
Abstract
Neurodegenerative diseases, such as Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, and Huntington’s disease, affect millions of people worldwide. Tremendous efforts have been put into disease-related research, but few breakthroughs have been made in diagnostic and therapeutic approaches. Extracellular vesicles (EVs) are heterogeneous cell-derived membrane structures that arise from the endosomal system or are directly separated from the plasma membrane. EVs contain many biomolecules, including proteins, nucleic acids, and lipids, which can be transferred between different cells, tissues, or organs, thereby regulating cross-organ communication between cells during normal and pathological processes. Recently, EVs have been shown to participate in various aspects of neurodegenerative diseases. Abnormal secretion and levels of EVs are closely related to the pathogenesis of neurodegenerative diseases and contribute to disease progression. Numerous studies have proposed EVs as therapeutic targets or biomarkers for neurodegenerative diseases. In this review, we summarize and discuss the advanced research progress on EVs in the pathological processes of several neurodegenerative diseases. Moreover, we outline the latest research on the roles of EVs in neurodegenerative diseases and their therapeutic potential for the diseases.
Similar content being viewed by others


Introduction
Neurodegenerative disorders such as Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS) and Huntington’s disease (HD) are a heterogeneous group of diseases characterized by gradual progression and selective loss of anatomically or physiologically related neurons, which significantly impair cognitive or behavioral abilities [1,2,3]. The primary hallmark of neurodegenerative disorders is the accumulation of misfolded proteins into insoluble aggregates (or inclusions) in the central nervous system (CNS), accompanied by progressive neuronal degeneration in the affected regions [4]. The mortality and morbidity associated with these disorders are rapidly increasing owing to the aging of the global population [5].
However, both AD and PD are associated with low detection efficiency due to the lack of available biomarkers [6]. The existing biomarkers allow diagnosis only at advanced stages of disease [7]. Until now, proteomic studies using complete blood, cerebrospinal fluid (CSF), saliva, and urine samples have identified several biomarkers for neurodegenerative diseases [8]. However, the biofluid-based methods have limitations, such as the extremely low concentrations of the protein biomarkers (estimated to account for less than one-millionth of the total CSF proteins and one ten-billionth of the total blood proteins) [9]. The blood–brain barrier (BBB) prevents the free passage of molecules between the CNS and blood, leading to a difference between CSF and blood. However, CSF collection is invasive in nature and is unacceptable when early symptoms of disease are not apparent [10]. In addition, the biomarkers are also expressed in other tissues in addition to the brain, which may confound their measurement in biofluids. Currently, the gold standard for the diagnosis of neurodegenerative diseases remains brain imaging, either magnetic resonance imaging or positron emission tomography [11]. Although imaging is highly sensitive, its accuracy depends on the experience and skill of the operator [12]. Given these clinical challenges, the discovery of specific in vivo biomarkers, including biofluid and molecular imaging biomarkers, is a major research priority for neurodegenerative disorders.
Extracellular vesicles (EVs) are essential mediators of communication between cells. In the CNS, EVs transmit signaling information between nerve cells and contribute to their development and function [13]. The EVs released from the brain and the spinal cord are proposed to be unique to the CNS [14]. In neurodegenerative disorders, pathological molecules are transferred to healthy tissues by EVs to perform pathological functions. Additionally, EVs are thought to play a protective role by expelling pathological molecules from cells [15]. In this review, we outline the evidence for the interaction of EVs with many of the specific proteins, nucleic acids and lipids implicated in neurodegenerative diseases, demonstrating that EVs are key regulators of neuronal dysfunction and death and play a central role in cell-to-cell communication and neurodegenerative disease progression. In addition, we discuss the latest research on the therapeutic potential of EVs for neurodegenerative diseases.
Classification of EVs
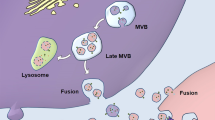
EVs are nanosized vesicles (30–2000 nm) with lipid bilayer membranes, which are actively secreted by almost all cells. The membrane structure of EVs protect the contents from destruction by the extracellular environment [16, 17]. EVs can be divided into small extracellular vesicles (sEVs), microvesicles (MVs), and apoptotic bodies based on their dimensions, as proposed by the MISEV guidelines [18] (Fig. 1).

Classification and biogenesis of extracellular vesicles. Cells can assimilate extracellular substances by plasma membrane invagination and endocytosis. The late sorting endosomes (LSEs) are transformed from vesicles fused with the early sorting endosomes (ESEs). Intraluminal vesicles (ILVs) are caused by a second invagination of the LSEs. Multivesicular bodies (MVBs), further transformed from LSEs, can fuse with lysosomes or autophagosomes for degradation, or with the plasma membrane to release ILVs, which are termed small extracellular vesicles. Microvesicles are produced from the outward budding and fission of the plasma membrane. Apoptotic bodies are large vesicles formed by apoptotic cells
sEVs (40–200 nm in diameter) are smallest EVs and are secreted by various living cells [19]. Fundamentally, sEVs are generated within cells through the endosomal pathway in three stages. First, the plasma membrane is invaginated to produce endocytic vesicles, some of which fuse to form early sorting endosomes (ESEs); this process involves the participation of proteins such as endosomal sorting complex required for transport (ESCRT) proteins, tetraspanin proteins (CD9, CD63, and CD81), apoptosis-linked gene 2-interacting protein X (Alix), and tumor susceptibility gene 101 (TSG101). Subsequently, these ESEs may exchange materials with other organelles or fuse with different ESEs to transform into late sorting endosomes (LSEs), which can further transform into multivesicular bodies (MVBs). MVBs generate many intraluminal vesicles (ILVs) to pack intracellular substances; this process involves RAB GTPase proteins and cytoskeletal proteins such as actin and tubulin. Next, the MVBs fuse with the plasma membrane and release ILVs into the extracellular space, where they are defined as sEVs; the secretion of sEVs requires the participation of the SNARE protein complex and the synaptotagmin family [20,21,22].
MVs (100–1000 nm) were previously called “the platelet dust” and are generated through outward budding and fission of the plasma membrane; therefore, the membrane composition of MVs is close to that of the donor cells [23]. The production of MVs is related to the asymmetric distribution of phospholipids in the cell membrane bilayer [24]. Calcium influx can disrupt the asymmetry of phospholipids by activating phospholipid scramblase to redistribute phospholipids in the cell membrane bilayer. Simultaneously, calcium-dependent proteolytic enzymes degrade the membrane-bound cytoskeleton and initiate the production of MVs [25]. Some researchers have shown that ARRDC1 (arrestin-domain-containing protein 1) can recruit the ESCRT proteins TSG101 and Vps4 to the cell membrane to initiate membrane budding [26].
Apoptotic bodies (500–2000 nm) are comparatively giant vesicles derived from apoptotic cells, and contain cytoplasm, organelles, and nuclear debris [23]. Blebbing from the plasma membrane during apoptosis leads to formation of apoptotic bodies in the form of MVs [27].
EV cargos
The application of mass spectrometry and high-throughput sequencing has enabled large-scale screening and characterization of EV contents [28]. EVs contain various bioactive molecules, including soluble proteins, nucleic acids, lipids and metabolites, all of which play crucial roles in cell-to-cell communication and are responsible for delivering various signaling molecules to both proximal and distant locations (Fig. 2) [29].

Structure of extracellular vesicle. The phospholipid bilayer encapsulates different types of membrane proteins, intracellular proteins, DNA, RNA, lipids, and metabolites to form EVs. Several membrane and intracellular proteins are used as EV markers, including TSG101, Alix, CD63, CD9, and CD81
The proteins in EVs are mainly divided into two types: (1) ubiquitous proteins, such as those participating in the formation of EV structure, including cytoskeleton components (tubulin, actin, and microfilament-associated protein); certain conserved proteins, including those involved in ESCRT-dependent biogenesis (Annexins, Alix, and TSG101); ESCRT-independent tetraspanin family proteins, such as CD9, CD63, and CD81; and heat shock proteins, such as HSP70 and HSP90 [30]; and (2) proteins related to the original cell. For example, EVs derived from antigen-presenting cells are abundant in major histocompatibility complex (MHC)-I (MHC-I), MHC-II, CD80 and CD86; platelet-derived EVs contain the factors of hemophilia and integrin CD41A; and proteins abundant in the EVs of tumor cells, such as Fas ligand and transforming growth factor-β, are frequently related to tumorigenesis [31]. These proteins, which are absent on other types of vesicles, can be considered “markers of EVs” [32].
Nucleic acid fragments in EVs are usually ~ 200 bp in length, and some of them can be translated into functional proteins that affect the biological function of the recipient cell [33]. EVs contain many nucleic acids, including genomic DNA [34] and mitochondrial DNA [35], as well as RNA (mRNA, microRNA [miRNA], lncRNA, and circRNA) [36, 37]. Considered as the primary regulator of recipient cell activity, RNA can regulate the gene expression and function of target cells by directly participating in transcription, post-transcriptional processing, as well as protein translation and modification [38], and play a regulatory role in the biological function of cells and the progression of diseases [39]. miRNAs are small, 22-nt-long, non-protein-coding RNAs that induce posttranscriptional gene silencing by binding to their complementary mRNA targets and inhibiting translation and/or inducing degradation of mRNA [40]. Under physiological conditions, miRNA-dependent gene regulation ensures precise protein output and minimal protein expression noise [41]. miRNAs in the CNS control gene expression in various cell types in a highly regulated temporospatial pattern and a neuronal activity-dependent manner [42]. In addition, the remarkable stability of miRNAs in the extracellular environment and hence in body fluids, together with the availability of sensitive methods for their detection and quantitation, has led to the wide use of circulating miRNAs as biomarkers for various human disorders [43]. Meanwhile, the miRNA-based therapeutics mainly comprise synthetic miRNAs to restore endogenous miRNA levels (e.g., miRNA mimics) or antisense inhibitor oligonucleotides to reduce functionally available endogenous miRNAs, such as antamiRNAs and antagomiRNAs.
EVs are generally enriched with lipids, including cholesterol, sphingomyelin, ceramides, sphingolipids, glycerophospholipids, and glycosphingolipids [44]. Lipids including phosphatidylcholine, phosphatidylserines and phosphatidylinositols are found in lower quantities [45]. The lipid composition of EVs generally represents that of the donor cell. The membranes of EVs contain lysophosphatidic acid, which is central to the formation of ILVs from multivesicular endosomes [46]. These lipids not only participate in the biosynthesis and uptake of EVs, but also act as a class of bioactive molecules in various biological processes, including immunological surveillance, modification of the tumor microenvironment, and regulation of inflammation [47, 48]. The lipids in EVs may also be used as biomarkers for disease diagnosis and treatment [49].
Functions of EVs in the CNS
A common feature of neurodegenerative diseases is the misfolding, aggregation and accumulation of pathological amyloids inside or outside the brain cells. Accordingly, detection of these pathological proteins in body fluids and tissues may be a powerful tool for early diagnosis of these diseases. Effective therapies to delay or prevent the onset and progression of neurodegenerative diseases have not been established to date. In the past decade, EVs have been reported as novel and important carriers of signaling molecules in vivo. A growing body of literature has highlighted an important role of EVs in the cell-to-cell transmission of pathogenic protein aggregates, thereby contributing to the pathological and clinical progression of neurodegenerative diseases [50]. These EVs can carry and protect a range of proteins, lipids, and nucleic acids from degradation in the extracellular space [51]. After being delivered to target cells, EVs can influence the physiology of the recipient cells [52]. It has been observed that EVs are released by neurons, oligodendrocytes, microglia, and astrocytes in the CNS [53]. Recently, several studies have revealed the physiological roles of EVs in the CNS, including regulation of glutamatergic synaptic activity during nerve cell development. Astrocytes maintain brain homeostasis by internalizing miR-124 from microglia-derived EVs to regulate levels of glutamate transporter 1 and glutamate uptake [54]. Non-neuronal cells promote neurite growth and neuron survival through the EV-mediated release of neuroactive substances such as Hsp70 and synaptophysin I [55, 56]. Another study showed that stimulation of serotonin receptors increases the release of insulin-degrading enzymes from microglia via EVs, which are capable of degrading the neurotoxic peptide amyloid β (Aβ) [57]. EVs from human bone marrow-derived endothelial progenitor cells are able to repair the damaged microvasculature in the CNS of symptomatic SOD1-G93A mutant mice [58]. Additionally, some EVs play a key role in the pathological processes of neurological diseases. Sardar Sinha et al. [59] found that the EVs from AD brains contain abundant toxic Aβ and promote the progression of AD by spreading Aβ between neurons. Moreover, Guo et al. [60] found that microglial exosomes promote the intercellular transmission of α-synuclein (α-syn), and induce neurodegeneration in the substantia nigra and striatum, a key mechanism of PD pathogenesis. Therefore, EVs not only play an important role in CNS development, neuroprotection, repair, and further regulation of neuronal activity, but are also involved in the occurrence and development of CNS diseases.
As EVs can cross the BBB into the blood, neurally derived EVs are present in both the bloodstream and CSF [61]. Although EVs can be isolated from CSF, plasma, and serum, the neurally derived EVs are more concentrated in the CSF and have more specific diagnostic and research value for neurological diseases. However, isolation of EVs from CSF is currently not very feasible due to the complicated and difficult process of CSF collection. Therefore, detection of EVs in human blood (plasma and serum) is a relatively simple and powerful approach, although it may be complicated by the presence of admixtures from multiple sources in blood, including serum proteins or a mixture of EVs from other organs [62]. In general, detection of EVs from both CSF and blood each have their advantages, and the roles of these EVs in the pathology and diagnosis of neurodegenerative disorders are summarized below.
EVs and AD
AD is clinically characterized by progressive cognitive decline, and pathologically by plaques comprising Aβ peptide and nerve fiber tangles containing the hyperphosphorylated Tau protein [63, 64]. The Aβ peptide is produced from cleavage of amyloid precursor protein (APP), and the excessive phosphorylation of tau protein can lead to the separation of tau from microtubules and mutual aggregation, leading to formation of neurofibrillary tangles and deposition in axons and dendrites [65]. About 10% of AD cases occur in an early-onset autosomal dominant manner; they are called familial cases. The following three proteins are associated with familial AD cases: APP, which is sequentially cleaved by β and γ secretases to produce Aβ, and presenilins 1 and 2 (PS1 and PS2) which are subunits of the γ-secretase [66]. A recent study showed that the protease-containing plasma EVs may be part of the communication axis between the brain and the periphery, and they accelerate the pathogenesis of AD in transgenic mouse models by splitting APP or other substrates in target neurons, providing evidence for the pathogenic role of plasma EVs in AD [67, 68]. Therefore, the transmission of EVs between cells is an important factor in the pathogenesis and development of AD (Table 1).
Growing evidence has shown that small EVs (for example, exosomes) can serve as biomarkers for neurodegenerative diseases and that they are more reliable than conventional specimens such as pure CSF, blood and urine [92]. Initial evidence for a link between EVs and AD came from a study showing the accumulation of EV proteins such as flotillin-1 and Alix around amyloid plaques in the brains of AD patients, and Aβ peptide release in association with EVs [93]. Recently, Aβ has been shown to be stored either in the lumen or on the surface of microglial EVs [94]. More importantly, Joshi et al. reported in 2014 the first evidence for the neurotoxicity of EVs carrying Aβ[95]. Later, several in vivo studies provided further evidence that Aβ in association with EVs is associated with AD. The CSF EVs of patients with AD contain high levels of Aβ, which can cause damage to neurons in vitro and in animal models in vivo [59, 96]. A recent study has indicated that large microglial EVs containing Aβ are capable of escalating and propagating early synaptic dysfunction in AD between entorhinal cortex and dentate gyrus in the mouse brain by moving at the surface of neurons [69]. Additionally, the Aβ levels in neurally derived EVs in plasma or serum are gradually elevated during disease progression in AD patients compared to asymptomatic individuals, and can thus be used as a biomarker for disease diagnosis and to define disease progression [70]. It has been proposed that the EVs may stimulate Aβ aggregation and interfere with the uptake of Aβ aggregates by astrocytes and microglia, leading to Aβ aggregation in AD [71, 73].
The plasma or serum EVs of AD patients also contain tau protein, which can be transferred to neurons, causing tau accumulation in neurons — another major feature of AD [70, 72]. Additionally, total tau (t-tau) and hyperphosphorylated tau (p-tau) have been detected in the EVs of primary neuron cultures and neuroblastoma cells overexpressing tau [97]. In neurodegenerative diseases, activated microglia release higher levels of EVs than inactivated microglia, and in AD, these activated microglia secrete EVs containing tau protein, which can propagate to neurons and promote the progression of AD [74, 98]. However, the rapid tau propagation from the entorhinal cortex to the dentate gyrus can be dramatically suppressed by depletion of microglia and inhibition of EV synthesis [99]. The EVs secreted by induced pluripotent stem cells carrying mutant A246E PS1 contain high levels of APP and induce tau deposition in mouse brains after in vivo injection [75]. A recent study compared the diagnostic capabilities of neuronal-derived plasma EV and CSF Aβ1-42, p-tau181, and t-tau and found that the combination of these biomarkers in either EVs or CSF had superior diagnostic performance than each single biomarker [76]. Meanwhile, other studies have shown that the levels of soluble Aβ1-42, Aβ oligomer, p-tau181, and p-tau396 in EVs isolated from brain tissue, blood, and CSF of AD patients are significantly increased before the clinical diagnosis of AD, suggesting that these biomarkers from EVs can be used for AD diagnosis [77, 78]. The above studies indicate that EVs are closely related to the pathological process of AD. Although EVs are involved in AD, their role in the pathological process remains controversial. Aβ and p-tau proteins are neurotoxic proteins that can cause AD, and EVs have been shown to promote their tansfer and diffusion [79]. However, other studies have shown that microglia can improve the pathological phenotype of AD by uptaking Aβ through EVs and transforming it into neuroprotective substances [100].
In addition to proteins, many miRNAs involved in AD progression have been found in AD-derived EVs and can also serve as biomarkers. miRNA profiles in the brains of AD patients are altered compared to healthy controls, often in a stage- and/or region-specific manner. How these alterations impact disease onset and progression and whether they act as a cause or an effect along the disease trajectory remains unclear. Nevertheless, the specific early miRNA aberrations in human brains indicate that disruption of miRNA homeostasis may act as a (co-)driver of certain pathological cascades [101].
In AD, changes in EV miRNAs that target APP processing, tau phosphorylation, and mitochondrial- and apoptosis-related genes that regulate neurodegenerative events in AD, have received much attention [102]. A recent study has shown that SNORDs–a group of Box C/D small nucleolar RNAs – are enriched differently in EVs isolated from the plasma of AD patients compared to controls [103]. McKeever et al. [80] demonstrated that EVs containing miR-16-5p, miR-451a, and miR-605-5p are decreased, and those containing miR-125b-5p are increased in the CSF of patients with early-onset AD compared to healthy controls. Analysis of mRNA targets of miR-16-5p, miR-451a, miR-125b-5p, and miR-605-5p revealed that these miRNAs are related to neuronal projection, synaptic signaling, metabolism, apoptosis, and the immune system. By comparing EVs miR-135a in CSF vs serum, Liu et al. [81] reported that the increased level of miR-135a in ABCA1-labeled EVs in CSF is more effective for the early diagnosis of AD. However, compared to CSF, EVs in peripheral blood are easier to obtain and detect. Li et al. [82] reported that Aβ42/40 and miR-384 in NCAM/ABCA1 dual-labeled plasma EVs have potential advantages in diagnosing subjective cognitive decline (SCD), i.e., the early stage of AD. The EVs with dual‐specific biomarkers can be obtained through a combination of magnetic bead method and the microtiter plate method, and used to achieve ideal AD diagnostic performance. This provides a new direction for future EV research, although these findings need to be further confirmed in future studies. Another study using the same approach showed a potential advantage of miR-29c-3p in NCAM/amphiphysin 1 dual-labeled EVs in plasma in the diagnosis of SCD [83]. It has been shown that double-labeled EVs from plasma have promising applications in the diagnosis of AD, and could potentially serve as a substitute for CSF markers. Aharon et al. [84] found that the let-7 g-5p, miR126-3p, miR142-3p, miR-146a-5p, and miR-223-3p levels in plasma EVs are correlated with disease severity and could be used as biomarkers to reflect the severity of AD. One study suggested that miR-193b in ABCA1-labeled serum EVs contributes to the early diagnosis of AD [85], although the use of ABCA1-labeled EVs from serum for AD diagnosis needs to be confirmed in future studies.
For most diseases, combined biomarkers are likely to have better diagnostic performance than a single one. Yang et al. [86] analyzed miR-135a, miR-193b, and miR-384 in serum EVs and demonstrated that the combination of miR-135a, miR-193b, and miR-384 had an outstanding diagnostic performance with an area under the curve (AUC) value of 0.997 and can be used for early diagnosis of AD. They found that miR-135a and miR-384 were upregulated and miR-193b was downregulated in the serum EVs of patients with AD. Another study showed that the combination of miR-125b and miR-361 in serum EVs had a high diagnostic efficacy, with a sensitivity of 91.67%, selectivity of 95.00%, and accuracy of 99.52% [87]. Moreover, a combination of miR-30b-5p, miR-22-3p and miR-378a-3p in serum EVs has good diagnostic capabilities, with AUC of 0.880 [88]. These data suggest that the combination of miRNAs from EVs in peripheral blood has potentials to distinguish AD from healthy controls. Nakano et al. [91] showed that the level of miR-146a was increased in the hippocampus of APP/PS1 AD model mice injected with bone marrow mesenchymal stem cells (BM-MSCs), and this upregulation was caused by the secretion of exosomal miR-146a from BM-MSCs and involved in the correction of cognitive impairment. Through a method of isolating neuron-derived EVs from plasma based on neuronal expression of L1 cell adhesion molecule (L1CAM), one study showed that the levels of miR-23a-3p, miR-223-3p, and miR-190a-5p in neuron-derived EVs isolated from the plasma of AD patients were significantly increased, whereas the level of miR-100-3p was significantly decreased [89]. Another study showed that the neuron-derived EVs in AD patients induce neuroinflammatory responses in microglia, and that the neuron-derived EV let-7e is a potential biomarker for AD [90]. However, a recent study reported that L1CAM may not be a good marker for neuron-derived EVs, as L1CAM is not associated with EVs in human CSF or plasma, disputing the use of this marker to isolate neuron-derived EVs [104]. To overcome these limitations, there is an urgent need to develop better separation methods or alternative markers.
In conclusion, we have noticed that numerous potential biomarkers have been identified by bioinformatics analyses and tested in experimental studies of EV miRNAs. However, similar to the current status of research on protein contents of EV, these studies were focused on the discovery of new biomarkers without clinical validation. There is a lack of consensus among research groups, and more practical work is needed for translation to clinical application.
EVs and PD
PD is clinically characterized by progressive rigidity, bradykinesia, and tremor [105]. Significant pathological changes associated with PD include the degenerative death of dopaminergic (DA) neurons in the substantia nigra, leading to a significant reduction of DA in the striatum and the presence of Lewy bodies in residual nigrostriatal neurons [106]. The aggregated α-syn plays a role in neurodegeneration, and the latter is a cause of the symptoms associated with PD. EVs have received much interest as a potential player in PD and in vitro studies have shown the α-syn-carrying potential of EVs since as early as 2010, paving the way for the extracellular seeding theory. Other studies have shown that the CSF EVs from PD patients can cause α-syn aggregates in target cells and could lead to the disease pathology [107].
The correlation between EVs and PD was first confirmed by the in vitro transmission of EVs and in vivo experiments of misfolded toxic proteins in PD (Table 2). Lee et al. [108] confirmed that both primary cortical neurons of rats and RA (all-trans-retinoic acid)-differentiated SH-SY5Y neurons can secrete EVs containing α-syn. These results suggest that vesicle-mediated release of the monomeric and oligomeric forms of α-syn contributes to proteasome defects and mitochondrial dysfunction in the pathogenesis of PD. The propagation of α-syn through EVs along multiple brain regions represents one of the central mechanisms of PD progression. Other studies have confirmed that lysosomal dysfunction may be one of the main factors accelerating PD pathology, as the release of EVs containing α-syn is increased when intracellular protein transport through lysosomes is blocked [109, 110]. Emerging evidence suggests that EVs play a role in the intercellular diffusion of aggregating α-syn, resulting in prion-like diffusion of the aggregates [111]. Neuroblastoma cells express α-syn protein in EVs and release it into the medium for extracellular transport [112]. These observations suggest that although α-syn may be released independently of EVs, it can also be released via EVs.
Other evidence also supports this hypothesis, including the fact that lysosomal dysfunction (a PD-related stress state) increases α-syn release through EVs. Emmanouilidou et al. reported that impairment of lysosomal acidification increased the levels of secreted α-syn. In addition, BFA, an effective inhibitor of the classical ER/Golgi-dependent pathway that induces disruption of the classical secretory pathway, did not alter the levels of secreted α-syn; in contrast, α-syn was released by externalized vesicles in a calcium-dependent manner, suggesting a non-classic secretory pathway for α-syn [126]. Additionally, the presence of EVs has been shown to increase the tendency of α-syn aggregation, and cultured cells have been found to be more likely to absorb EV-associated α-syn than free α-syn oligomers, suggesting that α-syn is transferred between cells via EVs [113]. Grey et al. [111] further demonstrated that pro-inflammatory cytokines, such as TNF-α, IL-1β and IL-6, increase the aggregation of α-syn in neuron-derived EVs. Additionally, direct injection of EVs from α-syn-treated microglia into the mouse striatum resulted in α-syn phosphorylation and aggregation, degeneration of DA neurons in several brain regions associated with the striatum, and dyskinesia. It has also been found that the α-syn oligomer in the microglia-derived EVs in the CSF of PD patients could induce α-syn aggregation in neurons [60]. In addition to α-syn diffusion, EVs have also been implicated in the transfer of mutated leucine-rich repeat kinase 2 (LRRK2) acting as a risk factor for developing PD [127].
To date, no reliable, clinically applicable biomarkers have been established for PD. One of the pathological hallmarks of PD is the presence of Lewy bodies in surviving neurons, which consist of insoluble aggregated proteins, with α-syn being the major component [128]. However, α-syn is easily secreted into the extracellular spaces and has been identified in CSF, blood, and saliva [129]. Although the mechanisms of α-syn secretion are not fully understood, Shi et al. have demonstrated that the diagnostic sensitivity and specificity of plasma exosomal α-syn are comparable to those of CSF α-syn [130]. Another study showed that the concentration of α-syn in plasma EVs may serve as a potential diagnostic biomarker for PD [114]. Furthermore, Jiang et al. showed that the combined serum neuronal exosome-associated α-syn and clusterin outperform any previously reported blood-based assay or CSF total or pathogenic α-syn in predicting PD from atypical parkinsonism in clinical and prodromal PD [131], while studies by Ohmichi et al. demonstrated the quantification of brain-derived EVs in plasma as a biomarker to diagnose PD [115].
In addition, proteins are not the only bioactive content of EVs studied in the context of PD. miRNAs, through their epigenetic control of recipient cells, have shown great impacts on the pathological mechanisms of numerous diseases. Previous studies have found that circulating miRNAs are closely related to the pathophysiological processes of PD and can be easily collected using non-invasive or minimally invasive techniques, making them promising biomarker candidates for PD [132]. miRNAs are highly stable and resistant to degradation in EVs, and recent studies have shown that EV miRNAs play an important role in both physiological and pathological status of PD and can be used as biomarkers of PD [133]. Dos Santos et al. [116] analyzed EV miRNAs from CSF samples of 40 early PD and 40 healthy controls in a cross-sectional cohort, and conducted small RNA sequencing, protein-binding ligand assays, and machine learning. The results showed that the expression levels of let-7f-5p and miR-125a-5p were increased, while those of miR-27a-3p, miR-423-5p and miR-151a-3p were decreased. The combination of miR-22-3p, miR-10b-5p, miR-151a-3p and α-syn had the best diagnostic performance for PD with a sensitivity of 97%, specificity of 90%, and AUC of 96%. Gui et al. [117] found that the expression of miR-1 and miR-19b-3p was significantly decreased, while the expression of miR-153, miR-409-3p, miR-10a-5p and let-7 g-3p was significantly increased in CSF EVs of PD patients. Manna et al. [118] reported that a set of miR-21-3p, miR-22-3p and miR-223-5p in serum EVs can discriminate PD from healthy controls with an AUC of 0.75. Additionally, it was found that the combination of miR-425-5p, miR-21-3p and miR-199a in serum EVs had a good performance in discriminating between progressive supranuclear palsy and PD, with an AUC of 0.86. He et al. [119] reported that six serum-derived EV miRNAs, including miR-374a-5p, miR-374b-5p, miR-199a-3p, miR-28-5p, miR-22-5p and miR-151a-5p, may be used as biomarkers for early diagnosis and progression of PD. The biological functions of these miRNAs in the occurrence and development of PD need to be further studied. The expression levels of let-7d, miR-22* (asterisk indicates anti-sense miR), miR-23a, miR-24, miR-142-3p and miR-222 were found to be significantly increased in serum EVs of PD patients, which can improve clinical diagnosis of PD [120]. A comparison of 24 miRNAs in serum EVs between 109 patients with PD and healthy controls showed that the levels of miR-24 (AUC 0.908) and miR-195 (AUC 0.697) were increased, whereas miR-19b (AUC 0.753) was decreased in PD. Therefore, they may represent novel biomarkers [121]. Ozdilek et al. [122] compared the expression levels of miR-19a, miR-19b, miR-29a, miR-29c, miR-181, miR-195 and miR-221 in serum EVs between 51 PD patients and 20 healthy controls. The results showed that the expression level of miR-29c was significantly increased in PD with an AUC of 0.689. In another study, RT-qPCR results showed that the expression level of miR-331-5p in plasma EVs of PD patients was significantly increased, while that of miR-505 was significantly decreased, with AUCs of 0.849 and 0.898, respectively, suggesting that they have potential value for early diagnosis of PD [123]. In another study, Nie et al. [124] pointed out that in plasma EVs from 34 normal controls, 5 donors with AD and 7 donors with PD, miR-125a-5p, miR-1468-5p, miR-204-5p, let-7e-5p, miR-375, miR-369-5p, miR-423-5p and miR-23a-3p were significantly increased/decreased. Among them, let-7e-5p expression was increased in patients with PD. Besides, they found that the level and quality of miRNAs in EVs were better than those in plasma, suggesting that biomarkers in plasma EVs have better diagnostic efficiency. Xie et al. [125] suggested that miR-15b-5p, miR-30c-2-3p, miR-138-5p, miR-106b-3p, miR-338-3p and miR-431-5p in plasma EVs represent potential biomarkers for PD diagnosis. Studies on SH-SY5Y cells treated by MPP+ demonstrated that the target genes of these miRNAs may be enriched in KEGG dopaminergic synapse pathway and PD pathway. It is not difficult to see that due to the influence of other components in blood, the diagnostic performance of blood EVs is weaker than that of CSF. Efforts should be made to develop a method to perfectly mark neurally derived EVs in blood.
Taken together, these observations provide a proof-of-concept that modulating miRNA levels in PD brains may concomitantly modify multiple aspects of PD pathology, and miRNAs may be candidate targets for intervention as common downstream regulators of functionally diverse molecular pathways in PD. Although some of these miRNAs have been previously studied and discussed in this paper, experimental testing of newly identified miRNA correlations in appropriate model systems is critical for drug development.
EVs and ALS
ALS causes weakness and atrophy of the muscles of the extremities, trunk, and chest following motor neuron injury. The clinical manifestations of ALS are progressive muscle weakness, atrophy, and spasticity, reflecting the degeneration of upper and lower motor neurons in the cortex, brainstem, and spinal cord. The pathogenesis of ALS includes an imbalance of protein homeostasis in the nervous system, prion-like proliferation and reproduction of abnormal proteins, mitochondrial dysfunction, and inflammatory cascade responses. Approximately 90% of cases are sporadic and 10% are familial. Furthermore, about 20% of familial cases are caused by mutations in Cu/zinc superoxide dismutase (SOD) [134]. Mutations in the SOD1 gene lead to abnormal folding of SOD1 protein in vivo, ultimately leading to the formation of toxic aggregates [135]. Multiple ALS-related mutations have also been found in the Tar DNA binding protein-43 (TDP-43) gene [136]. TDP-43, a member of the heterogeneous nuclear ribonucleoprotein (hnRNP) family, is involved in RNA processing and can form insoluble aggregates in the brains of patients with ALS [137].
Over the years, many molecular targets have been suggested to be involved in the pathogenesis of ALS, and several proteins encoded by genes involved in the pathogenesis of ALS have been identified. Recent evidence suggests that many of these proteins are present or differentially expressed in EVs and spread between neurons and glial cells within different brain regions, contributing to their transmission and propagation (Table 3). These proteins include SOD1 [138], TDP-43 [139], Fused in sarcoma (FUS) [140], and Dipeptide repeating proteins (DPRs) [141]. The first ALS-related gene mutation was found in SOD1. SOD1 was first detected in EVs derived from murine motor neuron-like NSC-34 cell model expressing wild-type or mutant human SOD1 [142]. Subsequent studies found SOD1 secretion in association with purified EVs from different sources, including the finding that astrocytes expressing mutant SOD1 could induce selective death of motor neurons through EV transfer. Misfolded mutant SOD1 has also been shown to be able to transfer between NSC-34 cells via EVs, as well as between primary SOD1-overexpressing mouse spinal cord cells [143]. A recent study comparing levels of SOD1 and ALS-related biomolecules in plasma EVs between patients with ALS and healthy controls has provided support for further characterization of ALS-related SOD1 levels in various types of EVs and has implicated it as a biomarker for ALS [144]. Earlier studies have identified TDP-43 in CSF EVs from patients with ALS by western blotting and mass spectrometry [139]. TDP-43 has been found to be enriched in EVs in the conditioned media of neuroblastoma cells expressing TDP-43, as well as in EVs extracted from the CSF of patients with ALS and frontotemporal dementia [145, 146]. These results suggest that the transmission of TDP-43 is achieved by EV release. Moreover, a study published in 2015 using a luciferase fragment-TDP-43 fusion peptide showed that cells preferentially take up oligomer TDP-43 encapsulated in EVs, leading to greater toxicity. This study demonstrated the transfer of oligomer TDP-43 between neurons via EVs and the axonal transportation of TDP-43 after uptake from the medium [147]. Studies have also shown that the TDP-43 oligomers in EVs are more toxic than free TDP-43; thus, TDP-43 in EVs is considered a potential marker of ALS.
In addition to SOD1 and TDP-43, other ALS-related targets are also contained in secreted EVs, albeit with a lower concentration. ALS-related mutations in FUS can lead to varying degrees of mislocalization of FUS in the cytoplasm, possibly related to the formation of stress granulosa structures. A previous study in familial ALS demonstrated that FUS interacts with RNA-binding proteins Matrin-3 and hnRNPA1 to form mutant complexes. This study also confirmed the presence of FUS in EVs, providing evidence for the spread of FUS between pathological cells, which may allow for the diagnosis of ALS [148]. GGGGCC repeats in C9orf72 are the most common cause of ALS and the basis of ALS vesicle trafficking [155, 156]. RNA containing the GGGGCC repeat sequences is translated into DPRs which can form aggregates in the CNS of patients with ALS [157]. Intercellular diffusion of DPR can occur through EVs, and DPR-containing EVs have been isolated from ALS spinal motor neurons containing C9orf72 repeat expansions, suggesting biomarker opportunity for ALS [141]. The presence of other ALS-related protein mutants in EVs, including valin-containing protein [158], sequestosome 1 [159], and Tank-binding kinase 1 [160], may also be used to diagnose ALS.
Apart from proteins, EV miRNAs have become promising tools for better diagnosis of ALS because some miRNAs may alter the expression of proteins involved in ALS. Rizzuti et al. [149] found that miR-34a, miR-335 and miR-625-3p in CSF EVs may be used as biomarkers for ALS. Yelick et al. [150] provided preliminary evidence supporting the use of miR-124-3p in CSF EVs as an indicator for ALS disease staging. Freischmidt et al. [151] measured miRNA levels in EVs from the CSF and serum of 22 patients with sporadic ALS and 24 healthy controls. In patients with ALS, EV-encapsulated miR-132-5p, miR-132-3p and miR-143-3p were significantly reduced, while miR-143-5p and miR-574-5p were significantly increased, implicating their biomarker potential for ALS diagnosis. Xu et al. [152] indicated that miR-27a-3p in serum EVs was significantly reduced in patients with ALS, suggesting that it may be a potential diagnostic biomarker for ALS. Lo et al. [153] demonstrated that miR-342-3p, miR-1254, miR-587, miR-298, miR-4443 and miR-450a-2-3p in serum and brain-tissue EVs reflect the state of CNS disease in ALS, thus providing an opportunity for possible diagnosis. Gomes et al. [154] found that the inflammatory-associated miRNAs miR-155, miR-21, and miR-146a are depleted in EVs both originating from the spinal and from cortical astrocytes in ALS mice, and may be used as biomarkers for ALS.
In conclusion, EVs have potential applications in the pathological investigation, early (possibly pre-clinical) diagnosis, and treatment management of ALS. They play a role in disease pathogenesis through the transfer and subsequent intracellular accumulation of pathological proteins such as TDP-43, SOD1, and FUS. Studies have reported dysregulation of protein and microRNA cargos of EVs in cell and animal models of ALS and in patients. However, there are multiple difficulties in developing EVs as biomarkers. The different biofluids (CSF, plasma, and serum) used for investigation and the different methods for isolating EVs are among the multiple reasons for the lack of consensus among studies.
EVs and HD
HD is clinically characterized by progressive motor deficits (e.g., chorea, oculomotor abnormalities, verbal ataxia, and dysphagia), cognitive dysfunction (dementia) and psychiatric disorders (e.g., depression, anxiety, and apathy), and pathologically by the loss of long-projection neurons in the cortex and striatum [161]. Progressive motor failure is a major cause of complications, leading to death within 15 to 20 years of onset. HD displays autosomal dominant inheritance caused by CAG (cytosine-adenine-guanine) repeats (≥ 36) of the Huntington’s disease chorea gene (IT15) on chromosome 4, resulting in an abnormal number of N-terminal glutamine repeats (polyQ) in mutated huntingtin protein (mHTT) [162].
EVs can cross the BBB and cause aggregation of mHTT in HD, resulting in mitochondrial dysfunction and cell death (Table 4). In HD, the aggregation of mHTT in neurons and the spread of mHTT aggregates between cells were revealed by the internalization of synthetic peptide (44Q) into cells and the formation of cytoplasmic aggregates in vitro and in animal experiments [163,164,165]. Interestingly, in three patients with HD, mHTT aggregates were found in the allografts of striatal tissue, confirming the spread mHTT into genetically unrelated tissue [166]. EVs may be involved in the proliferation of mHTT protein by delivering proteins or nucleic acids, suggesting its potential as a diagnostic marker for HD. In a previous study, EVs released from the fibroblasts of patients with HD were injected into the ventricles of neonatal mice, and they led to HD-related pathology and behaviors [167]. In an HD model, Lee et al. [168] demonstrated that the adipose-derived stem cell (ASC)-derived EVs alleviated disease progression by reducing mHTT aggregation and apoptotic protein levels. Furthermore, immunocytochemistry and western blot confirmed that the ASC-derived EVs could release neurotrophic factors and significantly reduce mHTT aggregation, mitochondrial dysfunction, and apoptosis in neuronal cells. The density of mHTT aggregates has been shown to decrease following injection of astrocyte-derived EVs into the striatum of HD 140Q knock-in mice. Interestingly, the mHTT protein was not detected in the EVs secreted by the primary astrocytes, suggesting that the astrocyte-derived EVs may be used for treating HD [169]. The neuroprotective synaptic chaperone cysteine string protein α mediates the cellular export of polyglutamine expanded protein 72Q HTTexon 1 via EVs, again showing potential value for HD therapy [170].
The miRNA content in EVs has not been studied in HD, and the use of EV miRNAs as biomarkers for HD diagnosis has not been reported. Several studies have reported a decreased level of miR-124 in the brains of patients with HD. However, a study reported that treatment of R6/2 transgenic HD mice by EV-mediated delivery of miR-124 [171] did not improve motor symptoms. Nevertheless, the function of miR-124 in EVs and its possible association with HD deserve further investigation.
Therapeutic potential of EVs for neurodegenerative diseases
Various studies have suggested that EVs have several advantages over conventional synthetic carriers, such as their ability to cross the BBB and the low tendency to evoke an immune response, opening a new frontier for their usage in drug delivery and as therapeutics for neurodegenerative diseases. Research on EVs as a therapeutic vector is increasing. There have been at least dozens of phase 1/2 clinical trials registered for cancer, SARS-CoV-2 and AD, and treatment methods include stem cell-derived EVs, autologous EVs or drug-loaded EVs [172]. Some studies have confirmed that exosomes can be used as a promising drug delivery platform for target therapies against PD and other neurodegenerative diseases [173]. Therefore, cell-derived EV-based carrier systems have attracted considerable interest [174]. EVs have been used in murine models of PD and AD to reduce pathological protein accumulation. EVs containing BACE1 siRNAs have been used in C57BL/6 mice, resulting in an overall 60% reduction of BACE1 mRNA and 55% decrease of Aβ1-42 level [175]. In another study, EVs with α-syn siRNA were peripherally injected into S129D α-syn transgenic mice, which decreased the level of α-syn aggregates in brain regions pathologically affected in PD [176]. Bonafede et al. showed that exosomes derived from murine adipose-derived stromal cells are able to protect NSC-34 cells (which overexpress human SOD1 mutants) from oxidative damage [177], and similar results were reported by Lee et al. [178]. In R6/2 mouse-derived neuronal cell model of HD, EVs derived from ASCs slowed the progression of the disease and reduced the levels of mHTT aggregates and apoptotic proteins, showing the potential to treat HD [168]. Better therapeutic efficacy can also be achieved by modifying EVs. Research by Didiot et al. [179] showed that EVs loaded with hydrophobic siRNAs targeting HTT mRNA were efficiently internalized by mouse primary cortical neurons and promoted dose-dependent silencing of HTT mRNA and protein. Thus, both natural exosomes and modified EVs could play important roles in the treatment of HD.
The results of these studies suggest that EVs have great potential as a novel therapy for the treatment of neurodegenerative diseases. EVs can penetrate the BBB in a bidirectional manner, providing a means of communication to and from the CNS [180]. They are stable in the peripheral circulation and able to protect their cargos from degradation [181]. However, the wide variety of EV sources and isolation methods have limited the reproducibility and comparability across studies. There are many methods for EV isolation, each with distinct advantages and disadvantages. Therefore, it is crucial to follow the International Society for Extracellular Vesicles guideline for EV characterization to maximize the effectiveness and enable more reliable comparisons between studies. Further explorations in the clinical context are also needed. Moreover, while EVs are found in human CSF, urine and blood, it is unclear which source of EVs is better for treatment [182]. Therefore, when using EVs as a treatment plan, a full understanding of their modes of action in the disease and appropriate design of EVs are essential for improving the therapeutic effects (Table 5).
Conclusions and prospects
To sum up, EVs are involved in the development and progression of neurodegenerative diseases. EVs in the microenvironment carry and transmit oxidative and inflammatory signals (such as proteins and miRNAs) secreted by neurons and glial cells. EVs may also directly transfer pathogenic substances (such as protein aggregates) from one cell to another. Therefore, the physiology and pathology of brain cells and even the progression of neurodegenerative diseases such as AD, PD, ALS and HD may be affected by EVs [195]. In this review, we discuss current advances on the roles of EVs in the development of neurodegenerative diseases, as well as their biomarker and therapeutic potentials in neurodegenerative diseases. EVs have also been found to be involved in neuronal self-rescue, where neurons remove harmful substances by secreting EVs; this could promote or inhibit disease depending on their content and the intrinsic nature of the disease. However, whether neurons preserve or pass proteins to other neurons via EVs, leading to more serious consequences, needs to be further explored.
EVs have been used as experimental tools for the diagnosis and treatment of animals [96]. Importantly, EVs in blood, CSF, urine and saliva contain various biomarkers, thus being a non-invasive tool for the early detection of disease and development of treatment strategies. Moreover, an increasing number of clinical trial have investigated the clinical applications of EVs. For example, a clinical study at University of Alabama at Birmingham aimed to determine biomarkers for PD susceptibility and/or progression from exosome-proteomes derived from PD patients (ClinicalTrials.gov Identifier: NCT01860118). Another study at Ruijin Hospital, Shanghai, China evaluated the safety and efficacy of exosomes derived from allogenic adipose mesenchymal stem cells in subjects with AD (ClinicalTrials.gov Identifier: NCT04388982). Molecular biomarkers, such as EV miRNAs, may provide new insights into the diagnosis and treatment of neurodegenerative diseases such as AD, PD, ALS, and HD. As evidenced by the studies covered in this review, with the development of basic research, EVs have shown great potential in neurodegenerative disease research, especially as a target-drug-carrier for the treatment of neurodegenerative diseases. In this review, we also present the recent advances in the analysis of EVs, which may lead to the discovery of new biomarkers for neurodegenerative diseases and facilitate the identification of new therapeutic targets. It has recently been suggested that altering the release level of EVs may be beneficial for therapeutic approaches in some neurodegenerative diseases, particularly at the onset of the disease. The current dilemma is that there are no published clinical trials on the role of EVs in treating neurodegenerative disorders. Indeed, previous research on EVs has focused on the effectiveness, but which components are responsible for the observed efficacy has not been established yet; this has raised doubts on the safety and effectiveness of EVs. Finally, there remains a lack of strict standards for the quality management of EVs. Different tissue sources, donor cells and preparation methods may result in heterogeneous EVs. In addition, due to the different in vivo and in vitro models among laboratories, the effective concentrations of EVs and intervention methods in different diseases have not been finalized, thus hindering their clinical translation. As such, it is crucial to develop more efficient separation methods to deal with these issues. High-quality cohort design and development of high-tech hardware equipment and artificial intelligence can assist biomarker discovery/validation/clinical translation. The correlation between multi-omics data and imaging biomarkers is the future direction of research.
Some issues need to be addressed before using EVs for treatment of neurodegenerative diseases. For example, the precise content sorting and regulatory mechanisms of secreted EVs remain largely unknown. Additionally, advanced selection and isolation techniques are required to better distinguish EVs from other extracellular particles. Most importantly, more studies are needed to improve the performance of EV-carrying biomarkers for clinical diagnosis. Further studies are also needed to examine the relationship between abnormal upregulation or downregulation of EV biomarkers and disease progression. Despite these obstacles, the use of EVs as a potential biomarker and a treatment for neurodegenerative diseases is attractive and worthy of future research.
Availability of data and materials
Not applicable.
Abbreviations
- AD:
-
Alzheimer’s disease
- PD:
-
Parkinson’s disease
- ALS:
-
Amyotrophic lateral sclerosis
- HD:
-
Huntington’s disease
- EVs:
-
Extracellular vesicles
- CNS:
-
Central nervous system
- CSF:
-
Cerebrospinal fluid
- BBB:
-
Blood–brain barrier
- NSC:
-
Neural stem cell
- sEV:
-
Small extracellular vesicle
- MV:
-
Microvesicle
- ESCRT:
-
Endosomal sorting complex required for transport
- Alix:
-
Apoptosis-linked gene 2-interacting protein X
- TSG101:
-
Tumor susceptibility gene 101
- MVB:
-
Multivesicular body
- ILVs:
-
Intraluminal vesicles
- LSE:
-
Late sorting endosome
- ESE:
-
Early sorting endosome
- MHC:
-
Major histocompatibility complex
- L1CAM:
-
L1 cell adhesion molecule
- Aβ:
-
Amyloid β
- α-syn:
-
α-Synuclein
- APP:
-
Amyloid precursor protein
- t-tau:
-
Total tau
- p-tau:
-
Hyperphosphorylated tau
- SCD:
-
Subjective cognitive decline
- AUC:
-
Area under the curve
- SOD:
-
Cu/zinc superoxide dismutase
- TDP-43:
-
Tar DNA binding protein-43
- FUS:
-
Fused in sarcoma
- DPR:
-
Dipeptide repeating protein
- hnRNP:
-
Heterogeneous nuclear ribonucleoprotein
- mHTT:
-
Mutated huntingtin protein
- CSPα:
-
Cysteine string protein α
References
Chiti F, Dobson CM. Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade. Annu Rev Biochem. 2017;86:27–68.
Taylor JP, Brown RH Jr, Cleveland DW. Decoding ALS: from genes to mechanism. Nature. 2016;539:197–206.
Saudou F, Humbert S. The biology of huntingtin. Neuron. 2016;89:910–26.
Dugger BN, Dickson DW. Pathology of neurodegenerative diseases. Cold Spring Harb Perspect Biol. 2017;9:e028035.
Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, Ganguli M, et al. Global prevalence of dementia: a Delphi consensus study. Lancet. 2005;366:2112–7.
Poste G. Bring on the biomarkers. Nature. 2011;469:156–7.
Obrocki P, Khatun A, Ness D, Senkevich K, Hanrieder J, Capraro F, et al. Perspectives in fluid biomarkers in neurodegeneration from the 2019 biomarkers in neurodegenerative diseases course-a joint PhD student course at University College London and University of Gothenburg. Alzheimers Res Ther. 2020;12:20.
Carlyle BC, Trombetta BA, Arnold SE. Proteomic approaches for the discovery of biofluid biomarkers of neurodegenerative dementias. Proteomes. 2018;6:32.
Blennow K, Zetterberg H. Understanding biomarkers of neurodegeneration: ultrasensitive detection techniques pave the way for mechanistic understanding. Nat Med. 2015;21:217–9.
Saint-Pol J, Gosselet F, Duban-Deweer S, Pottiez G, Karamanos Y. Targeting and crossing the blood-brain barrier with extracellular vesicles. Cells. 2020;9:851.
Saeed U, Compagnone J, Aviv RI, Strafella AP, Black SE, Lang AE, et al. Imaging biomarkers in Parkinson’s disease and Parkinsonian syndromes: current and emerging concepts. Transl Neurodegener. 2017;6:8.
Garborg K, Holme Ø, Løberg M, Kalager M, Adami HO, Bretthauer M. Current status of screening for colorectal cancer. Ann Oncol. 2013;24:1963–72.
Sharma P, Mesci P, Carromeu C, McClatchy DR, Schiapparelli L, Yates JR 3rd, et al. Exosomes regulate neurogenesis and circuit assembly. Proc Natl Acad Sci U S A. 2019;116:16086–94.
Ramirez SH, Andrews AM, Paul D, Pachter JS. Extracellular vesicles: mediators and biomarkers of pathology along CNS barriers. Fluids Barriers CNS. 2018;15:19.
Howitt J, Hill AF. Exosomes in the pathology of neurodegenerative diseases. J Biol Chem. 2016;291:26589–97.
Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. 2020;367(6478):eaau6977.
Cheng L, Sharples RA, Scicluna BJ, Hill AF. Exosomes provide a protective and enriched source of miRNA for biomarker profiling compared to intracellular and cell-free blood. J Extracell Vesicles. 2014;3:23743.
Théry C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7(1):1535750.
Shao H, Im H, Castro CM, Breakefield X, Weissleder R, Lee H. New technologies for analysis of extracellular vesicles. Chem Rev. 2018;118:1917–50.
Lee Y, El Andaloussi S, Wood MJ. Exosomes and microvesicles: extracellular vesicles for genetic information transfer and gene therapy. Hum Mol Genet. 2012;21:R125-134.
Pegtel DM, Gould SJ. Exosomes. Annu Rev Biochem. 2019;88:487–514.
Gurung S, Perocheau D, Touramanidou L, Baruteau J. The exosome journey: from biogenesis to uptake and intracellular signalling. Cell Commun Signal. 2021;19:47.
Gurunathan S, Kang MH, Jeyaraj M, Qasim M, Kim JH. Review of the isolation, characterization, biological function, and multifarious therapeutic approaches of exosomes. Cells. 2019;8:307.
van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19:213–28.
Hessvik NP, Llorente A. Current knowledge on exosome biogenesis and release. Cell Mol Life Sci. 2018;75:193–208.
Maas SLN, Breakefield XO, Weaver AM. Extracellular vesicles: unique intercellular delivery vehicles. Trends Cell Biol. 2017;27:172–88.
Akers JC, Gonda D, Kim R, Carter BS, Chen CC. Biogenesis of extracellular vesicles (EV): exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J Neurooncol. 2013;113:1–11.
Gézsi A, Kovács Á, Visnovitz T, Buzás EI. Systems biology approaches to investigating the roles of extracellular vesicles in human diseases. Exp Mol Med. 2019;51:1–11.
Mathieu M, Martin-Jaular L, Lavieu G, Thery C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat Cell Biol. 2019;21:9–17.
Huang D, Chen J, Hu D, Xie F, Yang T, Li Z, et al. Advances in biological function and clinical application of small extracellular vesicle membrane proteins. Front Oncol. 2021;11:675940.
He C, Zheng S, Luo Y, Wang B. Exosome theranostics: biology and translational medicine. Theranostics. 2018;8:237–55.
Kowal J, Arras G, Colombo M, Jouve M, Morath JP, Primdal-Bengtson B, et al. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci U S A. 2016;113:E968-977.
Schulz-Siegmund M, Aigner A. Nucleic acid delivery with extracellular vesicles. Adv Drug Deliv Rev. 2021;173:89–111.
Thakur BK, Zhang H, Becker A, Matei I, Huang Y, Costa-Silva B, et al. Double-stranded DNA in exosomes: a novel biomarker in cancer detection. Cell Res. 2014;24:766–9.
Sansone P, Savini C, Kurelac I, Chang Q, Amato LB, Strillacci A, et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc Natl Acad Sci U S A. 2017;114:E9066-e9075.
Li S, Li Y, Chen B, Zhao J, Yu S, Tang Y, et al. exoRBase: a database of circRNA, lncRNA and mRNA in human blood exosomes. Nucleic Acids Res. 2018;46:D106–12.
Colombo M, Raposo G, Théry C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. 2014;30:255–89.
O’Brien K, Breyne K, Ughetto S, Laurent LC, Breakefield XO. RNA delivery by extracellular vesicles in mammalian cells and its applications. Nat Rev Mol Cell Biol. 2020;21:585–606.
de Voogt WS, Tanenbaum ME, Vader P. Illuminating RNA trafficking and functional delivery by extracellular vesicles. Adv Drug Deliv Rev. 2021;174:250–64.
Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet. 2015;16:421–33.
Schmiedel JM, Klemm SL, Zheng Y, Sahay A, Blüthgen N, Marks DS, et al. Gene expression. MicroRNA control of protein expression noise. Science. 2015;348:128–32.
Walgrave H, Zhou L, De Strooper B, Salta E. The promise of microRNA-based therapies in Alzheimer’s disease: challenges and perspectives. Mol Neurodegener. 2021;16:76.
Martinez B, Peplow PV. MicroRNAs as diagnostic and therapeutic tools for Alzheimer’s disease: advances and limitations. Neural Regen Res. 2019;14:242–55.
Skotland T, Sandvig K, Llorente A. Lipids in exosomes: current knowledge and the way forward. Prog Lipid Res. 2017;66:30–41.
Kreimer S, Belov AM, Ghiran I, Murthy SK, Frank DA, Ivanov AR. Mass-spectrometry-based molecular characterization of extracellular vesicles: lipidomics and proteomics. J Proteome Res. 2015;14:2367–84.
Skotland T, Hessvik NP, Sandvig K, Llorente A. Exosomal lipid composition and the role of ether lipids and phosphoinositides in exosome biology. J Lipid Res. 2019;60:9–18.
Beloribi S, Ristorcelli E, Breuzard G, Silvy F, Bertrand-Michel J, Beraud E, et al. Exosomal lipids impact notch signaling and induce death of human pancreatic tumoral SOJ-6 cells. PLoS ONE. 2012;7:e47480.
Tao L, Zhou J, Yuan C, Zhang L, Li D, Si D, et al. Metabolomics identifies serum and exosomes metabolite markers of pancreatic cancer. Metabolomics. 2019;15:86.
Record M, Silvente-Poirot S, Poirot M, Wakelam MJO. Extracellular vesicles: lipids as key components of their biogenesis and functions. J Lipid Res. 2018;59:1316–24.
Hornung S, Dutta S, Bitan G. CNS-derived blood exosomes as a promising source of biomarkers: opportunities and challenges. Front Mol Neurosci. 2020;13:38.
Chen C, Zong S, Wang Z, Lu J, Zhu D, Zhang Y, et al. Visualization and intracellular dynamic tracking of exosomes and exosomal miRNAs using single molecule localization microscopy. Nanoscale. 2018;10:5154–62.
van Niel G, Carter DRF, Clayton A, Lambert DW, Raposo G, Vader P. Challenges and directions in studying cell-cell communication by extracellular vesicles. Nat Rev Mol Cell Biol. 2022;23:369–82.
Lai CP, Breakefield XO. Role of exosomes/microvesicles in the nervous system and use in emerging therapies. Front Physiol. 2012;3:228.
Serpe C, Monaco L, Relucenti M, Iovino L, Familiari P, Scavizzi F, et al. Microglia-derived small extracellular vesicles reduce glioma growth by modifying tumor cell metabolism and enhancing glutamate clearance through miR-124. Cells. 2021;10:2066.
Taylor AR, Robinson MB, Gifondorwa DJ, Tytell M, Milligan CE. Regulation of heat shock protein 70 release in astrocytes: role of signaling kinases. Dev Neurobiol. 2007;67:1815–29.
Wang S, Cesca F, Loers G, Schweizer M, Buck F, Benfenati F, et al. Synapsin I is an oligomannose-carrying glycoprotein, acts as an oligomannose-binding lectin, and promotes neurite outgrowth and neuronal survival when released via glia-derived exosomes. J Neurosci. 2011;31:7275–90.
Glebov K, Löchner M, Jabs R, Lau T, Merkel O, Schloss P, et al. Serotonin stimulates secretion of exosomes from microglia cells. Glia. 2015;63:626–34.
Garbuzova-Davis S, Borlongan CV. Stem cell-derived extracellular vesicles as potential mechanism for repair of microvascular damage within and outside of the central nervous system in amyotrophic lateral sclerosis: perspective schema. Neural Regen Res. 2021;16:680–1.
Sardar Sinha M, Ansell-Schultz A, Civitelli L, Hildesjo C, Larsson M, Lannfelt L, et al. Alzheimer’s disease pathology propagation by exosomes containing toxic amyloid-beta oligomers. Acta Neuropathol. 2018;136:41–56.
Guo M, Wang J, Zhao Y, Feng Y, Han S, Dong Q, et al. Microglial exosomes facilitate alpha-synuclein transmission in Parkinson’s disease. Brain. 2020;143:1476–97.
García-Romero N, Carrión-Navarro J, Esteban-Rubio S, Lázaro-Ibáñez E, Peris-Celda M, Alonso MM, et al. DNA sequences within glioma-derived extracellular vesicles can cross the intact blood-brain barrier and be detected in peripheral blood of patients. Oncotarget. 2017;8:1416–28.
Jingushi K, Uemura M, Ohnishi N, Nakata W, Fujita K, Naito T, et al. Extracellular vesicles isolated from human renal cell carcinoma tissues disrupt vascular endothelial cell morphology via azurocidin. Int J Cancer. 2018;142:607–17.
2022 Alzheimer’s disease facts and figures. Alzheimers Dement. 2022;18:700–89.
Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1:a006189.
Mucke L, Selkoe DJ. Neurotoxicity of amyloid β-protein: synaptic and network dysfunction. Cold Spring Harb Perspect Med. 2012;2:a006338.
Haass C, Kaether C, Thinakaran G, Sisodia S. Trafficking and proteolytic processing of APP. Cold Spring Harb Perspect Med. 2012;2:a006270.
Lee JH, Ostalecki C, Oberstein T, Schierer S, Zinser E, Eberhardt M, et al. Alzheimer’s disease protease-containing plasma extracellular vesicles transfer to the hippocampus via the choroid plexus. EBioMedicine. 2022;77:103903.
Vincent B. Plasma extracellular vesicles from the periphery as spreading vectors of Alzheimer’s disease pathogenesis? EBioMedicine. 2022;78:103961.
Gabrielli M, Prada I, Joshi P, Falcicchia C, D’Arrigo G, Rutigliano G, et al. Microglial large extracellular vesicles propagate early synaptic dysfunction in Alzheimer’s disease. Brain. 2022;145:2849–68.
Fiandaca MS, Kapogiannis D, Mapstone M, Boxer A, Eitan E, Schwartz JB, et al. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: a case-control study. Alzheimers Dement. 2015;11(600–607):e601.
Dinkins MB, Enasko J, Hernandez C, Wang G, Kong J, Helwa I, et al. Neutral sphingomyelinase-2 deficiency ameliorates Alzheimer’s disease pathology and improves cognition in the 5XFAD mouse. J Neurosci. 2016;36:8653–67.
Villemagne VL, Doré V, Burnham SC, Masters CL, Rowe CC. Imaging tau and amyloid-β proteinopathies in Alzheimer disease and other conditions. Nat Rev Neurol. 2018;14:225–36.
Dinkins MB, Dasgupta S, Wang G, Zhu G, Bieberich E. Exosome reduction in vivo is associated with lower amyloid plaque load in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol Aging. 2014;35:1792–800.
Agosta F, Dalla Libera D, Spinelli EG, Finardi A, Canu E, Bergami A, et al. Myeloid microvesicles in cerebrospinal fluid are associated with myelin damage and neuronal loss in mild cognitive impairment and Alzheimer disease. Ann Neurol. 2014;76:813–25.
Podvin S, Jones A, Liu Q, Aulston B, Mosier C, Ames J, et al. Mutant presenilin 1 dysregulates exosomal proteome cargo produced by human-induced pluripotent stem cell neurons. ACS Omega. 2021;6:13033–56.
Jia L, Qiu Q, Zhang H, Chu L, Du Y, Zhang J, et al. Concordance between the assessment of Aβ42, T-tau, and P-T181-tau in peripheral blood neuronal-derived exosomes and cerebrospinal fluid. Alzheimers Dement. 2019;15:1071–80.
Gallart-Palau X, Guo X, Serra A, Sze SK. Alzheimer’s disease progression characterized by alterations in the molecular profiles and biogenesis of brain extracellular vesicles. Alzheimers Res Ther. 2020;12:54.
Muraoka S, DeLeo AM, Sethi MK, Yukawa-Takamatsu K, Yang Z, Ko J, et al. Proteomic and biological profiling of extracellular vesicles from Alzheimer’s disease human brain tissues. Alzheimers Dement. 2020;16:896–907.
Polanco JC, Scicluna BJ, Hill AF, Götz J. Extracellular vesicles isolated from the brains of rTg4510 mice seed tau protein aggregation in a threshold-dependent manner. J Biol Chem. 2016;291:12445–66.
McKeever PM, Schneider R, Taghdiri F, Weichert A, Multani N, Brown RA, et al. MicroRNA expression levels are altered in the cerebrospinal fluid of patients with young-onset Alzheimer’s disease. Mol Neurobiol. 2018;55:8826–41.
Liu CG, Meng S, Li Y, Lu Y, Zhao Y, Wang PC. MicroRNA-135a in ABCA1-labeled exosome is a serum biomarker candidate for Alzheimer’s disease. Biomed Environ Sci. 2021;34:19–28.
Li Y, Meng S, Di W, Xia M, Dong L, Zhao Y, et al. Amyloid-β protein and MicroRNA-384 in NCAM-Labeled exosomes from peripheral blood are potential diagnostic markers for Alzheimer’s disease. CNS Neurosci Ther. 2022;28:1093–107.
Li Y, Xia M, Meng S, Wu D, Ling S, Chen X, et al. MicroRNA-29c-3p in dual-labeled exosome is a potential diagnostic marker of subjective cognitive decline. Neurobiol Dis. 2022;171:105800.
Aharon A, Spector P, Ahmad RS, Horrany N, Sabbach A, Brenner B, et al. Extracellular vesicles of Alzheimer’s disease patients as a biomarker for disease progression. Mol Neurobiol. 2020;57:4156–69.
Liu CG, Zhao Y, Lu Y, Wang PC. ABCA1-labeled exosomes in serum contain higher microRNA-193b levels in Alzheimer’s disease. Biomed Res Int. 2021;2021:5450397.
Yang TT, Liu CG, Gao SC, Zhang Y, Wang PC. The serum exosome derived microRNA-135a, -193b, and -384 were potential Alzheimer’s disease biomarkers. Biomed Environ Sci. 2018;31:87–96.
Song S, Lee JU, Jeon MJ, Kim S, Sim SJ. Detection of multiplex exosomal miRNAs for clinically accurate diagnosis of Alzheimer’s disease using label-free plasmonic biosensor based on DNA-Assembled advanced plasmonic architecture. Biosens Bioelectron. 2022;199:113864.
Dong Z, Gu H, Guo Q, Liang S, Xue J, Yao F, et al. Profiling of serum exosome miRNA reveals the potential of a miRNA panel as diagnostic biomarker for Alzheimer’s disease. Mol Neurobiol. 2021;58:3084–94.
Serpente M, Fenoglio C, D’Anca M, Arcaro M, Sorrentino F, Visconte C, et al. MiRNA profiling in plasma neural-derived small extracellular vesicles from patients with Alzheimer’s disease. Cells. 2020;9:1443.
Durur DY, Tastan B, Ugur Tufekci K, Olcum M, Uzuner H, Karakülah G, et al. Alteration of miRNAs in small neuron-derived extracellular vesicles of alzheimer’s disease patients and the effect of extracellular vesicles on microglial immune responses. J Mol Neurosci. 2022;72:1182–94.
Nakano M, Kubota K, Kobayashi E, Chikenji TS, Saito Y, Konari N, et al. Bone marrow-derived mesenchymal stem cells improve cognitive impairment in an Alzheimer’s disease model by increasing the expression of microRNA-146a in hippocampus. Sci Rep. 2020;10:10772.
Younas N, Fernandez Flores LC, Hopfner F, Höglinger GU, Zerr I. A new paradigm for diagnosis of neurodegenerative diseases: peripheral exosomes of brain origin. Transl Neurodegener. 2022;11:28.
Rajendran L, Honsho M, Zahn TR, Keller P, Geiger KD, Verkade P, et al. Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc Natl Acad Sci U S A. 2006;103:11172–7.
Gouwens LK, Ismail MS, Rogers VA, Zeller NT, Garrad EC, Amtashar FS, et al. Aβ42 Protofibrils interact with and are trafficked through microglial-derived microvesicles. ACS Chem Neurosci. 2018;9:1416–25.
Joshi P, Turola E, Ruiz A, Bergami A, Libera DD, Benussi L, et al. Microglia convert aggregated amyloid-β into neurotoxic forms through the shedding of microvesicles. Cell Death Differ. 2014;21:582–93.
Elsherbini A, Qin H, Zhu Z, Tripathi P, Crivelli SM, Bieberich E. In vivo evidence of exosome-mediated Aβ neurotoxicity. Acta Neuropathol Commun. 2020;8:100.
Wang Y, Balaji V, Kaniyappan S, Krüger L, Irsen S, Tepper K, et al. The release and trans-synaptic transmission of Tau via exosomes. Mol Neurodegener. 2017;12:5.
Clayton K, Delpech JC, Herron S, Iwahara N, Ericsson M, Saito T, et al. Plaque associated microglia hyper-secrete extracellular vesicles and accelerate tau propagation in a humanized APP mouse model. Mol Neurodegener. 2021;16:18.
Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci. 2015;18:1584–93.
Pacheco-Quinto J, Clausen D, Pérez-González R, Peng H, Meszaros A, Eckman CB, et al. Intracellular metalloprotease activity controls intraneuronal Aβ aggregation and limits secretion of Aβ via exosomes. FASEB J. 2019;33:3758–71.
Lau P, Bossers K, Janky R, Salta E, Frigerio CS, Barbash S, et al. Alteration of the microRNA network during the progression of Alzheimer’s disease. EMBO Mol Med. 2013;5:1613–34.
Canseco-Rodriguez A, Masola V, Aliperti V, Meseguer-Beltran M, Donizetti A, Sanchez-Perez AM. Long non-coding RNAs, extracellular vesicles and inflammation in Alzheimer’s disease. Int J Mol Sci. 2022;23:13171.
Fitz NF, Wang J, Kamboh MI, Koldamova R, Lefterov I. Small nucleolar RNAs in plasma extracellular vesicles and their discriminatory power as diagnostic biomarkers of Alzheimer’s disease. Neurobiol Dis. 2021;159:105481.
Norman M, Ter-Ovanesyan D, Trieu W, Lazarovits R, Kowal EJK, Lee JH, et al. L1CAM is not associated with extracellular vesicles in human cerebrospinal fluid or plasma. Nat Methods. 2021;18:631–4.
Jurado-Coronel JC, Cabezas R, Ávila Rodríguez MF, Echeverria V, García-Segura LM, Barreto GE. Sex differences in Parkinson’s disease: features on clinical symptoms, treatment outcome, sexual hormones and genetics. Front Neuroendocrinol. 2018;50:18–30.
Singh PK, Muqit MMK. Parkinson’s: a disease of aberrant vesicle trafficking. Annu Rev Cell Dev Biol. 2020;36:237–64.
Gassama Y, Favereaux A. Emerging roles of extracellular vesicles in the central nervous system: physiology, pathology, and therapeutic perspectives. Front Cell Neurosci. 2021;15:626043.
Lee HJ, Patel S, Lee SJ. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J Neurosci. 2005;25:6016–24.
Alvarez-Erviti L, Seow Y, Schapira AH, Gardiner C, Sargent IL, Wood MJ, et al. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol Dis. 2011;42:360–7.
Hasegawa T, Konno M, Baba T, Sugeno N, Kikuchi A, Kobayashi M, et al. The AAA-ATPase VPS4 regulates extracellular secretion and lysosomal targeting of α-synuclein. PLoS ONE. 2011;6:e29460.
Grey M, Dunning CJ, Gaspar R, Grey C, Brundin P, Sparr E, et al. Acceleration of α-synuclein aggregation by exosomes. J Biol Chem. 2015;290:2969–82.
Fan RZ, Guo M, Luo S, Cui M, Tieu K. Exosome release and neuropathology induced by α-synuclein: new insights into protective mechanisms of Drp1 inhibition. Acta Neuropathol Commun. 2019;7:184.
Bliederhaeuser C, Grozdanov V, Speidel A, Zondler L, Ruf WP, Bayer H, et al. Age-dependent defects of alpha-synuclein oligomer uptake in microglia and monocytes. Acta Neuropathol. 2016;131:379–91.
Stuendl A, Kraus T, Chatterjee M, Zapke B, Sadowski B, Moebius W, et al. α-Synuclein in plasma-derived extracellular vesicles is a potential biomarker of Parkinson’s disease. Mov Disord. 2021;36:2508–18.
Ohmichi T, Mitsuhashi M, Tatebe H, Kasai T, Ali El-Agnaf OM, Tokuda T. Quantification of brain-derived extracellular vesicles in plasma as a biomarker to diagnose Parkinson’s and related diseases. Parkinsonism Relat Disord. 2019;61:82–7.
Dos Santos MCT, Barreto-Sanz MA, Correia BRS, Bell R, Widnall C, Perez LT, et al. miRNA-based signatures in cerebrospinal fluid as potential diagnostic tools for early stage Parkinson’s disease. Oncotarget. 2018;9:17455–65.
Gui Y, Liu H, Zhang L, Lv W, Hu X. Altered microRNA profiles in cerebrospinal fluid exosome in Parkinson disease and Alzheimer disease. Oncotarget. 2015;6:37043–53.
Manna I, Quattrone A, De Benedittis S, Vescio B, Iaccino E, Quattrone A. Exosomal miRNA as peripheral biomarkers in Parkinson’s disease and progressive supranuclear palsy: A pilot study. Parkinsonism Relat Disord. 2021;93:77–84.
He S, Huang L, Shao C, Nie T, Xia L, Cui B, et al. Several miRNAs derived from serum extracellular vesicles are potential biomarkers for early diagnosis and progression of Parkinson’s disease. Transl Neurodegener. 2021;10:25.
Barbagallo C, Mostile G, Baglieri G, Giunta F, Luca A, Raciti L, et al. Specific signatures of serum miRNAs as potential biomarkers to discriminate clinically similar neurodegenerative and vascular-related diseases. Cell Mol Neurobiol. 2020;40:531–46.
Cao XY, Lu JM, Zhao ZQ, Li MC, Lu T, An XS, et al. MicroRNA biomarkers of Parkinson’s disease in serum exosome-like microvesicles. Neurosci Lett. 2017;644:94–9.
Ozdilek B, Demircan B. Serum microRNA expression levels in Turkish patients with Parkinson’s disease. Int J Neurosci. 2021;131:1181–9.
Yao YF, Qu MW, Li GC, Zhang FB, Rui HC. Circulating exosomal miRNAs as diagnostic biomarkers in Parkinson’s disease. Eur Rev Med Pharmacol Sci. 2018;22:5278–83.
Nie C, Sun Y, Zhen H, Guo M, Ye J, Liu Z, et al. Differential expression of plasma exo-miRNA in neurodegenerative diseases by next-generation sequencing. Front Neurosci. 2020;14:438.
Xie S, Niu W, Xu F, Wang Y, Hu S, Niu C. Differential expression and significance of miRNAs in plasma extracellular vesicles of patients with Parkinson’s disease. Int J Neurosci. 2022;132:673–88.
Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, et al. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci. 2010;30:6838–51.
Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet. 2009;41:1303–7.
Shults CW. Lewy bodies. Proc Natl Acad Sci U S A. 2006;103:1661–8.
Marques O, Outeiro TF. Alpha-synuclein: from secretion to dysfunction and death. Cell Death Dis. 2012;3:e350.
Shi M, Liu C, Cook TJ, Bullock KM, Zhao Y, Ginghina C, et al. Plasma exosomal α-synuclein is likely CNS-derived and increased in Parkinson’s disease. Acta Neuropathol. 2014;128:639–50.
Jiang C, Hopfner F, Katsikoudi A, Hein R, Catli C, Evetts S, et al. Serum neuronal exosomes predict and differentiate Parkinson’s disease from atypical parkinsonism. J Neurol Neurosurg Psychiatry. 2020;91:720–9.
Roser AE, Caldi Gomes L, Schünemann J, Maass F, Lingor P. Circulating miRNAs as diagnostic biomarkers for Parkinson’s disease. Front Neurosci. 2018;12:625.
Dong X, Zheng D, Nao J. Circulating exosome microRNAs as diagnostic biomarkers of dementia. Front Aging Neurosci. 2020;12:580199.
Longinetti E, Fang F. Epidemiology of amyotrophic lateral sclerosis: an update of recent literature. Curr Opin Neurol. 2019;32:771–6.
Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62.
Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–72.
Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–11.
Pansarasa O, Bordoni M, Diamanti L, Sproviero D, Gagliardi S, Cereda C. SOD1 in amyotrophic lateral sclerosis: “ambivalent” behavior connected to the disease. Int J Mol Sci. 2018;19:1345.
Feneberg E, Steinacker P, Lehnert S, Schneider A, Walther P, Thal DR, et al. Limited role of free TDP-43 as a diagnostic tool in neurodegenerative diseases. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15:351–6.
Aoki Y, Manzano R, Lee Y, Dafinca R, Aoki M, Douglas AGL, et al. C9orf72 and RAB7L1 regulate vesicle trafficking in amyotrophic lateral sclerosis and frontotemporal dementia. Brain. 2017;140:887–97.
Westergard T, Jensen BK, Wen X, Cai J, Kropf E, Iacovitti L, et al. Cell-to-cell transmission of dipeptide repeat proteins linked to C9orf72-ALS/FTD. Cell Rep. 2016;17:645–52.
Gomes C, Keller S, Altevogt P, Costa J. Evidence for secretion of Cu, Zn superoxide dismutase via exosomes from a cell model of amyotrophic lateral sclerosis. Neurosci Lett. 2007;428:43–6.
Basso M, Pozzi S, Tortarolo M, Fiordaliso F, Bisighini C, Pasetto L, et al. Mutant copper-zinc superoxide dismutase (SOD1) induces protein secretion pathway alterations and exosome release in astrocytes: implications for disease spreading and motor neuron pathology in amyotrophic lateral sclerosis. J Biol Chem. 2013;288:15699–711.
Sproviero D, La Salvia S, Giannini M, Crippa V, Gagliardi S, Bernuzzi S, et al. Pathological proteins are transported by extracellular vesicles of sporadic amyotrophic lateral sclerosis patients. Front Neurosci. 2018;12:487.
Nonaka T, Masuda-Suzukake M, Arai T, Hasegawa Y, Akatsu H, Obi T, et al. Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep. 2013;4:124–34.
Porta S, Xu Y, Restrepo CR, Kwong LK, Zhang B, Brown HJ, et al. Patient-derived frontotemporal lobar degeneration brain extracts induce formation and spreading of TDP-43 pathology in vivo. Nat Commun. 2018;9:4220.
Feiler MS, Strobel B, Freischmidt A, Helferich AM, Kappel J, Brewer BM, et al. TDP-43 is intercellularly transmitted across axon terminals. J Cell Biol. 2015;211:897–911.
Kamelgarn M, Chen J, Kuang L, Arenas A, Zhai J, Zhu H, et al. Proteomic analysis of FUS interacting proteins provides insights into FUS function and its role in ALS. Biochim Biophys Acta. 2016;1862:2004–14.
Rizzuti M, Melzi V, Gagliardi D, Resnati D, Meneri M, Dioni L, et al. Insights into the identification of a molecular signature for amyotrophic lateral sclerosis exploiting integrated microRNA profiling of iPSC-derived motor neurons and exosomes. Cell Mol Life Sci. 2022;79:189.
Yelick J, Men Y, Jin S, Seo S, Espejo-Porras F, Yang Y. Elevated exosomal secretion of miR-124-3p from spinal neurons positively associates with disease severity in ALS. Exp Neurol. 2020;333:113414.
Freischmidt A, Müller K, Ludolph AC, Weishaupt JH. Systemic dysregulation of TDP-43 binding microRNAs in amyotrophic lateral sclerosis. Acta Neuropathol Commun. 2013;1:42.
Xu Q, Zhao Y, Zhou X, Luan J, Cui Y, Han J. Comparison of the extraction and determination of serum exosome and miRNA in serum and the detection of miR-27a-3p in serum exosome of ALS patients. Intractable Rare Dis Res. 2018;7:13–8.
Lo TW, Figueroa-Romero C, Hur J, Pacut C, Stoll E, Spring C, et al. Extracellular vesicles in serum and central nervous system tissues contain microRNA signatures in sporadic amyotrophic lateral sclerosis. Front Mol Neurosci. 2021;14:739016.
Gomes C, Sequeira C, Barbosa M, Cunha C, Vaz AR, Brites D. Astrocyte regional diversity in ALS includes distinct aberrant phenotypes with common and causal pathological processes. Exp Cell Res. 2020;395:112209.
Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 2012;11:323–30.
Farg MA, Sundaramoorthy V, Sultana JM, Yang S, Atkinson RA, Levina V, et al. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet. 2014;23:3579–95.
Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339:1335–8.
Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010;68:857–64.
Fecto F, Yan J, Vemula SP, Liu E, Yang Y, Chen W, et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol. 2011;68:1440–6.
Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Müller K, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci. 2015;18:631–6.
Bates GP, Dorsey R, Gusella JF, Hayden MR, Kay C, Leavitt BR, et al. Huntington disease. Nat Rev Dis Primers. 2015;1:15005.
McColgan P, Tabrizi SJ. Huntington’s disease: a clinical review. Eur J Neurol. 2018;25:24–34.
Ren PH, Lauckner JE, Kachirskaia I, Heuser JE, Melki R, Kopito RR. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat Cell Biol. 2009;11:219–25.
Pecho-Vrieseling E, Rieker C, Fuchs S, Bleckmann D, Esposito MS, Botta P, et al. Transneuronal propagation of mutant huntingtin contributes to non-cell autonomous pathology in neurons. Nat Neurosci. 2014;17:1064–72.
Yang W, Dunlap JR, Andrews RB, Wetzel R. Aggregated polyglutamine peptides delivered to nuclei are toxic to mammalian cells. Hum Mol Genet. 2002;11:2905–17.
Cicchetti F, Lacroix S, Cisbani G, Vallières N, Saint-Pierre M, St-Amour I, et al. Mutant huntingtin is present in neuronal grafts in Huntington disease patients. Ann Neurol. 2014;76:31–42.
Jeon I, Cicchetti F, Cisbani G, Lee S, Li E, Bae J, et al. Human-to-mouse prion-like propagation of mutant huntingtin protein. Acta Neuropathol. 2016;132:577–92.
Lee M, Liu T, Im W, Kim M. Exosomes from adipose-derived stem cells ameliorate phenotype of Huntington’s disease in vitro model. Eur J Neurosci. 2016;44:2114–9.
Hong Y, Zhao T, Li XJ, Li S. Mutant Huntingtin inhibits αb-crystallin expression and impairs exosome secretion from astrocytes. J Neurosci. 2017;37:9550–63.
Deng J, Koutras C, Donnelier J, Alshehri M, Fotouhi M, Girard M, et al. Neurons export extracellular vesicles enriched in cysteine string protein and misfolded protein cargo. Sci Rep. 2017;7:956.
Lee ST, Im W, Ban JJ, Lee M, Jung KH, Lee SK, et al. Exosome-based delivery of miR-124 in a Huntington’s disease model. J Mov Disord. 2017;10:45–52.
Herrmann IK, Wood MJA, Fuhrmann G. Extracellular vesicles as a next-generation drug delivery platform. Nat Nanotechnol. 2021;16:748–59.
Qu M, Lin Q, Huang L, Fu Y, Wang L, He S, et al. Dopamine-loaded blood exosomes targeted to brain for better treatment of Parkinson’s disease. J Control Release. 2018;287:156–66.
Elsharkasy OM, Nordin JZ, Hagey DW, de Jong OG, Schiffelers RM, Andaloussi SE, et al. Extracellular vesicles as drug delivery systems: Why and how? Adv Drug Deliv Rev. 2020;159:332–43.
Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. 2011;29:341–5.
Cooper JM, Wiklander PB, Nordin JZ, Al-Shawi R, Wood MJ, Vithlani M, et al. Systemic exosomal siRNA delivery reduced alpha-synuclein aggregates in brains of transgenic mice. Mov Disord. 2014;29:1476–85.
Bonafede R, Scambi I, Peroni D, Potrich V, Boschi F, Benati D, et al. Exosome derived from murine adipose-derived stromal cells: neuroprotective effect on in vitro model of amyotrophic lateral sclerosis. Exp Cell Res. 2016;340:150–8.
Lee M, Ban JJ, Kim KY, Jeon GS, Im W, Sung JJ, et al. Adipose-derived stem cell exosomes alleviate pathology of amyotrophic lateral sclerosis in vitro. Biochem Biophys Res Commun. 2016;479:434–9.
Didiot MC, Hall LM, Coles AH, Haraszti RA, Godinho BM, Chase K, et al. Exosome-mediated delivery of hydrophobically modified siRNA for Huntingtin mRNA silencing. Mol Ther. 2016;24:1836–47.
Ramos-Zaldívar HM, Polakovicova I, Salas-Huenuleo E, Corvalán AH, Kogan MJ, Yefi CP, et al. Extracellular vesicles through the blood-brain barrier: a review. Fluids Barriers CNS. 2022;19:60.
O’Brien K, Ughetto S, Mahjoum S, Nair AV, Breakefield XO. Uptake, functionality, and re-release of extracellular vesicle-encapsulated cargo. Cell Rep. 2022;39:110651.
Osorio-Querejeta I, Alberro A, Muñoz-Culla M, Mäger I, Otaegui D. Therapeutic potential of extracellular vesicles for demyelinating diseases; challenges and opportunities. Front Mol Neurosci. 2018;11:434.
Cone AS, Yuan X, Sun L, Duke LC, Vreones MP, Carrier AN, et al. Mesenchymal stem cell-derived extracellular vesicles ameliorate Alzheimer’s disease-like phenotypes in a preclinical mouse model. Theranostics. 2021;11:8129–42.
Reza-Zaldivar EE, Hernández-Sapiéns MA, Gutiérrez-Mercado YK, Sandoval-Ávila S, Gomez-Pinedo U, Márquez-Aguirre AL, et al. Mesenchymal stem cell-derived exosomes promote neurogenesis and cognitive function recovery in a mouse model of Alzheimer’s disease. Neural Regen Res. 2019;14:1626–34.
Ding M, Shen Y, Wang P, Xie Z, Xu S, Zhu Z, et al. Exosomes isolated from human umbilical cord mesenchymal stem cells alleviate neuroinflammation and reduce amyloid-beta deposition by modulating microglial activation in Alzheimer's disease. Neurochem Res. 2018;43:2165-2177
Elia CA, Tamborini M, Rasile M, Desiato G, Marchetti S, Swuec P, et al. Intracerebral injection of extracellular vesicles from mesenchymal stem cells exerts reduced aβ plaque burden in early stages of a preclinical model of Alzheimer’s disease. Cells. 2019;8:1059.
Wang SS, Jia J, Wang Z. Mesenchymal stem cell-derived extracellular vesicles suppresses iNOS expression and ameliorates neural impairment in Alzheimer’s disease mice. J Alzheimers Dis. 2018;61:1005–13.
Cui GH, Guo HD, Li H, Zhai Y, Gong ZB, Wu J, et al. RVG-modified exosomes derived from mesenchymal stem cells rescue memory deficits by regulating inflammatory responses in a mouse model of Alzheimer’s disease. Immun Ageing. 2019;16:10.
Yuyama K, Sun H, Sakai S, Mitsutake S, Okada M, Tahara H, et al. Decreased amyloid-β pathologies by intracerebral loading of glycosphingolipid-enriched exosomes in Alzheimer model mice. J Biol Chem. 2014;289:24488–98.
Yuyama K, Sun H, Usuki S, Sakai S, Hanamatsu H, Mioka T, et al. A potential function for neuronal exosomes: sequestering intracerebral amyloid-β peptide. FEBS Lett. 2015;589:84–8.
Katsuda T, Tsuchiya R, Kosaka N, Yoshioka Y, Takagaki K, Oki K, et al. Human adipose tissue-derived mesenchymal stem cells secrete functional neprilysin-bound exosomes. Sci Rep. 2013;3:1197.
Haney MJ, Klyachko NL, Zhao Y, Gupta R, Plotnikova EG, He Z, et al. Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J Control Release. 2015;207:18–30.
Jarmalavičiūtė A, Tunaitis V, Pivoraitė U, Venalis A, Pivoriūnas A. Exosomes from dental pulp stem cells rescue human dopaminergic neurons from 6-hydroxy-dopamine-induced apoptosis. Cytotherapy. 2015;17:932–9.
Chen HX, Liang FC, Gu P, Xu BL, Xu HJ, Wang WT, et al. Exosomes derived from mesenchymal stem cells repair a Parkinson’s disease model by inducing autophagy. Cell Death Dis. 2020;11:288.
Hill AF. Extracellular vesicles and neurodegenerative diseases. J Neurosci. 2019;39:9269–73.
Acknowledgements
Not applicable.
Funding
This review was supported by the National Natural Science Foundation of China (82260422), Key R&D Planning Project of Jiangxi Science and Technology Commission, China (20203BBGL73126), and the Distinguished Expert Project of Sichuan Province Tianfu Scholar (CW202002).
Author information
Authors and Affiliations
Contributions
ZT and WJ conceptualized this review. LZ and WX drafted the manuscript and created the figures. WX, YX, WYK, WJ, XF, HD and WQ performed the literature search and reviewed the content of this manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, Z., Wang, X., Wang, X. et al. Research progress on the role of extracellular vesicles in neurodegenerative diseases. Transl Neurodegener 12, 43 (2023). https://doi.org/10.1186/s40035-023-00375-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40035-023-00375-9