
Abstract
Bicuspid aortic valve (BAV) is the most common congenital heart defect in human beings, with an estimated prevalence in the general population of between 0.5 and 2%. Moreover, BAV is the most common cause of aortic stenosis in the pediatric population. Patients with BAV may have no symptoms for life, and some of them may progress to aortic stenosis. Genetic factors increase the susceptibility and development of BAV. However, the pathogenesis and BAV are still unclear, and more genetic variants are still needed for elucidating the molecular mechanism and stratification of patients. The present study carried out screening of variants implicated in disease in BAV patients. The whole-exome sequencing (WES) was performed in 20 BAV patients and identified 40 different heterozygous missense mutations in 36 genes (MIB2, FAAH, S100A1, RGS16, MAP3K19, NEB, TTN, TNS1, CAND2, CCK, KALRN, ATP10D, SLIT3, ROS1, FABP7, NUP205, IL11RA, NPR2, COL5A1, CUBN, JMJD1C, ANXA7, TRIM8, LGR4, TPCN2, APOA5, GPR84, LRP1, NCOR2, AKAP11, ESRRB, NGB, AKAP13, WWOX, KCNJ12, ARHGEF1). The mutations in these genes were identified as recurrent variants implicated in disease by in silico prediction tool analysis. Nine genes (MIB2, S100A1, TTN, CCK, NUP205, LGR4, NCOR2, ESRRB, and WWOX) among the 36 genes were identified as variants implicated in disease via unanimous agreement of in silico prediction tool analysis and sequenced in an independent cohort of 137 BAV patients to validate the results of WES. BAV patients carrying these variants demonstrated reduced left ventricular ejection fractions (LVEF) (63.8 ± 7.5% vs. 58.4 ± 5.2%, P < 0.001) and larger calcification volume [(1129.3 ± 154) mm3 vs. (1261.8 ± 123) mm3, P < 0.001]. The variants in TTN, NUP205 and NCOR2 genes are significantly associated with reduced LVEF, and the variants in S100A1, LGR4, ESRRB, and WWOX genes are significantly associated with larger calcification volume. We identified a panel of recurrent variants implicated in disease in genes related to the pathogenesis of BAV. Our data speculate that these variants are promising markers for risk stratification of BAV patients with increased susceptibility to aortic stenosis.
Similar content being viewed by others


Background
Bicuspid aortic valve (BAV) is common congenital heart disease, and the prevalence rate is about 1 to 2% in the population. Some patients with BAV showed a family aggregation tendency [1]. Genetic studies showed that BAV had the characteristics of autosomal dominant inheritance and incomplete penetrance [2]. Aortic stenosis is the most common complication in patients with BAV. The pathophysiological basis of its formation includes endothelial dysfunction, local inflammation, lipid deposition, and secondary valve leaf emaciation [3]. Compared with patients with the tricuspid aortic valve, the time of valve stenosis in BAV patients is 10 years before, the progress is faster and higher mortality [4]. Once the patients with BAV have chest pain, syncope, and other symptoms, the alternative treatment is only valve replacement. However, there are some limitations and complications in mechanical or biological valves. The survival period of the untreated BAV patients with severe aortic stenosis is usually less than 10 years, especially for patients with heart failure [5]. Therefore, it is urgent to explore the pathogenesis of early calcification of BAV, carry out risk stratification for patients with asymptomatic BAV, delay the progression of the disease, and avoid surgery.
The heart valves of healthy people are composed of valve endothelial cells (VECs), valve interstitial cells (VICs), and extracellular matrix (ECM). VECs cover the valve surface, contact with blood, and maintain valve homeostasis by regulating permeability and inflammatory cell adhesion [6, 7]. VECs participate in heart valve formation through EndMT: endothelial to mesenchymal transformation [8]. VICs are the main cell groups of valve stroma, which constitute the skeleton of valve structure and play a role through their proliferation, differentiation, and secretion of ECM components. ECM provides physical and mechanical support for maintaining a certain morphological structure of the valve. The pathological characteristics of BAV are inflammatory infiltration, the synthesis of the fibrotic matrix after activation of valve interstitial cells (VICs), thickening, calcified mineral deposition in extracellular matrix (ECM), and then the obstruction of valve movement and blood flow. Calcification is a key process of aortic valve stenosis. When calcification occurs, alpha smooth muscle actin (α-SMA) can be activated and expressed in VICs, which can be transdifferentiated into myofibroblasts and show an osteoblast-like phenotype, which leads to massive calcium deposition and ossification, and eventually aortic valve stenosis [9].
Previous epidemiological studies have described the familial pattern of bicuspid aortic valve consistent with heredity and pointed out that genetic factors contribute more to disease susceptibility than environmental factors [10]. Genomic methods have just begun to elucidate the genetic determinants of BAV and have identified several pathogenic variants, such as NOTCH1, GATA5, TGFBR1, and TGFBR2 [11]. However, the penetrance of BAV is low, and currently, reported genes are mostly a form of familial studies. On the other hand, BAV is a heterogeneous disease, and many unknown variations need to be identified in sporadic BAV patients.
The present study aims to find the possible characteristic mutation gene in BAV. Deployed a two-step strategy to evaluate the clinical significance of germline genetic markers in BAV patients. We carried out whole-exome sequencing (WES) in 20 BAV patients (WES cohort) to identify potential pathogenic genes by bioinformatics analysis and in silico prediction. Then we selected several candidate genes for sequencing in independent BAV patients (Validation cohort).
Materials and methods
Study population
Patients with bicuspid aortic valves were selected from the Department of Cardiology of our hospital from January 2018 to December 2020 and were diagnosed by transthoracic echocardiography. Inclusion criteria included: (1) age ≥ 18 years old; (2) echocardiographic results: Patients showed one or more punctate or annular echo enhancement of aortic valve with a diameter more than 1 mm.
Exclusion criteria included: (1) acute infection; (2) history of rheumatic disease; (3) infective endocarditis; (4) congenital aortic valve malformation, such as Marfan syndrome, Loeys-Dietz syndrome (LDS), and other congenital cardiac defects; (5) being treated with anti-osteoporosis drugs. Eventually, 157 BAV patients were collected in this study: 20 patients were part of the WES cohort for exon sequencing, and the other 137 were part of the validation cohort for Sanger sequencing on selected genes.
The validation cohort consisted of 137 BAV. We also collected 130 cases of physical examination in our hospital during the same period as the control cohort. They were all tricuspid aortic valves and excluded from heart valve disease by color Doppler echocardiography. The control cohort consisted of 76 males and 54 females with an average of 62.9 ± 10 years. This study was carried out by the principles of the Declaration of Helsinki and was approved by the ethics committee of Zhongshan Hospital. Informed consent was obtained from all patients.
DNA extraction
Genomic DNA was isolated from peripheral whole blood samples that were cryopreserved under − 80 °C using QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany) and was quantified using a fluorometer or a Microplate Reader (Qubit Fluorometer, Invitrogen, Carlsbad, CA), with 260/280 ratios ranging from 1.75 to 2.00 for all DNA samples. Agarose Gel Electrophoresis (concentration of agarose gel: 1%, voltage: 150 V, electrophoresis time: 40 min) detected sample integrity and purification.
Whole‑exome sequencing
Genomic DNA extracted from whole blood samples was fragmented into 150 BP-220 BP by covaries, and the library was constructed and captured by Agilent sure select Human ALL Exon V6 kit. Terminal repair, Ploya tail addition, sequencing adaptor addition, purification, magnetic bead capture, PCR amplification, and other steps (Agilent Technologies, Santa Clara, CA, USA) finally constructed the DNA fragments. OEbiotech (Shanghai, China) performed next-generation sequencing on the Illumina HiSeq-2500 platform by BGI (Shenzhen, China). The average coverage of 190 × on target regions, of targeted bases, 99.91% was covered by at least 1 ×, and 99.34% was covered by at least 10 × coverage. Using BWA (Burrows-Wheeler Aligner) software, short reads mapping and alignment were performed. Single nucleotide polymorphisms (SNPs) were detected using GATK (Genome Analysis Tool Kit) v3.3.0 HaplotypeCaller. All reference sequences were based on the NCBI37/hg19 assembly of the human genome.
Single nucleotide variant (SNV) analysis
We selected variants in exon or splicing sites. We only included nonsynonymous SNV, such as missense, nonsense, and splicing site with minor allele frequency (MAF) < 0.05 in both 1000Genome and 1000Genome East Asia databases. The potential impact of missense mutations on protein function was evaluated using SIFT and Polyphen, two computational methods. SIFT scores, ranging from 0 to 1. The SIFT score represents the probability of toleration for a particular amino acid substitution, ranging from 0 to 1, and a score below the cutoff value of 0.05 is generally considered harmful. Polyphen is used to calculate the posterior probability to predict the pathogenicity of mutation based on evolutionary conservatism and the protein's three-dimensional structure. The predicted results were D: potentially harmful (score = 0.957 ~ 1), P: possibly harmful (score = 0.453 ~ 0.956), B: benign (score = 0 ~ 0.452). The variants implicated in disease were assessed via in silico prediction tool analysis (SIFT and Polyphen). The recurrent pathogenic variant was defined as a variants implicated in disease that appeared at least in two patients in the WES cohort.
Molecule annotation and network analysis
Single nucleotide polymorphisms (SNPs) were predicted and annotated by comparison using National Center for Biotechnology Information (NCBI) dbSNP version 141. Each SNP was mapped on the genome, and the number of SNP on detailed regions, such as coding region, untranslational region, an intron, was annotated. Nonsynonymous SNP information was extracted by comparing UCSC reference gene information (http://genome.Ucsc.edu/). Gene Ontology (GO) and KEGG pathway enrichment were analyzed by STRING online tools (http://string-db.org/).
Statistical analysis
Quantitative variables were expressed as mean and standard deviation, and category variables were expressed as cases (percentage). Statistical analyses were carried out with Statistical Package for the Social Sciences (SPSS) 20.0. Continuous variables between two subgroups were compared using the unpaired two-sided t-test. Categorical variables were analyzed using Chi-square or Fisher’s exact tests. Patients whose data were missing were not included in the analysis. A P-value < 0.05 was considered statistically significant.
Results
General information of 20 BAV patients
WES analysis was performed on 20 BAV patients. There were 12 BAV male patients with an average age of 67 ± 12 years, among all had a mean aortic valve gradient ≥ 40 mmHg and aortic valve orifice area ≤ 0.8 mm2, and 3 (15%) had moderate or severe aortic valve regurgitation (Table 1).
General features of whole-exon sequencing
WES analysis revealed an average of 299,980 SNPs (272,788 to 342,694) in 20 BAV samples. There are an average of 12,347 synonymous mutations in the overall SNP and 12,009 missense mutations in the coding region, including 108 SNPs making a stop codon and 15 SNPs making the stop codon a non-stop codon (Table 2).
Gene Ontology (GO) and KEGG pathway
We then filtered the results of the SNPs from sequencing to obtain the mutation gene, which changes the function of a protein. We compared the sequencing results of all samples to the reference genome, extracted all SNPs loci data for subsequent analysis, and obtained 37,225 SNPs loci. This SNPs site contains the site that changes the protein function and contains known high-frequency mutation sites. The synonymous mutation and unknown function mutation sites were removed. Then the SNPs were selected so that the variants have a MAF < 0.05 in both the 1000G and 1000G East Asia database. In the end, 14,862 SNPs sites from 9674 genes were left.
Gene Ontology (GO) and KEGG pathway enrichment were performed to analyze the most common molecular function and biological processes categories, respectively. Using the David database, 9674 genes were analyzed for GO. This analysis describes the three major components of the gene. The biological process is the main biological function of the gene-encoded protein; cell components are the main rich cellular areas of gene products; molecular functions are the possible activities of gene products at the molecular level. The top three enriched GO categories of SNP were cellular process, biological regulation, and metabolic process, cell part, organelle and membrane in cell components, binding, catalytic activity, and molecular transducer activity in molecular functions (Fig. 1). Based on the KEGG database and kobas database, signal pathway enrichment analysis was performed. And the top three enriched pathways of SNP were Signal transduction, Global and overview maps, and Infectious diseases (Fig. 2). Furthermore, the mutant gene's top three enriched KEGG pathways were Focal adhesion, Rap1 signaling pathway, and Phagosome (Fig. 3).

Gene Ontology of candidate genes with SNPs. This SNPs change the amino acid of protein and have a MAF < 0.05 in both 1000G and 1000G East Asia database. Only top enriched GO categories are shown in aspect of biological process, cell components and molecular functions from 9674 genes with 14,862 SNPs sites

KEGG pathway analysis of candidate genes with SNPs. Only top enriched pathway categories were shown in aspect of cellular processes, environmental information processing, genetic information processing, human diseases, metabolism, and organismal systems

Enriched mutant genes by KEGG pathway analysis. Gene Ontology/pathway analysis of candidate genes. The top 20 pathways are shown
Candidate recurrent variants implicated in disease in the WES cohort
The filtered variants were assessed via in silico prediction tool analysis (SIFT and Polyphen) to identify 1070 pathogenic variants. Two hundred forty-five recurrent variants were selected that appeared in at least two patients in the WES cohort [10]. Combined with a literature review for the biological function of genes with these variants, we identified 40 different recurrent pathogenic from 36 candidate genes, including MIB2, FAAH, S100A1, RGS16, MAP3K19, NEB, TTN, TNS1, CAND2, CCK, KALRN, ATP10D, SLIT3, ROS1, FABP7, NUP205, IL11RA, NPR2, COL5A1, CUBN, JMJD1C, ANXA7, TRIM8, LGR4, TPCN2, APOA5, GPR84, LRP1, NCOR2, AKAP11, ESRRB, NGB, AKAP13, WWOX, KCNJ12, ARHGEF1 (Table 3).
Genetic markers in the validation cohort
We performed a retrospective study on 137 BAV patients and sequenced their frozen DNA in 9 genes to confirm the WES results. These genes are chosen from 40 candidate genes with recurrent variants implicated in disease via unanimous agreement of in silico prediction tool analysis and are mostly related to BAV, including MIB2, S100A1, TTN, CCK, NUP205, LGR4, NCOR2, ESRRB, and WWOX. The panel of 9 variants implicated in disease was found in a total of 87 patients who had at least one heterozygous mutation among these genes, including 13 with MIB2, 11 with S100A1, 12 with TTN, 10 with CCK, 11 with NUP205, 14 with LGR4, 13 with NCOR2, 25 with ESRRB, and 14 with WWOX. The frequency of these 9 variants was significantly higher compared to healthy subjects with tricuspid aortic valves (Table 4). We then investigated the influence of these variants on the characteristics of BAV patients. Compared to 50 patients without a genetic marker, those harboring germline mutation demonstrated reduced LVEF, Left Ventricular Ejection Fractions (63.8 ± 7.5% vs. 58.4 ± 5.2%, P < 0.001), and larger calcification volume [(1129.3 ± 154) mm3 vs. (1261.8 ± 123) mm3, P < 0.001] (Table 5). We also divided all 137 BAV patients into wide-type and variant groups according to one of the nine genes to compare the LVEF and calcification volume. LVEF was significantly smaller in patients with variant TTN, NUP205, and NCOR2 Compared to patients with wild-type alleles (Fig. 4). Furthermore, calcification volumes are significantly larger in patients with variant S100A1, LGR4, ESRRB, and WWOX than in patients with wide-type alleles (Fig. 5).

Comparison LVEF between normal and variant genes in BAV patients. Compared to patients with wide-type allele, patients with variant TTN, NUP205 and NCOR2 have significantly reduced LVEF. Values are expressed as Mean ± SD. *P < 0.05 versus wide-type group. LVEF, Left Ventricular Ejection Fractions

Comparison of calcification volume between normal and variant genes in BAV patients. Compared to patients with wide-type allele, patients with variant S100A1, LGR4, ESRRB and WWOX have significantly larger calcification volume. *P < 0.05, **P < 0.01 versus wide-type group
Discussion
In this study, we performed whole-exon sequencing on 20 sporadic BAV patients to explore the potential genetic variations that may contribute to the pathogenesis of BAV. We identified 40 different heterozygous missense mutations in 36 genes. These are recurrent variants implicated in disease in that they appeared in at least two patients and were selected by in silico prediction tool analysis from 14,826 nonsynonymous SNV in exons. Then nine genes (MIB2, S100A1, TTN, CCK, NUP205, LGR4, NCOR2, ESRRB, and WWOX) were selected for sequencing to validate the WES results in an independent cohort of 137 BAV patients. 87 patients carry at least one variant, and 50 patients do not have any variant among these nine genes. Patients with germline mutations showed reduced LVEF and larger calcification volume than patients with a wide-type allele in all nine genes. The data indicate that these genes with recurrent variants implicated in disease may involve the pathogenesis of BAV.
This study speculated the hypothesis that genetic variations increase the susceptibility to BAV. Here we rediscovered two genes, COL5A1 and KCNJ12, in BAV patients. COL5A1 is an ECM-related genes, and its variant (COL5A1: c.A3481T:p.I1161F) was identified as variants implicated in disease in BAV [12]. We identified a new variant of COL5A1 (c.G378T:p.Q126H; rs145178917) in BAV, a common SNP (1000G-EA: 0.0347). Another reported gene is KCNJ2, and its heterozygous missense mutation (R67W) was detected in Andersen syndrome with cardiovascular malformation of the bicuspid aortic valve [13]. Interestingly, a heterozygous mutation in KCNJ12 (p.Glu334del) was identified as a candidate mutation in dilated cardiomyopathy [14], whose mutation site is close to our results (p.G145S). Whether KCNJ12 plays a role in the pathological mechanism of BAV remains unclear.
Due to the low penetrance and heterogeneity of BAV, many unknown genes may influence the susceptibility and progression of BAV, especially in sporadic BAV. This study uncovered many variants of candidate genes that have not previously been implicated in BAV. These genes that carry recurrent variants implicated in disease can be divided into several main cellular and molecular mechanisms associated with BAV. Some mutated genes are related to atherosclerosis, such as FAAH [15], KALRN [16], ATP10D [17], CUBN [18], APOA5 [19], and LRP1 [20]. Atherosclerosis share several molecular mechanisms with BAV, including dyslipidemia and the activation of specific pro-inflammatory pathways (NLRP3 inflammasome and TRL4) [21]. The SNPs in KALRN (rs9289231), ATP10D (rs2351791), CUBN (rs2291521), and APOA5 (Rs662799) are all significantly associated with the risk of coronary artery disease (CAD). Cardiac hypertrophy is common in BAV patients with increased LV mass and reduced aortic elasticity [22]. Genes associate with cardiac hypertrophy included JMJD1C [23], ANXA7 [24], TRIM8 [25], NGB [26], and AKAP13 [27]. Cardiac fibrosis is another pathogenic process in BAV. BAV patients with left ventricular (LV) fibrosis were more likely to progress to aortic stenosis that needed aortic valve replacement [28]. We also detected variations in genes involving cardiac fibrosis, such as CCK [29], SLIT3 [30], IL11RA [31], and ARHGEF1 [32]. Some identified genes are involved in the osteogenesis process, including GPR84 [33] and AKAP11 [34]. Other candidate genes are a pathway of known genes in BAV. For instance, MAP3K19 is a regulator of TGF-β [35], and FABP7 is a target of Notch1 [36].
We also sequenced 9 recurrent pathogenic genes for validation, whose allele frequency was significantly higher than healthy subjects with the tricuspid aortic valve. Patients with variant TTN, NUP205, and NCOR2 had significantly smaller LVEF than patients with wide-type alleles. The finding indicates the mutations TTN, NUP205, and NCOR2 can enhance the severity of aortic valve stenosis, a consequence of BAV. TTN gene encodes Titin, and it is a giant sarcomeric protein that regulates passive myocardial stiffness. The expression of less Titin isoform (N2BA and N2B) was changed in left ventricular biopsies of patients with aortic stenosis [37]. This change in Titin is in response to pressure overload and might further promote myocardial fibrosis or severe aortic stenosis [38]. NUP205 can modulate cilia function, and its depletion leads to loss of cilia and abnormal cardiac morphology [39]. Cilia participate in aortic valve morphogenesis, and recently defects in the cilia machinery have been discovered as a causal factor in BAV and aortic stenosis [40, 41]. NCOR2 is related to the Notch signaling pathway [42], but its role in BAV is unclear.
We found 4 genes, including S100A1, LGR4, ESRRB, and WWOX, are associated with the calcification volume of BAV patients. S100A1 modulates the molecular pathways and signaling cascades in cardiomyocytes, endothelial cells, and cardiac fibroblasts [43]. It modulates the function of cardiomyocytes via TLR4/ROS/NF-κB pathway [44], which is involved in enhanced osteogenic responses in human aortic valve cells [45]. LGR4 protects against ischemic injury of cardiomyocytes by modulating mitochondrial function and oxidative stress [46]. GPR48 also is another receptor for RANKL modulating osteoclast differentiation [47]. ESRRB can decrease calcium sensitivity in cardiomyocytes and thus promote cardiomyocyte contractility [48]. WWOX can modulate cellular lipid homeostasis by increasing serum HDL cholesterol concentrations, which may affect the progression of atherosclerotic disease [49]. Genome-wide association study of the gene showed genetic variants in WWOX are correlated with coronary artery calcification [50].
Limitation
The current investigation did not provide any additional evidence that detected genetic variants were responsible for the clinical manifestations of BAV patients. The present study's limitations were surmounted by the in vitro confirmation of these variations' biological effects, which warranted further investigations. To see a complete picture of the variant interpretation, more recent prediction tools (e.g., CADD) and a more recent genome-scale database (e.g., gnomAD) could be used.
Conclusion
In sum, we performed whole-exon sequencing in 20 sporadic BAV patients. We found 40 recurrent variants implicated in disease in 36 genes, and 9 variants were validated in another cohort of BAV patients (Fig. 6). Recurrent missense mutations on TTN, NUP205, NCOR2, S100A1, LGR4, ESRRB, and WWOX could be identified as potential pathogenic genes and associated with an elevated allele frequency, reduced left ventricular ejection fractions, and larger calcification volume in BAV patients.

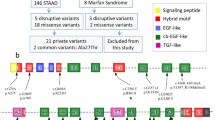
The selection process for WES data. It contains 37,225 total coding variants with nonsynonymous mutation. Then filtered 14,826 common variants for GO and KEGG analysis. One thousand and seventy variants implicated in disease are selected via in silico prediction tool analysis, 245 recurrent variants implicated in disease are selected that exist in at least two patients. Forty candidate variants among 36 genes are selected after the literature review that may be associated with phenotype of BAV. Finally, 9 genes were selected for validation in another cohort of 137 BAV patients by sequencing
Availability of data and materials
The data reported in this study are available upon a valid request from the corresponding author.
References
Verma S, Siu SC. Aortic dilatation in patients with bicuspid aortic valve. N Engl J Med. 2014;370(20):1920–9.
Bravo-Jaimes K, Prakash SK. Genetics in bicuspid aortic valve disease: Where are we? Prog Cardiovasc Dis. 2020;63(4):398–406.
Fishbein GA, Fishbein MC. Pathology of the aortic valve: aortic valve stenosis/aortic regurgitation. Curr Cardiol Rep. 2019;21(8):81.
Cotier P, Bruneval P, Amemiya K. Vascular malformation in a bicuspid aortic valve. Cardiovasc Pathol. 2019;38:39–41.
Yoon SH, Maeno Y, Kawamori H, Miyasaka M, Nomura T, Ochiai T, Nemanpour S, Raschpichler M, Sharma R, Chakravarty T, Makkar R. Diagnosis and outcomes of transcatheter aortic valve implantation in bicuspid aortic valve stenosis. Interv Cardiol. 2018;13(2):62–5.
Sakellaropoulos S, Mohammed M, Svab S, Lekaditi D, Sakellaropoulos P, Mitsis A. Causes, diagnosis, risk stratification and treatment of bicuspid aortic valve disease: an updated review. Cardiol Res. 2020;11(4):205–12.
Dahal S, Huang P, Murray BT, Mahler GJ. Endothelial to mesenchymal transformation is induced by altered extracellular matrix in aortic valve endothelial cells. J Biomed Mater Res A. 2017;105(10):2729–41.
Tao G, Kotick JD, Lincoln J. Heart valve development, maintenance, and disease: the role of endothelial cells. Curr Top Dev Biol. 2012;100:203–32.
Leopold JA. Cellular mechanisms of aortic valve calcification. Circ Cardiovasc Interv. 2012;5(4):605–14.
Galian-Gay L, CarroHevia A, Teixido-Turà G, et al. BICUSPID investigators. Familial clustering of bicuspid aortic valve and its relationship with aortic dilation in first-degree relatives. Heart. 2019;105(8):603–8.
Foffa I, Ait Alì L, Panesi P, et al. Sequencing of NOTCH1, GATA5, TGFBR1 and TGFBR2 genes in familial cases of bicuspid aortic valve. BMC Med Genet. 2013;14:44.
Wu B, Li J, Wang Y, et al. Recurrent germline mutations as genetic markers for aortic root dilatation in bicuspid aortic valve patients. Heart Vessels. 2021;36(4):530–40.
Andelfinger G, Tapper AR, Welch RC, et al. KCNJ2 mutation results in Andersen syndrome with sex-specific cardiac and skeletal muscle phenotypes. Am J Hum Genet. 2002;71(3):663–8.
Yuan HX, Yan K, Hou DY, et al. Whole exome sequencing identifies a KCNJ12 mutation as a cause of familial dilated cardiomyopathy. Medicine. 2017;96(33):e7727.
Lenglet S, Thomas A, Soehnlein O, et al. Fatty acid amide hydrolase deficiency enhances intraplaque neutrophil recruitment in atherosclerotic mice. Arterioscler Thromb Vasc Biol. 2013;33(2):215–23.
Shafiei A, Pilehvar-Soltanahmadi Y, Ziaee S, et al. Association between Serum Kalirin Levels and the KALRN gene rs9289231 polymorphism in early-onset coronary artery disease. J Tehran Heart Cent. 2018;13(2):58–64.
Kengia JT, Ko KC, Ikeda S, et al. A gene variant in the Atp10d gene associates with atherosclerotic indices in Japanese elderly population. Atherosclerosis. 2013;231(1):158–62.
Park HS, Kim IJ, Kim EG, et al. A study of associations between CUBN, HNF1A, and LIPC gene polymorphisms and coronary artery disease. Sci Rep. 2020;10(1):16294.
Chen H, Ding S, Zhou M, et al. Association of rs662799 in APOA5 with CAD in Chinese Han population. BMC Cardiovasc Disord. 2018;18(1):2.
Boucher P, Herz J. Signaling through LRP1: protection from atherosclerosis and beyond. Biochem Pharmacol. 2011;81(1):1–5.
Magni P. Bicuspid aortic valve, atherosclerosis and changes of lipid metabolism: Are there pathological molecular links? J Mol Cell Cardiol. 2019;129:231–5.
Grotenhuis HB, Ottenkamp J, Westenberg JJM, et al. Reduced aortic elasticity and dilatation are associated with aortic regurgitation and left ventricular hypertrophy in nonstenotic bicuspid aortic valve patients. J Am Coll Cardiol. 2007;49(15):1660–5.
Zhang S, Lu Y, Jiang C. Inhibition of histone demethylase JMJD1C attenuates cardiac hypertrophy and fibrosis induced by angiotensin II. J Recept Signal Transduct Res. 2020;40(4):339–47.
Voelkl J, Alesutan I, Pakladok T, et al. Annexin A7 deficiency potentiates cardiac NFAT activity promoting hypertrophic signaling. Biochem Biophys Res Commun. 2014;445(1):244–9.
Chen L, Huang J, Ji YX, et al. Tripartite motif 8 contributes to pathological cardiac hypertrophy through enhancing transforming growth factor β-activated kinase 1-dependent signaling pathways. Hypertension. 2017;69(2):249–58.
Liu ZF, Zhang X, Qiao YX, et al. Neuroglobin protects cardiomyocytes against apoptosis and cardiac hypertrophy induced by isoproterenol in rats. Int J Clin Exp Med. 2015;8(4):5351–60.
Johnson KR, Nicodemus-Johnson J, Spindler MJ, et al. Genome-wide gene expression analysis shows AKAP13-mediated PKD1 signaling regulates the transcriptional response to cardiac hypertrophy. PLoS ONE. 2015;10(7):e0132474.
Lluri G, Renella P, Finn JP, et al. Prognostic Significance of left ventricular fibrosis in patients with congenital bicuspid aortic valve. Am J Cardiol. 2017;120(7):1176–9.
Wang C, Zhang C, Wu D, et al. Cholecystokinin octapeptide reduces myocardial fibrosis and improves cardiac remodeling in post myocardial infarction rats. Int J Biochem Cell Biol. 2020;125:105793.
Gong L, Wang S, Shen L, et al. SLIT3 deficiency attenuates pressure overload-induced cardiac fibrosis and remodeling. JCI Insight. 2020;5(12):e136852.
Corden B, Adami E, Sweeney M, et al. IL-11 in cardiac and renal fibrosis: late to the party but a central player. Br J Pharmacol. 2020;177(8):1695–708.
Numaga-Tomita T, Kitajima N, Kuroda T, et al. TRPC3-GEF-H1 axis mediates pressure overload-induced cardiac fibrosis. Sci Rep. 2016;19(6):39383.
Park JW, Yoon HJ, Kang WY, et al. G protein-coupled receptor 84 controls osteoclastogenesis through inhibition of NF-κB and MAPK signaling pathways. J Cell Physiol. 2018;233(2):1481–9.
Park S, Daily JW, Song MY, et al. Gene-gene and gene-lifestyle interactions of AKAP11, KCNMA1, PUM1, SPTBN1, and EPDR1 on osteoporosis risk in middle-aged adults. Nutrition. 2020;79–80:110859.
Boehme SA, Franz-Bacon K, DiTirro DN, et al. MAP3K19 Is a novel regulator of TGF-β signaling that impacts bleomycin-induced lung injury and pulmonary fibrosis. PLoS ONE. 2016;11(5):e0154874.
Xie M, Wu X, Zhang J, et al. The prognostic significance of Notch1 and fatty acid binding protein 7 (FABP7) expression in resected tracheobronchial adenoid cystic carcinoma: a multicenter retrospective study. Cancer Res Treat. 2018;50(4):1064–73.
Williams L, Howell N, Pagano D, et al. Titin isoform expression in aortic stenosis. Clin Sci (Lond). 2009;117(6):237–42.
Gotzmann M, Grabbe S, Schöne D, et al. Alterations in titin properties and myocardial fibrosis correlate with clinical Phenotypes in hemodynamic subgroups of severe aortic stenosis. JACC Basic Transl Sci. 2018;3(3):335–46.
Marquez J, Bhattacharya D, Lusk CP, et al. Nucleoporin NUP205 plays a critical role in cilia and congenital disease. Dev Biol. 2021;469:46–53.
Toomer KA, Fulmer D, Guo L, et al. A role for primary cilia in aortic valve development and disease. Dev Dyn. 2017;246(8):625–34.
Dang Y, Su Y, Fogelgren B, et al. Defects in the exocyst-cilia machinery cause bicuspid aortic valve disease and aortic stenosis. Circulation. 2019;140(16):1331–41.
Zhang W, Liu H, Liu Z, et al. Functional variants in notch pathway genes NCOR2, NCSTN, and MAML2 predict survival of patients with cutaneous melanoma. Cancer Epidemiol Biomarkers Prev. 2015;24(7):1101–10.
Rohde D, Busch M, Volkert A, et al. Cardiomyocytes, endothelial cells and cardiac fibroblasts: S100A1’s triple action in cardiovascular pathophysiology. Future Cardiol. 2015;11(3):309–21.
Yu J, Lu Y, Li Y, et al. Role of S100A1 in hypoxia-induced inflammatory response in cardiomyocytes via TLR4/ROS/NF-κB pathway. J Pharm Pharmacol. 2015;67(9):1240–50.
Zeng Q, Song R, Ao L, et al. Augmented osteogenic responses in human aortic valve cells exposed to oxLDL and TLR4 agonist: a mechanistic role of Notch1 and NF-κB interaction. PLoS ONE. 2014;9(5): e95400.
Chen T, Qiao X, Cheng L, et al. LGR4 silence aggravates ischemic injury by modulating mitochondrial function and oxidative stress via ERK signaling pathway in H9c2 cells. J Mol Histol. 2021;52(2):363–71.
Jang Y, Sohn HM, Ko YJ, et al. Inhibition of RANKL-induced osteoclastogenesis by Novel Mutant RANKL. Int J Mol Sci. 2021;22(1):434.
Rowe GC, Asimaki A, Graham EL, et al. Development of dilated cardiomyopathy and impaired calcium homeostasis with cardiac-specific deletion of ESRRβ. Am J Physiol Heart Circ Physiol. 2017;312(4):662–71.
Iatan I, Choi HY, Ruel I, et al. The WWOX gene modulates high-density lipoprotein and lipid metabolism. Circ Cardiovasc Genet. 2014;7(4):491–504.
Polfus LM, Smith JA, Shimmin LC, et al. Genome-wide association study of gene by smoking interactions in coronary artery calcification. PLoS ONE. 2013;8(10): e74642.
Acknowledgements
We sincere gratitude to all patients and their relatives who participated in this study.
Funding
This study was supported by 1) Scientific research plan project of Shanghai science and technology committee (No.19DZ1930202); 2) Shanghai Clinical Research Center for Interventional Medicine(No.19MC1910300).
Author information
Authors and Affiliations
Contributions
SC, QJ and DZ designed the project, performed the WES data analysis and in silico analysis. SC and QJ wrote the first draft of the Manuscript. SH, ML, YZ, LG, WP, and JG were collected patients data and performed intellectual discussion. DZ supervised the project and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethics Committee of Zhongshan hospital, Fudan University and conducted in accordance with the 1964 Declaration of Helsinki and its later revisions.
Consent for publication
Written informed consent was obtained from all patients, their relatives and parents (if any).
Competing interests
All authors in this article declared that they do not have any competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chen, S., Jin, Q., Hou, S. et al. Identification of recurrent variants implicated in disease in bicuspid aortic valve patients through whole-exome sequencing. Hum Genomics 16, 36 (2022). https://doi.org/10.1186/s40246-022-00405-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40246-022-00405-z