Abstract
Acute myeloid leukemia (AML) is a clonal malignancy characterized by genetic heterogeneity due to recurrent gene mutations. Treatment with cytotoxic chemotherapy has been the standard of care for more than half of a century. Although much progress has been made toward improving treatment related mortality rate in the past few decades, long term overall survival has stagnated. Exciting developments of gene mutation-targeted therapeutic agents are now changing the landscape in AML treatment. New agents offer more clinical options for patients and also confer a more promising outcome. Since Midostaurin, a FLT3 inhibitor, was first approved by US FDA in 2017 as the first gene mutation-targeted therapeutic agent, an array of new gene mutation-targeted agents are now available for AML treatment. In this review, we will summarize the recent advances in gene mutation-targeted therapies for patients with AML.
Similar content being viewed by others

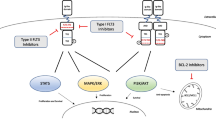
Introduction
Acute myeloid leukemia (AML) is a clonal malignancy originating from hematopoietic stem cells, characterized by heterogeneous chromosomal abnormalities, recurrent gene mutations, epigenetic modifications affecting chromatin structure, and microRNAs deregulations. Genomic heterogeneity, patients’ individual variability, and gene mutations are few major obstacles among the many factors that impact treatment efficacy for AML patients [1, 2].
Different strategies have been used to treat various types of cancer in preclinical models [3, 4]. Traditional chemotherapy using cytotoxic agents in AML treatment had been the main modality for decades. New molecular techniques, however, such as next-generation sequencing (NGS) identifying important genetic alterations, have paved the path for new drug development targeting those specific gene mutations. Since the past few years, the state-of-the-art treatment for AML has evolved rapidly: cytogenetic and molecular interactions being more individualized, the state of minimal residual disease (MRD) detected by flow cytometry and NGS, and incorporation of gene mutation-targeted novel therapies. In combination with precise clinical diagnosis and detailed risk stratification, gene mutation-targeted new drug therapies have made breakthrough and promising progresses for patients with AML [5, 6].
In April 2017, the US Food and Drug Administration (FDA) approved Midostaurin, a FMS-like tyrosine kinase 3 (FLT3) inhibitor, for AML patients with FLT3 mutations. Midostaurin is the first tyrosine kinase inhibitor (TKI) approved for AML; and it is also the first drug approved in a mutation-specific and non–acute promyelocytic leukemia (APL) subtype. Since then, many gene mutation-targeted therapies for AML have emerged, such as Enasidenib, an isocitrate dehydrogenase (IDH)2 inhibitor, for relapsed/refractory (R/R) AML with IDH2 mutations [7,8,9]. The “one-size-fits-all” cytotoxic chemotherapy regimen will soon be enhanced or replaced by more specific targeted treatment in AML.
Targeted therapy in AML can be divided into 3 groups: Group 1: agents that act on oncogenic effectors of recurrent AML associated mutations, which include FLT3 and IDH inhibitors. Group 2: agents that act on disrupting key cell metabolic or maintenance pathways without directly damaging DNA or its repair. These include epigenetic modifiers and agents that directly target apoptosis. Group 3: agents that act by targeted delivery of cytotoxic agents, such as ADCs [10]. In this review article, we will focus on the advances in the gene mutation-targeted agents, including FLT3 inhibitors, IDH inhibitors and Smoothened (SMO) inhibitors.
FLT3 inhibitors
FLT3 is a transmembrane ligand-activated receptor tyrosine kinase (RTK) which plays an important role in the early stages of both myeloid and lymphoid lineage development. FLT3 ligand binds and activates FLT3 through various signaling pathways, such as PI3K, RAS, and STAT5 [11]. FLT3 mutations are found in approximately 30–35% of newly diagnosed AML cases with either internal tandem duplications (FLT3-ITD) within the juxtamembrane domain coding region (exons 14 and 15, [12]) or missense mutations in the tyrosine kinase domain (FLT3-TKD) in the activation loop (exon20) [13]. FLT3-ITD and FLT3-TKD type mutations occur in about 25% and 7–10% of AML patients, respectively [14,15,16,17]. Data have suggested that there are racial and ethnic disparities in genetic alteration between Caucasian and Eastern Asian population. Lower proportion of FLT3-ITD mutation and more AML patients with core binding factor leukemia have been found in Eastern Asian cohorts [18]. FLT3-ITD mutation had been considered as a negative prognostic marker, used for AML risk stratification and disease monitoring via MRD, with the clinical importance of early detection at diagnosis and again at relapse [2].
As progresses have been made in understanding the mechanism of FLT3 gene mutation, TKI agents have been developed by targeting different points of the ATP binding site in the intracellular domain of the FLT3 RTK: Type 1 inhibitors, which include Sunitinib, Lestaurtinib, Midostaurin, Crenolanib, and Gilteritinib [19], bind to the RTK ATP-binding site in the active conformation and the inactive state; Type 2 inhibitors, which include Sorafenib, Quizartinib and Ponatinib [19, 20], bind to the hydrophobic region in juxtaposition to ATP-binding domain when RTK is in the inactive state and prevent receptor activation.
Midostaurin
Midostaurin was approved by the US FDA for AML induction and consolidation based on the RATIFY trial, which took 13 years to complete [7]. The RATIFY trial was the first large multicenter study investigating the addition of Midostaurin to induction and consolidation and continued as maintenance therapy for 1 year in patients not proceeding to allogeneic transplant [9, 21, 22]. Patients with FLT3 mutations, either ITD or TKD, had a 4-year survival of 51.4% on Midostaurin versus 44.2% on placebo (P = .0074), and the benefit was most pronounced in Nucleophosmin 1 (NPM1) wt and FLT3high patients [9]. In another study, Midostaurin was added to intensive induction chemotherapy, consolidation and continued as maintenance in FLT3-ITD AML patients complete remission (CR) plus complete remission with incomplete hematologic recovery (CRi) after induction therapy was observed in 76.4% patients. Event free survival (EFS) and overall survival (OS) at 2 years were 39 and 34% in younger and 53 and 46% in older patients, respectively. Propensity score-weighted analysis revealed a significant improvement of EFS by Midostaurin overall and in older patients [23].
In a new retrospective exploratory study, multivariate Cox model for OS using allogeneic hematopoietic stem cell transplantation (allo-HSCT) in first complete remission (CR1) as a time-dependent variable revealed treatment with Midostaurin, allo-HSCT, European Leukemia Net (ELN) favorable-risk group, and lower WBC counts as significant favorable factors. There was a consistent beneficial effect of Midostaurin across ELN risk groups [24]. Midostaurin has been recommended as frontline therapy for the FLT3 gene mutated AML patients with either FLT3-ITD or FLT3-TKD [19, 25]. It’s also been proved cost-effectiveness when Midostaurin was combined with standard chemotherapy in the treatment of newly diagnosed FLT3-mutated AML patients [26].
Midostaurin is among the least potent FLT3 inhibitors. More potent FLT3 inhibitors are Gilteritinib, Quizartinib, and Crenolanib [20]. Early phase trials combining these newer generation FLT3 TKIs with 7 + 3 induction chemotherapy in the frontline setting have been reported recently with meaningfully higher response rate [25].
Gilteritinib
Gilteritinib is an orally available small molecule receptor TKI for the treatment of AML harboring FLT3 mutations. Gilteritinib inhibits FLT3 signaling in cells expressing FLT3-ITD, TKD mutation FLT3-D835Y and the double mutant FLT3-ITD-D835Y, thereby inducing apoptosis. Gilteritinib also binds to and inhibits the wild-type and mutated forms of anaplastic lymphoma kinase (ALK), resulting in reduced tumor cell proliferation in cancer cell types that overexpress the mutation [27, 28].
In a phase 3 trial with R/R FLT3-mutated AML, the median OS for the group with single agent Gilteritinib was significantly longer than that of the group with chemotherapy (9.3 months vs. 5.6 months). The median EFS was 2.8 months in the Gilteritinib group and 0.7 months in the chemotherapy group. The percentage of patients who achieved complete remission with full or partial hematologic recovery was 34.0% in the Gilteritinib group and 15.3% in the chemotherapy group. Gilteritinib resulted in significantly longer survival and higher percentages of CR than salvage chemotherapy among patients with R/R FLT3-mutated AML [29]. These findings confirm the superior efficacy of Gilteritinib over chemotherapy for patients with FLT3-mutant AML. Currently, multiple clinical trials are ongoing to evaluate the combination of Gilteritinib with other agents and regimens [25, 30]. These clinical studies supported Gilteritinib’s approval by US FDA in 2018 as the new standard therapy for R/R FLT3-mutated AML [31].
Sunitinib
Sunitinib (SU11248) is a small-molecule FLT3 inhibitor with selectivity for FLT3 and others, such as platelet-derived growth factor receptors (PDGFR), vascular endothelial growth factor receptor (VEGFR1) 1, VEGFR2, and KIT [32]. It has both direct anti-tumor and anti-angiogenic properties [33]. One study found that the signal transducer and activator of transcription 5 (STAT5) phosphorylation in patients with FLT3-ITD was also reduced [34]. Furthermore, Sunitinib induces G1 phase arrest, increases pro-apoptotic molecule expression, and decreases anti-apoptotic molecule expression in AML cells [35].
Intriguingly, Sunitinib shows synergistic effects with Cytarabine or Daunorubicin in inhibiting proliferation and survival of primary AML myeloblasts expressing mutant FLT3-ITD, FLT3-D835V, or FLT3-WT [36]. Several early clinical trials evaluating Sunitinib in combination with chemotherapy had shown some promising results in phase I/II clinical trials [37, 38], however, due to high incidence of adverse effects such as blood and lymphatic system disorders, cardiac disorders, gastrointestinal disorders and others, no clinical trials in the realm of hematological malignancy are actively going on. Sunitinib has been approved and widely used for treatment of several solid tumors, such as renal cell cancer, gastrointestinal stromal cell tumor, and neuroendocrine tumors.
Lestaurtinib
Lestaurtinib (CEP-701) is a multi-targeted TKI that potently inhibits FLT3 tyrosine kinase and induces hematological remission in AML patients harboring FLT3-ITD. However, the majority of patients in clinical trials developed resistance to CEP-701. Although restoration of SHP-1 expression induces sensitivity towards CEP-701 and could serve as a target in the treatment of AML [39], Lestaurtinib failed to demonstrate any overall clinical benefit in a phase III trial when combined with intensive chemotherapy in patients with newly diagnosed FLT3-ITD-mutated AML [25, 40, 41].
Crenolanib
Crenolanib, a potent type I pan-FLT3 inhibitor, is effective against both ITD and resistance-conferring TKD mutations. While Crenolanib monotherapy has demonstrated clinical benefit in heavily pretreated R/R AML patients, responses are transient and relapse eventually occurs [42]. Study on the mechanisms of Crenolanib resistance has been done by performing whole exome sequencing of AML patient samples before and after Crenolanib treatment. Unlike other FLT3 inhibitors, Crenolanib does not induce FLT3 secondary mutations, and mutations of the FLT3 gatekeeper residue are infrequent. Instead, mutations of NRAS and IDH2 arise mostly as FLT3-independent subclones. Meanwhile TET2 and IDH1 predominantly co-occur with FLT3-mutant clones and are enriched in Crenolanib poor-responders. The other patients have exhibited post-Crenolanib expansion of mutations associated with epigenetic regulators, transcription factors, and cohesion factors, suggesting diverse genetic/epigenetic mechanisms of Crenolanib resistance. Drug combinations in experimental models can restore Crenolanib sensitivity [42].
Crenolanib was well tolerated in a phase II trial in combination with 7 + 3 induction therapy in newly diagnosed FLT3-mutated AM patients. Addition of Crenolanib to induction chemotherapy in patients with concurrent FLT3 and other mutations, such as NPM1, DNA methyltransferase 3A (DNMT3A), Runt-related transcription factor 1 (RUNX1), or Wilms’ tumour 1 (WT1), can overcome the poor prognostic implication of adverse mutations co-occurring with mutated FLT3 [12].
Ongoing clinical trials are assessing the efficacy of Crenolanib in combination with intensive salvage chemotherapy for patients with R/R FLT3 mutant AML (NCT2626338). Incorporation of Crenolanib into frontline intensive chemotherapy regimens have resulted in higher response rates and may eventually replace Midostaurin in the upfront setting [43]. Currently, there are 6 registered on-going clinical studies of Crenolanib for AML patients as of February 2020 (Table 1).
Quizartinib
Quizartinib is a potent and selective type 2 FLT3 inhibitor and has been used as an effective therapy for patients with FLT3-ITD AML. Quizartinib inhibits FLT3 thereby dampen oncogenic drive, leading to apoptosis of tumor cells. Phase 1 study demonstrated efficacy when combined with induction chemotherapy, and when used as monotherapy for maintenance therapy in AML patients after allo-HSCT [44, 45]. Another phase 1 multicenter dose-escalation study assessing the safety/tolerability of Quizartinib maintenance post-HSCT in FLT3+ AML demonstrated the safety, with promising results in the first 13 patients treated and no increase in graft versus host disease (GvHD) [46]. A phase 2 study demonstrated the potency and efficacy of Quizartinib amongst FLT3+ R/R AML patients within 1 year of induction therapy, or who had undergone salvage chemotherapy, or allo-HSCT [47, 48].
A phase III QUANTUM-R study further substantiated Quizartinib’s efficacy. Quizartinib could be considered a new standard of care for patients with rapidly proliferative disease and very poor prognosis [49]. Multiple clinical trials have proved its efficacy in R/R AML with FLT3-ITD mutation. Quizartinib resistance has been observed in clinical treatment. Further clinical studies are ongoing aiming to reduce toxicity, increase efficacy by combining with a targeted drug for RUNX1, and identify a predictive response biomarker in patients [47, 48, 50].
Strategies on combining Quizartinib with other TKI agents like Crenolinib, PIM kinase, and MEK inhibitors should be further explored [51]. When managing patients on Quizartinib, some special situations need to be considered for adequate scheduling and tolerability, bridging to allo-HSCT, and durable remission on maintenance therapy [52]. Currently, there are 15 registered on-going clinical studies of Quizartinib for leukemia patients as of February 2020 (Table 2).
Sorafenib
The multi-kinase inhibitor Sorafenib has demonstrated modest efficacy in FLT3+ AML as monotherapy. Sorafenib’s effect against AML is similar to that of Sunitinib [33]. However, resistance limited its use as a single agent [20, 53]. In combination with standard chemotherapy in patients under the age of 60, Sorafenib prolongs survival with modestly increased toxicity [54]. The survival benefit is less clear in patients over the age of 60 when added to standard induction chemotherapy [55, 56]. Sorafenib maintenance following allo-HSCT resulted in improved OS and EFS [57,58,59]. Sorafenib before transplantation, Sorafenib maintenance after transplantation, and their combined application all could improve the outcomes for patients with FLT3-ITD AML. Sorafenib’s effect on induction therapy and maintenance following allo-HSCT lead to superior 3-year EFS and 3-year OS as induction, re-induction therapy, or post-transplant as maintenance therapy when compared to no Sorafenib use. There was superior LFS with any use of Sorafenib with most benefit seeing in the group receiving both pre-transplant and post-transplant [60]. A phase 2 clinical trial showed Sorafenib and Omacetaxine Mepesuccinate as a safe and effective treatment for AML with FLT3-ITD mutation [61].
Allo-HSCT plus Sorafenib maintenance was an effective strategy to improve recurrence free survival and decrease relapse probability in FLT3-ITD AML patients. It had benefits to AML patients regardless of ITD mutant ratio, and to those with long ITD length instead of the short ITD length [62]. A prospective study of patients with FLT3-ITD AML undergoing allo-HSCT was conducted to evaluate the safety, tolerability, and outcome of Sorafenib administered peritransplant. Sorafenib dosing was individualized in the post-transplantation setting according to patient tolerability. Results indicate that Sorafenib is effective in vivo FLT3 inhibition and yields encouraging survival results [63].
Another study showed Sorafenib plus intensive chemotherapy improves survival in patients with newly diagnosed FLT3-ITD mutated AML regardless of whether they undergo allo-HSCT [64]. Addition of Sorafenib to chemotherapy not only nullifies the negative prognostic impact of higher allele burden, but also improves outcome of FLT3-ITD mutated AML patients regardless of the allele burden [65]. Sorafenib therapy is associated with improved outcomes for FLT3-ITD AML relapsing after allo-HSCT. Sorafenib combined with chemotherapy followed by donor lymphocyte infusion reveals an optimal efficacy [66]. Combination of Sorafenib with hypomethylating agents (azacitidine or decitabine) has resulted in high response rates in patients with FLT3 mutant AML inappropriate for intensive chemotherapy. FLT3 inhibitors are being explored in combination with other targeted agents [67]. Sorafenib has been approved and widely used in solid tumors, such as renal cell cancer, hepatocellular cancer, etc. [68, 69].
Ponatinib
As the second generation TKI, Ponatinib has been indicated for patients with TKI resistant chronic myeloid leukemia (CML) [70]. Recent results showed Ponatinib also comprises a high capability to inhibit constitutively activated FLT3. Ponatinib is able to overcome resistance to other TKI (e.g., Sorafenib) if it is conferred by additional point mutations of FLT3-ITD. It represents a promising compound in FLT3-ITD positive AML as well [71]. No systematic investigation of Ponatinib in AML patients has yet presented [72]. As of February 2020, there are 10 on-going clinical studies for Ponatinib mainly in CML patients from different research centers (Table 3).
IDH inhibitors
Mutations in the IDH gene, specifically R132 in IDH1, R140 and R172 in IDH2, are substrates for targeted therapy [73]. IDH1 and IDH2 are commonly mutated in cytogenetically normal AML (IDH1 6–16%, IDH2 8–19%). They impart a critical role in cellular metabolism by catalyzing the conversion of alpha-ketoglutarate to the oncometabolite R enantiomer of 2 hydroxyglutarate (R-2HG) [7, 73,74,75]. R-2HG inhibits cellular differentiation and promotes proliferation via TET2 inhibition and downstream effects of demethylation in vitro. It has a pivotal role of IDH mutations in leukemogenesis [76, 77]. Though currently not a component of the ELN guidelines for prognostication, IDH1/2 assessment should now be routinely done in AML patients, due to the availability of targeted therapy with the IDH1 and IDH2 inhibitors, Ivosidenib and Enasidenib [2].
IDH inhibitors (IDH-i) are used in patients with AML who have mutations in either IDH1 or IDH2 genes, causing abnormal maturation patterns in white blood cells, thus leading to leukemia [78]. Two targeted IDH-i: Ivosidenib and Enasidenib, blocking the proteins IDH1 and IDH2, respectively. The inhibition leads the leukemic cells to normal maturation and differentiation, thereby reducing immature blast counts and increasing the percentage of mature myoblasts [79, 80]. A safety concern with IDH-i is the possible side effect known as differentiation syndrome, the release of inflammatory cytokines from cancerous promyelocytes, referred to “cytokine storm”. Cytokine storm is serious and potentially fatal but can be reversed by stopping the offending agent [81].
Ivosidenib
IDH1 inhibitor Ivosidenib demonstrated overall safety and efficacy amongst patients with IDH1-mutated R/R AML leading to FDA approval [8]. In a phase 1 clinical trial, Ivosidenib monotherapy was well tolerated and induced durable remissions and transfusion independence in patients with newly diagnosed AML. IDH1 mutation clearance was seen in 9/14 patients achieving CR + CRh (5/10 CR, 4/4 CRh) [82]. Ivosidenib benefits a group of patients with poor prognosis and limited options. Although reports have revealed acquired resistance for these mutant IDH inhibitors, combination treatment can overcome this problem [83, 84]. Interestingly, amongst IDH1-mutated myelodysplastic syndrome (MDS) patients who were refractory to therapy with hypomethylating agents, Ivosidenib appeared to have a substantial efficacy, though the subgroup was small (n = 12) [8]. Accumulating data has indicated that targeted therapy using Ivosidenib may represent an encouraging therapeutic option in patients with acute undifferentiated leukemia and IDH1 mutations [85]. Currently, there are 10 registered active clinical studies for Ivosidenib in AML patients at different research centers (Table 4).
Enasidenib
Enasidenib is a FDA approved agent in the treatment of R/R AML. As the first-in-class mutant IDH2 inhibitor, Enasidenib has demonstrated safety and efficacy in phase 1/2 dose escalation and dose-expansion study [7]. Enasidenib was well tolerated and induced molecular remissions and hematologic responses in patients with AML for whom prior treatments had failed [86]. In clinical trials, Enasidenib has demonstrated remarkable activity in patients with mutated IDH2 [87]. Enasidenib has shown clinical activity in patients with R/R AML. Inducing differentiation of myeloblasts, not cytotoxicity, seems to drive the clinical efficacy of Enasidenib [8]. Recent research results have demonstrated Enasidenib motivated human erythroid differentiation independent of IDH2 and proved as a promising therapeutic agent for improving anemia. These results provided the basis for clinical trials using Enasidenib to decrease transfusion dependence in a wide array of clinical contexts [88]. Enasidenib is currently approved for the treatment of R/R AML at a dose of 100 mg oral daily. Study demonstrated that Enasidenib induces durable remissions in older patients with newly diagnosed AML. Oral, outpatient targeted treatment with Enasidenib may benefit older adults with newly diagnosed IDH2-mutant AML who are not candidates for cytotoxic regimens [89].
Older patients with AML are less likely to benefit from intensive chemotherapy. Instead, they benefit more from lower-intensity therapies and from newly available targeted AML treatments including Enasidenib [90]. Furthermore, even in the absence of a conventional CR, lower-intensity therapies may provide meaningful clinical benefit, including improved survival and quality of life, by inducing hematologic improvement and transfusion independence [90]. Adverse effects including indirect hyperbilirubinemia and IDH inhibitor induced cytokine storm which can be life threatening and should be identified and treated promptly [80, 87].
. Given the fact that Enasidenib is highly specific inhibitor acting on an early stable mutation, it is conceivable that this agent could be of more value if used in combination with other targeted agents. The proper role for single mutation targeting in AML therapy needs to be carefully considered [91].
Olutasidenib
Another IHD inhibitor named Olutasidenib, originally designed for glioma and glioblastoma, has been explored for AML and MDS treatment recently. Preclinical data showed Olutasidenib as a potent, orally bioavailable, brain penetrant, and selective IDH1 inhibitor. It has excellent ADME/PK properties and reduces 2-hydroxyglutarate levels in IDH1 mutation xenograft tumor model [92]. The Phase 1 study (NCT02719574) assessed the safety, PK/PD, and clinical activity of in AML or MDS patients with IDH1 mutation. The Olutasidenib study results have shown favorable safety and clinical activity in IDH1 mutant R/R AML as single agent with ORR of 41% and in a combination regimen with ORR of 46%, and durable disease control. Olutasidenib induces deep responses with IDH1 mutation clearance in a subset of treated patients [93]. Meanwhile, Olutasidenib has shown favorable safety profile and clinical activity in IDH1 mutant MDS, with an ORR rate of 59% and durable disease control. Phase 2 trial is ongoing as single agent and in combination with Azacitidine [94]. This agent may have good potential for AML and MDS treatment; however more clinical researches need to be explored.
Hedgehog signalling pathway inhibitors
The Hedgehog (Hh) signalling pathway is activated in many types of cancers including AML and naturally a promising target for therapeutic development. SMO plays very important role in the Hh signalling pathway and has been shown to be critical for acute leukemia disease progression. Approaches to inhibit Hh signalling for therapeutic benefit have focused primarily on SMO inhibitors. As a SMO antagonist, Glasdegib, an oral inhibitor of the Hedgehog signalling pathway, has been developed in clinical trials [95] in combination with standard chemotherapy for patients with AML or high-risk MDS [96, 97].
Glasdegib was approved in USA in November 2018 for use with low-dose cytarabine for treatment of patients with newly-diagnosed AML over age of 75 years or with comorbidities that precluding intensive induction chemotherapy. It is currently undergoing clinical development for use in other malignancies, including MDS, in various countries worldwide [98]. More recently, similar SMO inhibitors such as Vismodegib [99, 100], Sonidegib [100, 101] and Erismodegib [99, 102] are under-development in clinical trials for patients with AML and MDS, in combination with chemotherapies such as Azacytidine, a hypomethylating agent. Early results demonstrated promising responses in patients with AML and MDS. As of March 2020, there are 10 registered recruiting/completed clinical trials with Glasdegib, Vismodegib, Sonidegib and Erismodegib from the www.ClinicalTrials.gov website (Table 5).
Conclusion and future perspectives
Advances in molecular characterization of AML have provided important information for diagnosis, risk stratification, disease monitoring, and optimization of therapeutic strategies. Novel therapies for AML, including refinements of conventional cytotoxic chemotherapies, genetic and epigenetic targeted drugs, as well as immunotherapies, have significantly improved patient outcomes in recent years [103,104,105]. Newer generation of TKIs, such as Cabozantinib, Sel24-B489, G-749, AMG 925, TTT-3002, and FF-10101 may overcome disease resistance, and likely will further improve patients’ outcomes [105]. While the genomic complexity and the interplay of the many different molecular abnormalities in AML poses a huge challenge to successful translation into more accurate risk stratification and targeted therapy [18, 104, 105], opportunities do arise; various new agents, such as SMO inhibitors, immune checkpoint inhibitors, metabolic and pro-apoptotic agents, monoclonal or bispecific T-cell engager antibodies, antibody-drug conjugates and chimeric antigen receptor- engineered T cells, have been developed or under investigation as new therapies for AML [106, 107]. Molecularly, targeted therapies have changed the landscape of AML treatment and benefited patients with improved survival and quality of life. Yet, more needs to be done to make our patients live better and longer.
Availability of data and materials
Not Applicable.
Abbreviations
- ALK:
-
Anaplastic lymphoma kinase
- Allo-HSCT:
-
Allogeneic hematopoietic stem cell transplantation
- AML:
-
Acute myeloid leukemia
- AR:
-
Allele ratio
- ASXL1:
-
Additional sex combs-like 1
- biCEBPA:
-
Biallelic mutations of CCAAT/enhancer binding protein alpha
- CEBPA:
-
CCAAT/enhancer binding protein alpha
- CML:
-
Chronic myeloid leukemia
- CR:
-
Complete remission
- CR1:
-
First complete remission
- Cri:
-
Complete remission with incomplete hematologic recovery
- DNMT3A:
-
DNA methyltransferase 3A
- EFS:
-
Event-free survival
- ELN:
-
European Leukemia Net
- FLT3:
-
FMS-like tyrosine kinase 3
- FLT3-ITD:
-
FLT3 mutations
internal tandem duplication
- FLT3-TKD:
-
FLT3 mutations: tyrosine kinase domain
- GvHD:
-
Graft versus host disease
- Hh:
-
Hedgehog
- IDH:
-
Isocitrate dehydrogenases
- KMT2A:
-
Lysine (K)-specific Methyltransferase 2A
- moCEBPA:
-
Monoallelic mutations of CCAAT/enhancer binding protein alpha
- MRD:
-
Minimal residual disease
- NGS:
-
Next-generation sequencing
- NPM1:
-
Nucleophosmin
- OS:
-
Overall survival
- PDGFR:
-
Platelet-derived growth factor receptors
- R/R:
-
Relapsed/refractory
- RTK:
-
Receptor tyrosine kinase
- RUNX1:
-
Runt-related transcription factor 1
- SMO:
-
Smoothened
- STAT5:
-
Signal transducer and activator of transcription 5
- TKI:
-
Tyrosine kinase inhibitor
- TP53:
-
Tumor protein p53
- VAF:
-
Variant allele frequency
- VEGFR:
-
Vascular endothelial growth factor receptor
- WBC:
-
White blood cells
- WT1:
-
Wilms’ tumour 1
References
Chen J, Odenike O, Rowley JD. Leukaemogenesis: more than mutant genes. Nat Rev Cancer. 2010;10(1):23–36.
Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424–47.
Singh SV, Ajay AK, Mohammad N, Malvi P, Chaube B, Meena AS, et al. Proteasomal inhibition sensitizes cervical cancer cells to mitomycin C-induced bystander effect: the role of tumor microenvironment. Cell Death Dis. 2015;6(10):e1934.
Mohammad N, Singh SV, Malvi P, Chaube B, Athavale D, Vanuopadath M, et al. Strategy to enhance efficacy of doxorubicin in solid tumor cells by methyl-β-cyclodextrin: involvement of p53 and Fas receptor ligand complex. Sci Rep. 2015;5:11853.
Tallman M. Prognostic significance of molecular markers and targeted regimens in the Management of Acute Myeloid Leukemia. J Natl Compr Cancer Netw. 2018;16(5S):656–9.
Tallman MS, Wang ES, Altman JK, Appelbaum FR, Bhatt VR, Bixby D, et al. Acute myeloid leukemia, version 3.2019, NCCN clinical practice guidelines in oncology. J Natl Compr Cancer Netw. 2019;17(6):721–49.
Stein EM, DiNardo CD, Pollyea DA, Fathi AT, Roboz GJ, Altman JK, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017 Aug 10;130(6):722–31.
DiNardo CD, Stein EM, de Botton S, Roboz GJ, Altman JK, Mims AS, et al. Durable remissions with Ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med. 2018;378(25):2386–98.
Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med. 2017;377(5):454–64.
Perl AE. The role of targeted therapy in the management of patients with AML. Hematol Am Soc Hematol Educ Program. 2017;2017(1):54–65.
Grafone T, Palmisano M, Nicci C, Storti S. An overview on the role of FLT3-tyrosine kinase receptor in acute myeloid leukemia: biology and treatment. Oncol Rev. 2012;6(1):e8.
Nakao M, Yokota S, Iwai T, Kaneko H, Horiike S, Kashima K, et al. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia. 1996;10:1911–8.
Yamamoto Y, Kiyoi H, Nakano Y, Suzuki R, Kodera Y, Miyawaki S, et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood. 2001;97(8):2434–9.
O'Donnell MR, Tallman MS, Abboud CN, Altman JK, Appelbaum FR, Arber DA, et al. Acute myeloid leukemia, version 3.2017, NCCN clinical practice guidelines in oncology. J Natl Compr Cancer Netw. 2017;15(7):926–57.
Levis M. FLT3 mutations in acute myeloid leukemia: what is the best approach in 2013? Hematol Am Soc Hematol Educ Program. 2013;2013:220–6.
Nagel G, Weber D, Fromm E, Erhardt S, Lübbert M, Fiedler W, et al. Epidemiological, genetic, and clinical characterization by age of newly diagnosed acute myeloid leukemia based on an academic population-based registry study (AMLSG BiO). Ann Hematol. 2017;96(12):1993–2003.
Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209–21.
Wei H, Wang Y, Zhou C, Lin D, Liu B, Liu K, et al. Distinct genetic alteration profiles of acute myeloid leukemia between Caucasian and eastern Asian population. J Hematol Oncol. 2018;11(1):18.
Shimada A. Hematological malignancies and molecular targeting therapy. Eur J Pharmacol. 2019;862:172641.
Larrosa-Garcia M, Baer MR. FLT3 inhibitors in acute myeloid leukemia: current status and future directions. Mol Cancer Ther. 2017;16(6):991–1001.
Daver N, Schlenk RF, Russell NH, Levis MJ. Targeting FLT3 mutations in AML: review of current knowledge and evidence. Leukemia. 2019;33(2):299–312.
Stone RM, Manley PW, Larson RA, Capdeville R. Midostaurin: its odyssey from discovery to approval for treating acute myeloid leukemia and advanced systemic mastocytosis [published correction appears in blood Adv. 2018 Apr 10;2(7):787]. Blood Adv. 2018;2(4):444–53.
Schlenk RF, Weber D, Fiedler W, Salih HR, Wulf G, Salwender H, et al. Midostaurin added to chemotherapy and continued single-agent maintenance therapy in acute myeloid leukemia with FLT3-ITD. Blood. 2019;133(8):840–51.
Döhner K, Thiede C, Jahn N, Panina E, Gambietz A, Larson RA, et al. Impact of NPM1/FLT3-ITD genotypes defined by the 2017 European LeukemiaNet in patients with acute myeloid leukemia. Blood. 2020;135(5):371–80.
Antar AI, Otrock ZK, Jabbour E, Mohty M, Bazarbachi A. FLT3 inhibitors in acute myeloid leukemia: ten frequently asked questions. Leukemia. 2020. https://doi.org/10.1038/s41375-019-0694-3.
Tremblay G, Cariou C, Recher C, Dolph M, Brandt P, Blanc AS, et al. Cost-effectiveness of midostaurin in the treatment of newly diagnosed FLT3-mutated acute myeloid leukemia in France. Eur J Health Econ. 2020. https://doi.org/10.1007/s10198-019-01149-9.
Gilteritinib SN, Changes AML. Landscape. Oncology (Williston Park). 2019;33(8):683729.
Dhillon S. Gilteritinib: first global approval. Drugs. 2019;79(3):331–9.
Perl AE, Martinelli G, Cortes JE, Neubauer A, Berman E, Paolini S, et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. N Engl J Med. 2019;381(18):1728–40.
Zhao J, Song Y, Liu D. Gilteritinib: a novel FLT3 inhibitor for acute myeloid leukemia. Biomark Res. 2019;7:19.
Sidaway P. Gilteritinib improves outcomes in AML. Nat Rev Clin Oncol. 2020;17(2):69.
Mendel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G, et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9(1):327–37.
O'Farrell AM, Abrams TJ, Yuen HA, Ngai TJ, Louie SG, Yee KW, et al. SU11248 is a novel FLT3 tyrosine kinase inhibitor with potent activity in vitro and in vivo. Blood. 2003;101(9):3597–605.
O'Farrell AM, Foran JM, Fiedler W, Serve H, Paquette RL, Cooper MA, et al. An innovative phase I clinical study demonstrates inhibition of FLT3 phosphorylation by SU11248 in acute myeloid leukemia patients. Clin Cancer Res. 2003;9(15):5465–76.
Teng CL, Yu CT, Hwang WL, Tsai JR, Liu HC, Hwang GY, et al. Effector mechanisms of sunitinib-induced G1 cell cycle arrest, differentiation, and apoptosis in human acute myeloid leukaemia HL60 and KG-1 cells. Ann Hematol. 2013;92(3):301–13.
Yee KW, Schittenhelm M, O'Farrell AM, Town AR, McGreevey L, Bainbridge T, et al. Synergistic effect of SU11248 with cytarabine or daunorubicin on FLT3 ITD-positive leukemic cells. Blood. 2004;104(13):4202–9.
Fiedler W, Kayser S, Kebenko M, Janning M, Krauter J, Schittenhelm M, et al. A phase I/II study of sunitinib and intensive chemotherapy in patients over 60 years of age with acute myeloid leukaemia and activating FLT3 mutations. Br J Haematol. 2015;169(5):694–700.
Fiedler W, Serve H, Döhner H, Schwittay M, Ottmann OG, O'Farrell AM, et al. A phase 1 study of SU11248 in the treatment of patients with refractory or resistant acute myeloid leukemia (AML) or not amenable to conventional therapy for the disease. Blood. 2005;105(3):986–93.
Al-Jamal HA, Mat Jusoh SA, Hassan R, Johan MF. Enhancing SHP-1 expression with 5-azacytidine may inhibit STAT3 activation and confer sensitivity in lestaurtinib (CEP-701)-resistant FLT3-ITD positive acute myeloid leukemia. BMC Cancer. 2015;15:869.
Knapper S, Russell N, Gilkes A, Hills RK, Gale RE, Cavenagh JD, et al. A randomized assessment of adding the kinase inhibitor lestaurtinib to first-line chemotherapy for FLT3-mutated AML. Blood. 2017;129(9):1143–54.
Levis M, Ravandi F, Wang ES, Baer MR, Perl A, Coutre S, et al. Results from a randomized trial of salvage chemotherapy followed by lestaurtinib for patients with FLT3 mutant AML in first relapse. Blood. 2011;117(12):3294–301.
Zhang H, Savage S, Schultz AR, Bottomly D, White L, Segerdell E, et al. Clinical resistance to crenolanib in acute myeloid leukemia due to diverse molecular mechanisms. Nat Commun. 2019;10(1):244.
Wang ES. Incorporating FLT3 inhibitors in the frontline treatment of FLT3 mutant acute myeloid leukemia. Best Pract Res Clin Haematol. 2019;32(2):154–62.
Sandmaier BM, Khaled S, Oran B, Gammon G, Trone D, Frankfurt O. Results of a phase 1 study of quizartinib as maintenance therapy in subjects with acute myeloid leukemia in remission following allogeneic hematopoietic stem cell transplant. Am J Hematol. 2018;93(2):222–31.
Altman JK, Foran JM, Pratz KW, Trone D, Cortes JE, Tallman MS. Phase 1 study of quizartinib in combination with induction and consolidation chemotherapy in patients with newly diagnosed acute myeloid leukemia. Am J Hematol. 2018;93(2):213–21.
Sengsayadeth SM, Jagasia M, Engelhardt BG, Kassim A, Strickland SA, Goodman S, et al. Allo-SCT for high-risk AML-CR1 in the molecular era: impact of FLT3/ITD outweighs the conventional markers. Bone Marrow Transplant. 2012;47(12):1535–7.
Cortes JE, Tallman MS, Schiller GJ, Trone D, Gammon G, Goldberg SL, et al. Phase 2b study of 2 dosing regimens of quizartinib monotherapy in FLT3-ITD-mutated, relapsed or refractory AML. Blood. 2018;132(6):598–607.
Cortes J, Perl AE, Döhner H, Kantarjian H, Martinelli G, Kovacsovics T, et al. Quizartinib, an FLT3 inhibitor, as monotherapy in patients with relapsed or refractory acute myeloid leukaemia: an open-label, multicentre, single-arm, phase 2 trial. Lancet Oncol. 2018;19(7):889–903.
Cortes JE, Khaled S, Martinelli G, Perl AE, Ganguly S, Russell N, et al. Quizartinib versus salvage chemotherapy in relapsed or refractory FLT3-ITD acute myeloid leukaemia (QuANTUM-R): a multicentre, randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2019;20(7):984–97.
Zhou F, Ge Z, Chen B. Quizartinib (AC220): a promising option for acute myeloid leukemia. Drug Des Devel Ther. 2019;13:1117–25.
Naqvi K, Ravandi F. FLT3 inhibitor quizartinib (AC220). Leuk Lymphoma. 2019;60(8):1866–76.
Martínez-Cuadrón D, Rodríguez-Macías G, Rodríguez-Veiga R, Boluda B, Montesinos P. Practical considerations for treatment of relapsed/refractory FLT3-ITD acute myeloid leukaemia with quizartinib: illustrative case reports. Clin Drug Investig. 2020. https://doi.org/10.1007/s40261-019-00881-7.
Man CH, Fung TK, Ho C, Han HH, Chow HC, Ma AC, et al. Sorafenib treatment of FLT3-ITD(+) acute myeloid leukemia: favorable initial outcome and mechanisms of subsequent nonresponsiveness associated with the emergence of a D835 mutation. Blood. 2012;119(22):5133–43.
Röllig C, Serve H, Hüttmann A, Noppeney R, Müller-Tidow C, Krug U, et al. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): a multicentre, phase 2, randomised controlled trial. Lancet Oncol. 2015;16(16):1691–9.
Serve H, Krug U, Wagner R, Sauerland MC, Heinecke A, Brunnberg U, et al. Sorafenib in combination with intensive chemotherapy in elderly patients with acute myeloid leukemia: results from a randomized, placebo-controlled trial. J Clin Oncol. 2013;31(25):3110–8.
Uy GL, Mandrekar SJ, Laumann K, Marcucci G, Zhao W, Levis MJ, et al. A phase 2 study incorporating sorafenib into the chemotherapy for older adults with FLT3-mutated acute myeloid leukemia: CALGB 11001. Version 2. Blood Adv. 2017;1(5):331–40.
Battipaglia G, Ruggeri A, Massoud R, El Cheikh J, Jestin M, Antar A, et al. Efficacy and feasibility of sorafenib as a maintenance agent after allogeneic hematopoietic stem cell transplantation for Fms-like tyrosine kinase 3-mutated acute myeloid leukemia. Cancer. 2017;123(15):2867–74.
Chen YB, Li S, Lane AA, Connolly C, Del Rio C, Valles B, et al. Phase I trial of maintenance sorafenib after allogeneic hematopoietic stem cell transplantation for fms-like tyrosine kinase 3 internal tandem duplication acute myeloid leukemia. Biol Blood Marrow Transplant. 2014;20(12):2042–8.
Brunner AM, Li S, Fathi AT, Wadleigh M, Ho VT, Collier K, et al. Haematopoietic cell transplantation with and without sorafenib maintenance for patients with FLT3-ITD acute myeloid leukaemia in first complete remission. Br J Haematol. 2016;175(3):496–504.
Xuan L, Wang Y, Huang F, Jiang E, Deng L, Wu B, et al. Effect of sorafenib on the outcomes of patients with FLT3-ITD acute myeloid leukemia undergoing allogeneic hematopoietic stem cell transplantation. Cancer. 2018;124(9):1954–63.
Zhang C, Lam SSY, Leung GMK, Tsui SP, Yang N, Ng NKL, et al. Sorafenib and omacetaxine mepesuccinate as a safe and effective treatment for acute myeloid leukemia carrying internal tandem duplication of Fms-like tyrosine kinase 3. Cancer. 2020;126(2):344–53.
Chen F, Sun J, Yin C, Cheng J, Ni J, Jiang L, et al. Impact of FLT3-ITD allele ratio and ITD length on therapeutic outcome in cytogenetically normal AML patients without NPM1 mutation. Bone Marrow Transplant. 2019. https://doi.org/10.1038/s41409-019-0721-z.
Pratz KW, Rudek MA, Smith BD, Karp J, Gojo I, et al. A prospective study of Peritransplant Sorafenib for patients with FLT3-ITD acute myeloid leukemia undergoing allogeneic transplantation. Biol Blood Marrow Transplant. 2020;26(2):300–6.
Sasaki K, Kantarjian HM, Kadia T, Patel K, Loghavi S, Garcia-Manero G, et al. Sorafenib plus intensive chemotherapy improves survival in patients with newly diagnosed, FLT3-internal tandem duplication mutation-positive acute myeloid leukemia. Cancer. 2019;125(21):3755–66.
Yalniz F, Abou Dalle I, Kantarjian H, Borthakur G, Kadia T, Patel K, et al. Prognostic significance of baseline FLT3-ITD mutant allele level in acute myeloid leukemia treated with intensive chemotherapy with/without sorafenib. Am J Hematol. 2019. https://doi.org/10.1002/ajh.25553.
Xuan L, Wang Y, Chen J, Jiang E, Gao L, Wu B, et al. Sorafenib therapy is associated with improved outcomes for FMS-like tyrosine kinase 3 internal tandem duplication acute myeloid leukemia relapsing after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2019;25(8):1674–81.
Wang ES. Beyond midostaurin: which are the most promising FLT3 inhibitors in AML? Best Pract Res Clin Haematol. 2019;32(4):101103.
Rini BI, Pal SK, Escudier BJ, Atkins MB, Hutson TE, Porta C, et al. Tivozanib versus sorafenib in patients with advanced renal cell carcinoma (TIVO-3): a phase 3, multicentre, randomised, controlled, open-label study. Lancet Oncol. 2020;21(1):95–104.
Liu L, Gong Y, Zhang Q, Cai P, Feng L. Prognostic roles of blood inflammatory markers in hepatocellular carcinoma patients taking Sorafenib. a systematic review and meta-analysis. Front Oncol. 2020;9:1557.
Cortes JE, Kim DW, Pinilla-Ibarz J, le Coutre PD, Paquette R, Chuah C, et al. Ponatinib efficacy and safety in Philadelphia chromosome-positive leukemia: final 5-year results of the phase 2 PACE trial. Blood. 2018;132(4):393–404.
Zirm E, Spies-Weisshart B, Heidel F, Schnetzke U, Böhmer FD, Hochhaus A, et al. Ponatinib may overcome resistance of FLT3-ITD harbouring additional point mutations, notably the previously refractory F691I mutation. Br J Haematol. 2012;157(4):483–92.
Fleischmann M, Schnetzke U, Schrenk KG, Schmidt V, Sayer HG, Hilgendorf I, et al. Outcome of FLT3-ITD-positive acute myeloid leukemia: impact of allogeneic stem cell transplantation and tyrosine kinase inhibitor treatment. J Cancer Res Clin Oncol. 2017;143(2):337–45.
Im AP, Sehgal AR, Carroll MP, Smith BD, Tefferi A, Johnson DE, et al. DNMT3A and IDH mutations in acute myeloid leukemia and other myeloid malignancies: associations with prognosis and potential treatment strategies. Leukemia. 2014;28(9):1774–83.
Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2010;465(7300):966.
Medeiros BC, Fathi AT, DiNardo CD, Pollyea DA, Chan SM, Swords R. Isocitrate dehydrogenase mutations in myeloid malignancies. Leukemia. 2017;31(2):272–81.
Losman JA, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, et al. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science. 2013;339(6127):1621–5.
Chan SM, Majeti R. Role of DNMT3A, TET2, and IDH1/2 mutations in pre-leukemic stem cells in acute myeloid leukemia. Int J Hematol. 2013;98(6):648–57.
Stone RM. Which new agents will be incorporated into frontline therapy in acute myeloid leukemia? Best Pract Res Clin Haematol. 2017;30(4):312–6.
Myers RA, Wirth S, Williams S, Kiel PJ. Enasidenib: an Oral IDH2 inhibitor for the treatment of acute myeloid leukemia. J Adv Pract Oncol. 2018;9(4):435–40.
Abou Dalle I, DiNardo CD. The role of enasidenib in the treatment of mutant IDH2 acute myeloid leukemia. Ther Adv Hematol. 2018;9(7):163–73.
DiNardo CD, Stone RM, Medeiros BC. Novel therapeutics in acute myeloid leukemia. Am Soc Clin Oncol Educ Book. 2017;37:495–503.
Roboz GJ, DiNardo CD, Stein EM, de Botton S, Mims AS, Prince GT, et al. Ivosidenib induces deep durable remissions in patients with newly diagnosed IDH1-mutant acute myeloid leukemia. Blood. 2020;135(7):463–71.
Sidaway P. Ivosidenib effective in IDH1-mutant AML. Nat Rev Clin Oncol. 2018;15(8):472.
Norsworthy KJ, Luo L, Hsu V, Gudi R, Dorff SE, Przepiorka D, et al. FDA approval summary: Ivosidenib for relapsed or refractory acute myeloid leukemia with an Isocitrate Dehydrogenase-1 mutation. Clin Cancer Res. 2019;25(11):3205–9.
Patel SH, Vasu S, Guo L, Lemaster O, Byrd JC, Walker A. Molecular complete remission following Ivosidenib in a patient with an acute undifferentiated leukemia. J Natl Compr Cancer Netw. 2020;18(1):6–10.
Stein EM, DiNardo CD, Fathi AT, Pollyea DA, Stone RM, Altman JK, et al. Molecular remission and response patterns in patients with mutant-IDH2 acute myeloid leukemia treated with enasidenib. Blood. 2019;133(7):676–87.
Reed DR, Elsarrag RZ, Morris AL, Keng MK. Enasidenib in acute myeloid leukemia: clinical development and perspectives on treatment. Cancer Manag Res. 2019;11:8073–80 Published 2019 Aug 30.
Dutta R, Zhang TY, Köhnke T, Thomas D, Linde M, Gars E, et al. Enasidenib drives human erythroid differentiation independently of isocitrate dehydrogenase 2. J Clin Invest. 2020;2:133344.
Pollyea DA, Tallman MS, de Botton S, Kantarjian HM, Collins R, Stein AS, et al. Enasidenib, an inhibitor of mutant IDH2 proteins, induces durable remissions in older patients with newly diagnosed acute myeloid leukemia. Leukemia. 2019;33(11):2575–84.
Stein EM, DiNardo CD, Pollyea DA, Schuh AC. Response kinetics and clinical benefits of nonintensive aml therapies in the absence of morphologic response. Clin Lymphoma Myeloma Leuk. S2152–2650(19):2019, 32168–32018.
Dugan J, Pollyea D. Enasidenib for the treatment of acute myeloid leukemia. Expert Rev Clin Pharmacol. 2018;11(8):755–60.
Caravella JA, Lin J, Diebold RB, Campbell AM, Ericsson A, Gustafson G, et al. Structure-based design and identification of FT-2102 (Olutasidenib), a potent mutant-selective IDH1 inhibitor. J Med Chem. 2020;63(4):1612–23.
Watts JM, Baer MR, Yang J, Prebet T, Lee S, Schiller GJ, et al. Olutasidenib (FT-2102), an IDH1m inhibitor as a single agent or in combination with azacitidine, induces deep clinical responses with mutation clearance in patients with acute myeloid leukemia treated in a phase 1 dose escalation and expansion study. Blood. 2019;134(Supplement_1):231.
Cortes JE, Wang ES, Watts JM, Lee S, Baer MR, Dao K-H, et al. Olutasidenib (FT-2102) induces rapid remissions in patients with IDH1-mutant myelodysplastic syndrome: results of phase 1/2 single agent treatment and combination with Azacitidine. Blood. 2019;134(Supplement_1):674.
Chaudhry P, Singh M, Triche TJ, Guzman M, Merchant AA. GLI3 repressor determines hedgehog pathway activation and is required for response to SMO antagonist glasdegib in AML. Blood. 2017;129(26):3465–75.
Cortes JE, Douglas Smith B, Wang ES, Merchant A, Oehler VG, et al. Glasdegib in combination with cytarabine and daunorubicin in patients with AML or high-risk MDS: phase 2 study results. Am J Hematol. 2018;93(11):1301–10.
Savona MR, Pollyea DA, Stock W, Oehler VG, Schroeder MA, Lancet J, et al. Phase Ib study of Glasdegib, a hedgehog pathway inhibitor, in combination with standard chemotherapy in patients with AML or high-risk MDS. Clin Cancer Res. 2018;24(10):2294–303.
Hoy SM. Glasdegib: First Global Approval. Drugs. 2019;79(2):207–13.
Tibes R, Al-Kali A, Oliver GR, Delman DH, Hansen N, Bhagavatula K, et al. The hedgehog pathway as targetable vulnerability with 5-azacytidine in myelodysplastic syndrome and acute myeloid leukemia. J Hematol Oncol. 2015;8:114.
Cortes JE, Gutzmer R, Kieran MW, Solomon JA. Hedgehog signaling inhibitors in solid and hematological cancers. Cancer Treat Rev. 2019;76:41–50.
Katoh M. Genomic testing, tumor microenvironment and targeted therapy of hedgehog-related human cancers. Clin Sci (Lond). 2019;133(8):953–70.
Khatra H, Bose C, Sinha S. Discovery of hedgehog antagonists for Cancer therapy. Curr Med Chem. 2017;24(19):2033–58.
Yang X, Wang J. Precision therapy for acute myeloid leukemia. J Hematol Oncol. 2018;11(1):3.
Gu R, Yang X, Wei H. Molecular landscape and targeted therapy of acute myeloid leukemia. Biomarker Research. 2018;6(1):32.
Wu M, Li C, Zhu X. FLT3 inhibitors in acute myeloid leukemia. J Hematol Oncol. 2018;11(1):133.
Zhang C, Liu J, Zhong JF, Zhang X. Engineering CAR-T cells. Biomark Res. 201724;5:22.
Liu D. Cancer biomarkers for targeted therapy. Biomark Res. 2019;7:25.
Acknowledgements
We thank Dr. Delong Liu of New York Medical College and Dr. Kongming Wu of Tongji Hospital of Tongji Medical College, Huazhong University of Science and Technology for helpful discussion and Eric Yu of University of California, Berkeley for language editing assistance.
Funding
This study was funded by the Key Scientific Research Project of Henan Provincial Education Department (20A320062 and 19A320046); National Natural Science Foundation of China (U1804192) and Special Talents Project Fund of the First Affiliated Hospital of Zhengzhou University, Zhengzhou, China. The funding bodies did not participate in study design, in data collection, analysis, and interpretation, and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
All authors contributed to drafting and revising the article and agree to be accountable for all aspects of the work. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not Applicable.
Consent for publication
Not Applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yu, J., Jiang, P.Y.Z., Sun, H. et al. Advances in targeted therapy for acute myeloid leukemia. Biomark Res 8, 17 (2020). https://doi.org/10.1186/s40364-020-00196-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-020-00196-2