
Abstract
Phenotypic and functional heterogeneity is one of the hallmarks of human cancers. Tumor genotype variations among tumors within different patients are known as interpatient heterogeneity, and variability among multiple tumors of the same type arising in the same patient is referred to as intra-patient heterogeneity. Subpopulations of cancer cells with distinct phenotypic and molecular features within a tumor are called intratumor heterogeneity (ITH). Since Nowell proposed the clonal evolution of tumor cell populations in 1976, tumor heterogeneity, especially ITH, was actively studied. Research has focused on the genetic basis of cancer, particularly mutational activation of oncogenes or inactivation of tumor-suppressor genes (TSGs). The phenomenon of ITH is commonly explained by Darwinian-like clonal evolution of a single tumor. Despite the monoclonal origin of most cancers, new clones arise during tumor progression due to the continuous acquisition of mutations. It is clear that disruption of the "epigenetic machinery" plays an important role in cancer development. Aberrant epigenetic changes occur more frequently than gene mutations in human cancers. The epigenome is at the intersection of the environment and genome. Epigenetic dysregulation occurs in the earliest stage of cancer. The current trend of epigenetic therapy is to use epigenetic drugs to reverse and/or delay future resistance to cancer therapies. A majority of cancer therapies fail to achieve durable responses, which is often attributed to ITH. Epigenetic therapy may reverse drug resistance in heterogeneous cancer. Complete understanding of genetic and epigenetic heterogeneity may assist in designing combinations of targeted therapies based on molecular information extracted from individual tumors.
Similar content being viewed by others


Background
Cellular heterogeneity is a well-recognized attribute of both normal and neoplastic tissue [1]. Tumor morphologic heterogeneity has long been recognized by pathologists and forms the basis of many tumor grading prognostic classification systems [2]. Within a tumor, there is diversity in tumor cell proliferation, immune infiltration, differentiation status, and necrosis that can differ between microscopy fields [2]. In healthy tissue, the stroma functions are the main barrier against tumorigenesis; however, the presence of transformed tumor cells initiates crucial changes that can convert this environment into one that supports cancer progression [3]. Regional differences in extracellular microenvironment such as hypoxia, acidity and the presence of growth factors exist within a tumor and actively shape its development [3]. Normal fibroblasts typically suppress tumor formation, while cancer-associated fibroblast (CAFs) can significantly promote tumorigenesis [4,5,6]. Compared to normal tissue fibroblasts, CAFs have increased proliferation, enhanced extracellular matrix production and unique cytokine secretion [7]. Other mesenchyme-derived cell types, such as adipocytes, vascular endothelial cells and immune cells, as well as extracellular matrix, can also contribute to tumor growth and progression [8]. These stromal components may be different in many tumors.
By analyzing normal esophageal mucosa, esophageal dysplasia and esophageal squamous cell carcinoma, our previous study found that accumulation of aberrant tumor suppressor gene promoter region methylation is similar to classic gene mutation accumulation that occurs during tumor progression [9, 10]. In 1953, Slaughter et al. proposed the concept of field cancerization (also known as field defect) to explain the occurrence of multiple primary tumors, local recurrence, abnormal tissue surrounding the cancer and multifocal areas of precancerous change [11]. Phenotypic and functional heterogeneity are hallmarks of human cancers [12]. Tumor genotype variations among tumors within different patients are known as interpatient heterogeneity [13], and variability among multiple tumors of the same type arising in the same patient is referred to as intra-patient heterogeneity [13]. Subpopulations of cancer cells with distinct phenotypic and molecular features within a tumor is called intratumor heterogeneity (ITH) [13]. ITH is characterized by substantial phenotypic cell-to-cell variability, including differences in motility, metabolism, angiogenesis, proliferation, immunogenesis, and metastatic potential [14, 15]. ITH also includes heterogeneity of the tumor microenvironment [16, 17].
Phenotype heterogeneity of cells within tumors was noted in the earliest days of cancer biology [18]. Since the discovery that formation of tumors is dependent on the acquisition of oncogenic mutations, the existence of heterogeneity in clinically important traits was attributed to genetic diversity. Current approaches for molecular biomarker testing and targeting therapy are mainly focused on interpatient tumor heterogeneity [13]. However, there is growing recognition that ITH within the same patient is clinically relevant because the status of predictive biomarkers used for making clinical decisions may evolve during tumor progression, in particular for metastatic dissemination of the primary tumor to a distant organ or for established metastatic disease under the selection pressure of treatment [13].
The phenomenon of ITH is commonly explained by Darwinian-like clonal evolution of a single tumor [18]. Despite the monoclonal origin of most cancers, new clones arise during tumor progression due to the continuous acquisition of mutations. This promotes division into subclones and causes an increase in ITH [19]. Mutations that occur early in tumor evolution are present in all regions and almost all tumor cells harbor them. While, mutations that occur later or in the latest tumor progression are present in only some regions or only one subclone. These later occurring mutations are the basis for genetic ITH [2]. Heterogeneity in this field results in differences in features of subclones within a tumor, including different proliferation rates and different responses to treatment. However, the dominance of gene-centric views has been challenged with the rapid development of research within the cancer stem cell hypothesis, thus bringing non-genetic sources of phenotypic variability into focus [20]. In this review, we discuss the contributions of epigenetics to tumor phenotypic heterogeneity, mainly focused on the disruption of “epigenetic machinery”.
Genetic heterogeneity in cancer
Historically, research has focused on the genetic basis of cancer, particularly mutational activation of oncogenes or inactivation of tumor-suppressor genes (TSGs). Since Nowell proposed the clonal evolution of tumor cell populations in 1976, tumor heterogeneity, especially ITH, was actively studied [18]. However, many biological aspects of tumor heterogeneity remain unknown [21]. The analysis of multiple biopsies from the same tumor can reveal the spatial composition and evolutionary trajectory of subclones. The clonal and subclonal composition of each tumor can be used to construct distance-based phylogenetic trees. Mutations present in all samples of a tumor are inferred to be acquired by early precursor cells that clonally expanded (clonal mutations), represented by truncal events on the evolutionary tree, and mutations present in only a subset of samples are inferred to be later events, acquired at some point during or after the initial clonal expansion (subclonal mutations) [22, 23].
Gerlinger and colleagues obtained tumor samples from four patients with renal-cell cancer before and after treatment and took multiple samples from each parent’s primary and metastatic tumor sites. Analysis revealed that 63 to 69% of mutations in single biopsies were not detectable across every tumor region of the same patient [24]. Thus, a single tumor biopsy, the standard of tumor diagnosis and the cornerstone of personalized-medicine decisions, cannot be considered representative of the landscape of genomic abnormalities in a tumor. ITH is found in most, probably all, solid human tumors. Underestimation of tumor heterogeneity may lead to a serious flaw in cancer diagnosis and treatment selection.
Disruption of “epigenetic machinery” in cancer
In the nucleus of eukaryotic cells, chromatin provides the scaffold for the packaging of the entire genome. The basic functional unit of chromatin is the nucleosome, and it contains 147 base pairs of DNA wrapped around a histone octamer, with two copies each of histones H2A, H2B, H3 and H4. The epigenome consists of specific covalent modifications of chromatin components, including DNA methylation and histone modifications. These covalent modifications control the structure and function of chromatin. Epigenetic regulation of gene expression is mainly dependent on DNA methylation and histone modifications, without intrinsic changes in the DNA sequence, and epigenetic change is heritable [25]. Noncoding RNA, ubiquitylation and sumoylation are also included in the field of epigenetics [26]. The regulators of “epigenetic machinery” are divided into “writers” (enzymes that establish DNA methylation or histone modifications), “erasers” (proteins that remove these marks) and “readers” (proteins that bind to modifications and facilitate epigenetic effects). Protein complexes that position the nucleosomes across the genome are called “movers” [26].
In mammals, DNA methylation occurs predominantly at the 5′ position of cytosine forming cytosine guanine dinucleotides. This modification is carried out by DNA methyltransferases (DNMTs), enzymes that use S-adenosymethionine (SAM) as a methyl group donor. DNA methylation patterns are established and maintained by three DNMTs: DNMT1, DNMT3A and DNMT3B. Depending on the genomic location, DNA methylation may have different biological functions. Methylation in gene promoter regions is typically associated with gene repression, while methylation in the gene body is usually associated with active gene expression. Increasing evidence has shown that intergenic regions contain many regulatory elements, such as enhancers, silencers and non-coding RNAs, and their function may also be affected by DNA methylation. Early epigenetic research typically focused on gene promoter regions [26].
For a long time, 5-methylcyctosine (5mC) was considered to be a relatively permanent mark, but this view changed abruptly with the discovery of the function of the ten-eleven translocation (TET) proteins, TET1, TET2 and TET3. The TET gene family was initially identified as a result of a chromosomal rearrangement (t(10;11)), (q22;q23) involving TET1 and MLL, which encodes one of the histone H3 lysine 4 (H3K4) methyltransferases in acute myeloid and lymphocytic leukemias [27]. The TET family utilizes two key co-factors, Fe (II) and 2-oxoglutatate (2-OG), to successively oxidize the methyl group of 5mC to hydromethyl, forml or carboxyl groups, thus forming 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC), together termed ‘oxi-mC [28]. 5-hmC can actively facilitate DNA demethylation by inhibiting UHRF1/DNMT1 complex binding to DNA for methylation maintenance. 5fC and 5caC can be excised by the DNA repair enzyme thymine-DNA glycosylase (TDG), followed by replacement with unmodified cytosine through a base pair mechanism [29]. TET proteins bind preferentially to unmethylated CpGs within CpG-rich genomic regions (termed CpG islands), thus maintaining CpG islands (CGIs) in a hypomethylated state, and 5hmC is associated with active transcription [30]. Downregulation of TET proteins and loss of 5hmC are viewed as new epigenetic hallmarks of human cancer [31]. Isocitrate dehydrogenases (IDH) are key metabolic enzymes that function in the tricarboxylic acid (TCA) cycle; they convert isocitrate to 2OG using NADP+/NADPH as factors. 2OG is an essential cofactor for dioxygenases including TET proteins and the JmjC family of lysine demethylases. Among the three IDH enzymes, IDH1 and IDH2 are frequently mutated in glioma and hematological malignancies [32].
Histones are modified by different enzymes, including histone acetyltransferases (HATs), histone deacetylases (HDACs), histone methytransferases (HMTs) and histone demethylases (HDMs). HDACs are enzymes responsible for removing the acetyl group from lysine residues in histones [26, 33]. There are various types of histone tail modifications, such as acetylation, methylation, ubiquitination, among others. These modifications regulate gene expression through their interactions with chromatin-associated proteins in marking regions of transcriptionally active euchromatin and inactive heterochromatin, inducing transcriptional activation or repression. For example, in the promoter region, acetylated histone H3, and di- or tri-methylated histone H3 lysine 4 (H3K4me2, H3K4me3) represent activation of gene expression. Repressed promoters are usually marked with tri-methylated histone H3 lysine 27 (H3K27me3) and tri-methylated histone H3 lysine 9 (H3K9me3), which correlate with constitutive heterochromatin and DNA methylation [26].
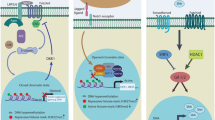
It is clear that disruption of the "epigenetic machinery" plays an important role in cancer development (Fig. 1). The recognition of an epigenetic component in tumorigenesis, or the existence of a cancer ‘epigenome’, has led to new opportunities for the understanding, detection, treatment, and prevention of cancer [33, 34]. DNA methylation is the most frequently found abnormal epigenetic change in human cancers. Global genomic DNA hypomethylation and promoter region hypermethylation have been extensively studied in human cancer [35, 36]. Aberrant epigenetic changes occur more frequently than gene mutations in human cancers. For example, epigenetic silencing of CDK2NA and MLH1 is much more common than mutational inactivation of either of these two well-recognized driver genes [26]. Beyond lifestyle determinants, the role of environmental factors as determinants of DNA methylation has gained considerable attention [35]. The epigenome is at the intersection of the environment and genome [37]. Epigenetic dysregulation occurs in the earliest stage of cancer. For example, DNA methylation was shown to be altered in the normal tissue of lung cancer patients [38]. A second example is that tumor suppressor genes were methylated in the early stage of esophageal squamous cell carcinogenesis, and accumulation of promoter region methylation was correlated with cancer progression [9, 39]. In addition, recently discovered mutations in the epigenetic apparatus likely contribute to epigenetic disruption in cancer [37]. DNA methylation is the most useful epigenetic marker for human disease studies because it is stable over a period of decades and is present in archival specimens, including paraffin blocks [40]. Aberrant DNA methylation is involved in the major components of cell cycle, DNA damage repair, Wnt, TGF-β, NF-kB and other cancer-related signaling pathways [41,42,43]. Additional information is provided in Table 1.

Disruption of the “epigenetic machinery” in cancer. Aberrant changes of major players of “epigenetic machinery” during cancer initiation, progression and metastasis. HATs, DOT1L, DNMT, EZH2, SUV39H1/2: representative writers (enzymes that establish DNA methylation or histone modifications); HDACs, JmjC–KDMs, LSDs, DNA demethylase: representative erasers (proteins that remove DNA methylation or histone modification marks)
The interplay between genetics and epigenetics
As originally defined by the embryologist Conrad Waddington, epigenetics is the branch of biology that studied the interactions between genes and their products that bring phenotype into being [109]. Epigenetic information is controlled by genome sequence, environmental exposure, and stochasticity, or random chance. Thus, epigenetics stands at the interface of the genome, development, and environmental exposure [35]. A major change in the epigenetic concept came from the realization that the environment has a profound effect on developmental plasticity, particularly with aging and susceptibility to common disease [110]. The vast majority of human cancers harbor both genetic and epigenetic abnormalities, with fascinating interplay between the two [33]. A key facet of epigenetics is that its modifications can be stably maintained yet adapt to changing developmental or environmental needs [39]. In contrast to the DNA sequence, the epigenome is relatively susceptible to modification by the environment as well as stochastic perturbations over time, adding to phenotypic diversity in the population [111]. A convincing example of intergeneration dietary epigenetic effects was an experiment involving mice with an insertional mutation in the Agouti locus that controls coat color and weight. These phenotypes are regulated by dietary methionine, the essential amino acid precursor for DNA methylation [112]. In humans, exposure to nicotine and other toxins causes substantial epigenetic changes in smokers, affecting genes involved in normal pulmonary function and cancer [113, 114]. A recent randomized trial showed that dietary fat composition affects DNA methylation in adipocytes [115].
Cancer has long been regarded as a genetic disease. Nevertheless, genetic events occur at low frequency and are thus not a particularly efficient means for malignant transformation [116, 117]. Genome-scale genomic and epigenomic analyses have only recently revealed the widespread occurrence of mutations in epigenetic regulators and the breadth of alterations to the epigenome in cancer cells [33]. It is now clear that genetic and epigenetic mechanisms interact with each other to enable the acquisition of the hallmarks of cancer during tumorigenesis [33, 116, 117]. Disruption of a key epigenetic regulator by mutation leads to an altered transcriptome, multiplying the effect of the single genetic alteration [116].
DNMT3A is recurrently mutated in acute myeloid leukemia (AML) and other myeloid malignancies [118, 119]. TET1 and TET3 are rarely mutated in hematological malignancies. By contrast, large-scale whole-exome sequencing studies by many groups have confirmed that TET2 is one of the most frequently mutated genes in chronic myelomonocytic leukemia (~ 50%), acute myeloid leukemia (~ 20%), and myelodysplastic syndromes (~ 20%) [120]. The majority of missense mutations impair the enzymatic activity of TET2, resulting in decreased 5hmC levels and aberrant DNA methylation [121]. The prognostic value of TET2 mutations in cancer patients remains unclear [121]. Since the initial discovery of IDH mutations in cancer in 2008, recurrent somatic mutations in IDH1 and IDH2 have been identified in different malignancies, including gliomas, thyroid carcinomas, cholangiocarcinomas, sarcomas, and AML [122]. The value of IDH mutations is under debate [123]. Mutations in histone variants H3.3 (H3F3A) and H3.1 (HIST1H3B) have been found in pediatric and adult brain tumors with K27 M and G34R or G34 V mutation hot spots [124, 125]. Mutations were also observed in the ATRS and DAXX genes, which encode proteins responsible for loading of the H3.3 variant into the telomere region [124]. The MLL gene, which encodes one of the H3K4 methyltransferases, has more than 50 translocation function partners in different lineages of leukemia. These rearrangements account for 80% of the cases of infant leukemia and 5–10% of adult leukemia cases and are generally associated with poor prognosis [126]. Loss-of-function mutations of MLL3 have been reported in many different types of cancer. MLL2 is mutated at a very high frequency in B cell follicular lymphoma and diffuse large B cell lymphoma [127]. More information about mutations in epigenetic regulator genes is listed in Table 2.
In addition to genetic disruption of epigenetic machinery, aberrant epigenetic changes may cause genetic abnormality. Epigenetic silencing of DNA repair genes such as MLH1, MGMT, BRCA1, FANCF, CHFR and SLFN11 can lead to gene mutation and genomic instability in cancer cells [181, 187,188,189]. Microsatellite instability (MSI) in Lynch syndrome results from germline mutations in mismatch repair genes, mainly MLH1 and MSH2. Approximately 15% of sporadic colorectal cancer patients with MSI were caused by epigenetic silencing of the MLH1 promoter region [190]. MSI caused by epigenetic silencing of MLH1 has also been reported in other types of cancer, including about a quarter of sporadic endometrial cancers [191]. Methylation of MGMT in colorectal cancer is associated with G-to-A mutations in the KRAS gene [192]. Additional epigenetically silenced DNA damage repair genes are listed in Table 2.
Heterogeneity of cancer epigenetics
Although researchers are still at the very beginning of understanding the full context of tumor heterogeneity, models of tumor evolution, or tumor phylogenies, derived from ITH have improved our understanding of tumorigenesis [2, 24]. A majority of cancer therapies fail to achieve durable responses, which is often attributed to ITH. Importantly, most strategies for cancer therapy still do not assess ITH and miss an opportunity to examine the prognostic value of ITH. ITH has been assessed with somatic mutation and copy-number alteration. The causal relevance of epigenetic changes in cancer has been recognized and the concept of epigenetic silencing being involved in Knudson’s two-hit theory has been accepted [60]. Thus, epigenetic mechanisms play an important role in tumor heterogeneity.
Some studies were not designed a priori for the purpose of analyzing heterogeneity; however, they provided information on epigenetic intratumor heterogeneity (eITH) and linked to clinical outcome. The “field defect” is one example [193]. In many cancers, cells have been shown to acquire pro-tumorigenic mutations that are not able to produce morphological change but predispose cells to subsequent malignant transformation [194]. These cells can expand, creating patches of mucosa that have an increased risk of developing into cancer. This process has been described as “field cancerization” or “field defect” [11, 195]. Epigenetic abnormalities may serve as a marker of a “field defect”, such as MGMT, p16 and RASSF1A promoter region methylation in normal-appearing mucosa of colorectal cancer patients [193, 196].
eITH can be examined at the level of histone modifications, chromatin conformation, or DNA methylation. Nevertheless, epigenetic heterogeneity remains poorly explored. To date, DNA methylation has been the major measurement due to the quantitative nature of DNA methylation assays and the relative ease of obtaining sufficient genomic DNA compared to chromatin. Using human cutaneous melanoma as a model, Sigalotti et al. found that the expression of cancer/testis antigens (CTA) was highly heterogeneous in different clones, which were generated from a melanoma lesion metastasized to the lymph-node. In addition, the clonal heterogeneity of CTA expression was negatively correlated with promoter region hypermethylation [197]. By analyzing the promoter region methylation of five genes (RASSF1A, p16, DAPK, MGMT, and Rb) in 34 tumors (15 melanoma primaries, 19 metastases), Rastetter et al. found that 70% of the cases exhibited heterogeneous methylation patterns [198]. In another study, in nine MSI-positive primary endometrial cancers that lacked MLH1 expression based on immunohistochemical evaluation, eight of nine tumors were methylated in the promoter region. Among these, four tumors were homogeneously methylated and four cases were heterogeneously methylated [199]. Genomic sequencing of 28 chronic lymphocytic leukemia (CLL) patients where samples were taken at two or more time points, Okes et al. found that CLL cases that showed high levels of genetic heterogeneity also showed widespread methylation changes over time [200]. While, Pan et al. found that intratumor methylation heterogeneity does not clearly correlate with genetic clonal heterogeneity in diffuse large B-cell lymphomas according to enhanced reduced representation bisulfite sequencing [201]. Aryee et al. reported that cancer-related genes are heterogeneously hypermethylated across individuals in prostate cancer patients, while the methylation status is generally consistent across metastases within individuals. However, some regions showed intraindividual metastatic tumor heterogeneity in promoter methylation, and such methylation alterations were generally not correlated with gene expression. This is likely due to the complexity of tumor components and/or one allele methylation [202]. By analyzing 40 tissue samples from seven lung adenocarcinoma patients (including normal, tumor segments and lymph node metastases), a recent study found that methylation profiles within tumors from same individual were not more similar to each other than to those from others [203].
Using Illumina Human Methylation 450 k BeadChip arrays, Brocks et al. analyzed DNA methylation and copy number alterations from multiple topographically distinct tumor sites in 5 patients, including primary tumor sites, premalignant lesions, lymph node metastases and matched normal prostate epithelium. They demonstrated that both DNA methylation and copy-number heterogeneity consistently reflect the life history of the tumors [204]. In this study, specimens of the same patient were generally more similar to each other than those from different individuals, consistent with the previously described interindividual heterogeneity of prostate cancer metastases [201, 205, 206]. Further study suggested that intratumor heterogeneous DNA methylation presents in multiple subclonal cell populations. In addition, metastases always carried unique alterations not found in the primary tumor bulk, and metastases-specific aberrant methylation events frequently co-localized with genes involved in metastases-associated processes. The degree of intratumoral DNA methylation variability strongly depends on the genetic and epigenetic context of a locus [204]. By performing a multitude of analyses of the DNA methylation data in tumors and normal samples of 21 cancer types in TCGA, Liu et al. revealed that the variability of the DNA methylomes are highly enriched in the gene promoters of the DNA-binding proteins, especially the transcription factors (TFS) [207]. Combining single-cell profiling of expression and DNA methylation, Linker et al. found DNA methylation is locus-specific, and methylation heterogeneity across cell is associated with splicing variability [208]. Quek et al. analyzed methylation profiles of 48 spatially separated tumor regions from 11 localized lung adenocarcinomas and their matched normal lung tissues using the Illumina Infinium Human Methylation k450 BeadChip array. All tumor samples had at least 40% viable cancer cells, and only approximately 25% of all differentially methylated probes were clonal events shared by all regions of individual tumors, and a higher extent of DNA methylation ITH was associated with larger tumor size [209]. Martinez-Cardus et al. found that intratumor DNA methylation differences are more extensive than genetic diversity in primary colorectal cancer. They also revealed that those locoregional colorectal cancer tumors more homogeneous at the epigenetic level show poor clinical outcomes [210]. To determine the prevalence and character of epigenetic tumor heterogeneity in time and space, DNA methylation sequencing was performed on a large cohort of IDH wildtype glioblastoma patients (n = 112) with mathched samples from primary and recurring tumors (between 2 and 4 time points per patient), including multiple subregion samples for a set of these tumors. By comparing DNA methylation levels of 5-kilobase tiling regions between primary and recurring tumors, Klughammer et al. observed wide-spread epigenetic heterogeneity at individual loci [211]. An example is that the MGMT promoter was unmethylated in the majority of samples, and patients with a methylated MGMT promoter in their recurring tumors had significantly better progression-free survival (PFS) and overall survival (OS) compared to patients with unmethylated MGMT promoters. A demethylation of Wnt signaling gene promoters was associated with worse prognosis. Extensive heterogeneity existed between patients, but not strong trend between primary and recurring tumors. Increased epigenomic heterogeneity was associated with worse prognosis. Authors also found that DNA methylation data could predict various types of immune cells in the primary and recurring tumors [211]. Developing a novel single-cell technology, Pi-ATAC, which simultaneously measures protein epitopes and active DNA regulatory elements of the same individual cell, Chen et al. found epigenetic variability of tumor cells is linked to the hypoxic tumor microenvironment [212]. By genome-wide methylotyping analysis, Tanas et al. divided breast cancer into six breast cancer methylotypes, and found that the majority of CpG islands appeared to be more densely hypermethylated in breast cancer cell lines than in primary tumors [213]. Using an epigenome-wide sequencing approach, Grasse et al. observed that aberrantly methylated regions in the PDX tumors were reflected in the corresponding primary NSCLC tumors, albeit the levels of differential methylation of the PDX samples were much higher compared to the levels within the primary tumors [214]. Mutations in epigenetic modifier genes, such as SETD2 and DNMT3A, are strongest determinants of ITH amongst a panel of 17 distinct cellular pathways [215]. Epigenetic regulators such as histone modifying enzymes are critical for the establishment of cell-type-specific gene expression patterns, thus, they are also likely to play a role in modulating cell-to-cell variability in transcription. The distinct epigenetic state of the cells could determine cellular response to treatment [216]. Lysine demethylase 5 (KDM5) was found to be a regulator of cellular transcriptomic heterogeneity in ER+ luminal breast cancer, and inhibiting KDM5 activity could decrease resistance to cancer therapies [217]. Pastore et al. suggested that intratumoral epigenetic diversity may permit leukemic cells to stochastically activate alternate gene regulatory programs, facilitating the emergence of novel cell sates, ultimately fostering CLL’s ability to efficiently explore the fitness landscape for superior evolutionary trajectories during tumorigenesis and in response to therapy [218].
Genome-wide sequencing of three cases of primary melanoma and matched metastatic cell lines derived from the same patients showed global hypomethylation in metastatic melanoma cell lines compared to the matched primary melanoma cell lines [219]. A recent study found that the activation-induced cytidine deaminase (AICDA) is a key driving force in generating cytosine methylation heterogeneity in germinal center B cells and GC-derived lymphomas. AICDA-linked epigenetic heterogeneity is predominantly associated with relative loss of cytosine methylation. AICDA-induced epigenetic heterogeneity increases plasticity, permitting cancer cells a greater degree of population diversity and enhancing the adaptive capacity of the overall tumor. AICDA overexpression in mice was associated with both increased inter-tumor and intra-tumor methylation heterogeneity [220].
The strategies of “epigenetic precision medicine” based on cancer heterogeneity implications
In contrast to the “one-size-fits-all-approach”, the ultimate aim of precision medicine is to enable clinicians to accurately and efficiently identify the most effective preventive or therapeutic intervention for a specific patient. A variety of high-throughput methods for characterizing cancer biomarkers (proteomics, genomics, epigenetic, transcriptomics), coupled with significant advances in computational tools, may improve understanding precision medicine in cancer [221]. Epigenetic switches play an important role in cancer development, and epigenetic switches are reversible. Thus, aberrant epigenetic changes may serve as early detection, prognostic and chemo-sensitive markers in cancer. They may also become preventative and therapeutic targets in cancer [26, 35]. One example is that an “epigenetic field defect” is formed during chronic inflammation-associated carcinogenesis, and aberrant DNA methylation is induced by chronic inflammation. DNA methylation was induced in colonic epithelia cells as early as 8 weeks after dextran sulfate sodium (DSS) treatment when no macroscopic tumors appeared, and the methylation level gradually increased until macroscopic tumors developed. Our previous study and others suggest that “epigenetic field defect” may serve as an early detection marker in cancer [201, 222]. Elucidation of the specific epigenetic marker that underlies the epigenetic heterogeneity could enable specific chemo-preventative agents to be designed to target these early changes prior to the development of any precancerous lesions. Several studies noted that the width of the surgical margin is directly associated with the risk of local recurrence (or development of invasive cancer) following breast conserving surgery for ductal carcinoma in situ (DCIS) [223]. These findings are consistent with the idea that aberrant epigenetic changes may exist in histologically normal appearance epithelia cells around the lesions.
Heterogeneity of the tumor microenvironment may also result in diversity of tumor cell phenotypes, which decreases the sensitivity of the tumor to therapy. For instance, under conditions of hypoxia, tumor cells are more aggressive and their response to treatment is worse than in normally oxygenated regions [224]. Aryee et al. found that there is a considerable amount of interindividual tumor heterogeneity at both the genetic and epigenetic levels in prostate cancer [202]. This interindividual heterogeneity challenges “one-size-fits-all” approaches for cancer management and implies the need for specific treatment for different molecular lesions. The finding of metastases-specific aberrant methylation and identification of high levels of epigenetic heterogeneity at androgen-receptor-bound enhancer domains adds information about regulatory activity at important cis-regulatory elements and assists in making decisions for precision medicine strategies in prostate cancer [204]. Overexpression of AICDA, a driver of epigenetic heterogeneity, is associated with a more aggressive disease phenotype and decreased survival in BCL2-driven lymphoma [220]. Clonal evolution of multiple myeloma cells and heterogeneity of the bone marrow microenvironment results in a rapid acquisition of chemotherapy resistance.
The central role of epigenetics in regulating many of the hallmarks of cancer has garnered the interest and focus of scientists, clinicians, and the pharmaceutical industry with the aim of manipulating and resetting the cancer epigenome. In the past few years, plenty of small molecules have been developed to target specially epigenetic writers, readers, and erasers [225]. The DNA demethylating agents 5-azacytidine and 5-aza-2’deoxycytidine (decitabine) are inhibitors of DNMT1 and DNMT3B. Decitabine has been approved by US FDA for myelodysplasia and AML treatment, and 5-azacytidine has also been approved for myelodysplasia therapy. Guadecitabine (SGI-110) is a second-generation demethylating agent, which is more stable in aqueous solution, and more demethylating agents are being testing in solid tumors. Trichostatin A (TSA) is the first natural product discovered to inhibit HDACs. There are currently at least 20 HDAC inhibitors in clinical testing. Vorinostat (also known as suberoylanilide hydroxamic acid [SAHA]) and romidepsin (also known as depsipeptide or FK228) were approved by the FDA for treatment of cutaneous T-cell lymphoma. Enhancer of zeste homologue 2 (EZH2) is the catalytic core subunit of the polycomb repressive complex 2 (PRC2). It is responsible for catalyzing trimethylation of histone H3 at lysine 27, which serves as a docking site for DNA methyltransferases and HDAC. As the C-terminal SET domain of EZH2 exhibit methyltransferase activity, specific inhibitor has been designed by targeting the conserved SET domain. A batch of SET domain inhibitors is being selected to minimize the off-target effects. GSK126 and EPZ-6438 are being tested in phase I trial in solid tumors. Disruptor of telomeric silencing-1-like (DOT1L) is a methyltransferase responsible for catalyzing methylation of H3K79. MLL-fusion proteins gain the ability to recruit DOT1L to MLL target genes, leading to aberrant expression of these genes by methylating H3K79. EPZ00477 and EPZ-5676 are inhibitors of the human DOT1L. The phase I clinical trial of EPZ-5676 has been completed in MLL-rearranged leukemia [226]. The methylation status of histone lysine is controlled by KDMs and their counterparts of lysine methyltransferases (KMTs). Lysine specific histone demethylase-1 (LSD1, also known as KDM1) catalyzes the demethylation of mono- and dimethylated lysines, but not tri-methylated lysines from H3K4 and H3K9. LSD1 was found to be highly expressed in neuroblastoma, prostate, estrogen-negative breast, bladder and colorectal cancers. GSK2879552 and ORY-1001 are specific inhibitors of LSD1. They are currently in clinical trials for small cell lung carcinoma and relapsed or refractory AML, respectively. Based on the JmjC domain sequence homology and their demethylase activities, JmjC-KDMs have been categorized into seven KDM subfamilies (KDM2–8). KDM5 members are capable of removing H3K4me3 activating mark from histones to make them potential players in the downregulation of tumor suppressors. Inhibition of KDM5 demethylase activity reduces the number of surviving cells after lethal drug exposures in a number of cell culture models, what makes this enzyme family a promising target for novel cancer treatment [227]. The bromodomains (BRDs) may contribute to highly specific histone acetylation by tethering transcriptional HATs to specific chromosomal sites, or to the activity of multiprotein complexes in chromatin remodeling. The bromodomain and extra-terminal motif (BET) proteins act as scaffolds for the recruitment of transcription factors and chromatin organizers required in transcription initiation and elongation. Extensive studies have explored small-molecule inhibitors of BET family proteins for cancer therapy. I-BET762 is being tested in early phase clinical trials. More clinical trials are performing for BET family inhibitors, including RVX-208, I-BET 762, OTX 015, CPI-0610 and TEN-010. There are more epigenome-based targeting therapeutics, but they are beyond the scope of this review [26, 225].
Epigenetic heterogeneity is far more dynamic than genetic heterogeneity, and it is likely that transcriptional plasticity driven by epigenetic regulators responding to environmental and therapeutic pressures underpins the failure of many cancer drugs to induce durable disease remission in patients [225]. Several classes of epigenetic regulators have been implicated in drug resistance and intratumoral heterogeneity [228]. Epigenetic therapy may reverse drug resistance in heterogeneous multiple myeloma [229]. Combination of epigenetic therapy and chemotherapy improved the efficacy in refractory advanced non-small cell lung cancer [230]. Ideal treatment regimens would target all the different subpopulations of cancer cells present at the time of treatment, thus avoiding resistance and delaying relapse [228]. By detecting genetic and epigenetic heterogeneity and analyzing compensatory signaling in cancer, we may develop novel “synthetic lethality” strategies. As cancer epigenetic heterogeneity is in its infancy, little can be generalized from epigenetic heterogeneous therapy.
Conclusions & future perspectives
ITH may reflect the evolutionary history of tumors, and genetic or epigenetic marks can also reflect the potential of the tumor to respond to an environmental or therapeutic pressure. Understanding ITH may guide new therapeutic strategies [21]. The current trend of epigenetic therapy is to use epigenetic drugs to reverse and/or delay future resistance to cancer therapies. As epigenetic abnormalities are apparent early in cancer risk and premalignant states, we may be able to develop strategies for cancer prevention. One of the major issues in elucidating the “road map” of human development and disease epigenomes is technique limitation. However, a new generation of sequencing instrument is in development. Nanopore sequencing is a third generation sequencing technology that assesses single molecules of unmodified DNA by sensing alterations in electrical current that occur as different bases pass through a nanopore. Oxford Naonopore Technologies has recently released the first commercially-available sequencer based on this technology. This technology accepts samples as small as 10 pg and does not require PCR amplification prior to analysis. Nanopores are also capable of distinguishing between cytosine, 5mC, and 5hmC [231]. Ideally, in vivo and in vitro tumor models that recapitulate the nature, dynamics, and heterogeneity of successive tumorigenic epigenetic alternations are needed [232]. Epigenetics may lead us at last to an era of comprehensive medical understanding, unlocking the relationships among the patient’s genome, environment, prenatal exposure, and disease risk in time for us to prevent diseases.
Many questions about epigenetic heterogeneity in cancer remain to be answered. In nearly every study to date, the proportion of a tumor that is assayed is quite small relative to the full tumor mass in the patient. ITH may explain the difficulties encountered in the validation of oncology biomarkers and prediction of therapeutic resistance owing to sampling bias [24]. Current measures of eITH significantly underestimate the levels of ITH, and signals from bulk tumor samples are dominated by major subclones, rendering rare subpopulations undetectable [23]. eITH may reflect a mix of subclones with distinct genomic and epigenomic features. In addition, epigenome variability comes from a variety of other cells present in tumor tissues, including nontumor stromal and immune cells. A plethora of newly identified mutations in epigenetic regulators remain largely uncharacterized. It is necessary to identify them to be epigenetic drivers/passengers by functional experiments [23]. The tumor microenvironment may represent as much as 90% of some tumor samples and contribute proportionally to the RNA pool, which affects measures of heterogeneity and resulting transcriptional profiles. Thus, both the tumor and its microenvironment, including tumor-infiltrating leukocytes, should ideally be assayed. Epigenetic modifications are dynamic and responsive to environmental pressures, and they may reflect the potential of the tumor to respond to an environmental or therapeutic pressure [21]. Complete understanding of genetic and epigenetic heterogeneity may assist in designing combinations of targeted therapies based on molecular information extracted from individual tumors. Ideally, we could always target druggable trunk mutations/aberrant epigenetic changes, and then add drugs to target emerging subclones.
Availability of data and materials
The material supporting the conclusion of this review has been included within the article.
Abbreviations
- 5mC:
-
5-methylcyctosine
- AICDA:
-
Activation-induced cytidine deaminase
- ALL:
-
Acute lymphoblastic leukemia
- AML:
-
Acute myeloid leukemia
- BC:
-
Breast cancer
- BER:
-
Base excision repair
- BET:
-
The bromodomain and extra-terminal motif
- BRDs:
-
Bromodomains
- CAFs:
-
Cancer-associated fibroblast
- CLL:
-
Chronic lymphoblastic leukemia
- CRC:
-
Colorectal cancer
- DCIS:
-
Ductal carcinoma in situ
- DNMTs:
-
DNA methyltransferases
- DOT1L:
-
Disruptor of telomeric silencing-1-like
- DSS:
-
Dextran sulfate sodium
- ESCC:
-
Esophageal squamous cell carcinoma
- EZH2:
-
Enhancer of zeste homologue 2
- FA:
-
Fanconi anemia
- GC:
-
Gastric cancer
- HATs:
-
Histone acetyltransferases
- HCC:
-
Hepatocellular carcinoma
- HDACs:
-
Histone deacetylases
- HDMs:
-
Histone demethylases
- HMTs:
-
Histone methytransferases
- HR:
-
Homologous recombination
- ITH:
-
Intratumor heterogeneity
- KDMs:
-
Lysine demethylases
- KMTs:
-
Lysine methyltransferases
- LSD1:
-
Lysine specific histone demethylase-1
- MMR:
-
Mismatch repair
- MSI:
-
Microsatellite instability
- NER:
-
Nucleotide excision repair
- NHEJ:
-
Non-homologous end-joining
- NSCLC:
-
Non-small cell lung cancer
- OS:
-
Overall survival
- PFS:
-
Progression-free survival;
- PRC2:
-
Polycomb repressive complex 2
- TDG:
-
Thymine-DNA glycosylase
- TET:
-
Ten-eleven translocation
- TFS:
-
Transcription factors
- TSA:
-
Trichostatin A
- TSGs:
-
Tumor-suppressor genes
- TSS:
-
Transcription start sites
References
Easwaran H, Tsai HC, Baylin SB. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell. 2014;54(5):716–27.
Swanton C. Intratumor heterogeneity: evolution through space and time. Cancer Res. 2012;72(19):4875–82.
Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;501(7467):346–54.
Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121(3):335–48.
Liu T, Han C, Wang S, Fang P, Ma Z, Xu L, et al. Cancer-associated fibroblasts: an emerging target of anti-cancer immunotherapy. J Hematol Oncol. 2019;12(1):86.
Yang L, Zhang Y. Tumor-associated macrophages: from basic research to clinical application. J Hematol Oncol. 2017;10(1):58.
Ohlund D, Elyada E, Tuveson D. Fibroblast heterogeneity in the cancer wound. J Exp Med. 2014;211(8):1503–23.
Bussard KM, Mutkus L, Stumpf K, Gomez-Manzano C, Marini FC. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016;18(1):84.
Guo M, Ren J, House MG, Qi Y, Brock MV, Herman JG. Accumulation of promoter methylation suggests epigenetic progression in squamous cell carcinoma of the esophagus. Clin Cancer Res. 2006;12(15):4515–22.
Vogelstein B, Kinzler KW. The multistep nature of cancer. Trends Genet. 1993;9(4):138–41.
Slaughter DP, Southwick HW, Smejkal W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer. 1953;6(5):963–8.
Visvader JE. Cells of origin in cancer. Nature. 2011;469(7330):314–22.
Bedard PL, Hansen AR, Ratain MJ, Siu LL. Tumour heterogeneity in the clinic. Nature. 2013;501(7467):355–64.
Almendro V, Marusyk A, Polyak K. Cellular heterogeneity and molecular evolution in cancer. Annu Rev Pathol. 2013;8:277–302.
Marusyk A, Polyak K. Tumor heterogeneity: causes and consequences. Biochim Biophys Acta. 2010;1805(1):105–17.
Place AE, Jin Huh S, Polyak K. The microenvironment in breast cancer progression: biology and implications for treatment. Breast Cancer Res. 2011;13(6):227.
Tlsty TD, Coussens LM. Tumor stroma and regulation of cancer development. Annu Rev Pathol. 2006;1:119–50.
Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194(4260):23–8.
Gerlinger M, Swanton C. How Darwinian models inform therapeutic failure initiated by clonal heterogeneity in cancer medicine. Br J Cancer. 2010;103(8):1139–43.
Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer. 2012;12(5):323–34.
Alizadeh AA, Aranda V, Bardelli A, Blanpain C, Bock C, Borowski C, et al. Toward understanding and exploiting tumor heterogeneity. Nat Med. 2015;21(1546-170X (Electronic)):846–53.
Jamal-Hanjani M, Quezada SA, Larkin J, Swanton C. Translational implications of tumor heterogeneity. Clin Cancer Res. 2015;21(6):1258–66.
Mazor T, Pankov A, Song JS, Costello JF. Intratumoral heterogeneity of the epigenome. Cancer Cell. 2016;29(4):440–51.
Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–92.
Baylin SB, Herman JG. DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trends Genet. 2000;16(4):168–74.
Yan W, Herman JG, Guo M. Epigenome-based personalized medicine in human cancer. Epigenomics. 2016;8(1):119–33.
Lorsbach RB, Moore J, Mathew S, Raimondi SC, Mukatira ST, Downing JR. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10;11)(q22;q23). Leukemia. 2003;17(3):637–41.
Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333(6047):1300–3.
Zhang L, Lu X, Lu J, Liang H, Dai Q, Xu GL, et al. Thymine DNA glycosylase specifically recognizes 5-carboxylcytosine-modified DNA. Nat Chem Biol. 2012;8(4):328–30.
Zhang H, Zhang X, Clark E, Mulcahey M, Huang S, Shi YG. TET1 is a DNA-binding protein that modulates DNA methylation and gene transcription via hydroxylation of 5-methylcytosine. Cell Res. 2010;20(12):1390–3.
Huang Y, Rao A. Connections between TET proteins and aberrant DNA modification in cancer. Trends Genet. 2014;30(10):464–74.
Dang L, Jin S, Su SM. IDH mutations in glioma and acute myeloid leukemia. Trends Mol Med. 2010;16(9):387–97.
You JS, Jones PA. Cancer genetics and epigenetics: two sides of the same coin? Cancer Cell. 2012;22(1):9–20.
Moran S, Martinez-Cardus A, Boussios S, Esteller M. Precision medicine based on epigenomics: the paradigm of carcinoma of unknown primary. Nat Rev Clin Oncol. 2017;14(11):682–94.
Feinberg AP. The key role of epigenetics in human disease prevention and mitigation. N Engl J Med. 2018;378(14):1323–34.
Feinberg AP, Koldobskiy MA, Gondor A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet. 2016;17(5):284–99.
Timp W, Feinberg AP. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat Rev Cancer. 2013;13(7):497–510.
Guo M, Akiyama Y, House MG, Hooker CM, Heath E, Gabrielson E, et al. Hypermethylation of the GATA genes in lung cancer. Clin Cancer Res. 2004;10(23):7917–24.
Guo M, Ren J, Brock MV, Herman JG, Carraway HE. Promoter methylation of HIN-1 in the progression to esophageal squamous cancer. Epigenetics. 2008;3(6):336–41.
Thirlwell C, Eymard M, Feber A, Teschendorff A, Pearce K, Lechner M, et al. Genome-wide DNA methylation analysis of archival formalin-fixed paraffin-embedded tissue using the Illumina Infinium HumanMethylation27 BeadChip. Methods (San Diego, Calif). 2010;52(3):248–54.
Cao B, Yang W, Jin Y, Zhang M, He T, Zhan Q, et al. Silencing NKD2 by promoter region hypermethylation promotes esophageal cancer progression by activating Wnt signaling. J Thorac Oncol. 2016;11(11):1912–26.
Yan W, Wu K, Herman JG, Brock MV, Zhou Y, Lu Y, et al. Epigenetic silencing of DACH1 induces the invasion and metastasis of gastric cancer by activating TGF-beta signalling. J Cell Mol Med. 2014;18(12):2499–511.
Yang W, Yang S, Zhang M, Gao D, He T, Guo M. ZNF545 suppresses human hepatocellular carcinoma growth by inhibiting NF-kB signaling. Genes Cancer. 2017;8(3–4):528–35.
Al-Kaabi A, van Bockel LW, Pothen AJ, Willems SM. p16INK4A and p14ARF gene promoter hypermethylation as prognostic biomarker in oral and oropharyngeal squamous cell carcinoma: a review. Dis Markers. 2014;2014:260549.
Das M, Saikia BJ, Sharma SK, Sekhon GS, Mahanta J, Phukan RK. p16 hypermethylation: a biomarker for increased esophageal cancer susceptibility in high incidence region of north East India. Tumour Biol. 2015;36(3):1627–42.
Derks S, Cleven AH, Melotte V, Smits KM, Brandes JC, Azad N, et al. Emerging evidence for CHFR as a cancer biomarker: from tumor biology to precision medicine. Cancer Metastasis Rev. 2014;33(1):161–71.
Grawenda AM, O'Neill E. Clinical utility of RASSF1A methylation in human malignancies. Br J Cancer. 2015;113(3):372–81.
Wang H, Wang S, Shen L, Chen Y, Zhang X, Zhou J, et al. Chk2 down-regulation by promoter hypermethylation in human bulk gliomas. Life Sci. 2010;86(5–6):185–91.
Kato N, Fujimoto H, Yoda A, Oishi I, Matsumura N, Kondo T, et al. Regulation of Chk2 gene expression in lymphoid malignancies: involvement of epigenetic mechanisms in Hodgkin's lymphoma cell lines. Cell Death Differ. 2004;11(Suppl 2):S153–61.
Zhang P, Wang J, Gao W, Yuan BZ, Rogers J, Reed E. CHK2 kinase expression is down-regulated due to promoter methylation in non-small cell lung cancer. Mol Cancer. 2004;3:14.
Cao X, Tang Q, Holland-Letz T, Gundert M, Cuk K, Schott S, et al. Evaluation of promoter methylation of RASSF1A and ATM in peripheral blood of breast cancer patients and healthy control individuals. Int J Mol Sci. 2018;19(3). https://doi.org/10.3390/ijms19030900.
Guo M, Jia Y, Yu Z, House MG, Esteller M, Brock MV, et al. Epigenetic changes associated with neoplasms of the exocrine and endocrine pancreas. Discov Med. 2014;17(92):67–73.
Yu J, Zhu T, Wang Z, Zhang H, Qian Z, Xu H, et al. A novel set of DNA methylation markers in urine sediments for sensitive/specific detection of bladder cancer. Clin Cancer Res. 2007;13(24):7296–304.
Harada H, Miyamoto K, Yamashita Y, Nakano K, Taniyama K, Miyata Y, et al. Methylation of breast cancer susceptibility gene 1 (BRCA1) predicts recurrence in patients with curatively resected stage I non-small cell lung cancer. Cancer. 2013;119(4):792–8.
Laskar RS, Talukdar FR, Choudhury JH, Singh SA, Kundu S, Dhar B, et al. Association of HPV with genetic and epigenetic alterations in colorectal adenocarcinoma from Indian population. Tumour Biol. 2015;36(6):4661–70.
Lee MN, Tseng RC, Hsu HS, Chen JY, Tzao C, Ho WL, et al. Epigenetic inactivation of the chromosomal stability control genes BRCA1, BRCA2, and XRCC5 in non-small cell lung cancer. Clin Cancer Res. 2007;13(3):832–8.
Hegi ME, Sciuscio D, Murat A, Levivier M, Stupp R. Epigenetic deregulation of DNA repair and its potential for therapy. Clin Cancer Res. 2009;15(16):5026–31.
Zhou C, Tang H, Yu J, Zhuang D, Zhang H. Blood-based DNA methylation of DNA repair genes in the non-homologous end-joining (NEHJ) pathway in patient with glioma. Int J Clin Exp Pathol. 2015;8(8):9463–7.
Agrelo R, Cheng WH, Setien F, Ropero S, Espada J, Fraga MF, et al. Epigenetic inactivation of the premature aging Werner syndrome gene in human cancer. Proc Natl Acad Sci U S A. 2006;103(23):8822–7.
Nagasaka T, Rhees J, Kloor M, Gebert J, Naomoto Y, Boland CR, et al. Somatic hypermethylation of MSH2 is a frequent event in lynch syndrome colorectal cancers. Cancer Res. 2010;70(8):3098–108.
Hinrichsen I, Kemp M, Peveling-Oberhag J, Passmann S, Plotz G, Zeuzem S, et al. Promoter methylation of MLH1, PMS2, MSH2 and p16 is a phenomenon of advanced-stage HCCs. PLoS One. 2014;9(1):e84453.
Peng B, Hodge DR, Thomas SB, Cherry JM, Munroe DJ, Pompeia C, et al. Epigenetic silencing of the human nucleotide excision repair gene, hHR23B, in interleukin-6-responsive multiple myeloma KAS-6/1 cells. J Biol Chem. 2005;280(6):4182–7.
Galamb O, Kalmar A, Peterfia B, Csabai I, Bodor A, Ribli D, et al. Aberrant DNA methylation of WNT pathway genes in the development and progression of CIMP-negative colorectal cancer. Epigenetics. 2016;11(8):588–602.
Jung YS, Jun S, Lee SH, Sharma A, Park JI. Wnt2 complements Wnt/beta-catenin signaling in colorectal cancer. Oncotarget. 2015;6(35):37257–68.
Ding Z, Qian YB, Zhu LX, Xiong QR. Promoter methylation and mRNA expression of DKK-3 and WIF-1 in hepatocellular carcinoma. World J Gastroenterol. 2009;15(21):2595–601.
Yoda Y, Takeshima H, Niwa T, Kim JG, Ando T, Kushima R, et al. Integrated analysis of cancer-related pathways affected by genetic and epigenetic alterations in gastric cancer. Gastric Cancer. 2015;18(1):65–76.
Wu D, Wong P, Li W, Vogel CF, Matsumura F. Suppression of WIF-1 through promoter hypermethylation causes accelerated proliferation of the aryl hydrocarbon receptor (AHR) overexpressing MCF10AT1 breast cancer cells. Toxicology. 2011;285(3):97–103.
Aguilera O, Fraga MF, Ballestar E, Paz MF, Herranz M, Espada J, et al. Epigenetic inactivation of the Wnt antagonist DICKKOPF-1 (DKK-1) gene in human colorectal cancer. Oncogene. 2006;25(29):4116–21.
Suzuki M, Shigematsu H, Nakajima T, Kubo R, Motohashi S, Sekine Y, et al. Synchronous alterations of Wnt and epidermal growth factor receptor signaling pathways through aberrant methylation and mutation in non small cell lung cancer. Clin Cancer Res. 2007;13(20):6087–92.
Suzuki H, Toyota M, Carraway H, Gabrielson E, Ohmura T, Fujikane T, et al. Frequent epigenetic inactivation of Wnt antagonist genes in breast cancer. Br J Cancer. 2008;98(6):1147–56.
Wang H, Duan XL, Qi XL, Meng L, Xu YS, Wu T, et al. Concurrent hypermethylation of SFRP2 and DKK2 activates the Wnt/beta-catenin pathway and is associated with poor prognosis in patients with gastric cancer. Mol Cells. 2017;40(1):45–53.
Berx G, van Roy F. Involvement of members of the cadherin superfamily in cancer. Cold Spring Harb Perspect Biol. 2009;1(6):a003129.
Dumitrescu RG. Epigenetic markers of early tumor development. Methods Mol Biol (Clifton, NJ). 2012;863:3–14.
Silva AL, Dawson SN, Arends MJ, Guttula K, Hall N, Cameron EA, et al. Boosting Wnt activity during colorectal cancer progression through selective hypermethylation of Wnt signaling antagonists. BMC Cancer. 2014;14:891.
Chuang LS, Ito Y. RUNX3 is multifunctional in carcinogenesis of multiple solid tumors. Oncogene. 2010;29(18):2605–15.
Katsuno Y, Qin J, Oses-Prieto JA, Wang H, Jackson-Weaver O, Zhang T, et al. Arginine methylation of Smad7 by PRMT1 in TGF-beta-induced epithelial-mesenchymal transition and epithelial stem cell generation. J Biol Chem. 2018;293(34):13059–72.
Wu L, Herman JG, Brock MV, Wu K, Mao G, Yan W, et al. Silencing DACH1 promotes esophageal cancer growth by inhibiting TGF-beta signaling. PLoS One. 2014;9(4):e95509.
Feng Y, Wang L, Wang M. Alteration of DACH1 methylation patterns in lung cancer contributes to cell proliferation and migration. Biochem Cell Biol. 2018;96(5):602–9.
Dalgin GS, Drever M, Williams T, King T, DeLisi C, Liou LS. Identification of novel epigenetic markers for clear cell renal cell carcinoma. J Urol. 2008;180(3):1126–30.
Guo W, Zhang M, Shen S, Guo Y, Kuang G, Yang Z, et al. Aberrant methylation and decreased expression of the TGF-beta/Smad target gene FBXO32 in esophageal squamous cell carcinoma. Cancer. 2014;120(16):2412–23.
Guo W, Zhang M, Guo Y, Shen S, Guo X, Dong Z. FBXO32, a new TGF-beta/Smad signaling pathway target gene, is epigenetically inactivated in gastric cardia adenocarcinoma. Neoplasma. 2015;62(4):646–57.
Wei H, Wang B, Miyagi M, She Y, Gopalan B, Huang DB, et al. PRMT5 dimethylates R30 of the p65 subunit to activate NF-kappaB. Proc Natl Acad Sci U S A. 2013;110(33):13516–21.
Levy D, Kuo AJ, Chang Y, Schaefer U, Kitson C, Cheung P, et al. Lysine methylation of the NF-kappaB subunit RelA by SETD6 couples activity of the histone methyltransferase GLP at chromatin to tonic repression of NF-kappaB signaling. Nat Immunol. 2011;12(1):29–36.
Cheng Y, Geng H, Cheng SH, Liang P, Bai Y, Li J, et al. KRAB zinc finger protein ZNF382 is a proapoptotic tumor suppressor that represses multiple oncogenes and is commonly silenced in multiple carcinomas. Cancer Res. 2010;70(16):6516–26.
Cao B, Yang Y, Pan Y, Jia Y, Brock MV, Herman JG, et al. Epigenetic silencing of CXCL14 induced colorectal cancer migration and invasion. Discov Med. 2013;16(88):137–47.
Chen L, Tang J, Feng Y, Li S, Xiang Q, He X, et al. ADAMTS9 is silenced by epigenetic disruption in colorectal cancer and inhibits cell growth and metastasis by regulating Akt/p53 signaling. Cell Physiol Biochem. 2017;44(4):1370–80.
Yan W, Wu K, Herman JG, Xu X, Yang Y, Dai G, et al. Retinoic acid-induced 2 (RAI2) is a novel tumor suppressor, and promoter region methylation of RAI2 is a poor prognostic marker in colorectal cancer. Clin Epigenetics. 2018;10:69.
Yu Y, Yin D, Hoque MO, Cao B, Jia Y, Yang Y, et al. AKT signaling pathway activated by HIN-1 methylation in non-small cell lung cancer. Tumour Biol. 2012;33(2):307–14.
Jin Y, Cao B, Zhang M, Zhan Q, Herman JG, Yu M, et al. RASSF10 suppresses hepatocellular carcinoma growth by activating P53 signaling and methylation of RASSF10 is a docetaxel resistant marker. Genes Cancer. 2015;6(5–6):231–40.
Fan C, Wang W, Jin J, Yu Z, Xin X. RASSF10 is epigenetically inactivated and suppresses cell proliferation and induces cell apoptosis by activating the p53 Signalling pathway in papillary thyroid carcinoma cancer. Cell Physiol Biochem. 2017;41(3):1229–39.
Li X, Yu J, Brock MV, Tao Q, Herman JG, Liang P, et al. Epigenetic silencing of BCL6B inactivates p53 signaling and causes human hepatocellular carcinoma cell resist to 5-FU. Oncotarget. 2015;6(13):11547–60.
Hu S, Cao B, Zhang M, Linghu E, Zhan Q, Brock MV, et al. Epigenetic silencing BCL6B induced colorectal cancer proliferation and metastasis by inhibiting P53 signaling. Am J Cancer Res. 2015;5(2):651–62.
Li L, Xu J, Qiu G, Ying J, Du Z, Xiang T, et al. Epigenomic characterization of a p53-regulated 3p22.2 tumor suppressor that inhibits STAT3 phosphorylation via protein docking and is frequently methylated in esophageal and other carcinomas. Theranostics. 2018;8(1):61–77.
Yokoi K, Yamashita K, Ishii S, Tanaka T, Nishizawa N, Tsutsui A, et al. Comprehensive molecular exploration identified promoter DNA methylation of the CRBP1 gene as a determinant of radiation sensitivity in rectal cancer. Br J Cancer. 2017;116(8):1046–56.
Mizuiri H, Yoshida K, Toge T, Oue N, Aung PP, Noguchi T, et al. DNA methylation of genes linked to retinoid signaling in squamous cell carcinoma of the esophagus: DNA methylation of CRBP1 and TIG1 is associated with tumor stage. Cancer Sci. 2005;96(9):571–7.
He D, Zhang YW, Zhang NN, Zhou L, Chen JN, Jiang Y, et al. Aberrant gene promoter methylation of p16, FHIT, CRBP1, WWOX, and DLC-1 in Epstein-Barr virus-associated gastric carcinomas. Med Oncol (Northwood, London, England). 2015;32(4):92.
Esteller M, Guo M, Moreno V, Peinado MA, Capella G, Galm O, et al. Hypermethylation-associated inactivation of the cellular retinol-binding-protein 1 gene in human cancer. Cancer Res. 2002;62(20):5902–5.
Jeronimo C, Henrique R, Hoque MO, Mambo E, Ribeiro FR, Varzim G, et al. A quantitative promoter methylation profile of prostate cancer. Clin Cancer Res. 2004;10(24):8472–8.
Eissa S, Swellam M, El-Khouly IM, Kassim SK, Shehata H, Mansour A, et al. Aberrant methylation of RARbeta2 and APC genes in voided urine as molecular markers for early detection of bilharzial and nonbilharzial bladder cancer. Cancer Epidemiol Biomarkers Prev. 2011;20(8):1657–64.
Swellam M, Abdelmaksoud MD, Sayed Mahmoud M, Ramadan A, Abdel-Moneem W, Hefny MM. Aberrant methylation of APC and RARbeta2 genes in breast cancer patients. IUBMB Life. 2015;67(1):61–8.
Zuo Y, Song Y. Detection and analysis of the methylation status of PTCH1 gene involved in the hedgehog signaling pathway in a human gastric cancer cell line. Exp Ther Med. 2013;6(6):1365–8.
Wolf I, Bose S, Desmond JC, Lin BT, Williamson EA, Karlan BY, et al. Unmasking of epigenetically silenced genes reveals DNA promoter methylation and reduced expression of PTCH in breast cancer. Breast Cancer Res Treat. 2007;105(2):139–55.
Heitzer E, Bambach I, Dandachi N, Horn M, Wolf P. PTCH promoter methylation at low level in sporadic basal cell carcinoma analysed by three different approaches. Exp Dermatol. 2010;19(10):926–8.
Wang LH, Choi YL, Hua XY, Shin YK, Song YJ, Youn SJ, et al. Increased expression of sonic hedgehog and altered methylation of its promoter region in gastric cancer and its related lesions. Mod Pathol. 2006;19(5):675–83.
Cui W, Wang LH, Wen YY, Song M, Li BL, Chen XL, et al. Expression and regulation mechanisms of sonic hedgehog in breast cancer. Cancer Sci. 2010;101(4):927–33.
Lin EH, Kao YR, Lin CA, Kuo TY, Yang SP, Hsu CF, et al. Hedgehog pathway maintains cell survival under stress conditions, and drives drug resistance in lung adenocarcinoma. Oncotarget. 2016;7(17):24179–93.
Song Y, Zhang J, Tian T, Fu X, Wang W, Li S, et al. SET7/9 inhibits oncogenic activities through regulation of Gli-1 expression in breast cancer. Tumour Biol. 2016;37(7):9311–22.
Loh M, Liem N, Vaithilingam A, Lim PL, Sapari NS, Elahi E, et al. DNA methylation subgroups and the CpG island methylator phenotype in gastric cancer: a comprehensive profiling approach. BMC Gastroenterol. 2014;14:55.
Waddington CH. The epigenotype. 1942. Int J Epidemiol. 2012;41(1):10–3.
Teschendorff AE, West J, Beck S. Age-associated epigenetic drift: implications, and a case of epigenetic thrift? Hum Mol Genet. 2013;22(R1):R7–r15.
Feinberg AP, Fallin MD. Epigenetics at the crossroads of genes and the environment. JAMA. 2015;314(11):1129–30.
Dolinoy DC, Weidman JR, Waterland RA, Jirtle RL. Maternal genistein alters coat color and protects Avy mouse offspring from obesity by modifying the fetal epigenome. Environ Health Perspect. 2006;114(4):567–72.
Joubert BR, Haberg SE, Nilsen RM, Wang X, Vollset SE, Murphy SK, et al. 450K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ Health Perspect. 2012;120(10):1425–31.
Richmond RC, Simpkin AJ, Woodward G, Gaunt TR, Lyttleton O, McArdle WL, et al. Prenatal exposure to maternal smoking and offspring DNA methylation across the lifecourse: findings from the Avon longitudinal study of parents and children (ALSPAC). Hum Mol Genet. 2015;24(8):2201–17.
Erratum for Perfilyev, et al. Impact of polyunsaturated and saturated fat overfeeding on the DNA-methylation pattern in human adipose tissue: a randomized controlled trial. Am J Clin Nutr. 2017;105:991–1000 The American Journal of Clinical Nutrition. 2017;106(1):325.
Shen H, Laird PW. Interplay between the cancer genome and epigenome. Cell. 2013;153(1):38–55.
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546–58.
Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363(25):2424–33.
Yan XJ, Xu J, Gu ZH, Pan CM, Lu G, Shen Y, et al. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat Genet. 2011;43(4):309–15.
Inoue S, Lemonnier F, Mak TW. Roles of IDH1/2 and TET2 mutations in myeloid disorders. Int J Hematol. 2016;103(6):627–33.
Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468(7325):839–43.
Yang H, Ye D, Guan KL, Xiong Y. IDH1 and IDH2 mutations in tumorigenesis: mechanistic insights and clinical perspectives. Clin Cancer Res. 2012;18(20):5562–71.
DiNardo CD, Ravandi F, Agresta S, Konopleva M, Takahashi K, Kadia T, et al. Characteristics, clinical outcome, and prognostic significance of IDH mutations in AML. Am J Hematol. 2015;90(8):732–6.
Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482(7384):226–31.
Gallo M, Coutinho FJ, Vanner RJ, Gayden T, Mack SC, Murison A, et al. MLL5 orchestrates a cancer self-renewal state by repressing the histone variant H3.3 and globally reorganizing chromatin. Cancer Cell. 2015;28(6):715–29.
Tan J, Jones M, Koseki H, Nakayama M, Muntean AG, Maillard I, et al. CBX8, a polycomb group protein, is essential for MLL-AF9-induced leukemogenesis. Cancer Cell. 2011;20(5):563–75.
Rao RC, Dou Y. Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat Rev Cancer. 2015;15(6):334–46.
Kanai Y, Ushijima S, Nakanishi Y, Sakamoto M, Hirohashi S. Mutation of the DNA methyltransferase (DNMT) 1 gene in human colorectal cancers. Cancer Lett. 2003;192(1):75–82.
Hamidi T, Singh AK, Chen T. Genetic alterations of DNA methylation machinery in human diseases. Epigenomics. 2015;7(2):247–65.
Scourzic L, Mouly E, Bernard OA. TET proteins and the control of cytosine demethylation in cancer. Genome Med. 2015;7(1):9.
Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360(22):2289–301.
Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18(6):553–67.
Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483(7390):479–83.
Neta G, Brenner AV, Sturgis EM, Pfeiffer RM, Hutchinson AA, Aschebrook-Kilfoy B, et al. Common genetic variants related to genomic integrity and risk of papillary thyroid cancer. Carcinogenesis. 2011;32(8):1231–7.
Bueno R, Stawiski EW, Goldstein LD, Durinck S, De Rienzo A, Modrusan Z, et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat Genet. 2016;48(4):407–16.
Walter DM, Venancio OS, Buza EL, Tobias JW, Deshpande C, Gudiel AA, et al. Systematic in vivo inactivation of chromatin-regulating enzymes identifies Setd2 as a potent tumor suppressor in lung adenocarcinoma. Cancer Res. 2017;77(7):1719–29.
Parker H, Rose-Zerilli MJ, Larrayoz M, Clifford R, Edelmann J, Blakemore S, et al. Genomic disruption of the histone methyltransferase SETD2 in chronic lymphocytic leukaemia. Leukemia. 2016;30(11):2179–86.
Moffitt AB, Ondrejka SL, McKinney M, Rempel RE, Goodlad JR, Teh CH, et al. Enteropathy-associated T cell lymphoma subtypes are characterized by loss of function of SETD2. J Exp Med. 2017;214(5):1371–86.
Malinowska-Ozdowy K, Frech C, Schonegger A, Eckert C, Cazzaniga G, Stanulla M, et al. KRAS and CREBBP mutations: a relapse-linked malicious liaison in childhood high hyperdiploid acute lymphoblastic leukemia. Leukemia. 2015;29(8):1656–67.
Beije N, Helmijr JC, Weerts MJA, Beaufort CM, Wiggin M, Marziali A, et al. Somatic mutation detection using various targeted detection assays in paired samples of circulating tumor DNA, primary tumor and metastases from patients undergoing resection of colorectal liver metastases. Mol Oncol. 2016;10(10):1575–84.
Ge S, Li B, Li Y, Li Z, Liu Z, Chen Z, et al. Genomic alterations in advanced gastric cancer endoscopic biopsy samples using targeted next-generation sequencing. Am J Cancer Res. 2017;7(7):1540–53.
Juskevicius D, Jucker D, Klingbiel D, Mamot C, Dirnhofer S, Tzankov A. Mutations of CREBBP and SOCS1 are independent prognostic factors in diffuse large B cell lymphoma: mutational analysis of the SAKK 38/07 prospective clinical trial cohort. J Hematol Oncol. 2017;10(1):70.
Winters AC, Bernt KM. MLL-rearranged Leukemias-an update on science and clinical approaches. Front Pediatr. 2017;5:4.
Andersen CL, Hasselbalch H, Gronbaek K. Lack of somatic mutations in the catalytic domains of CREBBP and EP300 genes implies a role for histone deacetylase inhibition in myeloproliferative neoplasms. Leuk Res. 2012;36(4):485–7.
Kasaian K, Wiseman SM, Walker BA, Schein JE, Zhao Y, Hirst M, et al. The genomic and transcriptomic landscape of anaplastic thyroid cancer: implications for therapy. BMC Cancer. 2015;15:984.
Donner I, Kiviluoto T, Ristimaki A, Aaltonen LA, Vahteristo P. Exome sequencing reveals three novel candidate predisposition genes for diffuse gastric cancer. Familial Cancer. 2015;14(2):241–6.
Zhang X, Liu D, Li M, Cao C, Wan D, Xi B, et al. Prognostic and therapeutic value of disruptor of telomeric silencing-1-like (DOT1L) expression in patients with ovarian cancer. J Hematol Oncol. 2017;10(1):29.
Yu H, Ma M, Yan J, Xu L, Yu J, Dai J, et al. Identification of coexistence of BRAF V600E mutation and EZH2 gain specifically in melanoma as a promising target for combination therapy. J Transl Med. 2017;15(1):243.
Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and pathogenesis of diffuse large B-cell lymphoma. N Engl J Med. 2018;378(15):1396–407.
Booth CAG, Barkas N, Neo WH, Boukarabila H, Soilleux EJ, Giotopoulos G, et al. Ezh2 and Runx1 mutations collaborate to initiate lympho-myeloid leukemia in early thymic progenitors. Cancer Cell. 2018;33(2):274–91.e8.
Rinke J, Muller JP, Blaess MF, Chase A, Meggendorfer M, Schafer V, et al. Molecular characterization of EZH2 mutant patients with myelodysplastic/myeloproliferative neoplasms. Leukemia. 2017;31(9):1936–43.
Vougiouklakis T, Hamamoto R, Nakamura Y, Saloura V. The NSD family of protein methyltransferases in human cancer. Epigenomics. 2015;7(5):863–74.
Lai J, Zhou Z, Tang XJ, Gao ZB, Zhou J, Chen SQ. A tumor-specific neo-antigen caused by a frameshift mutation in BAP1 is a potential personalized biomarker in malignant peritoneal mesothelioma. Int J Mol Sci. 2016;17(5). https://doi.org/10.3390/ijms17050739.
Wang L, Yamaguchi S, Burstein MD, Terashima K, Chang K, Ng HK, et al. Novel somatic and germline mutations in intracranial germ cell tumours. Nature. 2014;511(7508):241–5.
Liu XY, Gerges N, Korshunov A, Sabha N, Khuong-Quang DA, Fontebasso AM, et al. Frequent ATRX mutations and loss of expression in adult diffuse astrocytic tumors carrying IDH1/IDH2 and TP53 mutations. Acta Neuropathol. 2012;124(5):615–25.
Jiao Y, Shi C, Edil BH, de Wilde RF, Klimstra DS, Maitra A, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science (New York, NY). 2011;331(6021):1199–203.
Ogura K, Hosoda F, Arai Y, Nakamura H, Hama N, Totoki Y, et al. Integrated genetic and epigenetic analysis of myxofibrosarcoma. Nat Commun. 2018;9(1):2765.
Chen X, Bahrami A, Pappo A, Easton J, Dalton J, Hedlund E, et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep. 2014;7(1):104–12.
Kolla V, Zhuang T, Higashi M, Naraparaju K, Brodeur GM. Role of CHD5 in human cancers: 10 years later. Cancer Res. 2014;74(3):652–8.
Choi YJ, Yoo NJ, Lee SH. Mutation of HELLS, a chromatin remodeling gene, gastric and colorectal cancers. Pathol Oncol Res. 2015;21(3):851–2.
Yano M, Ouchida M, Shigematsu H, Tanaka N, Ichimura K, Kobayashi K, et al. Tumor-specific exon creation of the HELLS/SMARCA6 gene in non-small cell lung cancer. Int J Cancer. 2004;112(1):8–13.
Masliah-Planchon J, Bieche I, Guinebretiere JM, Bourdeaut F, Delattre O. SWI/SNF chromatin remodeling and human malignancies. Annu Rev Pathol. 2015;10:145–71.
Jelinic P, Schlappe BA, Conlon N, Tseng J, Olvera N, Dao F, et al. Concomitant loss of SMARCA2 and SMARCA4 expression in small cell carcinoma of the ovary, hypercalcemic type. Mod Pathol. 2016;29(1):60–6.
Kolin DL, Dong F, Baltay M, Lindeman N, MacConaill L, Nucci MR, et al. SMARCA4-deficient undifferentiated uterine sarcoma (malignant rhabdoid tumor of the uterus): a clinicopathologic entity distinct from undifferentiated carcinoma. Mod Pathol. 2018;31(9):1442–56.
Wu HC, Delgado-Cruzata L, Flom JD, Perrin M, Liao Y, Ferris JS, et al. Repetitive element DNA methylation levels in white blood cell DNA from sisters discordant for breast cancer from the New York site of the breast cancer family registry. Carcinogenesis. 2012;33(10):1946–52.
Park SY, Yoo EJ, Cho NY, Kim N, Kang GH. Comparison of CpG island hypermethylation and repetitive DNA hypomethylation in premalignant stages of gastric cancer, stratified for helicobacter pylori infection. J Pathol. 2009;219(4):410–6.
Neale RE, Clark PJ, Fawcett J, Fritschi L, Nagler BN, Risch HA, et al. Association between hypermethylation of DNA repetitive elements in white blood cell DNA and pancreatic cancer. Cancer Epidemiol. 2014;38(5):576–82.
Lee HS, Kim BH, Cho NY, Yoo EJ, Choi M, Shin SH, et al. Prognostic implications of and relationship between CpG island hypermethylation and repetitive DNA hypomethylation in hepatocellular carcinoma. Clin Cancer Res. 2009;15(3):812–20.
Cadieux B, Ching TT, VandenBerg SR, Costello JF. Genome-wide hypomethylation in human glioblastomas associated with specific copy number alteration, methylenetetrahydrofolate reductase allele status, and increased proliferation. Cancer Res. 2006;66(17):8469–76.
Luo Y, Lu X, Xie H. Dynamic Alu methylation during normal development, aging, and tumorigenesis. Biomed Res Int. 2014;2014:784706.
Ardeljan D, Taylor MS, Ting DT, Burns KH. The human long interspersed Element-1 retrotransposon: an emerging biomarker of neoplasia. Clin Chem. 2017;63(4):816–22.
Howard JH, Frolov A, Tzeng CW, Stewart A, Midzak A, Majmundar A, et al. Epigenetic downregulation of the DNA repair gene MED1/MBD4 in colorectal and ovarian cancer. Cancer Biol Ther. 2009;8(1):94–100.
Esteller M, Herman JG. Generating mutations but providing chemosensitivity: the role of O6-methylguanine DNA methyltransferase in human cancer. Oncogene. 2004;23(1):1–8.
Peng B, Hurt EM, Hodge DR, Thomas SB, Farrar WL. DNA hypermethylation and partial gene silencing of human thymine- DNA glycosylase in multiple myeloma cell lines. Epigenetics. 2006;1(3):138–45.
Slyskova J, Korenkova V, Collins AR, Prochazka P, Vodickova L, Svec J, et al. Functional, genetic, and epigenetic aspects of base and nucleotide excision repair in colorectal carcinomas. Clin Cancer Res. 2012;18(21):5878–87.
Chaisaingmongkol J, Popanda O, Warta R, Dyckhoff G, Herpel E, Geiselhart L, et al. Epigenetic screen of human DNA repair genes identifies aberrant promoter methylation of NEIL1 in head and neck squamous cell carcinoma. Oncogene. 2012;31(49):5108–16.
Do H, Wong NC, Murone C, John T, Solomon B, Mitchell PL, et al. A critical re-assessment of DNA repair gene promoter methylation in non-small cell lung carcinoma. Sci Rep. 2014;4:4186.
Fleischer T, Edvardsen H, Solvang HK, Daviaud C, Naume B, Borresen-Dale AL, et al. Integrated analysis of high-resolution DNA methylation profiles, gene expression, germline genotypes and clinical end points in breast cancer patients. Int J Cancer. 2014;134(11):2615–25.
Yang J, Xu Z, Li J, Zhang R, Zhang G, Ji H, et al. XPC epigenetic silence coupled with p53 alteration has a significant impact on bladder cancer outcome. J Urol. 2010;184(1):336–43.
Sabatino MA, Marabese M, Ganzinelli M, Caiola E, Geroni C, Broggini M. Down-regulation of the nucleotide excision repair gene XPG as a new mechanism of drug resistance in human and murine cancer cells. Mol Cancer. 2010;9:259.
Li Y, Yang Y, Lu Y, Herman JG, Brock MV, Zhao P, et al. Predictive value of CHFR and MLH1 methylation in human gastric cancer. Gastric Cancer. 2015;18(2):280–7.
V S, Bhagat R, C SP, V RP, Krishnamoorthy L. Microsatellite instability, promoter methylation and protein expression of the DNA mismatch repair genes in epithelial ovarian cancer. Genomics. 2014;104(4):257–63.
House MG, Herman JG, Guo MZ, Hooker CM, Schulick RD, Cameron JL, et al. Prognostic value of hMLH1 methylation and microsatellite instability in pancreatic endocrine neoplasms. Surgery. 2003;134(6):902–8 discussion 9.
Gonzalez-Ramirez I, Ramirez-Amador V, Irigoyen-Camacho ME, Sanchez-Perez Y, Anaya-Saavedra G, Granados-Garcia M, et al. hMLH1 promoter methylation is an early event in oral cancer. Oral Oncol. 2011;47(1):22–6.
Wang YC, Lu YP, Tseng RC, Lin RK, Chang JW, Chen JT, et al. Inactivation of hMLH1 and hMSH2 by promoter methylation in primary non-small cell lung tumors and matched sputum samples. J Clin Invest. 2003;111(6):887–95.
Matsushita M, Takeuchi S, Yang Y, Yoshino N, Tsukasaki K, Taguchi H, et al. Methylation of the MLH1 gene in hematological malignancies. Oncol Rep. 2005;14(1):191–4.
Toyota M, Suzuki H. Epigenetic drivers of genetic alterations. Adv Genet. 2010;70:309–23.
He T, Zhang M, Zheng R, Zheng S, Linghu E, Herman JG, et al. Methylation of SLFN11 is a marker of poor prognosis and cisplatin resistance in colorectal cancer. Epigenomics. 2017;9(6):849–62.
Guo M, Alumkal J, Drachova T, Gao D, Marina SS, Jen J, et al. CHFR methylation strongly correlates with methylation of DNA damage repair and apoptotic pathway genes in non-small cell lung cancer. Discov Med. 2015;19(104):151–8.
Herman JG, Umar A, Polyak K, Graff JR, Ahuja N, Issa JP, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci U S A. 1998;95(12):6870–5.
Simpkins SB, Bocker T, Swisher EM, Mutch DG, Gersell DJ, Kovatich AJ, et al. MLH1 promoter methylation and gene silencing is the primary cause of microsatellite instability in sporadic endometrial cancers. Hum Mol Genet. 1999;8(4):661–6.
Esteller M, Toyota M, Sanchez-Cespedes M, Capella G, Peinado MA, Watkins DN, et al. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is associated with G to A mutations in K-ras in colorectal tumorigenesis. Cancer Res. 2000;60(9):2368–71.
Guo M, House MG, Hooker C, Han Y, Heath E, Gabrielson E, et al. Promoter hypermethylation of resected bronchial margins: a field defect of changes? Clin Cancer Res. 2004;10(15):5131–6.
Leedham SJ, Graham TA, Oukrif D, McDonald SA, Rodriguez-Justo M, Harrison RF, et al. Clonality, founder mutations, and field cancerization in human ulcerative colitis-associated neoplasia. Gastroenterology. 2009;136(2):542–50.e6.
Braakhuis BJ, Tabor MP, Kummer JA, Leemans CR, Brakenhoff RH. A genetic explanation of Slaughter’s concept of field cancerization: evidence and clinical implications. Cancer Res. 2003;63(8):1727–30.
Shen L, Kondo Y, Rosner GL, Xiao L, Hernandez NS, Vilaythong J, et al. MGMT promoter methylation and field defect in sporadic colorectal cancer. J Natl Cancer Inst. 2005;97(18):1330–8.
Sigalotti L, Fratta E, Coral S, Tanzarella S, Danielli R, Colizzi F, et al. Intratumor heterogeneity of cancer/testis antigens expression in human cutaneous melanoma is methylation-regulated and functionally reverted by 5-aza-2′-deoxycytidine. Cancer Res. 2004;64(24):9167–71.
Rastetter M, Schagdarsurengin U, Lahtz C, Fiedler E, Marsch W, Dammann R, et al. Frequent intra-tumoural heterogeneity of promoter hypermethylation in malignant melanoma. Histol Histopathol. 2007;22(9):1005–15.
Varley KE, Mutch DG, Edmonston TB, Goodfellow PJ, Mitra RD. Intra-tumor heterogeneity of MLH1 promoter methylation revealed by deep single molecule bisulfite sequencing. Nucleic Acids Res. 2009;37(14):4603–12.
Oakes CC, Claus R, Gu L, Assenov Y, Hullein J, Zucknick M, et al. Evolution of DNA methylation is linked to genetic aberrations in chronic lymphocytic leukemia. Cancer Discov. 2014;4(3):348–61.
Pan H, Jiang Y, Boi M, Tabbo F, Redmond D, Nie K, et al. Epigenomic evolution in diffuse large B-cell lymphomas. Nat Commun. 2015;6:6921.
Aryee MJ, Liu W, Engelmann JC, Nuhn P, Gurel M, Haffner MC, et al. DNA methylation alterations exhibit intraindividual stability and interindividual heterogeneity in prostate cancer metastases. Sci Transl Med. 2013;5(169):169ra10.
Dietz S, Lifshitz A, Kazdal D, Harms A, Endris V, Winter H, et al. Global DNA methylation reflects spatial heterogeneity and molecular evolution of lung adenocarcinomas. Int J Cancer. 2019;144(5):1061–72.
Brocks D, Assenov Y, Minner S, Bogatyrova O, Simon R, Koop C, et al. Intratumor DNA methylation heterogeneity reflects clonal evolution in aggressive prostate cancer. Cell Rep. 2014;8(3):798–806.
Liu W, Laitinen S, Khan S, Vihinen M, Kowalski J, Yu G, et al. Copy number analysis indicates monoclonal origin of lethal metastatic prostate cancer. Nat Med. 2009;15(5):559–65.
Drake JM, Graham NA, Lee JK, Stoyanova T, Faltermeier CM, Sud S, et al. Metastatic castration-resistant prostate cancer reveals intrapatient similarity and interpatient heterogeneity of therapeutic kinase targets. Proc Natl Acad Sci U S A. 2013;110(49):E4762–9.
Liu Y, Huang R, Liu Y, Song W, Wang Y, Yang Y, et al. Insights from multidimensional analyses of the pan-cancer DNA methylome heterogeneity and the uncanonical CpG-gene associations. Int J Cancer. 2018;143(11):2814–27.
Linker SM, Urban L, Clark SJ, Chhatriwala M, Amatya S, McCarthy DJ, et al. Combined single-cell profiling of expression and DNA methylation reveals splicing regulation and heterogeneity. Genome Biol. 2019;20(1):30.
Quek K, Li J, Estecio M, Zhang J, Fujimoto J, Roarty E, et al. DNA methylation intratumor heterogeneity in localized lung adenocarcinomas. Oncotarget. 2017;8(13):21994–2002.
Martinez-Cardus A, Moran S, Musulen E, Moutinho C, Manzano JL, Martinez-Balibrea E, et al. Epigenetic homogeneity within colorectal tumors predicts shorter relapse-free and overall survival times for patients with locoregional cancer. Gastroenterology. 2016;151(5):961–72.
Klughammer J, Kiesel B, Roetzer T, Fortelny N, Nemc A, Nenning KH, et al. The DNA methylation landscape of glioblastoma disease progression shows extensive heterogeneity in time and space. Nat Med. 2018;24(10):1611–24.
Chen X, Litzenburger UM, Wei Y, Schep AN, LaGory EL, Choudhry H, et al. Joint single-cell DNA accessibility and protein epitope profiling reveals environmental regulation of epigenomic heterogeneity. Nat Commun. 2018;9(1):4590.
Tanas AS, Sigin VO, Kalinkin AI, Litviakov NV, Slonimskaya EM, Ibragimova MK, et al. Genome-wide methylotyping resolves breast cancer epigenetic heterogeneity and suggests novel therapeutic perspectives. Epigenomics. 2019;11(6):605–17.
Grasse S, Lienhard M, Frese S, Kerick M, Steinbach A, Grimm C, et al. Epigenomic profiling of non-small cell lung cancer xenografts uncover LRP12 DNA methylation as predictive biomarker for carboplatin resistance. Genome Med. 2018;10(1):55.
de Matos MR, Posa I, Carvalho FS, Morais VA, Grosso AR, de Almeida SF. A systematic pan-cancer analysis of genetic heterogeneity reveals associations with epigenetic modifiers. Cancers. 2019;11(3). https://doi.org/10.3390/cancers11030391.
Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14(10):611–29.
Hinohara K, Wu HJ, Sebastien V, McDonald TO, Igarashi KJ, Yamamoto KN, et al. KDM5 histone demethylase activity links cellular transcriptomic heterogeneity to therapeutic resistance. Cancer Cell. 2019;35(2):330–2.
Pastore A, Gaiti F, Lu SX, Brand RM, Kulm S, Chaligne R, et al. Corrupted coordination of epigenetic modifications leads to diverging chromatin states and transcriptional heterogeneity in CLL. Nat Commun. 2019;10(1):1874.
Chatterjee A, Stockwell PA, Ahn A, Rodger EJ, Leichter AL, Eccles MR. Genome-wide methylation sequencing of paired primary and metastatic cell lines identifies common DNA methylation changes and a role for EBF3 as a candidate epigenetic driver of melanoma metastasis. Oncotarget. 2017;8(4):6085–101.
Teater M, Dominguez PM, Redmond D, Chen Z, Ennishi D, Scott DW, et al. AICDA drives epigenetic heterogeneity and accelerates germinal center-derived lymphomagenesis. Nat Commun. 2018;9(1):222.
Reitz C. Toward precision medicine in Alzheimer’s disease. Ann Transl Med. 2016;4(6):107.
Patel A, Tripathi G, Gopalakrishnan K, Williams N, Arasaradnam RP. Field cancerisation in colorectal cancer: a new frontier or pastures past? World J Gastroenterol. 2015;21(13):3763–72.
Rivenbark AG, Coleman WB. Field cancerization in mammary carcinogenesis - implications for prevention and treatment of breast cancer. Exp Mol Pathol. 2012;93(3):391–8.
Terry S, Buart S, Chouaib S. Hypoxic stress-induced tumor and immune plasticity, suppression, and impact on tumor heterogeneity. Front Immunol. 2017;8:1625.
Dawson MA. The cancer epigenome: concepts, challenges, and therapeutic opportunities. Science. 2017;355(6330):1147–52.
Stein EM, Garcia-Manero G, Rizzieri DA, Tibes R, Berdeja JG, Savona MR, et al. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood. 2018;131(24):2661–9.
Plch J, Hrabeta J, Eckschlager T. KDM5 demethylases and their role in cancer cell chemoresistance. Int J Cancer. 2019;144(2):221–31.
Pribluda A, de la Cruz CC, Jackson EL. Intratumoral heterogeneity: from diversity comes resistance. Clin Cancer Res. 2015;21(13):2916–23.
Issa ME, Takhsha FS, Chirumamilla CS, Perez-Novo C, Vanden Berghe W, Cuendet M. Epigenetic strategies to reverse drug resistance in heterogeneous multiple myeloma. Clin Epigenetics. 2017;9:17.
Juergens RA, Wrangle J, Vendetti FP, Murphy SC, Zhao M, Coleman B, et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov. 2011;1(7):598–607.
Pisanic TR 2nd, Athamanolap P, Wang TH. Defining, distinguishing and detecting the contribution of heterogeneous methylation to cancer heterogeneity. Semin Cell Dev Biol. 2017;64:5–17.
Flavahan WA, Gaskell E, Bernstein BE. Epigenetic plasticity and the hallmarks of cancer. Science. 2017;357(6348). https://doi.org/10.1126/science.aal2380.
Acknowledgements
Not applicable.
Funding
This work was supported by grants from National Key Research and Development Programme of China (2018YFA0208902); National Science Foundation of China (NSFC No. U1604281, 81672138); National Basic Research Program of China (973 Program No. 2012CB934002, 863 Program No. 2012AA02A203, 2012AA02A209); National Key Scientific Instrument Special Programme of China (Grant No. 2011YQ03013405); Beijing Science Foundation of China (BJSFC No. 7171008).
Author information
Authors and Affiliations
Contributions
MG, YP, AG and CD drafted the manuscript. JGH discussed and revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Guo, M., Peng, Y., Gao, A. et al. Epigenetic heterogeneity in cancer. Biomark Res 7, 23 (2019). https://doi.org/10.1186/s40364-019-0174-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-019-0174-y