
Abstract
Background
Recent studies have shown the implication of the ROBO-SLIT pathway in heart development. Within this study, we aimed to further assess the implication of the ROBO and SLIT genes mainly in bicuspid aortic valve (BAV) and other human congenital heart defects (CHD).
Methods
We have analyzed a cohort of singleton exome sequencing data comprising 40 adult BAV patients, 20 pediatric BAV patients generated by the Pediatric Cardiac Genomics Consortium, 10 pediatric cases with tetralogy of Fallot (ToF), and one case with coarctation of the aorta. A gene-centered analysis of data was performed. To further advance the interpretation of the variants, we intended to combine more than 5 prediction tools comprising the assessment of protein structure and stability.
Results
A total of 24 variants were identified. Only 4 adult BAV patients (10%) had missense variants in the ROBO and SLIT genes. In contrast, 19 pediatric cases carried variants in ROBO or SLIT genes (61%). Three BAV patients with a severe phenotype were digenic. Segregation analysis was possible for two BAV patients. For the homozygous ROBO4: p.(Arg776Cys) variant, family segregation was consistent with an autosomal recessive pattern of inheritance. The ROBO4: c.3001 + 3G > A variant segregates with the affected family members. Interestingly, these variants were also found in two unrelated patients with ToF highlighting that the same variant in the ROBO4 gene may underlie different cardiac phenotypes affecting the outflow tract development.
Conclusion
Our results further reinforce the implication of the ROBO4 gene not only in BAV but also in ToF hence the importance of its inclusion in clinical genetic testing. The remaining ROBO and SLIT genes may be screened in patients with negative or inconclusive genetic tests.
Similar content being viewed by others

Introduction
The secreted SLIT glycoproteins and their Roundabout (ROBO) receptors were initially known for axon guidance and dendritic branching in the developing central nervous system [1,2,3]. Subsequently, several studies have expanded the functional spectrum of the ROBO-SLIT pathway by reporting other functions, such as cell migration and proliferation, angiogenesis, and vascularization in different organs and tissues [3,4,5]. Recently, a pivotal role of the SLIT ligands and their ROBO receptors has been reported in animal heart morphogenesis and development [6,7,8]. These findings have been reinforced by the identification of genetic variations in ROBO1 and ROBO4 genes in patients with tetralogy of Fallot (ToF) and bicuspid aortic valve (BAV) disease, respectively [9, 10]. Indeed, Kruszka et al. identified loss of function variants in ROBO1 gene in three unrelated patients with ToF and ventricular septal defects (VSD) [9]. More recently, Jaouadi et al. identified a ROBO1 variant in a BAV family with three affected members [11].
In 2019, Gould et al. reported variants in the ROBO4 gene in patients with BAV and ascending aortic aneurysms (AscAA). The phenotypes observed in Robo4 animal models were consistent with patients’ phenotypes with a novel endothelial etiology supporting a causative role of ROBO4 [10]. Thereafter, additional variants in ROBO4 have been linked to human BAV [12]. The authors have concluded that variants in ROBO4 along with NOTCH1, GATA4 and SMAD6 are enriched in BAV-patients with early onset complications [12].
Albeit human genetic variations have been identified in ROBO genes, mainly ROBO4 and ROBO1, data from several animal models point out the implication of the remaining ROBO and SLIT genes in CHD pathogenesis [6, 13, 14]. Moreover, Zhao et al. (2022) have underlined the clinical relevance of SLIT3 as a promising candidate gene for further screening in patients [13].
In the present study, we aimed firstly to screen adult and pediatric patients with BAV in order to identify genetic variants in ROBO and SLIT genes using exome sequencing data combined to a thorough in silico analysis. Based on the results of this analysis, we sought to expand the pediatric cohort to include other CHD phenotypes (10 patients with ToF and one case with coarctation of the aorta (CoA)) in order to determine whether variants in ROBO and SLIT genes may be implicated in CHD other than BAV.
Of note, the study includes CHD patients with no relevant variants in known CHD-related genes such as NOTCH1, NOTCH2, GATA5, GATA4, ACTA2, SMAD6, NKX2-5, FLT4, TGFBR1, and TGFBR2.
Patients and methods
This study was performed according to the principles of the Declaration of Helsinki and to the ethical standards of the first author’s institutional review board. The patients provided their written informed consent to participate in this study (approved by the Marseille ethic committee n°13.061 and 2016-A00958-53). Personal health data and DNA from two pediatric BAV patients and their related are part of the CARREG study (http://carreg.fr/en/), which was declared to the French national committee for informatics and liberties (France; CNIL; No. 1734573V0). The CARREG study is a prospective monocenter study promoted by the “Centre de Référence des Malformations Cardiaques Congénitales Complexes (M3C)” located at the Pediatric cardiology department of the Necker-Enfants Malades Hospital, Paris, France. Clinical records were reviewed by cardiologist or pediatric-cardiologist before recruitment and cardiovascular diagnosis was obtained by echocardiography mainly. Patients with 22q11.2 deletion or other recognized syndromes were excluded.
Patients
The starting study cohort includes a total of 71 patients with clinical diagnosis of bicuspid aortic valve (40 adult and 20 pediatric patients), tetralogy of Fallot (10 pediatric cases) and one pediatric case with coarctation of the aorta (CoA). No other defects are associated with the main clinical diagnosis with confirmed absence of structural myocardial and syndromic diseases.
Exome sequencing
Germline DNA was extracted from blood samples and subjected to exome sequencing. Whole exome sequencing (WES) was performed by the Genomics and Bioinformatics Platform (GBiM) of the INSERM U1251 Marseille Medical Genetics facility using the NimbleGen SeqCap EZ MedExome kit (total design size 47 Mb) according to the manufacturer’s protocol (Roche Sequencing Solutions, Madison, USA). All DNA and libraries preparations (KAPA HyperPrep Kits (Roche)) were performed according to the manufacturers’ instructions. The DNA libraries were subjected to paired-end sequencing using the Illumina NextSeq500 sequencing platform (Illumina, San Diego, CA, USA). Raw fastQ files were aligned to the hg19 reference human genome (University of California Santa Cruz, UCSC) using BWA software [15]. Variant calling workflow was performed according to the GATK best practices [16]. Both HaplotypeCaller and BaseRecalibration tools have been used for variant calling and quality score recalibration. The output files were annotated using ANNOVAR software [17]. On average, a depth of 125X and a coverage of 97.7% of the bases at 30X have been obtained per sample.
Variant annotation and prioritization
Variant annotation process and exome data analysis were performed using VarAFT software version 2.17–2 (http://varaft.eu/) [18]. Firstly, a patient- centered approach was applied. Thus, we excluded variants with a minor allele frequency (MAF) > 1% in gnomAD (Genome Aggregation Database) (http://gnomad.broadinstitute.org/). Then, we removed non-coding and synonymous variants with no impact on splicing with HSF-Pro tool. Subsequently, the remaining variants were filtered based on their in silico pathogenicity prediction with UMD_Predictor, SIFT and PolyPhen tools [19,20,21]. The prioritized variants were finally interpreted according to their clinical relevance. Indeed, patients with likely pathogenic and/or causative variants in genes linked to the NOTCH or TGFβ pathways or in cardiac transcription factors such as GATA4/5, NKX2-5, and TBX-5 have been selected for further analysis and excluded from the present study.
As a second step, patients with no-relevant variants in CHD-related genes were re-analyzed as following: a gene-centered approach was applied to the remaining patients toward identifying variants in the ROBO-SLIT pathway. Thus, we used a gene list including ROBO1, ROBO2, ROBO3, ROBO4, SLIT1, SLIT2, and SLIT3 genes to run the same prioritization strategy as above. The main functions of ROBO and SLIT genes are summarized in Table 1.
Combined Annotation Dependent Depletion (CADD)
Given the lack of detailed clinical description for some patients and the family history that would allow for segregation analysis, we used the CADD computational algorithm to further assess variants pathogenicity.
For annotation, CADD used the Ensembl Variant Effect Predictor, data from the ENCODE project and information from UCSC genome browser tracks. These annotations span a wide range of data types including conservation metrics such as GERP, phastCons, and phyloP; functional genomic data like DNase hypersensitivity and transcription factor binding; transcript information like distance to exon–intron boundaries or expression levels in commonly studied cell lines; and protein-level scores like Grantham, SIFT, and PolyPhen [22, 23]. Thus, CADD algorithm simulate neutral and deleterious variants from multiple species alignments, annotate variants based on the conservation among species, genetic context and epigenetics, rank the variants by a logistic regression model and finally generate a CADD score for each variant in the human genome.
A scaled C-score of greater of equal 10 indicates that these are predicted to be the 10% most deleterious substitutions that you can do to the human genome, a score of greater or equal 20 indicates the 1% most deleterious and so on.
To identify potentially pathogenic variants, a cutoff between 10 and 20 can be set. A cutoff of 15 is recommended as it is the median value for all possible canonical splice site changes and non-synonymous variants in CADD v1.0.
In silico assessment of protein stability and interactions
I-mutant
In order to aid the annotation process, an in silico prediction of protein stability free energy change (DDG) was performed using I-Mutant3.0 software (http://gpcr.biocomp.unibo.it/cgi/predictors/I-Mutant3.0/I-Mutant3.0.cgi) [24]. The substitutions are ranked according to a three-state classification system: destabilizing mutations (DDG < − 0.5 kcal/mol), stabilizing mutations (DDG > 0.5 kcal/mol) and neutral mutations (− 0.5 < = DDG < = 0.5 kcal/mol).
Project HOPE
The project HOPE tool (https://www3.cmbi.umcn.nl/hope) is a web service that analyses the structural and physicochemical effects of point mutations in a protein sequence using PDB file when the corresponding protein structure has been solved experimentally (95–100% match). Whenever this is not the case, HOPE will build a homology model using an existing template (between 30 and 95% match). As an estimation, HOPE uses information obtained from the 3D-structure in 60–70% of the cases [25].
Results
From a cohort of 40 BAV adult patients [26] and 20 BAV pediatric cases, we sought to determine the implication of the ROBO-SLIT pathway in patients with no relevant variants in known BAV-related genes. Interestingly, the yield of rare variants in ROBO and SLIT genes was greater in the pediatric cohort (13/20, 65%) compared to only 4 out 40 BAV cases (10%) from the adult cohort (Table 2).
Family segregation was performed for two BAV patients only, both with ROBO4 variants.
The first patient (BAV-PED-10) is a male pediatric case with BAV. His medical records include small aortic insufficiency in the posterior commissure, fusion of the anterior commissure, right anterior leaflet prolapse, aortic annulus dilatation and dilated ascending aorta (z score 3.2 and 3.3). The patient had a positive family history of aortic valve defects. His paternal and maternal grand-mothers underwent aortic valve replacement.
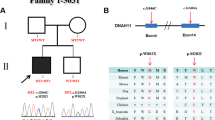
The patient (BAV-PED-10) carried a homozygous ROBO4 variant (p. Arg776Cys) (Table 3). His parents were found heterozygous for the variant (Fig. 1). The MAF of this variant (rs138481093) is 0.004699 in gnomAD with a total number of homozygotes equal to 8. Of note, our patient is of white European non-Finnish ethnic group which represents the highest MAF population (0.007781).

Family segregation of the ROBO4: p.Arg776Cys variant. Darkened left upper quadrant: Affected child with BAV
The second case (BAV-PED-12) is a male pediatric case with BAV. The analysis of his WES data allowed us to identify a splice site heterozygous variant in ROBO4 (c.3001 + 3G > A). The patient’s father was found to have hypoplastic left coronary artery and his brother had VSD. His mother and sister are healthy.
The ROBO4: c.(3001 + 3G > A) variant was found in the patient’s father (I-1) and brother (II-2). The mother (I-2) and sister (II-3) do not carry the variant.
Family pedigree and segregation are shown in Fig. 2.

Family segregation of the ROBO4: c.3001 + 3G > A variant. The index-case (BAV-PED-12) is marked with a star
As for the first patient (BAV-PED-10), this case is European non-Finnish also. The MAF of the ROBO4: c.3001 + 3G > A variant in this population is 0.01448. The highest population MAF of this variant (rs145918924) is 0.02831 in the Ashkenazi Jewish population.
Of note, Gould et al. reported a heterozygous splice site variant ROBO4: c.2056 + 1G > T in a multigenerational BAV-family. Interestingly, seven of eight affected cases were male [10]. These findings underline the intrafamilial variability as well as the phenotypic pleiotropy of ROBO4 variants.
As we mentioned above, the identification of more ROBO and SLIT variants in the pediatric BAV cases than in the adult cohort prompted us to investigate the implication of this pathway in another CHD phenotype. Thus, we analyzed 10 ToF patients and one CoA case. Five out 10 ToF patients carried variants in ROBO and SLIT genes. Three patients carried variants in the ROBO3 gene and strikingly, the two other patients (ToF-PED-9 and ToF-PED11) were carrying the aforementioned ROBO4 variants (p.(Arg776Cys) and c.3001 + 3G > A) at a heterozygous state (Table 2).
The clinical resume of the ToF-patient (ToF-PED-9) carrying the ROBO4: p.Arg776Cys variant is as following: Pregnancy was complicated by gestational diabetes. Pulmonary atresia and VSD as well as partial corpus callosum agenesis were prenatally diagnosed. Amniocentesis was refused by the parents. The anatomy was confirmed after birth. Pulmonary arteries were noted to be extremely hypoplastic (2 mm, z-value -5, birth weight 4 kg). A malformation of the arterial duct was noted, with no signs of spontaneous closure. At the age of 27 months, a total cardiac repair was performed.
The clinical resume of the ToF-patient (ToF-PED-11) carrying the ROBO4: c.3001 + 3G > A splicing variant is as following: Severe ToF and thoraco-abdominal situs inversus was prenatally diagnosed. Birth weight was small for gestational age (2.4 kg, 38W). Anatomy was confirmed after birth. Pulmonary annulus was very hypoplastic (Z value − 3.7) as well as pulmonary arteries (Z value RPA − 2.4, LPA − 2). The complete cardiac repair (closure VSD and patch enlargement of pulmonary valve and artery) was performed 11 months later.
ToF is defined by the presence of four cardiac defects namely; ventricular septal defect (VSD), pulmonary valve stenosis, right ventricular hypertrophy and overriding aorta, which potentially arise from a misalignment of the great arteries [27, 28]. The identification of the same ROBO4 variants in BAV and ToF patients points out the pleiotropic role of this gene with its implication in several CHD entities with different pattern of inheritance. This pleiotropy can be explained by the potential contribution of ROBO4 gene in different cardiac cell populations [8, 13, 29], but also by the difference of the genetic background of each individual and epigenetics mechanisms acting during heart morphogenesis.
Three additional ROBO4 variants were identified in the present study. The ROBO4 variant (p. Arg908Gln) was identified in a BAV patient with aortic stenosis (BAV-PED-1). This patient carried a second missense variant in ROBO2 gene (p. Arg811Trp). The ROBO4: p.(Ala303Asp) has been identified in a pediatric BAV-case (BAV-PED-7) with aneurysm. Similarly, this patient carried a second variant in the SLIT1 gene (p.Glu1340Asp). The third ROBO4: p.(Ala446Asp) variant was found in BAV-PED-8 case (Table 2). No BAV-related complications were noted for this patient.
In regards to BAV adult patients with variants in ROBO1, ROBO2, SLIT1 and SLIT3 genes, the presence of BAV-related complications such as aortic regurgitation, aortic stenosis, and AscAA was checked. Only the patient (BAV-AD-1) with the ROBO1: p.(Val610Ile) variant had AscAA.
Within this study, we report two stop-gain variants in SLIT1 (p.Cys263Ter) and SLIT3 (p.Cys1355Ter) genes. The patient carrying the SLIT3 stop-gain variant had BAV with mitral regurgitation.
Collectively, a total of 24 rare variants were identified including 21 missense, 2 stop-gain, and 1 splice site variants (Table 2). The majority of variants were found in the pediatric cohort. Indeed, 19 pediatric cases carried variants in ROBO and SLIT genes (19/31 CHD-patients; 61%), whereas, only 4 adult patients (10%) had missense variants in ROBO1, ROBO2, SLIT1 and SLIT3 genes.
It should be noted that, all the patients carried heterozygous variants except a BAV- patient (BAV-PED-10) with the homozygous ROBO4 variant (p. Arg776Cys).
Overall, the in-silico predictions of variant pathogenicity are quite consistent among the different software tools, specifically, variants in ROBO1, ROBO3, and SLIT genes, were predicted to have a large decrease of protein stability and high CADD scores (Table 4). Indeed, except for SLIT1 (p.Cys263Ter) and SLIT3 (p.Cys1355Ter) stop-gain variant with a very high CADD-scores (36 and 42, respectively) which is mainly due to the truncating type of the variants, the highest scores (≥ 30) are attributed to variants located in the fibronectin type III-3 domain of ROBO genes. As an example, the ROBO2: p. (Arg811Trp) and the ROBO3: p.(Pro859Gln) variants, with CADD-scores 31 and 32, respectively, are located within the Fibronectin type-III 3 domain of each gene (Additional file 1).
A more detailed description of variant localization and their predicted impact on protein structure, interaction and physicochemical properties is provided in the Additional file 1. Sanger confirmation of the prioritized variants is provided in Additional file 2.
Discussion
ROBO receptors and their SLIT ligands play versatile roles during heart development across species and have been associated with congenital cardiac defects (CHD) in humans [3, 7, 30]. With the exception of the mammalian ROBO4 receptor, the extracellular domain of ROBO contains 5 Ig-like domains and 3 fibronectin repeats [3, 31]. SLIT are the main ligands of ROBO receptors, which bind through their LRR2 domain to the first Ig domain of ROBO proteins [3]. Of note, SLIT ligands bind also to a wide range of extracellular matrix molecules such as type IV collagens. On the other hand heparin sulfate proteoglycans binds to both SLIT and ROBO [3]. Moreover, ROBO and SLIT proteins are involved in heart tube development of Dosophila and zebrafish and in neural crest migration and adhesion in mice. The absence of ROBO1 receptor has been linked to septal and outflow tract defects [7, 29, 32]. The knockdown of Robo1 in zebrafish resulted in an inhibition of endocardial and myocardial migration leading to an unfused heart fields [7, 33].
In vertebrates, ROBO4 is selectively expressed in endothelial cells and plays a key role in angiogenesis and blood vessel permeability [34]. Similarly, ROBO1/2 receptors and SLIT are also expressed in endothelial cells and contribute to cell motility and polarity [35]. Functional studies have suggested that ROBO4 mutations disrupt endothelial cells performance and impair barrier function leading to abnormal aorta remodeling [10]. Furthermore, Robo4 knockout mice showed severe cardiovascular defects such as aortic valve thickening combined with, in some cases, BAV, aortic regurgitation, aortic stenosis and AscAA [10].
It has been shown that the SLIT-ROBO pathway is involved in the guidance of cranial neural crest cell migration [36]. Additionally, SLIT-ROBO signaling is crucial for organizing neural crest cells and placode derived neurons to form ganglion [37]. Neural crest cells contribute to aortic valve development as well as aortico-pulmonary septation [38,39,40]. Our previous results indicated that SLIT-ROBO signaling might be involved in regulating earlier events during cardiac neural crest cell migration that are associated to outflow tract and aortic valve development [8].
In zebrafish models, both Slit2 and Slit3 are expressed in the heart during chamber formation. Slit2 is particularly expressed in endocardial cells, while Robo1 and Slit3 are expressed in the myocardial, endocardial and endothelial cells [7]. Slit3 is the predominant ligand transcribed in the early mouse heart. Indeed, its expression is detected in the ventral wall of the linear heart tube and subsequently in the heart chamber but not in the atrioventricular canal myocardium [8].
Functional studies using Drosophila, zebrafish, and mouse models have reported a significant role of each Robo-Slit member in heart chamber, lumen, and valve formation [3, 7, 10, 13, 14, 41]. Indeed, in Robo1/Robo2 and Slit3 knockout mice, the ventricular septum is absent, whereas in Slit2 mutants septum anomalies were less severe [14]. Using zebrafish models, it has been shown that Slit3 plays a crucial role in vascular development. Similarly, in mice, Slit3 is the earliest gene to be expressed with a strong expression in the myocardium. It is also expressed in the outflow tract, atrial and sinus horn myocardium, cardiac neural crest, the second heart field and later in the epicardium [6, 7, 13, 14]. Moreover, it has been shown that Slit3 also still expressed in the adult ventricle [13, 14].
The phenotypic analysis of mice mutants showed that Robo1/Robo2 mutants have developed highly penetrant BAV with two entire leaflets and one partial or absent leaflet. However Slit2 mutants have displayed less penetrant BAV phenotype and Slit3 mutants have thickened atrioventricular valves and hypoplastic non-coronary aortic valve [13, 14].
Additionally, it has been shown that Robo–Slit are related to the Notch and vascular endothelial growth factor signaling pathways [6, 13]. Both pathways are known to be involved in heart formation and development. Furthermore, genetic variations in NOTCH and VEGF genes have been found in patients with CHD [42, 43]. In the present study, we sought to identify genetic variants in ROBO and SLIT genes in patients with different CHD. We have identified (i) several variants with a consistent in silico prediction of pathogenicity, (ii) patients with digenic variants who have a more severe phenotype and (iii) two segregating variants, one with an autosomal recessive pattern of inheritance and one segregating with the disease in the family.
Limitations
There may be some possible limitations in this study. The first is the limited access to detailed clinical data for the majority of patients. The second limitation concerns family segregation. Indeed, family co-segregation was possible for two cases only. Parental samples were not available for the other index-cases.
Conclusion
Although CHD newborns are treated as soon as the disease is diagnosed, CHD persists among the most leading causes of mortality in the developed world [44]. The specific causative genetic variant remains unknown for a significant number of patients. The identification of novel variants in the ROBO and SLIT genes, as a recent associated pathway with CHD, will aid to improve the genetic testing yield of CHD. The functional effect of variants of unknown or uncertain significance remains to be elucidated as well as genotype–phenotype correlations.
Our study contributes to expand the phenotypic and allelic heterogeneity of CHD by reporting several variants in the ROBO-SLIT signaling pathway. Albeit the majority of the prioritized variants are predicted pathogenic with a consistency across different in silico predictions tools and the identification of ROBO4 variants segregating in families, functional studies are needed to assess their clinical relevance.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its additional files.
References
Tong M, Jun T, Nie Y, Hao J, Fan D. The role of the Slit/Robo signaling pathway. J Cancer. 2019;10:2694–705.
Fujiwara M, Ghazizadeh M, Kawanami O. Potential role of the Slit/Robo signal pathway in angiogenesis. Vasc Med Lond Engl. 2006;11:115–21.
Blockus H, Chédotal A. Slit-Robo signaling. Dev Camb Engl. 2016;143:3037–44.
Jiang Z, et al. Targeting the SLIT/ROBO pathway in tumor progression: molecular mechanisms and therapeutic perspectives. Ther Adv Med Oncol. 2019;11:1758835919855238.
Dai CF, et al. Expression and roles of Slit/Robo in human ovarian cancer. Histochem Cell Biol. 2011;135:475–85.
Mommersteeg MTM, et al. Slit-roundabout signaling regulates the development of the cardiac systemic venous return and pericardium. Circ Res. 2013;112:465–75.
Zhao J, Mommersteeg MTM. Slit-Robo signalling in heart development. Cardiovasc Res. 2018;114:794–804.
Medioni C, et al. Expression of Slit and Robo genes in the developing mouse heart. Dev Dyn Off Publ Am Assoc Anat. 2010;239:3303–11.
Kruszka P, et al. Loss of function in ROBO1 is associated with tetralogy of Fallot and septal defects. J Med Genet. 2017;54:825–9.
Gould RA, et al. ROBO4 variants predispose individuals to bicuspid aortic valve and thoracic aortic aneurysm. Nat Genet. 2019;51:42–50.
Jaouadi H, et al. Identification of non-synonymous variations in ROBO1 and GATA5 genes in a family with bicuspid aortic valve disease. J Hum Genet. 2022;67:515–8.
Musfee FI, et al. Rare deleterious variants of NOTCH1, GATA4, SMAD6, and ROBO4 are enriched in BAV with early onset complications but not in BAV with heritable thoracic aortic disease. Mol Genet Genomic Med. 2020;8: e1406.
Zhao J, Bruche S, Potts HG, Davies B, Mommersteeg MTM. Tissue-specific roles for the Slit-Robo pathway during heart, caval vein, and diaphragm development. J Am Heart Assoc. 2022;11: e023348.
Mommersteeg MTM, Yeh ML, Parnavelas JG, Andrews WD. Disrupted Slit-Robo signalling results in membranous ventricular septum defects and bicuspid aortic valves. Cardiovasc Res. 2015;106:55–66.
Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–95.
Van der Auwera GA, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinform. 2013;11:11.10.1-11.10.33.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38: e164.
Desvignes J-P, et al. VarAFT: a variant annotation and filtration system for human next generation sequencing data. Nucleic Acids Res. 2018;46:W545–53.
Salgado D, et al. UMD-predictor: a high-throughput sequencing compliant system for pathogenicity prediction of any human cDNA substitution. Hum Mutat. 2016;37:439–46.
Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–4.
Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013;76(1):7–20.
Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47:D886–94.
Kircher M, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–5.
Capriotti E, Fariselli P, Casadio R. I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005;33:W306–10.
Venselaar H, Te Beek TAH, Kuipers RKP, Hekkelman ML, Vriend G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinform. 2010;11:548.
Théron A, et al. Clinical insights into a tertiary care center cohort of patients with bicuspid aortic valve. Int J Cardiovasc Imaging. 2022;38:51–9.
Bajolle F, et al. Rotation of the myocardial wall of the outflow tract is implicated in the normal positioning of the great arteries. Circ Res. 2006;98:421–8.
Apitz C, Webb GD, Redington AN. Tetralogy of fallot. Lancet Lond Engl. 2009;374:1462–71.
Santiago-Martínez E, Soplop NH, Kramer SG. Lateral positioning at the dorsal midline: Slit and Roundabout receptors guide Drosophila heart cell migration. Proc Natl Acad Sci U S A. 2006;103:12441–6.
Vogler G, Bodmer R. Cellular mechanisms of Drosophila heart morphogenesis. J Cardiovasc Dev Dis. 2015;2:2–16.
Dickson BJ, Gilestro GF. Regulation of commissural axon pathfinding by slit and its Robo receptors. Annu Rev Cell Dev Biol. 2006;22:651–75.
Santiago-Martínez E, Soplop NH, Patel R, Kramer SG. Repulsion by Slit and Roundabout prevents Shotgun/E-cadherin-mediated cell adhesion during Drosophila heart tube lumen formation. J Cell Biol. 2008;182:241–8.
Fish JE, et al. A Slit/miR-218/Robo regulatory loop is required during heart tube formation in zebrafish. Dev Camb Engl. 2011;138:1409–19.
Bedell VM, et al. roundabout4 is essential for angiogenesis in vivo. Proc Natl Acad Sci U S A. 2005;102:6373–8.
Dubrac A, et al. Targeting NCK-mediated endothelial cell front-rear polarity inhibits neovascularization. Circulation. 2016;133:409–21.
Li Y, et al. Robo signaling regulates the production of cranial neural crest cells. Exp Cell Res. 2017;361:73–84.
Jia L, Cheng L, Raper J. Slit/Robo signaling is necessary to confine early neural crest cells to the ventral migratory pathway in the trunk. Dev Biol. 2005;282:411–21.
Odelin G, et al. Krox20 defines a subpopulation of cardiac neural crest cells contributing to arterial valves and bicuspid aortic valve. Dev Camb Engl. 2018;145:dev151944.
Kirby ML. Cardiac morphogenesis—recent research advances. Pediatr Res. 1987;21:219–24.
Phillips HM, et al. Neural crest cells are required for correct positioning of the developing outflow cushions and pattern the arterial valve leaflets. Cardiovasc Res. 2013;99:452–60.
MacMullin A, Jacobs JR. Slit coordinates cardiac morphogenesis in Drosophila. Dev Biol. 2006;293:154–64.
Wang Y, Fang Y, Lu P, Wu B, Zhou B. NOTCH signaling in aortic valve development and calcific aortic valve disease. Front Cardiovasc Med. 2021;8: 682298.
Lambrechts D, Carmeliet P. Genetics in zebrafish, mice, and humans to dissect congenital heart disease: insights in the role of VEGF. Curr Top Dev Biol. 2004;62:189–224.
Zaidi S, Brueckner M. Genetics and genomics of congenital heart disease. Circ Res. 2017;120:923–40.
Acknowledgements
Part of this data was generated by the Pediatric Cardiac Genomics Consortium (PCGC), under the auspices of the National Heart, Lung, and Blood Institute's Bench to Bassinet Program <http://www.benchtobassinet.org/>. The Pediatric Cardiac Genomics Consortium (PCGC) program is funded by the National Heart, Lung, and Blood Institute, National Institutes of Health, U.S. Department of Health and Human Services through grants U01HL098123, U01HL098147, U01HL098153, U01HL098162, U01HL098163, and U01HL098188. This manuscript was not prepared in collaboration with investigators of the PCGC, has not been reviewed and/or approved by the PCGC, and does not necessarily reflect the opinions of the PCGC investigators or the NHLBI. H.J. received postdoctoral fellowship from the AFM-Telethon. S.Z. is a Research Director at the INSERM.
Funding
This research received a grant from the Fédération Française de Cardiologie (FFC-Equipe 2019).
Author information
Authors and Affiliations
Contributions
Conceptualization, SZ and HJ; Methodology, HJ; Validation, SZ; CJ; Clinical Investigation of the patients and family members; AT; FB; AF; HG; SD; DB; CO; JFA; Analysis and interpretation of data: HJ; CO; CJ Molecular investigation and in silico analysis: HJ; writing—original draft preparation, HJ; Writing—Review and Editing, HJ; SZ. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Marseille ethic committee no 13.061 and the patients provided their written informed consent to participate.
Consent for publication
Informed consent for publication was obtained from all subjects involved in the study.
Competing interests
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Figure S1: Overview of ROBO1 protein in ribbon presentation. The protein is colored by element: α-helix=blue, β-strand = red, turn=green, 3/10helix=yellow, and random coil=cyan. Figure S2: Close-up of the ROBO1: p.Val610Ile variant. The protein is colored in grey, the side chain of the mutated residue is in magenta and shown as small balls. The protein is colored grey, the side chains of both the wild-type and the mutant residue are shown and colored green and red respectively Figure S3: Overview of ROBO2 protein in ribbon presentation. The protein is colored by element: α-helix=blue, β-strand = red, turn=green, 3/10 helix=yellow, and random coil=cyan. Other molecules in the complex are colored grey when present. Figure S4: Close-up of the ROBO2: p.Arg811Trp variant. The protein is colored grey, and the side chains of both the wild-type and the mutant residue are shown and colored green and red respectively. Figure S5: Overview of ROBO3 protein in ribbon presentation. The protein is colored by element: α-helix=blue, β-strand = red, turn=green, 3/10helix=yellow, and random coil=cyan. Figure S6: Close-up of the ROBO3: p.Thr323Met variant. The protein is colored grey, and the side chain of the mutated residue is colored magenta and shown as small balls. The side chains of both the wild-type and the mutant residue are shown and colored green and red respectively. Figure S7: Close-up of the ROBO3: p.Arg539Trp variant. Figure S8: Close-up of the ROBO3: p.Pro859Gln variant. Figure S9: Overview of ROBO4 protein in ribbon presentation. The protein is colored by element: α-helix=blue, β-strand = red, turn=green, 3/10helix=yellow, and random coil=cyan. Figure S10: Close-up of the ROBO4: p.Ala303Asp variant. The protein is colored grey and the side chains of both the wild-type and the mutant residue are shown and colored green and red respectively. The side chain of the mutated residue is colored magenta and shown as small balls. Figure S11: Overview of SLIT1 protein in ribbon presentation. The protein is colored by element: α-helix=blue, β-strand = red, turn=green, 3/10helix=yellow, and random coil=cyan. Figure S12: Close-up of the SLIT1: p.Pro149Leu variant. The side chain of the mutated residue is colored magenta and shown as small balls Figure S13: Close-up of the SLIT1: p.Arg455Ser variant. The side chain of the mutated residue is colored magenta and shown as small balls. Figure S14: Overview of SLIT3 protein in ribbon presentation. The protein is colored by element: α-helix=blue, β-strand = red, turn=green, 3/10helix=yellow, and random coil=cyan. Figure S15: Close-up of the SLIT3: p.Ser629Asn variant. The side chain of the mutated residue is colored magenta and shown as small balls.
Additional file 2:
Sanger sequencing of the prioritized variants.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Jaouadi, H., Jopling, C., Bajolle, F. et al. Expanding the phenome and variome of the ROBO-SLIT pathway in congenital heart defects: toward improving the genetic testing yield of CHD. J Transl Med 21, 160 (2023). https://doi.org/10.1186/s12967-023-03994-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-023-03994-y