
Abstract
Alzheimer’s disease (AD) is a globally common neurodegenerative disease, which is accompanied by alterations to various lifestyle patterns, such as sleep disturbance. The pineal gland is the primary endocrine organ that secretes hormones, such as melatonin, and controls the circadian rhythms. The decrease in pineal gland volume and pineal calcification leads to the reduction of melatonin production. Melatonin has been reported to have multiple roles in the central nervous system (CNS), including improving neurogenesis and synaptic plasticity, suppressing neuroinflammation, enhancing memory function, and protecting against oxidative stress. Recently, reduced pineal gland volume and pineal calcification, accompanied by cognitive decline and sleep disturbances have been observed in AD patients. Here, I review current significant evidence of the contribution of pineal dysfunction in AD to the progress of AD neuropathology. I suggest new insights to understanding the relationship between AD pathogenesis and pineal gland function.
Similar content being viewed by others


Background
Alzheimer’s disease (AD) is the most common neurodegenerative disease and is characterized by progressive loss of memory function and other neurobehavioral manifestations [1, 2]. The pathological hallmarks of AD have been reported, including extracellular senile plaques, which are mainly composed of β-amyloid (Aβ) and intracellular neurofibrillary tangles (NFTs) [1]. Current research reported that at present there are more than 47 million AD patients globally, and that this number will be projected to triple to nearly 150 million by 2050 [3]. The development of AD is accompanied by changes in lifestyle factors, such as sleep disturbance [3]. Unlike other neurodegenerative diseases, AD patients show sleep disturbances from an early phase [4]. Several studies suggest that the sleep disturbance in AD is an important diagnostic indicator for predicting AD progress [5, 6]. Due to this evidence, the relationship between pineal gland dysfunction and AD neuropathology is emerging as a new concept in understanding AD pathology, and suggests that circadian rhythms that control sleep disturbances are regulated by the pineal gland [7].
The pineal gland is a circumventricular organ that is derived from the embryonic forebrain, and it is the major part of the epithalamus, along with the habenular nuclei [7]. The pineal gland has been reported to secrete melatonin and directly control circadian rhythms in humans [8]. Melatonin is the main hormone produced by pineal gland, and is known to involved in antioxidant defense, immune responses, neuroprotective effects, anti-amyloid effects, and anti-apoptotic activity [9, 10]. Recently, studies have found lower levels of melatonin in AD, and noted that the decreased melatonin secretion triggers cognitive impairment [8, 11, 12]. The secretory capacity of the pineal gland is directly proportional to the pineal parenchymal volume and pineal gland function [13, 14]. Pineal calcification, also referred to as “brain sand”, is caused by hydroxyapatite deposition in the pineal gland [15, 16]. Certain studies have reported the reduced pineal volume and have found calcification in AD [17, 18]. Even though these relationship between pineal gland function and AD neuropathology were markedly found through various researches, the importance about that has not been highlighted until recent years.
Here, I review the recent evidences that how pineal dysfunction, by pineal volume reduction and pineal calcification, is involved in the AD pathogenesis.
Pineal gland dysfunction in Alzheimer’s disease
Pineal gland is an endocrine organ localized in the human brain, and is present in a variety of weights and sizes among individuals [19]. Several studies demonstrated that the morphology and function of pineal gland are influenced by various physiological conditions [20]. Generally, the pineal gland has been reported to synthesize and secrete melatonin as a neuroendocrine hormone, which can regulate circadian rhythms in humans [21, 22]. In order to produce melatonin, the transcription of aralkylamine N-acetyltransferase (Aanat) and phosphorylation of AANAT are controlled on a daily basis by the pineal gland, and its activity is modulated by photoperiod seasonal change [23]. Furthermore, the phosphorylation of AANAT by protein kinase A (PKA) is mediated by the stimulation of pinealocytes, and ultimately, contributes to the production of melatonin [24]. The pineal gland consists primarily of pinealocytes, a few microglia, and astrocytes [25]. A portion of the pineal gland is exposed into the cerebrospinal fluid (CSF) of the third ventricle [26]. A large number of canaliculi of the pineal gland open directly into the CSF of the third ventricle, resulting in high melatonin level in the CSF of the third ventricle [27, 28].
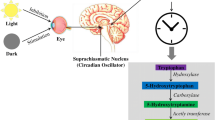
Melatonin production is directly controlled by the internal circadian timer, which is located in the suprachiasmatic nucleus (SCN) [29] and also known as the “pacemaker” [30]. Membrane receptors of melatonin have been identified in the SCN [31, 32], and the signal transduction pathways through melatonin receptor 1 and 2 (MT1 and MT2) increase the expression of clock genes, including Period circadian regulator 1 (Per1) [33, 34]. Therefore, the melatonin action through melatonin receptors contributes to the circadian rhythms. The volume of the pineal gland is correlated with the function of the pineal gland, because pinealocytes which produce melatonin mainly compose pineal gland. One study demonstrated that the volume of the pineal gland is also significantly reduced in patients with insomnia, and noted that lower pineal gland volumes contribute to sleep disorders [18].
In AD patients, the level of melatonin in CSF and blood serum was decreased compared to normal subjects and decreased level of melatonin finally leads to the aberrant diurnal rhythm [35,36,37]. Some studies demonstrated that reduced level of melatonin in AD brain contributes to the cognitive decline in AD patients and also is linked to the pineal gland volume [35, 37, 38]. Furthermore, the expression of melatonin receptor such as MT2 was decreased in the hippocampus of AD patients [39, 40]. The level of melatonin was observed the reduction of it in compared with normal subjects and also the aberrant size of pineal gland was found in AD patients. As mentioned, the reduction of melatonin level is important feature in AD patients. Some actions of melatonin in AD have been reported by various researchers. In AD, melatonin could efficiently inhibit tau hyperphosphorylation [41] and attenuate levels of secreted soluble amyloid beta precursor protein (APP) from neurons [42]. Melatonin administration attenuated Aβ generation and deposition in AD mice [43, 44]. Moreover, melatonin suppressed the peroxynitrite-induced inhibition of choline transport in neuronal proteins from synaptosomes and synaptic vesicles [45]. In AD, melatonin attenuated the accumulation of Aβ plaques that trigger pro-inflammatory responses and oxidative stress in the brain, causing cognitive impairment [46, 47]. Therefore, melatonin action may be critical to improve neuropathogenesis in AD based on upper findings.
Even though melatonin is secreted in pineal glands, the amount of melatonin synthesized by extrapineal organs was greater than the amount of melatonin secreted by the pineal gland [48]. However, the melatonin synthesized by extrapineal organs could not replace the function of melatonin produced by pineal gland, such as the regulation of circadian rhythms [49], the neuroprotection, and anti-inflammatory responses [50, 51]. Therefore, the melatonin produced by the pineal gland is important and irreplaceable in suppressing neuropathogenesis in AD brains. Regarding previous consequences, the dysfunction of pineal gland on neuropathology is an important issue to be investigated in AD brain.
Pineal calcification is calcium deposition in pineal gland, which has long been reported in humans [52, 53]. The occurrence of pineal calcification depends on environmental factors, such as sunlight exposure [54], and results in the decrease of melatonin production [55, 56]. Pineal calcification occurs when calcareous deposits form within the connective tissue of the pineal gland stroma, and it is similar to calcification observed in the habenular commissure and choroid plexus [57]. Unlike kidney stones, the main component of pineal calcification is hydroxyapatite [Ca10(PO4)6(OH)2], and the Ca/P molar ratio in pineal calcification is similar to that found in the enamel and dentine of teeth [58]. Morphological changes associated with pineal calcification include changes in the production of melatonin, due to the decreased function in the pineal gland parenchyma, and results in decreased pineal volume, reduced melatonin production in humans [14], and altered sleep patterns [59]. A few studies reported that pineal calcification [13, 14] and pineal cysts [60] trigger severe sleep disorders by disturbing melatonin secretion in the pineal gland. In clinical studies, patients with primary insomnia showed reduced plasma melatonin levels during the daytime [61].
Considering some studies, pineal calcification contributes to the reduction of melatonin production in humans that is directly associated with the development of neurodegenerative diseases, such as AD [54, 56, 62]. Previous researches demonstrated that the reduction of melatonin levels in CSF and serum leads to the aggravation of AD neuropathology [37, 38, 63]. In AD, reduced pineal size, pineal gland dysfunction, and pineal calcification have been reported [38], and decreased melatonin levels have been detected in serum [64] and urine [65]. A recent computed tomography study clearly observed pineal calcification in AD patients [56].
Regarding these observations, pineal dysfunction reduces melatonin production, and, ultimately, contributes to diverse AD neuropathologies (Fig. 1a). However, the detailed mechanisms on pineal gland calcification and pineal gland dysfunction in AD are not fully understood yet. The mechanism related with pineal gland dysfunction in AD should be investigated to find therapeutic solution to cure AD pathologies, given that the pineal gland dysfunction is strongly linked to the AD pathologies.

The schematic diagram of pineal gland dysfunction in AD, immune-pineal axis, and the role of melatonin in neurogenesis. a Pineal calcification and reduced pineal volume causes pineal dysfunction, which are commonly observed in AD brain. The pineal dysfunction leads to the reduction of melatonin level, subsequently results in sleep deficit. b Immune cells could regulate NF-κB activation and promote the production of melatonin in pinealocytes through CREB-AANAT signaling. c The decreased melatonin level leads to the impairment of neurogenesis in AD, because reduced melatonin level contributes the reduction of BDNF and GDNF expression, which are known as boosters in neurogenesis
Impaired immune-pineal axis by pineal gland dysfunction and AD
The pineal gland, as part of the circumventricular organ, interacts with various molecules present in the blood and CSF [66]. Inflammatory mediators, such as cytokines, regulate the function of the pineal gland, leading to the suppression (i.e., secretion of proinflammatory cytokines) or potentiation (i.e., secretion of glucocorticoids) of melatonin synthesis [67]. Melatonin, derived from serotonin (5-HT), is synthesized in a rhythmic manner by the pineal gland [67]. Melatonin acts as an antioxidant to protect cells, and it exerts chronobiotic functions [22]. Moreover, melatonin is an important free radical scavenger that suppresses deleterious oxidant activities and contributes to the control of the redox state of cells [68].
Induced extrapineal synthesis of melatonin does not contribute to pineal diurnal rhythms [67]. Extrapineal synthesis of melatonin is responsible for the circulation of the endocrine and paracrine-produced melatonin [67]. Several neurotransmitters, including glutamate, acetylcholine, vasoactive intestinal peptide, substance P, and pituitary adenylate cyclase-activating peptide are associated with direct central modulation of the melatonin synthesis process [21]. Melatonin synthesized in the gastrointestinal tract in a nonrhythmic manner has a protective role in the gastric mucosa against stress conditions [69].
With regard to one research, the regulatory role of melatonin contributes the immune defense response [70]. In addition, the synthesis of melatonin by extrapineal tissues has been clearly shown to be related to defense responses, such as activation of polymorphonuclear and mononuclear cells in the blood [71], peritoneum [72], and colostrum [73]. The immune-competent cells, such as phagocytes, are activated to produce melatonin upon injury stimuli [74, 75]. Activated mononuclear and polymorphonuclear cells synthesize melatonin, and subsequently contribute to the recovery process by suppressing oxidative stress and by boosting macrophage phagocytic activity [73]. In summary, immune cells could produce melatonin in blood and this melatonin could regulate immune responses against stress condition.
Innate immune responses require the recruitment of leukocytes to the site of the lesion. Moreover, the maintenance of circulating leukocytes and the termination of migration during the resolution of the immune response is an important process [76]. Melatonin contributes to the regulation of leukocyte migration to injury sites [76, 77]. Therefore, melatonin is able to control immune responses by inhibiting the activation of inflammatory signaling and by regulating the proliferation and activation of immune competent cells [78, 79]. Recent study reported that melatonin synthesized by macrophages and microglia could suppress their immune activity and increase their phagocytic capacity, characteristics associated with an anti-inflammatory, the M2-like phenotype [80]. As mentioned, melatonin may be an important anti- inflammatory mediator in inflammatory responses and immune actions.
Furthermore, melatonin inhibits the activation of nuclear factor kappa B (NF-κB) as a key transcription factor that mediates the inflammatory response [81]. In the central nervous system (CNS), NF-κB is associated with both innate and acquired immune responses, and it is necessary for neuronal survival [82]. NF-κB modulates neurite outgrowth [83], determines cell fate [84], circuit formation, and brain tissue homeostasis [85]. In the pineal gland, NF-κB is translocated into the nucleus of cells during the daytime [86]. The nocturnal production of melatonin is regulated by several cytokines that interfere with the NF-κB pathway [67]. Melatonin suppresses NF-κB activation in macrophages [87], T cells [88], and neuronal cells [89].
Additionally, some studies have shown that melatonin plays an anti-inflammatory role that is mediated by the inhibition of NF-κB nuclear translocation [90, 91]. Melatonin boosts the phagocytic activity of mononuclear cells, and the expression of the interleukin 2 (IL-2) through MT1 and MT2 membrane receptors [92]. Considering one study, the promoter of the gene that codes for AANAT includes κB sequences [67]. A current study demonstrated that the immune-pineal axis orchestrates the timing of leukocyte migration and alters monocyte phenotype through the NF-κB pathway [93]. The mechanisms of melatonin synthesis in the rat pineal gland has been known to be related with AANAT activation by regulating pCREB [94, 95], and by activating NF-κB [96]. There are some researches proving that the NF-κB transcription factor could modulate a phosphorylation of CREB in cellular mechanism [97, 98]. According to these evidences, the activation of NF-kB in pinealocytes through immune cells at AD brain results in the activation of Aanat gene, leading to the production of melatonin (Fig. 1b).
Melatonin reduces the rolling and adherence of leukocytes and neutrophils to the endothelial layer, decreasing vascular permeability [77, 99]. Melatonin is known to inhibit endothelial nitric oxide synthase activation in blood vessels and, subsequently, reduces vascular inflammation [100]. The adhesiveness of neutrophils to endothelial cells inversely correlates with the melatonin concentration in blood [101]. Endothelial cells decreased the expression of adhesion molecules, including platelet endothelial cell adhesion molecular-1 (PECAM-1) and intercellular cell adhesion molecular-1 (ICAM-1), when melatonin levels were increased at night [101]. One study demonstrated that the reduced expression of pro-inflammatory proteins and the increase in anti-inflammatory proteins, such as CD180, in endothelial cells was caused by the increased melatonin production at night [102]. In terms of chronobiotic pattern, melatonin also has chrobiotic antioxidant and anti-apoptotic properties [103]. Therefore, melatonin also could control the vascular homeostasis related with immune responses.
The melatonin synthesized by both pineal gland and extrapineal glands coordinate with each other to regulate immune molecules, including pathogen-associated molecular patterns (PAMPs), danger-associated molecular patterns (DAMPs), toxic Aβ, heat-shock proteins, and tissue debris [67]. Several studies described this bidirectional communication process between the pineal gland and the immune response as “the immune-pineal axis” [67, 104, 105].
In AD, pineal dysfunction leads to decreased melatonin production [37, 38], suggesting that melatonin production is positively linked to promoting neuroprotection [106, 107]. A current study reported that Aβ was observed to interact with toll like receptor (TLRs) in the pineal gland of AD patients, and that the interaction subsequently triggers the synthesis of pro-inflammatory cytokines, and inhibits the expression of Aanat and synthesis of melatonin through the NF-κB pathway [108]. The increase of the pro-inflammatory cytokine tumor necrosis factor (TNF), caused by reduced melatonin production is considered as biomarker of AD progression [109]. In AD, pineal gland calcification leads to reduced total melatonin excretion [54], and the resulting melatonin deficit aggravates the progress of AD [110].
Ultimately, the production of melatonin in immune cells is organically linked to neuropathogenesis organically the melatonin secreted from pineal gland. To sum up, pineal gland dysfunction triggers the reduction of melatonin, and leads to the aggravation of inflammation, the abnormal immune response, and the impairment of vascular homeostasis, involving in the neuropathology in AD.
Sleep disturbance by pineal gland dysfunction and AD
The effect of sleep in human has been reported to be beneficial in many aspects, such as tissue repair, improvement of memory consolidation, and the preservation of neuroimmune-endocrine integrity [111, 112]. Sleep is a vital phenomenon that is generally divided into two phases, sleep with rapid eye movements (REM) and sleep without rapid eye movements (non-REM) [113]. REM sleep is known to be important in memory function, neurogenesis, and the regulation of blood-brain barrier homeostasis [114], whereas non-REM sleep is associated with the release of diverse hormones and is characterized by decreased blood pressure [115].
Sleep disorders occur in 25–66% of AD patients [116]. Current studies showed that sleep disturbance leads to cognitive decline [117, 118], and increases the risk of AD by increasing Aβ burden. [119, 120]. Previous researches demonstrated that the increase of inflammation in the brain with chronic sleep deprivation could boost the risk of neurodegenerative disease onset [121, 122]. Other studies demonstrated that aggravated inflammation caused by sleep disturbance triggers cognitive decline and promotes the onset of AD [123, 124]. Further, sleep quality in AD patients worsens with AD progression [125]. The association between cognitive decline and impaired sleep quality has been reported in AD models with increased Aβ deposition [126, 127]. One brain positron-emission tomography (PET) study mentioned that sleep impairment was related with increased Aβ load in healthy subjects [128]. Furthermore, several studies found that lower sleep quality was associated with an increased brain Aβ load in normal brains [129, 130].
Several studies suggested that sleep dysfunction worsens AD pathology and increases the risk of developing dementia [119, 131]. Furthermore, a recent study demonstrated that reduced glucose uptake in the hypothalamus leads to sleep impairment and can be used as a CSF biomarker of AD [132]. Another study reported that transgenic amyloid precursor protein/presenilin 1 (APP/PS1) mouse model of AD showed significant hypothalamic abnormalities prior to memory loss [133]. In AD, sleep disturbance is linked to physiological changes in the suprachiasmatic nucleus (SCN) and pineal gland [12]. Considering previous reports, the sleep impairment is commonly observed in AD patients and is regarded as the strong booster related with the aggravation of AD pathologies.
Recently, reduced melatonin due to lower pineal gland volume has been observed in AD brains, establishing the relationship between lower pineal gland volume and cognitive impairment in AD patients [134]. Several studies suggested that reduced pineal volume results in insomnia, and is significantly associated with sleep disturbances in AD [18, 134, 135]. Therefore, the pineal gland dysfunction by lower pineal gland volume contributes directly to the sleep deficit in AD patients. Taken together, the decrease in melatonin secretion due to pineal gland dysfunction triggers insomnia, sleep disturbance, and poor sleep quality, and ultimately, results in memory loss in AD.
Impaired neurogenesis by pineal gland dysfunction and AD
Neural plasticity is an important feature of brain function, because continuous adaptation in changing conditions is essential to preserve homeostasis [136]. Neurogenesis is a major component of plasticity in response to CNS damage and occurs at sites of brain injury [136]. Neural stem cells (NSCs) are multipotent and are present in the sub-ventricular zone (SVZ) of the forebrain and the subgranular zone (SGZ) of the hippocampus [137]. Neurogenesis in the hippocampus is important for the maintenance and recovery of cognitive function [138], suggesting that the circuitry of the hippocampal dendrite gyrus outputs to the dorsal CA3, is associated with the encoding of time in new memories [139].
The latest study reported that melatonin contributes to structural plasticity in axons of the hippocampal dentate gyrus [140]. Judging from some studies, melatonin induces the proliferation and survival of NSCs in the midbrain and the hippocampus [141, 142]. A couple of studies also reported that melatonin promotes neurogenesis in the hippocampus of C57BL/6 mice [143, 144]. It was also assumed that the melatonin effect on NSCs was mediated by neurotrophic factors, such as brain-derived neurotrophic factor (BDNF) and glial cell-derived neurotrophic factor (GDNF) [145]. In brief, melatonin has a cardinal effect in the improvement of neurogenesis and synaptic plasticity (Fig. 1c).
Recent research demonstrated that the thalamus, including the pineal gland, has emerged as a host site for such a neurogenic niche [146, 147]. A number of studies report that the hypothalamus has the ability for neurogenesis [148, 149]. Latest studies suggest that NSC proliferation and neurogenesis in the thalamus are increased when the day length decreases [150, 151]. It was found that hypothalamic neurogenesis is more activated depending on the day length [147], and neurogenesis is related to daylight and melatonin secretion [151]. NSC proliferation in the SVZ was independent of seasons and was shown to be influenced by reduced melatonin secretion through pinealectomy in animal models of AD [152].
In AD brains, neurogenesis in hippocampal areas is attenuated compared to the normal brains [153]. Decreased neurogenesis and neuronal loss was observed in the hippocampus and cortex areas of AD mice [154, 155], and the positive correlation between memory loss and level of neurogenesis in AD has been reported [155, 156]. Additionally, the impaired neurogenesis in the dentate gyrus of AD mice has been reported by decreased numbers of doublecortin, a new neuronal marker, positive cells [155, 157]. Neurogenesis is impaired by reduced melatonin secretion in AD. Therefore, the pineal gland dysfunction is related with the impaired neurogenesis, leading to memory loss in AD brain.
Ultimately, pineal gland dysfunction, which reduces melatonin secretion, may be one of the crucial factors that causes impaired neurogenesis in AD. Finding the way to improve pineal dysfunction may be a key to circumvent impaired neurogenesis in AD brain.
Conclusion
The progression and onset of AD is complicated, as the disease is related with other organs and is affected by various factors. Here, I have summarized the recent findings of pineal dysfunction in AD pathogenesis.
I highlight three points in this review (Fig. 1). First, pineal dysfunction involving reduced pineal volume and pineal calcification in AD contributes to the impaired immune-pineal axis. Melatonin secreted from the pineal gland and extrapineal organs are important mediators of communication between pineal gland and the immune system in AD. Second, pineal dysfunction in AD results in reduced melatonin production, which finally triggers sleep disturbance and poor sleep quality in AD. Third, pineal dysfunction in AD is a critical factor in the inhibition of neurogenesis in AD brains. Pineal dysfunction leads to the reduced hippocampal neurogenesis and hypothalamic neurogenesis. Finally, that these alterations due to pineal dysfunction are linked to memory loss in AD.
Recently, there are some interesting approaches to overcome pineal gland dysfunction. Epigenetic trial may be a solution to solve the pineal dysfunction in AD, suggesting from recent study that the function of pineal gland could be epigenetically regulated by valproic acid (VPA) that up-regulates the expression of melatonin receptor in brain [158]. Moreover, the regulation of noncoding RNAs such as miR-325-3p may be a solution to control the secretion of melatonin by modulating the expression of Aanat genes in pineal gland, suggesting that the interaction between miR-325-3p and the 3’UTR of Aanat mRNA 3′-UTR control the activation of Aanat gene expression in pinealocytes [159].
Based on the significant consequences stated above, I highlight the need for the further study of pineal gland function in AD and to find more effective solution to overcome pineal gland dysfunction in AD with various points. Hence, I assume that this review may provide new concepts in understanding the importance of pineal gland function on diverse AD neuropathology.
Availability of supporting data
Not applicable.
Abbreviations
- AD:
-
Alzheimer’s disease
- APP/PS1:
-
Amyloid precursor protein/presenilin 1
- Aβ:
-
β-amyloid
- BDNF:
-
Brain-derived neurotrophic factor
- CNS:
-
Central nervous system
- CSF:
-
Cerebrospinal fluid
- DAMPs:
-
Danger-associated molecular patterns
- GDNF:
-
Glial cell-derived neurotrophic factor
- ICAM-1:
-
Intercellular cell adhesion molecular-1
- IL-2:
-
Interleukin 2
- MT1:
-
Melatonin receptor 1
- MT2:
-
Melatonin receptor 2
- NFTs:
-
Intracellular neurofibrillary tangles
- NF-κB:
-
Nuclear factor kappa B
- NSCs:
-
Neural stem cells
- PAMPs:
-
Pathogen-associated molecular patterns
- PECAM-1:
-
Platelet endothelial cell adhesion molecular-1
- REM:
-
Rapid eye movements
- SCN:
-
Suprachiasmatic nucleus
- SGZ:
-
Subgranular zone
- SVZ:
-
Sub-ventricular zone
- TLRs:
-
Toll like receptor
References
Jack CR Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 2010;9:119–28.
Bhat SA, Kamal MA, Yarla NS, Ashraf GM. Synopsis on Managment strategies for neurodegenerative disorders: challenges from bench to bedside in successful drug discovery and development. Curr Top Med Chem. 2017;17:1371–8.
Livingston G, Sommerlad A, Orgeta V, Costafreda SG, Huntley J, Ames D, Ballard C, Banerjee S, Burns A, Cohen-Mansfield J, et al. Dementia prevention, intervention, and care. Lancet. 2017;390:2673–734.
dos Santos AB, Kohlmeier KA, Barreto GE. Are sleep disturbances preclinical markers of Parkinson's disease? Neurochem Res. 2015;40:421–7.
Lim AS, Kowgier M, Yu L, Buchman AS, Bennett DA. Sleep fragmentation and the risk of incident Alzheimer's disease and cognitive decline in older persons. Sleep. 2013;36:1027–32.
Spalletta G, Long JD, Robinson RG, Trequattrini A, Pizzoli S, Caltagirone C, Orfei MD. Longitudinal neuropsychiatric predictors of death in Alzheimer's disease. J Alzheimer's Dis. 2015;48:627–36.
Ekstrom P, Meissl H. Evolution of photosensory pineal organs in new light: the fate of neuroendocrine photoreceptors. Philos Trans R Soc Lond Ser B Biol Sci. 2003;358:1679–700.
Srinivasan V, Kaur C, Pandi-Perumal S, Brown GM, Cardinali DP. Melatonin and its agonist ramelteon in Alzheimer's disease: possible therapeutic value. Int J Alzheimers Dis. 2010;2011:741974.
Rosales-Corral SA, Acuna-Castroviejo D, Coto-Montes A, Boga JA, Manchester LC, Fuentes-Broto L, Korkmaz A, Ma S, Tan DX, Reiter RJ. Alzheimer's disease: pathological mechanisms and the beneficial role of melatonin. J Pineal Res. 2012;52:167–202.
He H, Dong W, Huang F. Anti-amyloidogenic and anti-apoptotic role of melatonin in Alzheimer disease. Curr Neuropharmacol. 2010;8:211–7.
Skene DJ, Swaab DF. Melatonin rhythmicity: effect of age and Alzheimer's disease. Exp Gerontol. 2003;38:199–206.
Wu YH, Swaab DF. Disturbance and strategies for reactivation of the circadian rhythm system in aging and Alzheimer's disease. Sleep Med. 2007;8:623–36.
Mahlberg R, Kienast T, Hadel S, Heidenreich JO, Schmitz S, Kunz D. Degree of pineal calcification (DOC) is associated with polysomnographic sleep measures in primary insomnia patients. Sleep Med. 2009;10:439–45.
Nolte I, Lutkhoff AT, Stuck BA, Lemmer B, Schredl M, Findeisen P, Groden C. Pineal volume and circadian melatonin profile in healthy volunteers: an interdisciplinary approach. J Magn Reson Imaging. 2009;30:499–505.
Yalcin A, Ceylan M, OF B, Sonkaya AR, Yuce I. Age and gender related prevalence of intracranial calcifications in CT imaging; data from 12,000 healthy subjects. J Chem Neuroanat. 2016;78:20–4.
Admassie D, Mekonnen A. Incidence of normal pineal and chroids plexus calcification on brain CT (computerized tomography) at Tikur Anbessa teaching hospital Addis Ababa, Ethiopia. Ethiop Med J. 2009;47:55–60.
Bumb JM, Brockmann MA, Groden C, Nolte I. Microstructural analysis of pineal volume using trueFISP imaging. World J Radiol. 2013;5:166–72.
Bumb JM, Schilling C, Enning F, Haddad L, Paul F, Lederbogen F, Deuschle M, Schredl M, Nolte I. Pineal gland volume in primary insomnia and healthy controls: a magnetic resonance imaging study. J Sleep Res. 2014;23:274–80.
Tan DX, Manchester LC, Fuentes-Broto L, Paredes SD, Reiter RJ. Significance and application of melatonin in the regulation of brown adipose tissue metabolism: relation to human obesity. Obes Rev. 2011;12:167–88.
Grosshans M, Vollmert C, Vollstaedt-Klein S, Nolte I, Schwarz E, Wagner X, Leweke M, Mutschler J, Kiefer F, Bumb JM. The association of pineal gland volume and body mass in obese and normal weight individuals: a pilot study. Psychiatr Danub. 2016;28:220–4.
Simonneaux V, Ribelayga C. Generation of the melatonin endocrine message in mammals: a review of the complex regulation of melatonin synthesis by norepinephrine, peptides, and other pineal transmitters. Pharmacol Rev. 2003;55:325–95.
Tan DX, Hardeland R, Manchester LC, Paredes SD, Korkmaz A, Sainz RM, Mayo JC, Fuentes-Broto L, Reiter RJ. The changing biological roles of melatonin during evolution: from an antioxidant to signals of darkness, sexual selection and fitness. Biol Rev Camb Philos Soc. 2010;85:607–23.
Pawlak J, Golab M, Markowska M, Majewski P, Skwarlo-Sonta K. Photoperiod-related changes in hormonal and immune status of male Siberian hamsters, Phodopus sungorus. Comp Biochem Physiol A Mol Integr Physiol. 2009;152:299–303.
Mortani Barbosa EJ, Ferreira ZS, Markus RP. Purinergic and noradrenergic cotransmission in the rat pineal gland. Eur J Pharmacol. 2000;401:59–62.
Ibanez Rodriguez MP, Noctor SC, Munoz EM. Cellular basis of pineal gland development: emerging role of microglia as phenotype regulator. PLoS One. 2016;11:e0167063.
Welsh MG. CSF-contacting pinealocytes in the pineal recess of the Mongolian gerbil: a correlative scanning and transmission electron microscope study. Am J Anat. 1983;166:483–93.
Reiter RJ, Tan DX, Kim SJ, Cruz MH. Delivery of pineal melatonin to the brain and SCN: role of canaliculi, cerebrospinal fluid, tanycytes and Virchow-Robin perivascular spaces. Brain Struct Funct. 2014;219:1873–87.
Krstic RV. Scanning electron microscopic study of the freeze-fractured pineal body of the rat. Cell Tissue Res. 1979;201:129–35.
McArthur AJ, Gillette MU, Prosser RA. Melatonin directly resets the rat suprachiasmatic circadian clock in vitro. Brain Res. 1991;565:158–61.
Gillette MU, Tischkau SA. Suprachiasmatic nucleus: the brain's circadian clock. Recent Prog Horm Res. 1999;54:33–58 discussion 58-39.
Waly NE, Hallworth R. Circadian pattern of melatonin MT1 and MT2 receptor localization in the rat suprachiasmatic nucleus. J Circadian Rhythms. 2015;13:1.
McMahon DG, Iuvone PM, Tosini G. Circadian organization of the mammalian retina: from gene regulation to physiology and diseases. Prog Retin Eye Res. 2014;39:58–76.
Kandalepas PC, Mitchell JW, Gillette MU. Melatonin signal transduction pathways require E-box-mediated transcription of Per1 and Per2 to reset the SCN clock at dusk. PLoS One. 2016;11:e0157824.
Gauer F, Masson-Pevet M, Skene DJ, Vivien-Roels B, Pevet P. Daily rhythms of melatonin binding sites in the rat pars tuberalis and suprachiasmatic nuclei; evidence for a regulation of melatonin receptors by melatonin itself. Neuroendocrinology. 1993;57:120–6.
Wu YH, Swaab DF. The human pineal gland and melatonin in aging and Alzheimer's disease. J Pineal Res. 2005;38:145–52.
Ferrari E, Arcaini A, Gornati R, Pelanconi L, Cravello L, Fioravanti M, Solerte SB, Magri F. Pineal and pituitary-adrenocortical function in physiological aging and in senile dementia. Exp Gerontol. 2000;35:1239–50.
Zhou JN, Liu RY, Kamphorst W, Hofman MA, Swaab DF. Early neuropathological Alzheimer's changes in aged individuals are accompanied by decreased cerebrospinal fluid melatonin levels. J Pineal Res. 2003;35:125–30.
Wu YH, Feenstra MG, Zhou JN, Liu RY, Torano JS, Van Kan HJ, Fischer DF, Ravid R, Swaab DF. Molecular changes underlying reduced pineal melatonin levels in Alzheimer disease: alterations in preclinical and clinical stages. J Clin Endocrinol Metab. 2003;88:5898–906.
Savaskan E, Olivieri G, Meier F, Brydon L, Jockers R, Ravid R, Wirz-Justice A, Muller-Spahn F. Increased melatonin 1a-receptor immunoreactivity in the hippocampus of Alzheimer's disease patients. J Pineal Res. 2002;32:59–62.
Savaskan E, Ayoub MA, Ravid R, Angeloni D, Fraschini F, Meier F, Eckert A, Muller-Spahn F, Jockers R. Reduced hippocampal MT2 melatonin receptor expression in Alzheimer's disease. J Pineal Res. 2005;38:10–6.
Deng YQ, Xu GG, Duan P, Zhang Q, Wang JZ. Effects of melatonin on wortmannin-induced tau hyperphosphorylation. Acta Pharmacol Sin. 2005;26:519–26.
Lahiri DK. Melatonin affects the metabolism of the beta-amyloid precursor protein in different cell types. J Pineal Res. 1999;26:137–46.
Lahiri DK, Chen D, Ge YW, Bondy SC, Sharman EH. Dietary supplementation with melatonin reduces levels of amyloid beta-peptides in the murine cerebral cortex. J Pineal Res. 2004;36:224–31.
Wang XC, Zhang YC, Chatterjie N, Grundke-Iqbal I, Iqbal K, Wang JZ. Effect of melatonin and melatonylvalpromide on beta-amyloid and neurofilaments in N2a cells. Neurochem Res. 2008;33:1138–44.
Guermonprez L, Ducrocq C, Gaudry-Talarmain YM. Inhibition of acetylcholine synthesis and tyrosine nitration induced by peroxynitrite are differentially prevented by antioxidants. Mol Pharmacol. 2001;60:838–46.
Rosales-Corral S, Tan DX, Reiter RJ, Valdivia-Velazquez M, Martinez-Barboza G, Acosta-Martinez JP, Ortiz GG. Orally administered melatonin reduces oxidative stress and proinflammatory cytokines induced by amyloid-beta peptide in rat brain: a comparative, in vivo study versus vitamin C and E. J Pineal Res. 2003;35:80–4.
Chuang JI, Mohan N, Meltz ML, Reiter RJ. Effect of melatonin on NF-kappa-B DNA-binding activity in the rat spleen. Cell Biol Int. 1996;20:687–92.
Acuna-Castroviejo D, Escames G, Venegas C, Diaz-Casado ME, Lima-Cabello E, Lopez LC, Rosales-Corral S, Tan DX, Reiter RJ. Extrapineal melatonin: sources, regulation, and potential functions. Cell Mol Life Sci. 2014;71:2997–3025.
Cardinali DP, Furio AM, Reyes MP. Clinical perspectives for the use of melatonin as a chronobiotic and cytoprotective agent. Ann N Y Acad Sci. 2005;1057:327–36.
Berkiks I, Benmhammed H, Mesfioui A, Ouichou A, El Hasnaoui A, Mouden S, Touil T, Bahbiti Y, Nakache R, El Hessni A. Postnatal melatonin treatment protects against affective disorders induced by early-life immune stimulation by reducing the microglia cell activation and oxidative stress. Int J Neurosci. 2018;128:495–504.
Leeboonngam T, Pramong R, Sae-Ung K, Govitrapong P, Phansuwan-Pujito P. Neuroprotective effects of melatonin on amphetamine-induced dopaminergic fiber degeneration in the hippocampus of postnatal rats. J Pineal Res. 2018;64(3). https://doi.org/10.1111/jpi.12456.
Goree JA, Wallace KK Jr, Bean RL. The pineal tomogram. Visualization of the faintly calcified pineal gland. Am J Roentgenol Radium Ther Nucl Med. 1963;89:1209–11.
Bojkowski CJ, Arendt J. Factors influencing urinary 6-sulphatoxymelatonin, a major melatonin metabolite, in normal human subjects. Clin Endocrinol. 1990;33:435–44.
Turgut AT, Karakas HM, Ozsunar Y, Altin L, Ceken K, Alicioglu B, Sonmez I, Alparslan A, Yurumez B, Celik T, et al. Age-related changes in the incidence of pineal gland calcification in Turkey: a prospective multicenter CT study. Pathophysiology. 2008;15:41–8.
Kunz D, Schmitz S, Mahlberg R, Mohr A, Stoter C, Wolf KJ, Herrmann WM. A new concept for melatonin deficit: on pineal calcification and melatonin excretion. Neuropsychopharmacology. 1999;21:765–72.
Mahlberg R, Walther S, Kalus P, Bohner G, Haedel S, Reischies FM, Kuhl KP, Hellweg R, Kunz D. Pineal calcification in Alzheimer's disease: an in vivo study using computed tomography. Neurobiol Aging. 2008;29:203–9.
Macpherson P, Matheson MS. Comparison of calcification of pineal, habenular commissure and choroid plexus on plain films and computed tomography. Neuroradiology. 1979;18:67–72.
Kodaka T, Mori R, Debari K, Yamada M. Scanning electron microscopy and electron probe microanalysis studies of human pineal concretions. J Electron Microsc. 1994;43:307–17.
Kunz D, Bes F, Schlattmann P, Herrmann WM. On pineal calcification and its relation to subjective sleep perception: a hypothesis-driven pilot study. Psychiatry Res. 1998;82:187–91.
Bumb JM, Brockmann MA, Groden C, Al-Zghloul M, Nolte I. TrueFISP of the pediatric pineal gland: volumetric and microstructural analysis. Clin Neuroradiol. 2012;22:69–77.
Riemann D, Klein T, Rodenbeck A, Feige B, Horny A, Hummel R, Weske G, Al-Shajlawi A, Voderholzer U. Nocturnal cortisol and melatonin secretion in primary insomnia. Psychiatry Res. 2002;113:17–27.
Friedland RP, Luxenberg JS, Koss E. A quantitative study of intracranial calcification in dementia of the Alzheimer type. Int Psychogeriatr. 1990;2:36–43.
Liu RY, Zhou JN, van Heerikhuize J, Hofman MA, Swaab DF. Decreased melatonin levels in postmortem cerebrospinal fluid in relation to aging, Alzheimer's disease, and apolipoprotein E-epsilon4/4 genotype. J Clin Endocrinol Metab. 1999;84:323–7.
Ohashi Y, Okamoto N, Uchida K, Iyo M, Mori N, Morita Y. Daily rhythm of serum melatonin levels and effect of light exposure in patients with dementia of the Alzheimer's type. Biol Psychiatry. 1999;45:1646–52.
Luboshitzky R, Shen-Orr Z, Tzischichinsky O, Maldonado M, Herer P, Lavie P. Actigraphic sleep-wake patterns and urinary 6-sulfatoxymelatonin excretion in patients with Alzheimer's disease. Chronobiol Int. 2001;18:513–24.
Duvernoy HM, Risold PY. The circumventricular organs: an atlas of comparative anatomy and vascularization. Brain Res Rev. 2007;56:119–47.
Markus RP, Ferreira ZS, Fernandes PA, Cecon E. The immune-pineal axis: a shuttle between endocrine and paracrine melatonin sources. Neuroimmunomodulation. 2007;14:126–33.
Tan DX, Manchester LC, Terron MP, Flores LJ, Reiter RJ. One molecule, many derivatives: a never-ending interaction of melatonin with reactive oxygen and nitrogen species? J Pineal Res. 2007;42:28–42.
Bubenik GA. Gastrointestinal melatonin: localization, function, and clinical relevance. Dig Dis Sci. 2002;47:2336–48.
Guerrero JM, Reiter RJ. Melatonin-immune system relationships. Curr Top Med Chem. 2002;2:167–79.
Carrillo-Vico A, Calvo JR, Abreu P, Lardone PJ, Garcia-Maurino S, Reiter RJ, Guerrero JM. Evidence of melatonin synthesis by human lymphocytes and its physiological significance: possible role as intracrine, autocrine, and/or paracrine substance. FASEB J. 2004;18:537–9.
Martins E Jr, Ferreira AC, Skorupa AL, Afeche SC, Cipolla-Neto J, Costa Rosa LF. Tryptophan consumption and indoleamines production by peritoneal cavity macrophages. J Leukoc Biol. 2004;75:1116–21.
Pontes GN, Cardoso EC, Carneiro-Sampaio MM, Markus RP. Injury switches melatonin production source from endocrine (pineal) to paracrine (phagocytes) - melatonin in human colostrum and colostrum phagocytes. J Pineal Res. 2006;41:136–41.
Ferreira ZS, Fernandes PA, Duma D, Assreuy J, Avellar MC, Markus RP. Corticosterone modulates noradrenaline-induced melatonin synthesis through inhibition of nuclear factor kappa B. J Pineal Res. 2005;38:182–8.
Fernandes PA, Cecon E, Markus RP, Ferreira ZS. Effect of TNF-alpha on the melatonin synthetic pathway in the rat pineal gland: basis for a 'feedback' of the immune response on circadian timing. J Pineal Res. 2006;41:344–50.
Bertuglia S, Colantuoni A, Intaglietta M. Effect of leukocyte adhesion and microvascular permeability on capillary perfusion during ischemia-reperfusion injury in hamster cheek pouch. Int J Microcirc Clin Exp. 1993;13:13–26.
Lotufo CM, Lopes C, Dubocovich ML, Farsky SH, Markus RP. Melatonin and N-acetylserotonin inhibit leukocyte rolling and adhesion to rat microcirculation. Eur J Pharmacol. 2001;430:351–7.
Carrillo-Vico A, Lardone PJ, Alvarez-Sanchez N, Rodriguez-Rodriguez A, Guerrero JM. Melatonin: buffering the immune system. Int J Mol Sci. 2013;14:8638–83.
Radogna F, Diederich M, Ghibelli L. Melatonin: a pleiotropic molecule regulating inflammation. Biochem Pharmacol. 2010;80:1844–52.
Pires-Lapa MA, Tamura EK, Salustiano EM, Markus RP. Melatonin synthesis in human colostrum mononuclear cells enhances dectin-1-mediated phagocytosis by mononuclear cells. J Pineal Res. 2013;55:240–6.
Tamura EK, Cecon E, Monteiro AW, Silva CL, Markus RP. Melatonin inhibits LPS-induced NO production in rat endothelial cells. J Pineal Res. 2009;46:268–74.
Bhakar AL, Tannis LL, Zeindler C, Russo MP, Jobin C, Park DS, MacPherson S, Barker PA. Constitutive nuclear factor-kappa B activity is required for central neuron survival. J Neurosci. 2002;22:8466–75.
de Freitas MS, Spohr TC, Benedito AB, Caetano MS, Margulis B, Lopes UG, Moura-Neto V. Neurite outgrowth is impaired on HSP70-positive astrocytes through a mechanism that requires NF-kappaB activation. Brain Res. 2002;958:359–70.
Luningschror P, Stocker B, Kaltschmidt B, Kaltschmidt C. miR-290 cluster modulates pluripotency by repressing canonical NF-kappaB signaling. Stem Cells. 2012;30:655–64.
Imielski Y, Schwamborn JC, Luningschror P, Heimann P, Holzberg M, Werner H, Leske O, Puschel AW, Memet S, Heumann R, et al. Regrowing the adult brain: NF-kappaB controls functional circuit formation and tissue homeostasis in the dentate gyrus. PLoS One. 2012;7:e30838.
Cecon E, Fernandes PA, Pinato L, Ferreira ZS, Markus RP. Daily variation of constitutively activated nuclear factor kappa B (NFKB) in rat pineal gland. Chronobiol Int. 2010;27:52–67.
Gilad E, Wong HR, Zingarelli B, Virag L, O'Connor M, Salzman AL, Szabo C. Melatonin inhibits expression of the inducible isoform of nitric oxide synthase in murine macrophages: role of inhibition of NFkappaB activation. FASEB J. 1998;12:685–93.
Pedrosa AM, Weinlich R, Mognol GP, Robbs BK, Viola JP, Campa A, Amarante-Mendes GP. Melatonin protects CD4+ T cells from activation-induced cell death by blocking NFAT-mediated CD95 ligand upregulation. J Immunol. 2010;184:3487–94.
Chang CC, Tien CH, Lee EJ, Juan WS, Chen YH, Hung YC, Chen TY, Chen HY, Wu TS. Melatonin inhibits matrix metalloproteinase-9 (MMP-9) activation in the lipopolysaccharide (LPS)-stimulated RAW 264.7 and BV2 cells and a mouse model of meningitis. J Pineal Res. 2012;53:188–97.
Huang SH, Cao XJ, Wei W. Melatonin decreases TLR3-mediated inflammatory factor expression via inhibition of NF-kappa B activation in respiratory syncytial virus-infected RAW264.7 macrophages. J Pineal Res. 2008;45:93–100.
Fang Q, Chen G, Zhu W, Dong W, Wang Z. Influence of melatonin on cerebrovascular proinflammatory mediators expression and oxidative stress following subarachnoid hemorrhage in rabbits. Mediat Inflamm. 2009;2009:426346.
Carrillo-Vico A, Garcia-Maurino S, Calvo JR, Guerrero JM. Melatonin counteracts the inhibitory effect of PGE2 on IL-2 production in human lymphocytes via its mt1 membrane receptor. FASEB J. 2003;17:755–7.
Markus RP, Cecon E, Pires-Lapa MA. Immune-pineal axis: nuclear factor kappaB (NF-kB) mediates the shift in the melatonin source from pinealocytes to immune competent cells. Int J Mol Sci. 2013;14:10979–97.
Maronde E, Pfeffer M, Olcese J, Molina CA, Schlotter F, Dehghani F, Korf HW, Stehle JH. Transcription factors in neuroendocrine regulation: rhythmic changes in pCREB and ICER levels frame melatonin synthesis. J Neurosci. 1999;19:3326–36.
Villela D, de Sa LL, Peres R. Peliciari-Garcia RA, do Amaral FG, Cipolla-Neto J, Scavone C, Afeche SC: norepinephrine activates NF-kappaB transcription factor in cultured rat pineal gland. Life Sci. 2014;94:122–9.
Muxel SM, Pires-Lapa MA, Monteiro AW, Cecon E, Tamura EK, Floeter-Winter LM, Markus RP. NF-kappaB drives the synthesis of melatonin in RAW 264.7 macrophages by inducing the transcription of the arylalkylamine-N-acetyltransferase (AA-NAT) gene. PLoS One. 2012;7:e52010.
Bajova H, Nelson TE, Gruol DL. Chronic CXCL10 alters the level of activated ERK1/2 and transcriptional factors CREB and NF-kappaB in hippocampal neuronal cell culture. J Neuroimmunol. 2008;195:36–46.
Zou J, Crews F. CREB and NF-kappaB transcription factors regulate sensitivity to excitotoxic and oxidative stress induced neuronal cell death. Cell Mol Neurobiol. 2006;26:385–405.
Lotufo CM, Yamashita CE, Farsky SH, Markus RP. Melatonin effect on endothelial cells reduces vascular permeability increase induced by leukotriene B4. Eur J Pharmacol. 2006;534:258–63.
Tamura EK, Silva CL, Markus RP. Melatonin inhibits endothelial nitric oxide production in vitro. J Pineal Res. 2006;41:267–74.
Marcola M, da Silveira C-MS, Fernandes PA, Monteiro AW, Markus RP, Tamura EK. Endothelial cell adhesiveness is a function of environmental lighting and melatonin level. J Pineal Res. 2013;54:162–9.
Karper JC, Ewing MM, de Vries MR, de Jager SC, Peters EA, de Boer HC, van Zonneveld AJ, Kuiper J, Huizinga EG, Brondijk TH, et al. TLR accessory molecule RP105 (CD180) is involved in post-interventional vascular remodeling and soluble RP105 modulates neointima formation. PLoS One. 2013;8:e67923.
Srinivasan V, Pandi-Perumal SR, Maestroni GJ, Esquifino AI, Hardeland R, Cardinali DP. Role of melatonin in neurodegenerative diseases. Neurotox Res. 2005;7:293–318.
Papaioannou V, Mebazaa A, Plaud B, Legrand M. 'Chronomics' in ICU: circadian aspects of immune response and therapeutic perspectives in the critically ill. Intensive Care Med Exp. 2014;2:18.
Skwarlo-Sonta K, Majewski P, Markowska M, Oblap R, Olszanska B. Bidirectional communication between the pineal gland and the immune system. Can J Physiol Pharmacol. 2003;81:342–9.
Mack JM, Schamne MG, Sampaio TB, Pertile RA, Fernandes PA, Markus RP, Prediger RD. Melatoninergic system in Parkinson's disease: from neuroprotection to the management of motor and nonmotor symptoms. Oxidative Med Cell Longev. 2016;2016:3472032.
Noseda AC, Rodrigues LS, Targa AD, Aurich MF, Vital MA, Da Cunha C, Lima MM. Putative role of monoamines in the antidepressant-like mechanism induced by striatal MT2 blockade. Behav Brain Res. 2014;275:136–45.
Cecon E, Chen M, Marcola M, Fernandes PA, Jockers R, Markus RP. Amyloid beta peptide directly impairs pineal gland melatonin synthesis and melatonin receptor signaling through the ERK pathway. FASEB J. 2015;29:2566–82.
Lehallier B, Essioux L, Gayan J, Alexandridis R, Nikolcheva T, Wyss-Coray T, Britschgi M. Alzheimer's disease neuroimaging I: combined plasma and cerebrospinal fluid signature for the prediction of midterm progression from mild cognitive impairment to Alzheimer disease. JAMA neurol. 2016;73:203–12.
Maslinska D, Laure-Kamionowska M, Deregowski K, Maslinski S. Association of mast cells with calcification in the human pineal gland. Folia Neuropathol. 2010;48:276–82.
Gomez-Gonzalez B, Dominguez-Salazar E, Hurtado-Alvarado G, Esqueda-Leon E, Santana-Miranda R, Rojas-Zamorano JA, Velazquez-Moctezuma J. Role of sleep in the regulation of the immune system and the pituitary hormones. Ann N Y Acad Sci. 2012;1261:97–106.
Westermann J, Lange T, Textor J, Born J. System consolidation during sleep - a common principle underlying psychological and immunological memory formation. Trends Neurosci. 2015;38:585–97.
Everson CA, Gilliland MA, Kushida CA, Pilcher JJ, Fang VS, Refetoff S, Bergmann BM, Rechtschaffen A. Sleep deprivation in the rat: IX. Sleep. 1989;12:60–7.
Gomez-Gonzalez B, Hurtado-Alvarado G, Esqueda-Leon E, Santana-Miranda R, Rojas-Zamorano JA, Velazquez-Moctezuma J. REM sleep loss and recovery regulates blood-brain barrier function. Curr Neurovasc Res. 2013;10:197–207.
Schmid DA, Wichniak A, Uhr M, Ising M, Brunner H, Held K, Weikel JC, Sonntag A, Steiger A. Changes of sleep architecture, spectral composition of sleep EEG, the nocturnal secretion of cortisol, ACTH, GH, prolactin, melatonin, ghrelin, and leptin, and the DEX-CRH test in depressed patients during treatment with mirtazapine. Neuropsychopharmacology. 2006;31:832–44.
Guarnieri B, Adorni F, Musicco M, Appollonio I, Bonanni E, Caffarra P, Caltagirone C, Cerroni G, Concari L, Cosentino FI, et al. Prevalence of sleep disturbances in mild cognitive impairment and dementing disorders: a multicenter Italian clinical cross-sectional study on 431 patients. Dement Geriatr Cogn Disord. 2012;33:50–8.
Mander BA, Winer JR, Walker MP. Sleep and Human Aging. Neuron. 2017;94:19–36.
Krause AJ, Simon EB, Mander BA, Greer SM, Saletin JM, Goldstein-Piekarski AN, Walker MP. The sleep-deprived human brain. Nat Rev Neurosci. 2017;18:404–18.
Mander BA, Winer JR, Jagust WJ, Walker MP. Sleep: a novel mechanistic pathway, biomarker, and treatment target in the pathology of Alzheimer's disease? Trends Neurosci. 2016;39:552–66.
Van Erum J, Van Dam D, De Deyn PP. Sleep and Alzheimer's disease: a pivotal role for the suprachiasmatic nucleus. Sleep Med Rev. 2018;40:17–27.
Irwin MR, Olmstead R, Carroll JE. Sleep disturbance, sleep duration, and inflammation: a systematic review and meta-analysis of cohort studies and experimental sleep deprivation. Biol Psychiatry. 2016;80:40–52.
Irwin MR, Opp MR. Sleep health: reciprocal regulation of sleep and innate immunity. Neuropsychopharmacology. 2017;42:129–55.
Lo JC, Groeger JA, Cheng GH, Dijk DJ, Chee MW. Self-reported sleep duration and cognitive performance in older adults: a systematic review and meta-analysis. Sleep Med. 2016;17:87–98.
Spira AP, Gottesman RF. Sleep disturbance: an emerging opportunity for Alzheimer's disease prevention? Int Psychogeriatr. 2017;29:529–31.
Liguori C, Romigi A, Nuccetelli M, Zannino S, Sancesario G, Martorana A, Albanese M, Mercuri NB, Izzi F, Bernardini S, et al. Orexinergic system dysregulation, sleep impairment, and cognitive decline in Alzheimer disease. JAMA neurol. 2014;71:1498–505.
Sethi M, Joshi SS, Webb RL, Beckett TL, Donohue KD, Murphy MP, O'Hara BF, Duncan MJ. Increased fragmentation of sleep-wake cycles in the 5XFAD mouse model of Alzheimer's disease. Neuroscience. 2015;290:80–9.
Kincheski GC, Valentim IS, Clarke JR, Cozachenco D, Castelo-Branco MTL, Ramos-Lobo AM, Rumjanek V, Donato J Jr, De Felice FG, Ferreira ST. Chronic sleep restriction promotes brain inflammation and synapse loss, and potentiates memory impairment induced by amyloid-beta oligomers in mice. Brain Behav Immun. 2017;64:140–51.
Shokri-Kojori E, Wang GJ, Wiers CE, Demiral SB, Guo M, Kim SW, Lindgren E, Ramirez V, Zehra A. Freeman C, et al: beta-amyloid accumulation in the human brain after one night of sleep deprivation. Proc Natl Acad Sci U S A. 2018;115:4483–8.
Branger P, Arenaza-Urquijo EM, Tomadesso C, Mezenge F, Andre C, de Flores R, Mutlu J, de La Sayette V, Eustache F, Chetelat G, Rauchs G. Relationships between sleep quality and brain volume, metabolism, and amyloid deposition in late adulthood. Neurobiol Aging. 2016;41:107–14.
Spira AP, Gamaldo AA, An Y, Wu MN, Simonsick EM, Bilgel M, Zhou Y, Wong DF, Ferrucci L, Resnick SM. Self-reported sleep and beta-amyloid deposition in community-dwelling older adults. JAMA neurol. 2013;70:1537–43.
Westwood AJ, Beiser A, Jain N, Himali JJ, DeCarli C, Auerbach SH, Pase MP, Seshadri S. Prolonged sleep duration as a marker of early neurodegeneration predicting incident dementia. Neurology. 2017;88:1172–9.
Liguori C, Chiaravalloti A, Nuccetelli M, Izzi F, Sancesario G, Cimini A, Bernardini S, Schillaci O, Mercuri NB, Fabio P. Hypothalamic dysfunction is related to sleep impairment and CSF biomarkers in Alzheimer's disease. J Neurol. 2017;264:2215–23.
Zheng H, Zhou Q, Du Y, Li C, Xu P, Lin L, Xiao J, Gao H. The hypothalamus as the primary brain region of metabolic abnormalities in APP/PS1 transgenic mouse model of Alzheimer's disease. Biochim Biophys Acta Mol Basis Dis. 1864;2018:263–73.
Matsuoka T, Imai A, Fujimoto H, Kato Y, Shibata K, Nakamura K, Yokota H, Yamada K, Narumoto J. Reduced pineal volume in Alzheimer disease: a retrospective cross-sectional MR imaging study. Radiology. 2018;286:239–48.
Zhang W, Chen XY, Su SW, Jia QZ, Ding T, Zhu ZN, Zhang T. Exogenous melatonin for sleep disorders in neurodegenerative diseases: a meta-analysis of randomized clinical trials. Neurol Sci. 2016;37:57–65.
Migaud M, Batailler M, Segura S, Duittoz A, Franceschini I, Pillon D. Emerging new sites for adult neurogenesis in the mammalian brain: a comparative study between the hypothalamus and the classical neurogenic zones. Eur J Neurosci. 2010;32:2042–52.
Rojczyk-Golebiewska E, Palasz A, Wiaderkiewicz R. Hypothalamic subependymal niche: a novel site of the adult neurogenesis. Cell Mol Neurobiol. 2014;34:631–42.
Lazarov O, Marr RA. Of mice and men: neurogenesis, cognition and Alzheimer's disease. Front Aging Neurosci. 2013;5:43.
Lazarov O, Hollands C. Hippocampal neurogenesis: learning to remember. Prog Neurobiol. 2016;138-140:1–18.
Ramirez-Rodriguez GB, Olvera-Hernandez S, Vega-Rivera NM, Ortiz-Lopez L. Melatonin influences structural plasticity in the axons of granule cells in the dentate gyrus of Balb/C mice. Int J Mol Sci. 2018;20(1). https://doi.org/10.3390/ijms20010073.
Tocharus C, Puriboriboon Y, Junmanee T, Tocharus J, Ekthuwapranee K, Govitrapong P. Melatonin enhances adult rat hippocampal progenitor cell proliferation via ERK signaling pathway through melatonin receptor. Neuroscience. 2014;275:314–21.
Kong X, Li X, Cai Z, Yang N, Liu Y, Shu J, Pan L, Zuo P. Melatonin regulates the viability and differentiation of rat midbrain neural stem cells. Cell Mol Neurobiol. 2008;28:569–79.
Ramirez-Rodriguez G, Klempin F, Babu H, Benitez-King G, Kempermann G. Melatonin modulates cell survival of new neurons in the hippocampus of adult mice. Neuropsychopharmacology. 2009;34:2180–91.
Ramirez-Rodriguez G, Ortiz-Lopez L, Dominguez-Alonso A, Benitez-King GA, Kempermann G. Chronic treatment with melatonin stimulates dendrite maturation and complexity in adult hippocampal neurogenesis of mice. J Pineal Res. 2011;50:29–37.
Niles LP, Armstrong KJ, Rincon Castro LM, Dao CV, Sharma R, McMillan CR, Doering LC, Kirkham DL. Neural stem cells express melatonin receptors and neurotrophic factors: colocalization of the MT1 receptor with neuronal and glial markers. BMC Neurosci. 2004;5:41.
Migaud M, Butrille L, Batailler M. Seasonal regulation of structural plasticity and neurogenesis in the adult mammalian brain: focus on the sheep hypothalamus. Front Neuroendocrinol. 2015;37:146–57.
Batailler M, Chesneau D, Derouet L, Butruille L, Segura S, Cognie J, Dupont J, Pillon D, Migaud M. Pineal-dependent increase of hypothalamic neurogenesis contributes to the timing of seasonal reproduction in sheep. Sci Rep. 2018;8:6188.
Kokoeva MV, Yin H, Flier JS. Evidence for constitutive neural cell proliferation in the adult murine hypothalamus. J Comp Neurol. 2007;505:209–20.
McNay DE, Briancon N, Kokoeva MV, Maratos-Flier E, Flier JS. Remodeling of the arcuate nucleus energy-balance circuit is inhibited in obese mice. J Clin Invest. 2012;122:142–52.
Batailler M, Droguerre M, Baroncini M, Fontaine C, Prevot V, Migaud M. DCX-expressing cells in the vicinity of the hypothalamic neurogenic niche: a comparative study between mouse, sheep, and human tissues. J Comp Neurol. 2014;522:1966–85.
Batailler M, Derouet L, Butruille L, Migaud M. Sensitivity to the photoperiod and potential migratory features of neuroblasts in the adult sheep hypothalamus. Brain Struct Funct. 2016;221:3301–14.
Migaud M, Batailler M, Pillon D, Franceschini I, Malpaux B. Seasonal changes in cell proliferation in the adult sheep brain and pars tuberalis. J Biol Rhythm. 2011;26:486–96.
Jin K, Peel AL, Mao XO, Xie L, Cottrell BA, Henshall DC, Greenberg DA. Increased hippocampal neurogenesis in Alzheimer's disease. Proc Natl Acad Sci U S A. 2004;101:343–7.
Demars M, Hu YS, Gadadhar A, Lazarov O. Impaired neurogenesis is an early event in the etiology of familial Alzheimer's disease in transgenic mice. J Neurosci Res. 2010;88:2103–17.
Wang JM, Singh C, Liu L, Irwin RW, Chen S, Chung EJ, Thompson RF, Brinton RD. Allopregnanolone reverses neurogenic and cognitive deficits in mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2010;107:6498–503.
Aizawa K, Ageyama N, Terao K, Hisatsune T. Primate-specific alterations in neural stem/progenitor cells in the aged hippocampus. Neurobiol Aging. 2011;32:140–50.
Rodriguez JJ, Jones VC, Tabuchi M, Allan SM, Knight EM, LaFerla FM, Oddo S, Verkhratsky A. Impaired adult neurogenesis in the dentate gyrus of a triple transgenic mouse model of Alzheimer's disease. PLoS One. 2008;3:e2935.
Bahna SG, Niles LP. Epigenetic regulation of melatonin receptors in neuropsychiatric disorders. Br J Pharmacol. 2018;175:3209–19.
Yang Y, Sun B, Huang J, Xu L, Pan J, Fang C, Li M, Li G, Tao Y, Yang X, et al. Up-regulation of miR-325-3p suppresses pineal aralkylamine N-acetyltransferase (Aanat) after neonatal hypoxia-ischemia brain injury in rats. Brain Res. 1668;2017:28–35.
Acknowledgments
I appreciate Prof. Young-Kook Kim about a technical assistance in figure illustration process.
Ethical approval and consent to participate
Not applicable.
Funding
This study was supported by funded by grants 2016R1D1A1B03930394 and 2019R1F1A1054111 from the Basic Science Research Program through the National Research Foundation of Korea (NRF).
Author information
Authors and Affiliations
Contributions
Juhyun Song wrote and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Consent for publication
Not applicable.
Competing interests
The author has no conflicts of interest to declare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Song, J. Pineal gland dysfunction in Alzheimer’s disease: relationship with the immune-pineal axis, sleep disturbance, and neurogenesis. Mol Neurodegeneration 14, 28 (2019). https://doi.org/10.1186/s13024-019-0330-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13024-019-0330-8