
Abstract
Neurological disorders such as stroke, multiple sclerosis, as well as the neurodegenerative diseases Parkinson's or Alzheimer's disease are accompanied or even powered by danger associated molecular patterns (DAMPs), defined as endogenous molecules released from stressed or damaged tissue. Besides protein-related DAMPs or “alarmins”, numerous nucleic acid DAMPs exist in body fluids, such as cell-free nuclear and mitochondrial DNA as well as different species of extracellular RNA, collectively termed as self-extracellular nucleic acids (SENAs). Among these, microRNA, long non-coding RNAs, circular RNAs and extracellular ribosomal RNA constitute the majority of RNA-based DAMPs. Upon tissue injury, necrosis or apoptosis, such SENAs are released from neuronal, immune and other cells predominantly in association with extracellular vesicles and may be translocated to target cells where they can induce intracellular regulatory pathways in gene transcription and translation. The majority of SENA-induced signaling reactions in the brain appear to be related to neuroinflammatory processes, often causally associated with the onset or progression of the respective disease. In this review, the impact of the diverse types of SENAs on neuroinflammatory and neurodegenerative diseases will be discussed. Based on the accumulating knowledge in this field, several specific antagonistic approaches are presented that could serve as therapeutic interventions to lower the pathological outcome of the indicated brain disorders.
Similar content being viewed by others
Background
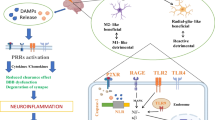
Neuroinflammation as a multifactorial process substantially contributes to several neurological diseases such as ischemic stroke, bacterial/viral infections, traumatic brain injury (TBI), and neurodegenerative diseases such as Alzheimer's disease (AD), Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS), or multiple sclerosis (MS) [169, 244, 264, 413]. In the early defense stage, the innate immune system protects against sterile hyperinflammation and microbial infections by the recognition of endogenous alarmins or danger-associated molecular patterns (DAMPs) as well as microbial pathogen-associated molecular patterns (PAMPs), respectively. DAMPs include cytosolic, mitochondrial, or nuclear components such as proteins (e.g. heat shock proteins, histones, high mobility group box protein 1 (HMGB1), cold-inducible RNA-binding protein), carbohydrates (e.g. hyaluronan), nucleic acids (various types of RNA and DNA) and low molecular weight components (e.g. uric acid crystals, ATP, heme). PAMPs include different types of microbial molecules such as bacterial cell wall components as well as viral nucleic acids [26, 114, 276, 482]. These diverse agonists contain specific recognition epitopes that are sensed by pattern recognition receptors (PRRs), which are expressed by a variety of host immune and non-immune cells [26, 132, 482].
A major class of PRRs is defined by cell membrane-expressed or intracellular endosomal Toll-like receptors (TLRs) or cytosolic nucleic acid sensors, the latter being activated predominantly by PAMPs but also by some DAMPs, exhibiting some overlapping specificity and selectivity [132, 199, 262, 459, 482]. Following the binding of a particular agonist at the cell membrane or after endocytosis and degradation in endosomes (particularly relevant for nucleic acid-based PAMPs), the activation of respective PRRs together with the intracellular recruitment of adapter proteins such as myeloid differentiation factor 88 (MyD88) initiate inflammatory immune responses via intracellular signaling pathways. Primarily, these pathways involve the nuclear factor-kappa B (NF-κB) but also other transcription factors that induce the expression and release of cytokines, chemokines and antiviral interferons (IFNs) in immune and non-immune cells [187, 222, 228]. As a consequence, the inflammatory tissue environment is sensitized to help recruiting inflammatory cells (such as neutrophils and monocytes/macrophages) that will remove the pathogens or damaging factors and thereby contribute to tissue repair, resolution of inflammation and cellular homeostasis. Hyperinflammatory conditions may be caused if the defense system remains insufficiently controlled [19, 186].
Numerous self-extracellular nucleic acids (SENAs), including nuclear (nuc) and mitochondrial (mt)DNA, messenger RNA (mRNA), transfer RNA (tRNA), ribosomal RNA (rRNA) and other non-coding RNA species, have been identified as potential DAMPs in a variety of pathophysiological situations [36, 46, 114, 375, 406]. During the previous decade, self-extracellular rRNA (rexRNA), which is predominantly liberated under conditions of tissue damage or cell injury, has been characterized as the primary RNA-type alarmin, but it can also be considered as a damaging factor that contributes to disease progression in ischemic stroke, thrombosis, myocardial infarction, atherosclerosis, rheumatoid arthritis or cancer [37, 110,111,112, 114, 342]. Besides its multifunctional and disease-promoting potential in sterile inflammatory diseases of several organ systems, the ubiquitous DAMP rexRNA has previously been recognized as a potent adjuvant for PAMPs as well, particularly inducing the activation of TLR2 on macrophages and astrocytes in a synergistic manner [112, 286], thereby serving as relevant sensitizer during microbial infections.
While a variety of non-nucleic acid DAMPs have been recognized and their functional role being partially characterized in the central nervous system (CNS), the contribution of SENAs in neuroinflammatory pathologies remains greatly unexplored, particularly regarding their mechanisms of action. With this review we aim to compile the current knowledge on the involvement of SENAs for the pathophysiology of various neuroinflammatory diseases and their clinical implications. Pertinent open questions in the field such as “Which cell types in the brain do contribute to the release of SENAs with high inflammatory or degenerative potential?” and “Which factors or stress situations can trigger the active or passive release of SENAs in the brain?” will be addressed in the first section with various SENAs and their principal reactions being presented. In the second section we will focus on different receptor types and signaling pathways used by SENAs to transmit their functions. In the third, and major part of this review, we will then collate current scientific knowledge on the role of SENAs in selected neuroinflammatory diseases, namely ischemic stroke, MS, AD, and PD. A further open question: “Which regulatory or antagonistic mechanisms are required or available to dampen or even prevent the pro-inflammatory activation of brain-resident cells and peripheral immune cells?” will be addressed in the perspectives part, where potential therapeutic options to modulate the activity of both adverse and favorable SENAs will be discussed. Finally, in the conclusion section, we put up the question what we can learn from the presented data, and point out some hypotheses and possible directions for future translational research.
Major types of self-extracellular nucleic acids
Cell-free circulating forms of nucleic acids such as nuclear (nuc)DNA, mitochondrial (mt)DNA, and various species of RNA have been detected in all extracellular fluids, including blood plasma and cerebrospinal fluid (CSF) [272, 373, 383]. Under various pathological conditions such as hypoxia/ischemia, oxidative or metabolic stress, these nucleic acids can leak or are actively released from injured/damaged cells, tumor cells, monocytes/macrophages and other immune cells as well as they are liberated from mast cells during their degranulation reaction into the extracellular environment throughout the body, including the brain [64, 81, 99, 305, 306, 369]. The released exRNA includes several forms of RNA species such as microRNA (miRNA), long non-coding RNA (lncRNA), circular RNA (circRNA), as well as tRNA, rRNA, and mRNA that can be liberated from cells either in free form or in association with proteins, lipids as well as with extracellular vesicles (EVs) [114, 167, 382, 407]. EV-associated exRNAs such as miRNAs are thereby shuttled from one cell type to another to promote the regulation of gene expression at the transcriptional and translational level in the target cell [96, 192, 272]. Yet, a detailed discussion of exRNA-related aspects in this context is beyond the scope of this article, and thus will be only marginally addressed when discussing the role of exRNAs in inflammatory processes during the course of neurological diseases. In the following, the major types of SENAs will be introduced.
Ribosomal RNA
Circulating rexRNA in body fluids can be liberated in principle from all cell types upon mechanical stress (such as fluid shear stress in blood vessels), hypoxia/ischemia as well as under various cell stimulatory conditions and during tissue injury [58, 111, 203, 324, 407, 477]. RexRNA in isolated form or in complex with proteins or EVs is considered as a common damaging factor in a variety of cardiovascular and non-cardiovascular diseases [305, 306, 436, 446]. Quantitatively, the heterogeneous forms of rexRNA (as ribosomal fragments with a varying degree of ribosomal proteins) are by far the most abundant exRNA species, being detectable in blood plasma, CSF and other body fluids and are considered nucleic acid DAMPs [59, 66, 114].
As to its role as non-typical DAMP, no direct interactions of rexRNA with cell membrane PRRs or other receptors has been recognized so far. Yet, some observations point to the uptake of EV-associated rexRNA fragments that may become recognized by endosomal TLR3 [46], however, unequivocal experimental proofs are missing so far. Nevertheless, rexRNA is able to directly induce the expression and release of several cytokines such as tumor necrosis factor-α (TNF-α) or interleukin-6 (IL-6) in monocytes/macrophages [36, 111], and thereby promotes a robust pro-inflammatory circuit, involving NF-κB-mediated signal transduction. Moreover, rexRNA was found to enhance the activity of pro-inflammatory PAMPs (such as TLR2-ligands) in a synergistic manner both in macrophages and astrocytes [112, 286].
MicroRNAs
MiRNAs are small ribonucleic acids consisting of 21–25 nucleotides (often folded as hairpins), which primarily act intracellularly as transcriptional and translational regulators by targeting specific mRNAs via base pairing of their untranslated regions to promote e.g. their RNase-dependent degradation. About 70% of the so far identified miRNAs are expressed in the brain. Since various miRNAs are associated with EVs or exosomes that are released from cells upon various stimulations, EV-associated miRNAs can be easily translocated to target cells or tissues, where they are taken up [267]. In the narrower sense, however, miRNAs cannot be considered as an exRNA-DAMP that operates in the extracellular compartment, rather, they facilitate molecular communications between cells. Yet, the functional consequences of miRNA transfer between particular cell types in the CNS for the development of neuroinflammatory or neurodegenerative diseases remains a great challenge for further investigations.
Circular RNAs
CircRNAs are coding or non-coding RNA molecules, which are characterized by back-splicing and the formation of covalent closed continuous nucleotide loops [265]. These RNAs are defined as single-stranded RNA (ssRNA) formed by head-to-tail splicing of a linear mRNA fragment, which can regulate gene expression at multiple levels as they induce transcription and alternative splicing in the nucleus. Moreover, they serve (together with RNA-binding proteins) as sponges for miRNAs, thereby inhibiting their interactions with the respective mRNA targets in the cytoplasm. Thus, circRNAs are considered as post-transcriptional regulatory elements [313]. In line with these structural properties, including covalently closed loops with neither 5′-3′ polarity nor a poly-adenylated tail, circRNAs are much more stable than linear RNAs and insusceptible to degradation by RNA exonuclease or RNase R [358]. Under pathological conditions, particular circRNAs have been identified in the blood stream as potential biomarkers for certain inflammatory diseases [238].
Long non-coding RNAs
Another class of ncRNAs entails lncRNAs with a length of more than 200 nucleotides, which are transcribed by RNA polymerase II and processed like protein-coding RNAs [421, 437]. LncRNAs are described to promote apoptosis, angiogenesis, inflammation, or cell death through mechanisms of gene regulation, epigenetically as well as on transcriptional and post-transcriptional levels [28, 94, 131, 322, 437]. As such, lncRNAs are associated with chromatin-modifying enzymes or DNA-binding proteins and thereby mediate activation or silencing of gene transcription [28, 322, 421, 437]. Furthermore, lncRNAs influence nuclear transport mechanisms [20, 468], they modulate gene expression by interfering with the splicing of pre-mRNAs [421], they act upon miRNAs and thereby compete with mRNAs for the binding to their target miRNAs [94, 421, 437], and they influence the assembly of the translation initiation complex [421, 437]. Functional abnormalities in lncRNAs are strongly associated with the development of various inflammatory diseases [105].
Cell-free extracellular DNA and neutrophil extracellular traps (NETs)
Upon cellular stress, tissue injury or infection, different species of extracellular DNA, particularly cell-free DNA (cfDNA), are detectable in the blood. Under physiological conditions, the level of cfDNA is very low (1–50 ng/ml) due to their degradation, particularly by DNase1 and DNase1-like 3 [7, 403]. Due to mutations in such DNases or due to an impaired apoptotic clearance of cfDNA, autoimmune disorders such as systemic lupus erythematosus (SLE) can develop with the occurrence of anti-DNA antibodies followed by e.g. massive complement activation [349].
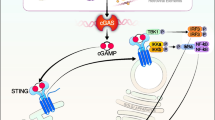
Mitochondrial dysfunction leads to the release of mtDAMPs, such as mtDNA, mitochondrial transcription factor A (TFAM), cardiolipin, cytochrome c and other mitochondrial-derived molecules, thereby activating specific inflammatory cascades, collectively referred to as mito-inflammation [293]. Among cfDNAs, mitochondrial exDNA (mexDNA) has been identified as a stable DAMP, being released under conditions of tissue damage and cell death, such as in myocardial infarction, TBI [389] or in response to increased oxidative or metabolic stress [237, 300]. Furthermore, mexDNA is a potent trigger of the innate immunity response due to its bacterial ancestry and the presence of hypo-methylated CpG motifs [300]. Once released into either the cytosol or the extracellular space, mtDNA fragments instigate inflammation via the interaction with PRRs, including TLRs, nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), or the cyclic GMP/AMP (cGAMP) synthase (cGAS)/stimulator of interferon genes (STING) pathway [123, 300, 462] (see below). In line with the higher resistance of mtDNA towards nuclease-dependent degradation compared with nucDNA, the circulating mexDNA is highly stable and can be detected in body fluids such as plasma and CSF [123].
ExDNA is also a major component of the extracellular decondensed chromatin, designated as NETs, which are released from neutrophils upon activation by various endogenous and exogenous (inflammatory) factors. NETs are generated by NADPH-oxidase-dependent and -independent pathways particularly in neutrophils and mast cells, whereby extrinsic (microbial) as well as intrinsic stimuli (e.g. hydrogen peroxide) or phorbol myristate acetate can induce NETosis [33, 87, 120]. Furthermore, the post-translational modification of histones by the peptidyl-arginine-deiminase-4 (PAD4) is required for loosening the DNA-histone interactions of the chromatin network to promote NETosis [412].
Two major functional areas of NETs have been described:
(a) Upon stimulation of neutrophils, the generated ultra-large scaffold of NETs (composed of the entire decondensed nucDNA, histones, and various antimicrobial proteins and enzymes), serves to catch and kill microbes in the initial immune response [33, 120]. Together with the phagocytic action of macrophages (intracellular killing), the extracellular killing function of NETs thereby serves to protect various organisms from invading pathogens [12, 280].
(b) Activated blood platelets also serve as inducer of NETosis by providing adhesive interactions with neutrophils, culminating in the immediate formation of cellular aggregates from which NETs are released to provoke prothrombotic functions. Besides fibrin, NETs appear to be a major component of the generated venous and arterial thrombi [259, 386]. In fact, in experimental models of thrombosis, the administration of DNase1 significantly prevented or reduced the outcome of thromboembolic diseases [121, 127, 259]. In essence, hardly any inflammatory, cardiovascular or chronic disease is devoid of the generation of NETs, which thereby not only function as a causal disease factor but may also serve as diagnostic or prognostic biomarkers [69, 253].
Meanwhile, not only neutrophils but also mast cells, eosinophils, basophils, macrophages and also microglial cells as the resident immune cell of the CNS have been described to release nucDNA-containing extracellular traps (ETs) in response to various stimuli. Yet, the mechanisms of formation and particular functions of ETs also in an organ-specific context such as the brain are still insufficiently understood and under intensive investigation [335, 416].
Pattern recognition receptors and self-nucleic acid-mediated inflammatory signal transduction
Pattern recognition receptors in inflammation
The functional activities of DAMPs and PAMPs in body defense related to the innate immune response are mediated by several receptor types in immune and other cells, designated as PRRs [132, 182, 276, 289, 291, 360]. PRRs were originally described only to recognize specific PAMPs [168], but it is now well accepted that these receptors are also involved in the signal transduction of different DAMPs, including HMGB1, histones, heat shock proteins, as well as SENAs alone or in complex with other components [132, 199, 262, 459, 482]. PRRs such as TLRs are not only expressed by various peripheral immune cells, but also by resident cells of the CNS such as microglia, astrocytes, oligodendrocytes, and neurons, which all participate in the initial immune response against bacterial/viral brain infections and acute CNS injuries (e.g. mechanical traumas, ischemic stroke) [75, 191, 200, 213, 352]. These PRR-expressing cells also play a crucial role in the generation of neuroinflammation in neurodegenerative chronic diseases such as AD, PD, Huntington's disease (HD), ALS, and MS [199, 352] (Table 1).
Toll-like receptors
Within the class of PRRs with a wide variety for recognition of DAMPs and PAMPs, the TLRs exert a key role in both body defense and hyperinflammatory diseases, if left uncontrolled [168, 262, 482]. Cell membrane-expressed TLRs including TLR2, TLR4, TLR5, TLR6, and TLR11 recognize microbial motifs of the pathogen cell wall, whereas TLR3, TLR7, TLR8, TLR9, and TLR13 are expressed on the endosomal compartments and are responsible for the recognition of pathogen-derived exogenous nucleic acids, following their endocytosis and degradation [224, 228, 312]. Since SENAs principally exhibit a much higher degree of post-transcriptional modification than nucleic acid PAMPs, they are largely prevented from recognition by TLRs in order to avoid hyperinflammation and autoimmunity [118].
Double-stranded RNAs (dsRNA) as contained in viruses but also artificial dsRNA such as poly (I:C) as well as fragments of mRNA or exRNA released from necrotic cells were described to activate endosomal TLR3 [42, 185, 241], whereas TLR7/TLR8 preferentially recognize ssRNA [146], and TLR9 shows a preference for recognizing bacterial or viral DNA [273]. The activation of these receptors by self-nucleic acids is very limited, whereas foreign nucleic acids are protected from the ribonucleolytic degradation among others by their capsids unless they are released within the endolysosomal compartment after their cellular uptake via phagocytosis or endocytosis [254].
TLRs belong to the Toll/IL-1 receptor (TIR) family of proteins, and binding of a respective ligand leads to the (hetero-) dimerization of a given TLR, followed by the intracellular recruitment of adaptor proteins. Here, the majority of TLRs use MyD88 as intracellular adaptor protein, which binds to the TIR domain of all TLRs (except endosomal TLR3) to activate downstream signaling pathways, involving IL-1R-associated kinases [146, 360]. The signaling reactions culminate in the phosphorylation of IκB, followed by its ubiquitination and proteasomal degradation, which enables the dissociation of the formerly bound transcription factor NF-κB. NF-κB then translocates into the nucleus and induces the expression/production of inflammatory cytokines such as TNF-α, IL-6, inducible nitric oxide synthase (iNOS), or pro IL-1β [146, 360]. In contrast, TLR3 signals through the TIR domain-containing adaptor inducing IFN-β (TRIF), finally leading to the production of type I IFNs and antiviral immunity-related proteins [360, 424, 425, 441].
Retinoic acid-inducible gene-1-like receptors
Besides TLRs, cytosolic RNA sensors such as retinoic acid-inducible gene-1 (RIG-1)-like receptors (RLRs), and certain NLRs, or DNA sensors such as absent in melanoma 2 (AIM2) and cGAS contribute to inflammatory responses by various cell types, also in the CNS [191, 323, 408]. The protein family of RLRs include RIG-1, melanoma differentiation-associated protein 5 (MDA5), and laboratory of genetics and physiology 2 (LGP2) [133, 149, 318]. RLRs preferentially recognize dsRNA with different structural features and they are key sensors of virus infections in mediating mainly the transcriptional activation of type I IFNs [318, 323]. All RLRs have a central helicase domain and a so-called carboxy-terminal domain. RIG-1 and MDA5 also contain two amino-terminal caspase activation and recruitment domains (CARD), which interact upon RNA binding, being present in mitochondrial antiviral-signaling protein (MAVS). MAVS serves as the essential adaptor protein initiating the activation of TANK-binding kinase (TBK1) and IκB kinase-ε, which together with NF-κB induce the transcription of genes encoding type I IFNs as well as other immuno-regulatory proteins [133, 149, 318].
Absent in melanoma 2
AIM2 is another cytosolic sensor that detects double-stranded DNA (dsDNA) of 50–80 bp from foreign species or of self-origin. It triggers the formation of an inflammasome complex resulting in the activation of caspase-1 [158, 159]. The inflammasome is another essential component of the innate immune system mainly involved in NF-κB-dependent expression and production of IL-1β and IL-18. Here, a DAMP- or PAMP-driven initial stimulus provokes a second signal, which is mediated via intracellular receptors such as AIM2-like receptors or NLRs. These events result in the activation of caspase-1, which ultimately drives the assembly of the ultra-large inflammasome protein complex and the maturation and release of IL-1β and IL-18 from their protein precursors [23, 30, 34, 139]. In the CNS, several cells including microglia, neurons, and astrocytes, express components of the inflammasome and may respond to extracellular nucleic acids as well [8]. Moreover, it has been reported that inflammasome proteins in CSF of brain-injured patients could serve as biomarkers of functional outcome, yet, any connection to self-nucleic acids is missing so far [3].
Cyclic GMP/AMP synthase
Moreover, models of TLR9-independent sterile inflammation indicate the existence of a further cytosolic DNA-sensing pathway [354], which contains cGAS as a cytosolic DNA-sensing PRR [124, 323]. cGAS is a cytoplasmic nucleotidyl-transferase that belongs to the class of template-independent polymerases. Upon dsDNA binding, cGAS catalyzes the conversion of GTP and ATP into 2′3′-cGAMP, followed by the activation of STING, thereby inducing the gene expression of type I IFNs, IFN-stimulated genes, and several other inflammatory mediators, proapoptotic genes and chemokines [74]. cGAS recognizes a broad repertoire of DNA species of both foreign and self-origin (sterile inflammation) [74]. Conclusively, blocking of the cGAS/STING pathway is discussed as a therapeutic regimen in the treatment of several inflammatory diseases [74, 124].
Receptor for advanced glycation end products
The receptor for advanced glycation end-products (RAGE) belongs to the immunoglobulin superfamily of proteins and was originally described to be recognized and activated by several ligands such as advanced glycated proteins (AGEs), S100 proteins, but also by nucleic acids [27, 163, 344]. In the healthy brain, RAGE is expressed by neurons, astrocytes, microglia, and vascular cells at a low level. However, during various pathological conditions its expression is strongly upregulated in a ligand-dependent manner, and propagates cellular dysfunction in inflammatory diseases [44, 163, 333]. Although AGEs accumulate in the aging brain and could be one of the reasons for age-related diseases like PD, a combined action of DAMPs together with AGEs to provoke engagement/activation of RAGE has not been studied so far.
Self-nucleic acid-mediated inflammatory signal transduction
Under conditions of cell injury and tissue damage or defects in the intracellular degradation or processing machinery of nucleic acids, substantial amounts of exDNA and exRNA accumulate, resulting in the activation of endosomal TLRs or the aforementioned cytosolic nucleic acid sensors with subsequent activation of the NF-κB signaling pathway [25, 190, 268, 269, 323, 344]. Yet, as mentioned above, several structural features of self-nucleic acids in comparison to nucleic acid-PAMPs prevent the former ones from inducing inflammatory signal transduction reactions. Conversely, due to the insufficient control by extracellular endonucleases or high concentrations of self-nucleic acids that may associate with proteins to induce the production of autoantibodies, autoimmune diseases such as SLE may develop [21, 198, 317]. Accordingly, the RAGE-dependent uptake of complexes containing DNA and the DAMP HMGB1 was demonstrated to stimulate cytokine production in plasmacytoid dendritic cells and B cells [367]. Furthermore, cytosolic RNA- and DNA-sensors might be involved in the recognition of immune responses as well, whereby the stimulation of such nucleic acid receptors are exploited for adjuvant therapies and treatment of non-neurological disorders such as cancer or allergy [273].
Endosomal TLRs also provide major recognition sites for complexes of self-exRNA or self-exDNA fragments with the neutrophil-derived antimicrobial peptide LL37, a C-terminal peptide of human cathelicidin [126, 201]. In fact, in such complexes, LL37 appears to prevent further degradation of exRNA and exDNA. As a consequence, the self-nucleic acid-LL37 complex, but not self-RNA or self-DNA alone, activates TLR7, TLR8, or TLR9, which initiates the autoimmune-inflammatory cascade in e.g. psoriasis, a chronic skin disease [126, 201, 455].
Moreover, SENAs were found to sensitize PRRs for their respective PAMP ligands. As a nucleic acid binding protein, HMGB1 appears to be crucial for the recognition of self-nucleic acids by TLR3, TLR7, and TLR9, whereas no receptor activation occurred in the absence of HMGB1 [427]. Furthermore, a synergistic activation of TLR2 by rexRNA together with TLR2-ligands resulted in the synergistically elevated expression of cytokines [286]. Similarly, complexes of rexRNA with canonical lipopeptide TLR2-ligands but also with HMGB1 caused a pro-inflammatory activation of astrocytes both in vitro and in vivo, indicative for the potential action of self-nucleic acids as sensitizer of brain dysfunction and damage [112]. A graphical illustration of DAMPs, PAMPs, and PRRs and their downstream signaling is presented in Fig. 1.

Signaling pathways induced by DAMPs/PAMPs and nucleic acids in the brain. AIM2: Absent in melanoma 2; cGAS: cyclic GMP/AMP (cGAMP) synthase; DAMPs: danger-associated molecular patterns; EV: extracellular vesicle; IRAK: IL-1 receptor-associated kinase; IRF3: interferon regulatory factor 3; LGP2: Laboratory of genetics and physiology 2; MAVS: mitochondrial antiviral-signaling protein; MDA5: melanoma differentiation-associated protein 5; MyD88: myeloid differentiation factor 88; NF-κB: nuclear factor-kappa B; NLR: nucleotide-binding oligomerization domain (NOD)-like receptor; NLRP3: NLR family pyrin domain containing protein 3; PAMPs: pathogen-associated molecular patterns; RAGE: receptor for advanced glycation end-products; RIG-1: retinoic acid-inducible gene-1; SENAs: self-extracellular nucleic acids; STING: stimulator of interferon genes; TLR: Toll-like receptor; TRIF: TIR-domain-containing adaptor-inducing interferon beta; TBK1: TANK-binding kinase
Self-extracellular nucleic acids in neuroinflammatory diseases
Ischemic stroke
Etiology and pathogenesis of ischemic stroke
Stroke is the major cause of adult physical disability and one of the leading causes of mortality worldwide, accounting for 7.08 million deaths in 2020. Ischemic stroke is responsible for approximately 87% of all strokes, while hemorrhagic stroke accounts for 13% [374]. Ischemic stroke occurs as a result of interruption of blood flow to the brain due to thrombotic and embolic events. Despite the huge global burden of ischemic stroke, intravenous thrombolysis with recombinant tissue plasminogen activator (rt-PA) and mechanical thrombectomy are the only evidence-based treatment options approved for acute ischemic stroke [321]. However, the uptake of intravenously administered rt-PA is limited by a clinically challenging diagnosis, short therapeutic time window and numerous contra-indications. Similarly, mechanical thrombectomy is only indicated for patients with acute ischemic stroke due to a large cerebral artery occlusion in the anterior circulation, and needs to be achieved within 6 to 24 h (for certain patients) after stroke onset [362]. Therefore, there is an urgent need to better understand pathological cellular and molecular mechanisms in ischemic stroke to develop novel therapeutic perspectives that can protect and recover salvageable brain tissue.
The onset of cerebral ischemia initiates a complex cascade of several interrelated and overlapping pathological mechanisms. The first event of the ischemic cascade is the reduction of oxygen and glucose, which leads to a failure to produce high-energy metabolites to maintain the cellular homeostasis. The involved processes include ionic imbalance, excitotoxicity, calcium overload, cytotoxic and vasogenic edema, peri-infarct depolarization, oxidative and nitrosative stress, cell death, BBB disruption, and inflammation [256]. Immediately after onset of cerebral ischemia, injured and dying neurons release DAMPs to be involved in the activation of brain-resident cells, including microglia, astrocytes, and endothelial cells. Upon M1-like polarization of microglia, reactive astrocytes and activated endothelial cells release pro-inflammatory cytokines, matrix metalloproteinases and reactive oxygen species (ROS), which cause the loss of the BBB integrity [169, 181]. Moreover, pericytes and astrocytic endfeets are lifted from the basement membrane, which further weakens the BBB allowing circulating leukocytes to infiltrate the cerebral parenchyma, where they produce pro-inflammatory factors and exacerbate tissue injury. In the delayed subacute phase, microglia/macrophage switch to an M2-like (anti-inflammatory) phenotype results in the clearance of cellular debris, and, by expressing anti-inflammatory mediators and neurotrophic factors, promotes glial scar formation as well as BBB repair, neurogenesis, oligodendrogenesis, and angiogenesis [169, 181].
The role of extracellular RNAs in the neuroinflammatory cascade after ischemic stroke
Various species of exRNA are expressed and become released from brain microvascular endothelial cells, thereby participating in the regulation of the blood–brain barrier (BBB) permeability [109, 431]. We have previously demonstrated in a rat stroke model that self-rexRNA did aggravate ischemic injury by inducing vascular permeability via VEGF [110, 113], and that pretreatment of animals with RNase1 resulted in vessel protection accompanied by reduced edema formation as well as a smaller infarct volume [110, 387] (Table 2). We also reported that neurons respond to hypoxia/ischemia or glutamate excitotoxicity with the release of rexRNA. Although most rexRNA is probably liberated into the extracellular space in a passive manner by necrotic cell death, an active calcium-dependent release of rexRNA by structurally intact neurons was observed as well [112]. While low-dose rexRNA alone had no pro-inflammatory activity on astrocytes, a prominent TLR2/NF-κB-dependent signaling mechanism was achieved in the presence of either Pam2CSK4 (a synthetic PAMP molecule that mimics bacterial infection) or HMGB1 (the most abundant DAMP, liberated in ischemic brain tissue) [112]. Conclusively, self-exRNA may act as an essential sensitizer or adjuvant to engage the binding of endogenous DAMPs to their cognate receptors to trigger sterile inflammation during the course of ischemic stroke.
Further experimental evidence has demonstrated that regulatory ncRNAs are involved in many aspects of the pathogenic mechanisms that underlie the tissue damage following stroke, including excitotoxicity, oxidative stress, neuroinflammation, BBB damage and apoptosis as well as aspects of post-stroke recovery including neurogenesis and angiogenesis. Apart from intracellular, functionally active ncRNAs, including housekeeping RNAs (rRNA, tRNA, small nuclear and nucleolar RNAs) as well as miRNAs, lncRNAs and circRNAs, circulating extracellular ncRNAs have been proposed as potential clinical stroke biomarkers with regard to diagnosis, prognosis or disease severity (ncRNAs with proven prognostic value in human stroke patients are summarized in Table 3). Several selected examples of regulatory ncRNAs that either augment or mitigate the neuroinflammatory response to ischemic stroke are discussed below. For a more comprehensive overview the reader is referred to recent review articles in the field [183, 211, 231, 381, 411].
MicroRNAs and stroke-associated neuroinflammation
Accumulating evidence indicates that particular miRNAs play an important role in post-ischemic inflammatory responses (for an overview see also [218]). Upon cerebral ischemia/reperfusion (I/R) injury in mice, the expression of miR-455-5p in the brain parenchyma and respective levels in peripheral blood are decreased [453]. Intracerebral pretreatment with agomir-455-5p, a miR-455-4p mimic, decreased the infarct volume, enhanced BBB integrity, and improved the neurological function, whereas administration of the miR-455-5p antagonist antagomiR-455-5p amplified these pathogenic processes [453]. Moreover, miR-455-5p agonism alleviated stroke-induced microglia activation and release of inflammatory factors at least partly by downregulation of C-C chemokine receptor type 5 [453]. Similarly, in a murine model of ischemic stroke, intracerebral application of miR-671-5p agomir alleviated tissue injury, functional deficits and neuroinflammatory processes by directly targeting the NF-κB mRNA expression [77].
Furthermore, let-7c-5p levels were demonstrated to be decreased in patients with acute stroke but also in mice that underwent I/R injury. Intracerebral let-7c-5p overexpression reduced neuroinflammation, infarct volume and functional deficits after ischemic stroke in mice as well [282]. Accordingly, overexpression in vitro of let-7c-5p suppressed the expression of pro-inflammatory mediators in microglia activated by either lipopolysaccharide (LPS), by oxygen–glucose deprivation/reoxygenation (OGD/R) or the exposure to conditioned medium obtained from OGD-treated neurons. Let-7c-5p inhibited the pro-inflammatory activation of microglia via the direct targeting of caspase-3 [282].
As another example of anti-inflammatory miRNAs in the context of ischemic stroke, the expression of miR-210 is substantially upregulated in astrocytes of human brain tissue from white matter stroke patients as well as in primary human fetal astrocytes, exposed to a combination of hypoxic and inflammatory stress in vitro [189]. The transfection with miR-210-mimics increased glycolysis, enhanced lactate export, and promoted an anti-inflammatory transcriptional and translational signature in human astrocytes [189]. In contrast, the pre- and post-stroke treatment with a miR-210 inhibitor in mice significantly decreased cerebral infarction, behavioral deficits, expression of pro-inflammatory cytokines, microglial activation, and macrophage infiltration [160].
Furthermore, the expression of pro-inflammatory miR-3473b is upregulated in the cortex and striatum of mice following experimental stroke. An intracerebroventricular injection of the miR-3473b antagomir prior to stroke remarkably attenuated the ischemia-induced expression of miR-3473b and pro-inflammatory factors, decreased infarct volume and sensorimotor impairment [398]. Complementary in vitro experiments revealed that miR-3473b triggers the pro-inflammatory activation of microglia via inhibition of suppressor of cytokine signaling 3 (SOCS3), an intracellular, cytokine-inducible protein that inhibits cytokine signaling in numerous cell types [398].
Finally, in a mouse model of ischemic stroke, global genetic ablation of pro-inflammatory miR-155 reduced the extent of brain tissue damage and improved neurobehavioral impairments [405]. Intracerebral overexpression of miR-155 further enhanced the expression of pro-inflammatory cytokines in the ischemic brain by upregulating TLR4 and NF-κB expression as well as downregulating SOCS1 and MyD88, whereas miR-155 knockout abrogated the effects of cerebral ischemia on the TLR4/NF-κB/MyD88/SOCS1 axis [405].
Conclusively, inducing anti-inflammatory miRNAs or suppressing pro-inflammatory miRNAs could be a therapeutic strategy to ameliorate brain tissue damage following ischemic stroke. However, much work remains to be done in deciphering disease-specific miRNA-mRNA interactions, developing efficient systems for the targeted delivery of miRNA-based therapeutics across the BBB and in determining therapeutic windows and modes of treatment.
Long non-coding RNAs and stroke-associated neuroinflammation
A large number of studies have illustrated that various lncRNAs are closely associated with the regulation of inflammation and microglial activation in cerebral ischemia (summarized in [292]). For example, the pro-inflammatory lncRNAs nuclear paraspeckle assembly transcript 1 (NEAT1), functional intergenic RNA repeat element (FIRRE), Gm4419, and small nucleolar RNA host gene 14 (SNHG14) are upregulated in microglia exposed to OGD/R, and promote microglial activation via different mechanisms [48]. NEAT1 promotes microglial activation via the Wnt/β-catenin signaling pathway [143], whereas the FIRRE and NF-κB pathway forms a positive feedback loop promoting activation of the NLR family pyrin domain containing protein 3 (NLRP3) inflammasome [442]. Similarly, Gm4419 facilitates microglial activation upon ischemic stress via activation of the NF-κB pathway [404]. SNHG14 increases the expression of cytosolic phospholipase A2 via competitively interacting with miR-145-5p, which contributes to activation of microglial cells in ischemic stroke [311].
In contrast, anti-inflammatory lncRNA SNHG8 is downregulated in brain tissue of mice that underwent experimental stroke as well as microglia exposed to OGD/R. SNHG8 overexpression attenuated the microglial inflammatory response by regulating the miR-425-5p/sirtuin 1 (Sirt1)/NF-κB axis [368].
The anti-inflammatory lncRNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) has been shown to be upregulated in microglia and neurons during ischemic stroke, resulting in enhanced sponging of the targeted miRNAs [40, 449]. For example, the MALAT-mediated decrease of miR-375 and miR-181c-5p causes enhanced expression of phosphodiesterase 4D and HMGB1, respectively, aggravating the extent of neuroinflammation during acute stroke [40, 449].
Compared with healthy controls, the level of lncRNA SNHG4 in CSF samples of patients with acute ischemic stroke as well as in microglia of mice subjected to I/R injury was remarkably downregulated, whereas the expression of miR-449c-5p went strongly up [461]. Both, overexpression of SNHG4 and knockdown of miR-449c-5p inhibited the expression of pro-inflammatory cytokines in microglia and promoted the expression of anti-inflammatory factors in microglia at least partly through activation of signal transducer and activator of transcription 6 (STAT6) [461].
Furthermore, lncRNAs also influence the polarization of microglia following ischemic stroke. Gain- and loss-of-function experiments provided convincing evidence that pro-inflammatory lncRNA taurine upregulated 1 (TUG1) and rhabdomyosarcoma 2-associated transcript (RMST) trigger microglial polarization towards a pro-inflammatory (M1-like) phenotype by activation of the NF-κB pathway via competitive interaction with miR-145a-5p and heterogeneous nuclear ribonucleoprotein K (hnRNP K), respectively [357, 391].
Similarly, lncRNA H19 is significantly increased in microglia exposed to I/R in vitro and in vivo and promotes neuroinflammation by driving histone deacetylase 1 (HDAC1)-dependent M1 microglial polarization [393].
In contrast, anti-inflammatory lncRNA Nesp-antisense (Nespas) reduces the polarization of microglia toward pro-inflammatory phenotype through direct interaction with transforming growth factor-β-activated kinase 1 (TAK1), which suppresses the TAK1-mediated activation of the NF-κB pathway [79].
A previous study demonstrated that lncRNA 1810034E14Rik is downregulated in OGD-exposed microglia. The overexpression of 1810034E14Rik decreased the infarct volume and production of pro-inflammatory factors in mice subjected to ischemic stroke in vivo, and promoted polarization of OGD-exposed microglia toward anti-inflammatory M2 phenotype in vitro via inhibiting the NF-κB pathway [463].
Upon cerebral ischemia rapid activation of brain-resident microglia, predominantly by DAMPs released from injured and dying cells, leads to a massive liberation of pro-inflammatory cytokines and chemokines, which substantially contribute to the recruitment and infiltration of circulating immune cells into the ischemic area, exhibiting both detrimental and beneficial effects on the outcome of stroke [169]. In this regard, macrophage contained LCP1 related pro-inflammatory lncRNA (Maclpil) was demonstrated to be highly expressed in pro-inflammatory monocyte-derived macrophages but not in microglia-derived macrophages purified from ischemic mouse brain three days after stroke [401]. Exposure of bone marrow-derived macrophages in vitro to either pro-inflammatory stimuli or OGD revealed that Maclpil triggers cell polarization towards a pro-inflammatory phenotype through lymphocyte cytosolic protein 1 (LCP1) [400, 401]. Also, adoptive transfer of Maclpil silenced macrophages or systemic silencing of Maclpil reduced ischemic brain infarction, improved functional deficits and attenuated the accumulation of monocyte-derived macrophages, CD4+ T cells, and CD8+ T cells in the ischemic hemisphere without affecting microglia cellularity [400, 401].
Despite the emerging importance of lncRNAs in ischemic stroke, being unraveled in a growing number of preclinical studies, further investigations are needed to elucidate lncRNA biological functions to accelerate the progress of lncRNA-based therapeutics against stroke.
Circular RNAs and stroke-associated neuroinflammation
Recent studies have proposed that circRNAs exert a central effect in neuroinflammation caused by acute cerebral ischemia. In blood samples from patients with acute stroke as well as in brain tissue from mice subjected to ischemic stroke, circ_0000831 levels were strongly decreased as compared to healthy controls [161]. Intracerebral overexpression of circ_0000831 in mice substantially ameliorated infarct volume, cell apoptosis, BBB dysfunction, vasogenic edema formation, oxidative stress and neuroinflammation [161]. Mechanistically, circ_0000831 overexpression repressed apoptosis and the release of pro-inflammatory factors induced by OGD in microglia via activation of the adiponectin receptor 2/peroxisome proliferator-activated receptor-γ (PPARγ) axis by downregulating miR-16-5p [161]. Consistently, the beneficial effects on the outcome of murine ischemic stroke evoked through circ_0000831 overexpression were almost completely prevented by the intracerebral knockdown of PPARγ [161].
In mice subjected to ischemic stroke, the expression of circ_CDC14A increased in circulating neutrophils within hours. Two to three days upon stroke, circ_CDC14A levels also increased in astrocytes colocalized with neutrophils that infiltrated into the peri-infarct cortex, indicating an intercellular transfer of circ_CDC14A from infiltrating neutrophils to resident astrocytes [485]. A selective knockdown of circ_CDC14A expression in peripheral blood cells, but not in brain tissue, evoked anti-inflammatory effects as it inhibited the activation of astrocytes in the peri-infarct cortex, increased the N2/N1 ratio of neutrophil populations in the ischemic brain, reduced the infarct size, and improved functional impairment und post-stroke survival [485].
Furthermore, in microglial cells exposed to pro-inflammatory stimuli, the overexpression of circ_Dlgap4 promoted the decay of pro-inflammatory cytokine mRNAs by interacting with AU-rich element/poly(U)-binding/degradation factor 1 (AUF1) [229]. Accordingly, the intracerebral overexpression of anti-inflammatory circ_Dlgap4 in a mouse model of ischemic stroke reduced neuroinflammation, brain tissue damage and neurobehavioral deficits, all of which were reversed by the intracerebral knockdown of AUF1 [229].
Taken together, although recent studies in animals provided the first evidence that certain circRNAs play a pivotal role in neuroinflammation and other pathological processes in the course of ischemic stroke, the clinical evaluation of circRNAs as potential diagnostic biomarker and therapeutic target for stroke is still in its early stage.
The importance of extracellular DNA for neuroinflammatory processes in ischemic stroke
Microglia, as the major resident immune cell in the CNS, has emerged as a key mediator of neuroinflammation in the course of ischemic stroke. Studies in vitro and in vivo have demonstrated that I/R injury causes a release of mtDNA into microglial cytoplasm, promoting the polarization of microglia towards the pro-inflammatory M1-like phenotype and restraining anti-inflammatory M2-type microglia polarization through activation of the STING pathway [193]. Accordingly, pharmacologic inhibition of STING with the low molecular weight inhibitor C-176 in mice, subjected to experimental ischemic stroke, reduced I/R-induced brain infarction, edema, neuronal injury/degeneration as well as sensorimotor and cognitive impairments, whereas the intracerebroventricular administration of mtDNA worsened brain tissue damage and functional deficits [193]. Moreover, treatment with C-176 was sufficient to prevent the detrimental effects of exogenous mtDNA on stroke outcomes [193]. Similarly, in a mouse model of ischemic stroke, the elevated occurrence of dsDNA in the cytoplasm of astrocytes and microglia across the penumbra as early as 6 h after onset of cerebral ischemia was shown [215]. However, the origin of cytoplasmic dsDNA is not fully clear. It may leak from dysfunctional mitochondria [298], but uptake of extracellular dsDNA, massively released from necrotic neurons upon I/R injury, might also play a role.
Accordingly, the exposure of in vitro cultured microglia to conditioned medium derived from neurons subjected to OGD caused substantial M1-like polarization, which was, however, attenuated by either addition of the mtDNA inhibitor dideoxycytidine or the knockdown of the key cytosolic dsDNA sensor cGAS, working upstream from STING [174]. It is worth mentioning that neurons and astrocytes can exchange damaged mitochondria with each other for disposal and recycling after stroke raising the possibility that the elevated mtDNA in microglial cytoplasm of ischemic brain may be partly derived from astrocytes and neurons through cell-to-cell communication [67, 152, 193].
Furthermore, experimental evidence was provided that pharmacological blockade of cGAS with the irreversible STING-inhibitor A-151 was sufficient to alleviate cerebral damage and functional defects [215]. The improved stroke recovery upon A-151 treatment was further accompanied by reversal of cGAS/STING-mediated upregulation of AIM2 inflammasome- and pyroptosis-associated molecules, neutrophil infiltration as well as the production of pro-inflammatory factors and pyroptosis in microglia [215]. Consistently, cell-specific genetic ablation of cGAS in microglia protected against brain damage, improved neurobehavioral performance, and reduced cell death after stroke to a similar extent as compared to the pharmacological inhibition of cGAS through A-151 [215].
Of note, circulating neutrophils, the first immune cells to be recruited into the brain tissue after stroke by excessive local release of pro-inflammatory cytokines and DAMPs, may further exert harmful effects by subsequent release of NETs, and NETs themselves directly allow dsDNA to be released into the microenvironment, thus forming a positive feedback loop of inflammation [215]. Indeed, numerous studies have localized NETs in the perivascular space of infarcted lesions in specimens from ischemic stroke patients and corresponding animal models [474]. CfDNA together with histones as major network structure of NETs, was firstly shown to act as a reaction platform for blood cell adhesion, platelet activation and the induction of blood coagulation promoting thrombosis and limiting the fibrinolytic effect of t-PA, the only approved pharmacological therapy for acute ischemic stroke in humans [474]. In fact, plasma levels of cfDNA and NETs (as biomarkers) are significantly increased in patients with acute stroke and are associated with t-PA-resistance as well as with increased disease severity and mortality (Table 3) [80, 210, 460]. Furthermore, treatment of blood clots obtained from ischemic stroke patients with DNase1 ex vivo substantially increased t-PA-induced thrombolysis in comparison to t-PA alone [93, 202, 253]. Moreover, either free or NET-associated extracellular histones provide a strong cytotoxic potential for different cell types that should not be neglected in searching for DNase-based therapeutic measures as mentioned above [327].
In murine ischemic stroke, the degradation of NETs by systemic application of DNase1 reduced BBB breakdown and increased neovascularization and vascular remodeling after stroke to a similar extent as compared to neutrophil depletion by injection of anti-Ly6G antibody or blockade of PAD4, an enzyme essential for NET formation, respectively [184]. PAD4 inhibition also reduced stroke-induced STING-mediated production of IFN-β. Consistently, STING knockdown and IFN receptor-neutralizing antibody treatment decreased BBB breakdown and increased vascular plasticity [184]. In a follow-up study using a mouse model of thrombotic middle cerebral artery occlusion, the same research group demonstrated that t-PA-induced neutrophil recruitment, NET formation, BBB breakdown and cerebral hemorrhage, a most feared clinical complication of t-PA-mediated therapy for acute stroke patients, were effectively alleviated by either DNase1 treatment or PAD4 deficiency [397].
Furthermore, NETs were revealed to be essential for the t-PA-induced upregulation of cGAS/STING and the downstream pro-inflammatory type 1 IFN signaling in microglia and infiltrating macrophages, as DNase1 and ablation of PAD4 substantially reduced the activation of the cGAS/STING pathway and the production of IFN-β and IL-6 in mice subjected to photo-thrombotic stroke and treatment with t-PA [397]. Accordingly, DNase1-mediated reversal of microglia activation, cerebrovascular protection and anti-hemorrhagic effects after ischemic stroke were abolished by co-administration of the cGAS product cGAMP, whereas cGAS deficiency rescued t-PA-associated BBB disruption and cerebral hemorrhage [397]. Beneficial effects of DNase1 treatment alone or in combination with t-PA on the outcome from ischemic and hemorrhagic stroke were confirmed in further preclinical animal studies [70, 80, 297, 361, 419, 444, 460] (detailed information is provided in Table 2).
Conclusively, recent experimental studies suggest that brain-resident microglia and infiltrating neutrophils may interact synergistically to coordinate dsDNA-induced inflammatory responses and culminate in the expansion of ischemic infarction. Thus, inhibitory targeting of NETosis and innate DNA-sensing signaling may be promising therapeutic interventions to treat ischemic stroke [215].
Multiple sclerosis
Etiology and pathogenesis of multiple sclerosis
MS is an inflammatory demyelinating disease of the CNS affecting mainly young people aged between 20 and 40 at disease onset. Initial symptoms are diverse but the most frequent ones are visual disturbances, paresthesias, ataxia and muscle weakness [98]. In 80–85% of patients, the course of the disease is associated with periods of increasing neurological symptoms (relapses) alternating with remissions (relapsing–remitting course of MS, RRMS). With time, for most patients the disease passes into a secondary progressive course (SPMS) characterized by continuous progression of symptoms. In 10–15% of patients the neurological deficit increases continuously from the moment of the disease manifestation (primary progressive form of MS, PPMS) [195]. Although incompletely understood, the etiology of MS presumably involves interaction between genetic, environmental, and other factors triggering an aberrant autoimmune attack resulting in damage to myelin and axons. In the pathogenesis of MS, two mutually complementary processes can be distinguished: the autoimmune neuroinflammation directed against the myelin sheath components that actively develops during the early stages of the disease, and neurodegeneration, which plays a leading role in the progression of MS [195]. At the cellular level, pathological hallmarks include peripheral activation of autoreactive myelin-specific T cells, their migration into the CNS and reactivation of self-reactive T cells by resident and infiltrating activated antigen-presenting cells (APCs), demyelination, remyelination, gliosis, and axonal/neuronal degeneration [98]. In the initial stages of MS development, autoreactive CD4+ T helper type 1 (Th1) and CD4+ T helper type 17 (Th17) cells are elevated in the CNS, where they initiate inflammation and neuronal cell death by producing IFN-γ and IL-17, respectively [439]. In addition, CD4+ regulatory T cells (Tregs), which normally prevent damage to host cells by limiting the immune response, are decreased in the frequency and suppressive function of MS [439].
The role of extracellular RNAs during neuroinflammation in multiple sclerosis
Over the past decade, many studies have identified a large set of circulating cell-free or cell-associated ncRNAs that are dysregulated in MS, particularly in a lineage-related manner or in specific cell populations as well as during particular stages/subtypes of MS, providing new MS-specific biomarkers to predict disease activity and progression (summarized in Table 4) or therapy response. Furthermore, some of these regulatory ncRNAs have been functionally characterized to play critical roles in MS pathogenesis (for a comprehensive overview see also [128, 439]).
MicroRNAs and neuroinflammation in multiple sclerosis
Among them are several miRNAs that influence the differentiation of pro-inflammatory Th1 cells and Th17 cells (e.g. miR-326, miR-448, let-7e), the development of Tregs (miR-106b, miR-25), and the alteration of the Th2 to Th1 response in MS (miR-128, miR-27b, miR-340) [439]. In addition to T cells, several important miRNAs regulating the activation and effector functions of APCs such as B cells (e.g. miR-320a, miR-132), blood-derived macrophages and microglia (e.g. miR-155, miR-124), have been identified to be differentially expressed in patients with MS [439]. Similar to immune cells, aberrant expression of miRNAs in resident CNS cells presumably contributes to the mechanisms underlying inflammation in MS [439]. In active human MS lesions, 20 miRNAs have been identified to be at least twice more abundant and 8 miRNAs at least twice less abundant than in normal white matter. Interestingly, astrocytes contained all 10 miRNA that were most strongly upregulated. Amongst, the local upregulation of the three miRNAs miR-34a, miR-155 and miR-326 is supposed to be linked to the local downregulation of CD47 (considered as a “Don’t eat me” signal) on brain-resident cells and myelin in active MS lesions, thereby unleashing macrophages for tissue destruction [180].
The pro-inflammatory miR-155 is highly expressed in the serum and in CNS lesions of MS patients [365]. Both, global or T cell-specific knockout of miR-155 in mice confers resistance to experimental autoimmune encephalomyelitis (EAE), a mouse model of MS, by reducing the encephalogenic potential of CNS-infiltrating Th17 T cells [365].
A recent study has designated miR-223-3p as a negative regulator of NLRP3 inflammasome engagement in activated macrophages/microglia, both in experimentally-induced demyelination and human MS lesions [122]. Systemic delivery of miR-223-3p mimics to mice following lysolecithin-induced demyelination suppressed NLRP3 inflammasome activity in both macrophages and microglia, and resulted in a significant reduction of axonal injury within demyelinated lesions [122].
Furthermore, miR-409-3p and miR-1896, upregulated in IL-17-activated astrocytes in vitro and in EAE mice in vivo, co-ordinately promoted the production of inflammatory cytokines in reactive astrocytes through the SOCS3/STAT3 pathway and enhanced astrocyte-directed chemotaxis of CD4+ T cells, aggravating demyelination in EAE mice [233].
Overexpression of miR-99a, another anti-inflammatory miRNA, alleviated EAE development by promoting Tregs and inhibiting Th1 cell differentiation through suppression of mechanistic target of rapamycin (mTOR)-regulated glycolysis in CD4+ T cells [137].
The systemic knockdown of pro-inflammatory miR-181c resulted in attenuated EAE clinical symptoms and decreased the spinal cord inflammation and demyelination, along with a decreased Th17 cell population [467]. MiR-181c knockdown rendered T cells less sensitive to TGF-β-induced Smad2/3, enhancing the expression of IL-2, which has been reported to inhibit Th17 cell differentiation [467].
Moreover, overexpression of anti-inflammatory miR-23b resulted in a strong resistance to EAE by inhibiting the migration of pathogenic T cells to the CNS through targeting C-C motif chemokine ligand 7 [464].
Altogether, numerous miRNAs which were found to be dysregulated either in blood-borne immune cells, brain-resident cells or body fluids of MS patients are predicted to regulate immune/inflammatory responses. As a consequence, in-depth in silico studies are needed to identify their target genes and related immune/inflammatory pathways. Further gain- and loss-of-function studies in animal MS models will also be necessary to evaluate the therapeutic potential of the most promising miRNA candidates.
Long non-coding RNAs and neuroinflammation in multiple sclerosis
Like miRNAs, lncRNAs play an important role in both innate and acquired immunity. In the last years, a continuously growing number of deregulated lncRNAs have been identified in serum, peripheral blood mononuclear cells (PBMCs) and blood samples of MS patients (summarized in [128, 285]). A higher abundance of three circulating lncRNAs in the serum of patients suffering from RRMS has been reported: NEAT1, RNA component of 7SK nuclear ribonucleoprotein (RN7SK) and TUG1 [329]. These three lncRNAs are involved in specific regulatory functions: NEAT1 promotes expression of the CXC motif chemokine ligand 8 gene encoding IL-8 via relocation of splicing factor proline- and glutamine-rich (SFPQ), RN7SK is involved in regulation of CD4+ T cells, and TUG1 is a component of the p53 regulatory network [285].
Similarly, higher levels of lncRNA growth arrest-specific 5 (GAS5) are found in amoeboid-shaped microglia in MS patients. Functional studies had demonstrated that GAS5 has pro-inflammatory properties as it suppressed microglia M2 polarization through repression of TRF4 transcription by recruiting the polycomb repressive complex 2 (PRC2). Consequently, intracerebroventricular transplantation of GAS5-depleted microglia attenuated disease progression and promoted re-myelination in animal models of MS [355].
Moreover, linc-MAF-4 levels were considerably higher in PBMCs from MS patients than in healthy controls. Linc-MAF-4 exacerbates MS pathogenesis by altering the Th1/Th2 ratio and by targeting musculo-aponeurotic fibrosarcoma (MAF), a Th2 cell transcription factor required for Th2 differentiation [439].
The lncRNA MALAT1 is downregulated in both the CNS of human MS patients and in spinal cords of EAE animals at the peak of disease [258]. The knockdown of MALAT1 in EAE mice exacerbated autoimmune neuroinflammation through changing the pattern of macrophage differentiation towards a M1-like phenotype as well as enhancing T cell differentiation towards pathogenic Th1 and Th17 cells, while impeding the differentiation of protective Treg cells, collectively pointing to a potential anti-inflammatory effect for MALAT1 in the context of MS [258].
Conclusively, several pieces of evidence have demonstrated a promising role of lncRNAs as potential diagnostic and prognostic biomarkers in MS patients. However, studies in this area have just begun, and further research is required to determine the specific molecular mechanisms and biological functions of these lncRNAs in the pathogenesis of MS.
Circular RNAs and neuroinflammation in multiple sclerosis
While the contribution of miRNAs and lncRNAs to the progression of MS is well accepted, the role of circRNAs in the pathogenesis of MS is still largely obscure and only a few reports have addressed this topic. More than 400 circRNAs that were differentially expressed in blood samples of RRMS patients have been identified [165]. From these, two circRNAs, circ_0005402 and circ_0035560, have been confirmed to be downregulated in the MS population upon several validation steps. Both of them are located inside the annexin A2 (ANXA2) gene, which had not been previously related to MS but other immune-mediated diseases. Moreover, ANXA2 has also been reported to be a target of miR-155, a critical miRNA in neuroinflammation at the BBB and relevant in Th1 and Th17 cell differentiation and myeloid cell polarization in MS as mentioned above [165]. Based on the fact that ANXA2 and miR-155 are inversely regulated in MS patients, a complex interaction between miRNA, mRNA and circRNAs can be anticipated in the course of MS [165].
Similarly, a recent study on circRNA expression profiles in PBMCs revealed more than 900 transcripts to be differentially expressed between patients with RRMS in relapse and healthy controls, and demonstrated the overexpression of circ_101348, circ_102611, and circ_104361 in MS patients [488]. Bioinformatic analysis revealed 15 miRNAs interacting with these circRNAs in a complementary manner and led to the discovery and validation of three protein-coding RNAs upregulated in patients with RRMS during relapse. Two of these, adenylate kinase 2 and Ikaros family zinc finger protein 3 (IKZF3), have previously been implicated in B cell function [488].
Moreover, circ_0000518 was shown to be upregulated in CSF and in the peripheral blood of MS patients as well as to exacerbate EAE by promoting macrophage/microglial M1 polarization through the fused in sarcoma (FUS)/calcium/calmodulin-dependent protein kinase kinase beta (CaMKKβ)/AMP-activated kinase (AMPK) pathway [173].
A previous study showed an upregulated expression of circINPP4B in Th17 cells from mice with EAE and during Th17 differentiation in vitro [144]. The silencing of circINPP4B inhibited Th17 differentiation and alleviated EAE, characterized by a reduced demyelination and Th17 infiltration in the spinal cord. Mechanistically, circINPP4B served as a sponge that directly targeted miR-30a to regulate Th17 differentiation [144].
Together, emerging recent data clearly support the notion of circRNAs involvement in the pathogenesis of MS. Several lines of evidence indicate that circRNAs may play a distinctive role in both adaptive and innate immune responses in MS by determining the availability of miRNAs for their known post-transcriptional regulation of genes related to immune cell polarization and immune effector functions [487]. In addition, contrary to other RNA species they are very stable in the blood and other biological fluids and thus might be considered as valuable biomarker candidates for MS [487].
The emerging roles of extracellular DNA in neuroinflammatory processes during multiple sclerosis
In recent years, impaired mitochondrial function is increasingly recognized as a key pathological hallmark of MS. Demyelination leads to an increase in energy demand in order to maintain an appropriate intra-axonal ion balance and could thereby affect mitochondria at multiple levels [209]. Among others, disturbances in mitochondrial dynamics may promote the release of mitochondrial DAMPs, particularly cf-mtDNA [71]. On entering the cytoplasm or the extracellular space, mtDNA can become pro-inflammatory and initiate innate and adaptive immune responses by activating cell surface and intracellular receptors in both resident and infiltrating cells [134]. In accordance with the possible role of mitochondrial dysfunction in the pathology of MS, increased levels of mexDNA were found in blood plasma and lumbar CSF samples of patients suffering from progressive forms of MS [115, 209, 277] (Table 4). Higher plasma levels of mexDNA were accompanied by increased plasma concentrations of pro-inflammatory cytokines [277]. Moreover, enhanced mexDNA concentrations in the CSF of patients with PMS correlated with high T2 lesion volumes and were inversely related to normal brain volume, indicating that the increased concentration of mexDNA is mostly due to ongoing neuro-axonal damage, which is known to be more extensive in progressive forms of MS [209].
Significantly higher levels of mexDNA were also found in lumbar CSF samples taken from patients with RRMS, which, however, declined over the disease duration [115, 380]. There is increasing evidence that the pathological mechanisms of PMS and RRMS are different. While relapses are thought to be caused by acute focal inflammation, relapse-independent progression is the clinical consequence of more diffuse inflammatory and neurodegenerative processes [150]. Thus, high levels of mexDNA in RRMS might be predominantly due to active release in response to a stimulus, and could reflect early inflammatory activity rather than neuronal loss. Accordingly, RRMS patients treated with Fingolimod, which limits autoreactive inflammation in the CNS by acting on sphingosine-1-phosphate receptors, which are present on peripheral immune cells as well as glial and nerve cells, had significantly lower mexDNA copy levels at follow-up compared to baseline [209]. In contrast, post-mortem ventricular CSF analysis revealed a decreased mexDNA abundance and integrity in patients suffering from PMS, which, however, did not correlate with protein markers of neurodegeneration [236].
Despite the potential of mexDNA as a reliable diagnostic and prognostic biomarker for MS (Table 4), only a few studies have addressed the mechanistic link between mtDNA release, inflammatory response, and progression of MS. In this regard, the stimulation of human microglia with mexDNA in vitro increased ROS production, but did not affect antigen presentation properties and expression of pro-inflammatory cytokines [278], implying that increased levels of mexDNA might contribute to the chronic and dysregulated activation of microglia, as demonstrated in MS and other neurodegenerative diseases. In a similar fashion, mtDNA-containing neuron-derived mitochondrial lysates, but not mitochondrial lysates from mtDNA-depleted cells, could activate inflammatory pathways in cultured neuronal and microglial cells [409].
Consistently, injection of mtDNA into mouse hippocampus increased NF-κB signaling, TNF-α expression and astrocyte proliferation [410]. Cultured microglial cells transfected with mtDNA revealed a pro-inflammatory microenvironment by activation of the cGAS/STING pathway as one of the primary aberrant cytoplasmic DNA sensors [221]. Moreover, control or oxidant-initiated degraded mtDNA triggered a pro-inflammatory response in mouse primary astrocytes [260]. Overall, emerging evidence point to mexDNA, and even more when degraded by oxidation, as an important DAMP in MS and other neurodegenerative diseases associated with inflammation and oxidative imbalance.
Neutrophils, as the most abundant circulating and first-responding innate myeloid cells, have been increasingly demonstrated to play crucial roles in the development and pathology of MS, among others, by the formation and release of mtDNA-containing NETs [68]. Circulating NETs were found to be elevated in the serum of RRMS patients compared to healthy controls [275]. NETs were, however, not detected in CSF samples of MS patients, corresponding with previous reports that pointed to the absence of neutrophils within the CNS of MS patients. Yet, it was suggested that cytotoxic components of NETs may contribute to BBB damage in this disease [251, 370]. Accordingly, in mice subjected to EAE the depletion of NET-associated proteins such as myeloperoxidase and neutrophil elastase caused an attenuated disease severity and BBB breakdown [440, 450]. Also, the transmigration of murine neutrophils through an activated cerebrovascular endothelium induced a pro-inflammatory, neurotoxic phenotype that subsequently leads to the release of NETs containing de-condensed DNA associated with proteases [10, 68]. The blockade of histone–DNA complexes attenuated transmigrated neutrophil-induced neuronal death, whereas the inhibition of key neutrophil proteases in the presence of transmigrated neutrophils rescued neuronal viability [10].
Furthermore, upon activation, CD4+ T lymphocytes have been shown to release extracellular oxidized DNA that provides autocrine costimulatory signals to T cells [57]. Pharmacological inhibition of mitochondrial ROS during the priming phase of EAE abolished the extrusion of DNA by CD4+ T cells and reduced T cell priming against myelin. Moreover, mitochondrial ROS blockade during established EAE markedly ameliorated the disease severity, thereby dampening autoimmune inflammation of the CNS [57].
Neuroinflammation is also associated with high levels of extracellular ATP, which is released from activated cells, mostly astrocytes, or leaking from injured or dead cells, to serve as a DAMP that activates pro-inflammatory responses [55]. Neurons, glia and infiltrated immune cells can sense ATP as well as other extracellular nucleotides (e.g., ADP, UTP, and UDP) via specific purinergic P2 receptors [55]. The P2X7 receptor, one of the most abundant P2 receptors in the CNS and activated by ATP, triggers a cascade of responses including the release of pro-inflammatory mediators and excitatory neurotransmitters, induction of cell proliferation but also cell death [55]. In line with the role of P2X7 in MS, the expression of this receptor is significantly elevated in neurons, astrocytes, and microglial cells/macrophages of MS patients and in brain samples from rodents subjected to EAE. Consistently, mice lacking P2X7 are less susceptible to EAE, while EAE is ameliorated by pharmacological blockade of P2X7 signaling [261, 339].
Conclusively, exDNAs and extracellular nucleotides that are accumulating in the brain during MS to promote inflammatory processes, are potential therapeutic targets for MS. Yet, upon MS-related injury conditions it remains to be studied whether direct interactions between polyanionic exRNAs or exDNAs with basic proteins in the myelin sheet might cause autoimmune reactions due to the generation of neo-antigen complexes. Such a pathomechanism has been uncovered for the autoimmune disease heparin-induced thrombocytopenia (HIT) where complexes between extracellular nucleic acids and the basic protein platelet factor 4 induce the formation of HIT-antibodies [167].
Alzheimer's disease
Etiology and pathogenesis of Alzheimer's disease
AD is the most prevalent neurodegenerative disorder related to age, which is clinically associated with a global cognitive decline and progressive loss of memory and reasoning [257]. The defining neuropathological features of AD comprise deposition of extracellular amyloid plaques and intraneuronal neurofibrillary tangles (NFTs), consisting of densely packed amyloid-β (Aβ) peptides, derived from the amyloid precursor protein (APP) via sequential proteolytic cleavage by β- and γ-secretases, and the hyper-phosphorylated microtubule-binding protein tau (tubulin associated unit), respectively [257]. According to the prevailing amyloid cascade hypothesis, the accumulation of these proteins appear to follow a temporal sequence, with Aβ accumulation triggering a cascade of events comprising NFT formation, synaptic and mitochondrial dysfunction, and neuronal loss [72, 257].
Chronic neuroinflammation is also a typical feature of AD pathogenesis. It is widely accepted that microglia-mediated neuroinflammatory responses may promote neurodegeneration in AD [443]. Microglial activation precedes neuronal loss in patients with AD, and recent genome-wide association studies have revealed that microglial genes such as CD33, triggering receptor expressed on myeloid cells 2 (TREM2) and human leukocyte antigen-DR isotype (HLA-DR) are associated with susceptibility to late-onset AD [443]. Aβ oligomers and fibrils are capable of priming microglial cells through interactions with various receptors, which enhance the production of inflammatory cytokines and chemokines, and make microglia more susceptible to secondary stimuli, thereby promoting chronic activation of primed microglia [156, 257]. In addition to microglia, astrocytes undergo complex, brain region- and disease stage-specific changes in the course of AD. Astrocytic atrophy and loss of function, preceding the formation of senile Aβ-plaques, can contribute to early AD pathophysiology, including synaptic dysfunction, impaired synaptogenesis and cognitive deficits [13].
In addition, hypertrophic astrocytes have been described to reside within the vicinity of senile Aβ-plaques, taking part in the proteolytic clearance of Aβ-peptide [257]. However, similar to microglia, astrocytes also sense Aβ-aggregates in a TLR/RAGE-dependent manner, which leads to increased production of neurotoxic factors, including ROS, NO, pro-inflammatory cytokines and chemokines. Excessive production of neurotoxic factors disturbs astrocyte's APP processing homeostasis, which leads to increased Aβ-peptide load and toxicity [343]. Although Aβ-peptide is probably the key inducer of neuroinflammation in AD, it does not exclude the possibility that other intrinsically generated molecules such as SENAs might also contribute.
Extracellular RNAs and their neuroinflammatory implications in Alzheimer's disease
Emerging evidence indicates that regulatory ncRNAs such as lncRNAs, miRNAs, and circRNAs exert crucial regulatory effects in the initiation and development of AD. Compared to healthy controls, the levels of certain ncRNAs and their target mRNAs are significantly altered in the CNS, CSF, and blood of patients affected by AD, highlighting circulating ncRNAs as promising biomarkers for early diagnosis and prediction of AD progression (Table 5). Although the mechanisms are still not fully elucidated, recent studies have further revealed that these highly conserved ncRNAs impact in a convergent as well as divergent manner on core pathophysiological processes underlying AD such as neuroinflammation and oxidative stress, aberrant generation of Aβ-peptide, anomalies in the production, cleavage and post-translational marking of tau, impaired clearance of Aβ-peptide and tau, perturbation of axonal organization, disruption of synaptic plasticity, endoplasmic reticulum stress and the unfolded protein response, mitochondrial dysfunction, aberrant induction of cell cycle re-entry, and apoptotic loss of neurons (systematically reviewed in [204, 266, 466]). According to the scope of this review, the role of certain members from different classes of ncRNAs in neuroinflammation during AD is summarized below.
MicroRNAs and neuroinflammation in Alzheimer's disease
Microglia exposed to pro-inflammatory conditions upregulate miR-155, which increases the production of pro-inflammatory cytokines and reduces the ability of microglia to catabolize fibrillar Aβ1-42 in vitro [11, 220]. Similarly, in astrocytes miR-155 is elevated in response to inflammatory stress, and is involved in the upregulation of pro-inflammatory cytokines by targeting SOCS1 mRNA [220]. In a murine AD model, pro-inflammatory miR-155 levels were strongly upregulated and coincided with an increase in microglia and astrocyte activation before the appearance of extracellular Aβ aggregates [138]. The inhibition of miR-155 expression attenuated the upregulation of TNF-α, IL-1β, IL-6, and their receptors, and substantially restored the impaired learning ability of AD rats [227]. Moreover, in neutrophils miR-155 promotes the generation of NETs by increasing the mRNA expression of PAD4 [151].
As another example, miR-146a is abundantly expressed in neurons, microglial cells, and astrocytes, where it acts as a negative feedback regulator of inflammation, whereby miR-146a is upregulated in the temporal cortex of AD patients and hippocampus of AD mice [220, 466]. Here, nasal administration of a miR-146a agomir relieved the progression of AD-associated neuroinflammation by inhibiting the expression of the TLR4 signaling pathway and its related inflammatory genes NF-κB, IL-1 receptor-associated kinase 1 (IRAK1), and TNF receptor-associated factor 6 (TRAF6) as well as reducing the release of inflammatory factors IL-1β, IL-6, and TNF-α [220, 466]. Similarly, microglia-specific miR-146a overexpression in AD mice reduced cognitive deficits in learning and memory, attenuated neuroinflammation, reduced Aβ levels, ameliorated plaque-associated neuritic pathology, and prevented neuronal loss mainly through downregulation of neuroinflammation-related pathways [219]. At the cellular level, anti-inflammatory miR-146a triggered microglial phenotype switching, reduced pro-inflammatory cytokines and enhanced phagocytic function to protect neurons under AD conditions in vitro and in vivo [219]. Autophagy has been proposed as a route of Aβ clearance by microglia that is halted in AD. Accordingly, primary microglia from adult AD mice has been demonstrated to fail to degrade Aβ and expresses low levels of autophagy cargo receptor xext to BRCA1 gene 1 (NBR1), which is required for Aβ proteolysis [100]. Interestingly, NBR1 expression in murine and human AD microglia was negatively correlated with the production of the Mirc1/Mir17-92a cluster member miR-17, which is known to downregulate autophagy proteins. Concordantly, the inhibition of elevated miR-17 in mouse AD microglia improves Aβ degradation, autophagy, and NBR1 expression [100]. Furthermore, in the peripheral blood of AD patients, miRNA-22 levels were found to be negatively correlated with the expression of pro-inflammatory factors [142]. The intracerebroventricular application of miRNA-22-mimic to AD mice inhibited the release of inflammatory cytokines by regulating the inflammatory pyroptosis of glial cells via targeting gasdermin D, and thereby improved the cognitive abilities [142]. In the hippocampus of AD mice the expression of miR-216a-5p was reduced, and the restoration of miR-216a-5p expression improved learning-memory ability and attenuated the inflammatory response of AD mice through targeted inhibition of the HMGB1/NF-κB pathway [338].
There are many other dysregulated miRNAs, which may be involved in AD-related neuroinflammation (for a comprehensive overview see also [220, 234]). However, further gain- and loss-of-function studies are needed to fully decipher the miRNA network in AD-associated neuroinflammation.
Long non-coding RNAs and neuroinflammation in Alzheimer's disease
In serum samples of AD patients, the levels of the lncRNA MAGI2-AS3 and its target miR-374b-5p were negatively correlated with disease severity [452]. Similarly, microglial cells exposed to Aβ-peptide in vitro showed elevated MAGI2-AS3 and reduced miR-374b-5p expression. Moreover, overexpression of miR-374b-5p or MAGI2-AS3 knockdown prevented the Aβ-induced upregulation of pro-inflammatory cytokines in microglia [452].
In a rat AD model, hippocampal lncRNA MEG3 levels were substantially reduced. In contrast, MEG3 overexpression improved cognitive impairment, alleviated neuronal damage, as well as reduced the proportion of pro-inflammatory astrocytes and inflammatory cytokine expression [435]. Moreover, lncRNA 4344 overexpression enhanced the expression of the NLRP3-inflammasome and its downstream genes caspase-1, IL-1β, and IL-18 in LPS-treated microglia in vitro, whereas lncRNA 4344 silencing attenuated the inflammatory response. Structural prediction analysis revealed that pro-inflammatory lncRNA 4344 mediates NLRP3 upregulation by negatively targeting miR-138-5p [107]. Detrimental impact of the lncRNA-4344/miR-138-5p/NLRP3 axis on neuronal viability, cognitive function and neuroinflammatory processes was also confirmed in LPS-treated rats in vivo [107].
In cell AD models, the overexpression of the lncRNA MALAT1 reduced IL-6 and TNF-α levels, and increased IL-10 level, while MALAT1 knockdown had the opposite effect. Additionally, MALAT1 reversely regulated miR-125b expression, and rescue experiments revealed that miR-125b prevented the anti-inflammatory effects due to MALAT1 overexpression in Aβ-treated neurons [246]. Similar to the negative correlation of MALAT1 and miR-125b levels determined in Aβ-treated neurons, the abundance of anti-inflammatory MALAT1 in CSF and plasma of AD patients was reduced, whereas miR-125b was increased as compared to healthy controls [246, 480].
Overall, numerous lncRNAs have been identified to be dysregulated in brain and body fluids of AD patients, or have been shown to play crucial roles in neuroinflammation and other processes related to AD pathogenesis in animal disease models. However, more research is required to further elucidate the functions of lncRNAs at molecular and cellular levels, and investigate the full potential of lncRNAs as diagnostic and therapeutic targets in AD.
Circular RNAs and neuroinflammation in Alzheimer's disease
In senescent astrocytes, circNF1-419 levels were enhanced and circNF1-419 overexpression promoted autophagy in astrocytes in vitro [83]. In a mouse AD model, circNF1-419 overexpression enhanced autophagy by binding dynamin-1 and adaptor-related protein complex 2 subunit β1 (AP2B1) protein, which was associated with a reduction of AD- and aging-related marker proteins as well as a decrease in the expression of pro-inflammatory mediators [83].
CircHDAC9 levels were lowered in both serum samples of AD patients or brains of AD mice [240]. As demonstrated in an animal model of AD, circHDAC9 acts as a miR-138 sponge, decreasing miR-138 expression, and thus reversing the Sirt1 suppression and excessive Aβ production induced by miR-138 in neurons [240]. Furthermore, circHDAC9 overexpression in neurons in vitro alleviated Aβ-induced pro-inflammatory response and apoptosis through miR-142-5p sequestration [458].
A previous study has shown that ciRS-7, which is downregulated in the brain of AD patients [469], attenuates generation of Aβ-peptide in neurons by promoting the degradation of APP and β-site APP cleaving enzyme-1 (BACE1) protein in a NF-κB-dependent manner [340]. CiRS-7 inhibits the translation of NF-κB and induces its cytoplasmic localization, thus de-repressing the expression of ubiquitin C-terminal hydrolase L1 (UCHL1), which promotes APP and BACE1 degradation [340].
In both, AD mice and Aβ-treated neurons in vitro the expression of circLPAR1 was elevated. The knockdown of circLPAR1 protected cells against Aβ-caused inflammation, oxidative stress, and neuronal apoptosis as well as improved AD-related pathological traits and ameliorated cognitive dysfunctions in vivo [422]. Mechanistically, circLPAR1 inhibits the Sirt1/Nrf-2/HO-1 axis through repression of growth differentiation factor 15 (GDF-15) [422].
As described above, growing evidence demonstrates that circRNAs have been implicated in the pathogenesis of AD. Further efforts are needed to uncover the regulatory roles of circRNAs and their contribution in the underlying mechanisms of AD pathology.
The impact of extracellular DNA on neuroinflammation in Alzheimer's disease
As shown in AD animal models and human post-mortem brains, accumulation of mutant APP and APP-derived fragments drives mitochondrial dysfunction and mitophagy failure in neurons [205, 316, 377]. An altered concentration of mexDNA in the CSF of AD patients compared with healthy control subjects was found, indicating that CSF mtDNA levels could serve as a biomarker of mitochondrial dysfunction in the etiology of AD and other neurodegenerative disorders [43, 303]. Neuronal and microglial cells exposed to neuron-derived mitochondrial lysates exhibited not only an increased inflammatory gene expression but also showed elevated mRNA and protein levels of APP, while mtDNA-depleted lysates failed to activate inflammatory pathways [409]. Yet, further experimental studies in animal models of AD are required to elucidate whether and how mexDNA contributes to the neuroinflammatory processes in AD.
NETosis is a pathological hallmark of various neurological diseases as already mentioned, and neutrophils together with NETs have also been identified in both human post-mortem brain tissue of AD patients as well as in murine models of AD [347, 443]. It is hypothesized that neutrophils migrate inside the parenchyma in areas with Aβ-plaques, where among others Aβ-peptide triggers the formation and release of NETs [302]. Neutrophil depletion or the inhibition of neutrophil intravascular adhesion in mouse AD models improved cognitive decline and neuroinflammation without interfering with the accumulation of Aβ-plaques [31, 61, 385, 443].
The function of exDNA extruded from neutrophils in AD remains unknown, nevertheless, the systemic application of DNase in animal models of stroke and TBI (summarized in Table 2) appears to be effective as an antagonistic treatment. Yet, further studies need to explore the relationship between NET-associated DNA and AD pathogenesis. Further evidence pointing to a therapeutic potential of DNase for treatment of AD comes from a previous clinical case report. A patient with severe dementia and behavioral disturbance secondary to late-onset AD was given 40 mg of recombinant human DNase1 (1500 KU/mg) three times a day in conjunction with continued Memantine therapy (10 mg daily), and apparently produced a rapid and lasting improvement of cognition [346, 363].
Parkinson’s disease
Etiology and pathogenesis of Parkinson’s disease
PD is the second most common neurodegenerative disorder after AD, affecting approximately 2% of the global population over the age of 65 years [14]. Currently, there are two known PD variants: idiopathic or sporadic and rare familial PD. Risk factors of idiopathic PD, most common in late-onset PD cases, include a combination of genetic (e.g. mutations in leucine rich repeat kinase 2” (LRRK2) or glucosylceramidase-β) and environmental factors (e.g. pesticide exposure, prior head injury, rural living, and intensive use of β-blockers). Early-onset PD is often associated with familial inheritance caused by gene mutations in 18 specific chromosomal regions/PD-related loci (i.e. PARK1-18), such as the SNCA gene (α-synuclein; PARK1 and 4), Parkin (ubiquitin protein ligase; PARK2), DJ-1 (PARK7) or LRRK2 (PARK8) [16].
The PD pathology includes two hallmarks: the progressive degeneration of dopaminergic neurons in the substantia nigra pars compacta and the formation of Lewy bodies, which largely consist of misfolded and fibrillary forms of α-synuclein in surviving neurons [470]. The main symptoms of PD patients involve bradykinesia, rigidity and resting tremor, whereas non-motor manifestations, such as dementia, depression and dysautonomia are also an integral part of the clinical phenotype [14]. So far, there is no efficient strategy for therapy of the disease, albeit the current dopamine replacement strategies and surgical interventions can provide symptom relief, but still fail to prevent or reverse the underlying pathology [475].
Multiple lines of evidence indicate that neuroinflammatory processes also contribute to PD progression. In particular, pro-inflammatory cytokines are elevated in serum and CSF from patients with PD [29]. From animal models of PD-like neurodegeneration we know that due to the continuous release and accumulation of misfolded α-synuclein, microglial cells are in a chronic or prolonged activation state that substantially contributes to the death of dopaminergic neurons in the midbrain via overproduction of pro-inflammatory cytokines and ROS [108, 470]. Microgliosis has also been demonstrated in the human brain by PD post-mortem studies and in vivo PET imaging analysis of diagnosed PD patients [108].
The role of extracellular RNAs in the neuroinflammatory cascade during Parkinson’s disease
Numerous studies have proclaimed the impact of regulatory ncRNAs in PD, which may help developing new ways to treat PD. Moreover, circulating ncRNAs have been identified as robust non-invasive prognostic and predictive biomarkers in human PD patients (summarized in Table 6). In line with the focus of the present review on the neuroinflammatory response during CNS diseases, we will introduce several examples out of the families of miRNAs, lncRNAs and circRNAs, highlighting the importance of regulatory ncRNAs for inflammatory processes in the course of PD.
MicroRNAs and neuroinflammation in Parkinson’s disease
Accumulating evidence demonstrates that aberrant expression of numerous miRNAs might be linked to PD pathogenesis (extensively reviewed in [22, 89, 432]). A total of 125 different miRNAs were significantly altered in the post-mortem analysis of the prefrontal cortex from PD patients compared to inconspicuous controls [284]. Among them, various miRNAs are known to modulate the expression of α-synuclein (miR-7, miR-153 and miR-203a-3p) and further PD-causing genes such as Parkin (miR-103a-3p, miR-146a, miR-181a and miR-218), LRRK2 (miR-205 and miR-599), DJ-1 (miR-494 and miR-4639) or PTEN-induced kinase 1 (PINK1) (miR-27a/b). Other miRNAs such as miR-34b/c, miR-126, miR-128, miR-200a, miR-216a, miR-221 or miR-326 are related to the survival and maintenance of midbrain dopaminergic neurons or alternatively affect the α-synuclein-induced neuroinflammation (miR-29c, miR-124, miR-135b, miR-155) [284]. Moreover, a previous study investigated the profile of a selected set of inflammatory miRNAs in the serum of idiopathic PD patients and patients carrying a mutation in the LRRK2 gene [287]. While miR-146a, miR-335-3p, and miR-335-5p supposed to have anti-inflammatory properties, were downregulated in idiopathic PD and LRRK2-PD patients as compared to control cohorts, pro-inflammatory miR-155 was upregulated in LRRK2 but not in idiopathic PD patients [287].
MiR-155, a key regulator of the mammalian immune system, induces neuroinflammation predominantly through the inhibition of endogenous anti-inflammatory molecules such as SOCS1, a negative regulator of pro-inflammatory cytokines, SH2 domain-containing inositol 5´-phosphatase 1 (SHIP1), a negative regulator of TNF-α, or IL-13 receptor-α1 [483]. Moreover, upregulation of miR-155 is believed to be crucial for the conversion of microglia from a quiescent state to a pro-inflammatory M1-like phenotype in the presence of strong inflammatory stimuli [483]. In a mouse model with AAV-mediated overexpression of α-synuclein in the substantia nigra pars compacta, miR-155 was found to be upregulated [364], whereas global genetic ablation of miR-155 reduced pro-inflammatory responses to α-synuclein and blocked α-synuclein-induced neurodegeneration. Moreover, miR-155-deficient microglia exhibited a markedly reduced inflammatory response to α-synuclein fibrils, whereas treatment with a synthetic mimic of miR-155 restored the inflammatory response [364].
A recent study demonstrated that miR-485-3p is upregulated in the serum of PD patients and LPS-treated microglia cells in vitro [223]. The release of pro-inflammatory cytokines by activated microglia was even higher in the presence of miR-485-3p mimic, while it was robustly declined after silencing miR-485-3p [223].
Furthermore, intracerebroventricular application of a miR-3473b antagomir to mice treated with 1-methyl-1-4-phenyl-1,2,3,6-tetrahydripyridine hydrochloride (MPTP) promoted autophagy and inhibited the expression of pro-inflammatory factors in microglia within the substantia nigra pars compacta [243]. Mechanistically, miR-3473b has been shown to prevent microglial autophagy by targeting TREM2/UNC51-like kinase-1 (ULK1) expression [243].
As an example for anti-inflammatory miRNA in the context of PD, miR-335 has been shown to be downregulated in LPS-treated or LRRK2-overexpressing microglia, in the MPTP-induced PD mouse model as well as in sera from patients with idiopathic PD and those harboring mutations in LRRK2 [288]. In microglia, miR-335 overexpression strongly counteracted the expression of pro-inflammatory genes triggered by either LPS or LRRK2 overexpression [288].
As a further example, miR-let-7a overexpression via injection of miR-7 mimics into the striatum of α-synuclein-induced PD mice suppressed microglia activation and reduced pro-inflammatory cytokine production, which were accompanied by relieved movement disorder and improved spatial memory deficits [451]. Mechanistic investigations revealed that miR-let-7a suppresses the α-synuclein-induced microglial inflammatory response through targeting STAT3 [451].
Similarly, overexpression of miR‐190 in an MPTP‐induced PD mouse model alleviated neuronal damage and inhibited inflammation via negatively regulating the expression and activation of NLRP3 in microglia [356].
These examples imply that deregulated miRNAs in biological fluids do not only represent reliable non-invasive biomarkers for PD diagnosis, prognosis, and treatment response, but could also serve as potential therapeutic targets for the regulation of neuroinflammation and neurodegeneration in PD.
Long non-coding RNAs and neuroinflammation in Parkinson’s disease
It is estimated that about 40% of lncRNAs are specifically expressed in brain tissue, and their number far exceeds that of protein-coding genes [475]. Aberrant expression of various lncRNAs has been determined in post-mortem brain specimens and biological fluids from PD patients (reviewed in [359, 432]). Compared to non-affected brain tissue, significant changes in the expression of 87 different lncRNAs were identified in the substantia nigra of PD patients, among which the significantly upregulated lncRNA AL049437 likely contributes to the risk of PD, whereas the dramatically downregulated lncRNA AK021630 probably leads to the inhibition of PD development [283]. In PBMCs of PD patients 13 lncRNAs exhibited differential expression as compared to healthy controls [351]. Moreover, animal and cell models of PD provide accumulating evidence that lncRNAs contribute to the following pathological processes that ultimately account for the pathological manifestations and clinical symptoms of PD: (i) protein misfolding and aggregation (e.g. SNHG1, long intergenic noncoding RNA-p21 (lincRNA-p21), HOX transcript antisense RNA (HOTAIR)), (ii) mitochondrial dysfunction, oxidative stress, autophagy and apoptosis (e.g. H19, NEAT1, HAGLR opposite strand (HAGLROS), MALAT1), and (iii) neuroinflammation (involving e.g. GAS5). For a more detailed description of the impact of lncRNAs on the pathogenesis of PD the reader is referred to several excellent reviews [242, 245, 420, 475].
With regard to neuroinflammatory processes, GAS5 appears to trigger a pro-inflammatory response of microglia through upregulation of NLRP3 expression via competitively sponging miR-223-3p [423]. Furthermore, lncRNA-p21 sponges miR-181 to promote the activation of microglia and exacerbate neuroinflammation and disease progression by upregulating the expression of protein kinase C-δ in PD models [434]. Gain- and loss-of function approaches in animal and cell models of PD further revealed that lncRNA SNHG1 contributes to neuroinflammation during PD by modulating the miR-7/NLRP3 pathway in microglia [39].
Albeit apparently under-represented, there are few studies on lncRNAs which exhibit remarkable anti-inflammatory properties in the context of PD. For example, overexpression of the lncRNA ZNFX1 antisense RNA 1 (ZFAS1) in 1-methyl-4-phenylpyridinium (MPP+)-treated neurons was demonstrated to prevent NLPR3 inflammasome activation by blocking the miR590-3p-mediated repression of E3 Ubiquitin ligase Mindbomb1 (MIB1)-triggered TXNIP ubiquitination [479].
During the past few years, our understanding of the role of lncRNAs in the development and progression of PD has made substantial progress. However, many lncRNAs are not yet functionally characterized, or we have only minimal information regarding their molecular mechanism of action. Thus, efforts are still needed to identify and functionally characterize lncRNA species involved in the complex pathogenesis of PD. Based on preclinical studies, the use of antisense oligonucleotide-based therapeutics for specific targeting of disease-promoting lncRNAs and their direct delivery across the BBB might represent a powerful and promising approach for PD treatment [371].
Circular RNAs and neuroinflammation in Parkinson’s disease
A growing number of studies indicate that circRNAs are implicated in neurological and cardiovascular diseases [197]. In post-mortem tissue samples from the substantia nigra of individuals without any signs of neuropathology, circRNAs have been shown to accumulate in an age-dependent manner, whereas in the substantia nigra of individuals with PD, this correlation is lost and the total number of circRNAs is reduced [145]. Interestingly, an opposite trend was observed in the amygdala and medial temporal gyrus of PD patients [145]. In peripheral blood of PD patients 139 differentially expressed circRNAs were identified. Of them, 78 circRNAs were upregulated, whereas 61 were downregulated [417]. A total of 10 candidate circRNAs (five upregulated and five downregulated) were retrieved for further verification in a larger cohort. Of these circRNAs, circ_103730, circ_101275, and circ_038416 were upregulated, while circ_102850 was downregulated in patients with PD when compared to healthy controls [417]. Similarly, a remarkably differently expressed circRNA landscape was found in plasma samples of PD patients and healthy controls [472]. Further validation revealed a high diagnostic ability of circ_0004381 and circ_0017204 in predicting the early stage of PD from healthy controls, while circ_0085869, circ_0004381, circ_0017204, and circ_0090668 also presented a high ability to distinguish the late stage of PD from early stage [472].
In cell and animal models of PD various circRNAs have been recently demonstrated to be involved in a set of PD's pathogenic processes (extensively reviewed in [88, 90, 456]). Just to give some examples, ciRS-7 and circSNCA have been shown to positively regulate SCNA expression through sponging miR-7 [88]. CircDLGAP4 exerts neuroprotective effects in PD models by preventing the inhibitory effect of miR-134-5p on the cAMP response element-binding (CREB) protein signaling pathway. Circzip-2 exerts modulatory effects on PD progression through targeting miR-60-3p, in a way that miR-60-3p could be upregulated in the absence of circzip-2, and thus, its target mRNAs, coding for PD-protective genes, become downregulated [88]. The circSAMD4A/miR-29c-3p axis modulates autophagy and apoptotic death of dopaminergic neurons through the AMPK/mTOR pathway [88]. CircSLC8A1, upregulated in the substantia nigra of PD individuals probably due to oxidative stress, regulates the activity of miR-128, indirectly leading to the deregulation of PD-associated miR-128 targets such as the neurodegeneration and aging-related B lymphoma Mo-MLV insertion region 1 homolog (BMI1), Sirt1, or axis inhibition protein 1 (AXIN1) transcripts [88, 145].
Several circRNAs have also been implicated in neuroinflammation but not in the precise context of PD as yet. For example, circHIPK2 acts as an endogenous sponge for miR-124, and blockage of circHIPK2 inhibited methamphetamine-induced astrocytic activation by lowering the expression of sigma non-opioid intracellular receptor 1 (SIGMAR1) [90]. Moreover, OGD-activated microglia-induced neuronal apoptosis is mediated by circPTK2 that sequesters miR-29b in microglia [90]. Forthcoming studies need to elucidate the circRNA-modulating neuroinflammatory processes during PD pathogenesis as well.
Taken together, circRNAs are differentially expressed in the brain of patients with PD, and growing evidence suggests that they regulate pathological processes in PD. However, it is worth noting that research in this field is still in the initial stage. More insights into the interplay of circRNAs with other regulatory networks involving ncRNAs and proteins may further improve our understanding of PD pathogenesis and could provide a valuable basis for the development of new diagnostic and therapeutic regimes.
Extracellular DNA and its impact on neuroinflammation during Parkinson’s disease
Recent studies have linked the amount of mexDNA to neurodegeneration in patients affected by PD. In detail, a low concentration of mexDNA was reported in the CSF of early-stage idiopathic PD patients, suggesting that reduced CSF mexDNA could serve as a biomarker for the onset of PD [123, 310]. The occurrence of reduced amounts of mexDNA may appear contradictory when considering the occurrence of cell death, which is expected to massively release mtDNA. This would elevate rather than depress mexDNA levels [123]. However, following early mitochondrial loss, it is likely that a suppression of baseline release of mtDNA occurs in a compensatory manner. This would explain why only low mexDNA levels, which may anticipate the occurrence of neuronal death, are detected at early stages of neurodegeneration [123]. Accordingly, in dopaminergic substantia nigra neurons of healthy individuals, the mtDNA copy number was demonstrated to increase with age in order to maintain the pool of wild-type mtDNA population in spite of accumulating deletions. Such an upregulation is not observed in idiopathic PD patients, resulting in depletion of wild-type mtDNA and an increase in mtDNA deletions [85, 123, 308].
PARK2, encoding the E3 ubiquitin ligase Parkin, is the second most common gene mutated in cases of early-onset familial PD. Upon recruitment and activation by PINK1, Parkin promotes the removal of dysfunctional mitochondria by mitophagy, a selective form of macro-autophagy [301]. Accordingly, in a mouse model that results in the accumulation of dysfunctional mitochondria due to an accelerated generation of mtDNA mutations, the loss of Parkin caused dopaminergic neurodegeneration and motor defects, indicating that the inability to remove mutated mtDNA via mitophagy might result in a PD-like pathology [281, 301]. Moreover, in the absence of Parkin, mtDNA mutational stress results in elevated circulating mtDNA levels, the activation of the DNA-sensing cGAS/STING pathway and an inflammatory phenotype. Remarkably, the genetic inactivation of STING prevented inflammation, motor defects and neurodegeneration in Parkin-deficient mice that had been subjected to mtDNA mutational stress, indicating a connection between mtDNA-induced inflammation and PD [281, 345].
In accordance with the murine model, patients with biallelic or heterozygous PARK2/PINK1 mutations exhibited elevated serum levels of mexDNA and IL-6 as compared to either healthy control subjects or idiopathic PD patients, indicating that mexDNA levels provide a good predictive potential to discriminate between idiopathic PD and PD linked to heterozygous PARK2/PINK1 mutations [29]. Consistently, induced pluripotent stem cell-derived midbrain neurons from PD patients with PARK2 mutations showed deficits in the mitochondrial biogenesis pathway, resulting in mtDNA dyshomeostasis potentially through downregulation of the energy sensor Sirt1, which controls mitochondrial biogenesis and clearance [402]. Moreover, mtDNA dyshomeostasis has been confirmed in post-mortem midbrain with PARK2 mutations and was accompanied by an upregulation of microglia overexpressing pro-inflammatory cytokines [402]. Accordingly, parkin-deficient neuron-microglia co-cultures elicited an enhanced immune response when exposed to mtDNA/LPS [402].
Contrary to a previous study demonstrating decreased mexDNA levels in CSF of early-stage patients with idiopathic PD [310], differences in serum mexDNA levels of idiopathic PD patients in an advanced disease state compared to healthy control subjects were not observed [29]. This discrepancy may be explained by the fact that mtDNA levels differ over the course of the disease and vary between tissues. Furthermore, the idiopathic PD group exhibited elevated serum IL-6 concentrations despite comparable mexDNA levels, thus implicating that additional molecular mechanisms independent of the mtDNA-cGAS/STING pathway, may contribute to neuroinflammation during PD progression [29]. Otherwise, the accumulation of oxidative modifications and somatic mutations of mtDNA during the progression of mitochondrial dysfunction might constitute additional parameters that would modulate cGAS/STING signaling and, in turn, IL-6 levels in idiopathic PD patients [29, 281].
Moreover, in addition to cGAS/STING, mtDNA activates other immune receptors as well. Previous studies demonstrated that, in addition to TLR9, newly synthesized oxidized mtDNA can activate the NLRP3 inflammasome [295, 473], whereas double-stranded mtRNA stimulates the RNA-sensing immune receptor MDA5 [82].
Overall, the available experimental evidence points to a close relationship between mitochondrial dysfunction, mtDNA release, inflammatory signaling and loss of substantia nigra dopaminergic neurons in PD. If true, then finding ways to reduce the release and accumulation of mtDNA into the cytoplasm or the circulation may be of therapeutic value for PD.
Perspectives
Based on the here presented translational approaches, several therapeutical regimens have been introduced: For example, specific oligonucleotide-based anti-miRNAs, known as antagomirs, have been designed and successfully used to silence endogenous miRNAs in this regard with potential benefit for therapeutic applications. A recent proof-of-principle study in healthy volunteers revealed that, following a single dose of anti-miR-92a, it efficiently inhibits miR-92a and de-represses its targets in a cell type-specific manner in the peripheral blood compartment as evidenced by single-cell RNA sequencing [1]. It is hoped that such approaches will also be available to target brain-specific miRNAs mentioned in the article, responsible for certain neuroinflammatory/neurodegenerative diseases.
Other types of exRNA such as rexRNA are less well characterized in their structure/function profiles, and considerable effort is needed to decipher the pathophysiological consequences of their appearance in the body. Although the patho-mechanistic insights for rexRNA in several inflammatory-based diseases have been acquired from in vitro and preclinical animal studies, these may not always be transferable to the human disease situation. In addition, as proposed by our group in several experimental studies, the tissue-specific as well as disseminated inflammatory potential of rexRNA can be limited or even inhibited by the application of RNase1, the natural antagonistic endonuclease, present in the circulation as well. Here, recent developments of an IgG1-Fc-fusion protein with RNase1, designated RSLV-132, proved its availability in phase-II studies of inflammatory diseases such as Sjögren's syndrome [35, 304]. Future studies will tell whether RNase1-based therapies may allow to tackle other cases of chronic neuroinflammation, disseminated intravascular coagulation, or even long-Covid syndrome as a scenario of “runaway inflammation”.
Another challenge for improved diagnostic as well as therapeutic measures with respect to exRNAs is related to the (patho-) physiological interplay between different organ systems, characterized by the brain–heart or brain-lung axis, thereby harboring disease-progressing exRNAs in a bidirectional manner. As an example, subarachnoid hemorrhage (SAH), caused by the rupture of an intracranial aneurysm, not only contributes to hemorrhagic strokes to a large degree [248], to microglia accumulation and activation, but may also lead to a cascade of systemic pathologies, including cardiac damage after SAH (Xu et al., 2021, unpublished). In which way different types of disseminating exRNAs may causally contribute to the underlying pathology of the interplay between cerebral hemorrhage and cardiac dysfunction is poorly understood and requires intensive future studies.
Likewise, different types of exDNA such cfDNA or NETs have been identified under various pathophysiological conditions (including hyperinflammation, tumor progression or neurodegeneration) in the brain and can contribute to disease onset and progression in various ways [52, 210, 447]. A common denominator in the pathogenesis is the release of mtDNA and cfDNA, the latter being particularly available in NETs, whereby both parameters may serve as disease biomarkers in plasma and CSF as well. As to MS, oxidized exDNA together with oxidized lipids are highly enriched in lesions and plaques, to be associated with active demyelination and axonal or neuronal injury [141]. Despite the fact that the mechanisms leading to demyelination and neurodegeneration are poorly understood, mitochondrial damage together with the release of mtDNA as well as complex formation of extracellular nucleic acids with basic myelin sheet proteins may contribute to the initial patho-mechanisms of neurodegeneration and -regeneration in the cortex of patients with MS.
With regard to ischemic stroke, particularly cfDNAs do correlate with worse prognosis, whereas the depletion of platelets or platelet-specific DAMPs (as agonists for NETosis) greatly improved stroke outcomes. Here, the administration of a novel endogenous poly-peptide, designated neonatal NET-inhibitory factor (nNIF) [438], in experimental stroke models improved long-term neurological and motor function as well as enhanced survival after stroke [80]. nNIF specifically blocked NET formation without affecting neutrophil recruitment after stroke. Together with the combined application of DNase1/t-PA, as described in this article, nNIF and related peptides may serve as new therapeutic regimen towards NETs as targets in ischemic stroke. Interestingly enough, the generation of NETs in association with SAH could be prevented by administration of either RNase A or DNase1, an observation that indicates a close association between different SENAs in situations of brain tissue damage [119, 444]. Despite the fact that DNase-related targeting also of other NET-associated pathologies such as cystic fibrosis, sepsis or subcutaneous lupus erythematosus may provide some benefit, the patho-mechanistic role of extracellular cytotoxic histones (as a major component of NETs) in these diseases as well as in brain-associated pathologies requires further engaged investigation to weaken the dark side of NETs [328].
Conclusions
In recent years, we have witnessed the appearance and molecular interplay of the class of diverse extracellular nucleic acid molecules, exRNAs and exDNAs, present in different body fluids and tissues, including their structural characterization, biological functions and possible medical applications. A plethora of basic research as well as clinical studies concerned with the profiling of these high and low molecular weight poly-anionic compounds, either in isolated form, as complexes with proteins or in association with EVs, has been undertaken. An emphasis was particularly laid on their definition as biomarkers such as for miRNAs, mtDNAs or NET fragments (summarized in Tables 3, 4, 5, 6) and their potential as causal disease factors, especially for neuroinflammatory and neurodegenerative disorders as discussed here.
Although we are far away from understanding all the mechanistic details in the biological systems we have touched upon in this review, we certainly appreciate the diversity of molecular interactions by which such nucleic acid DAMPs provide ways to provoke cellular signal transduction pathways, both under physiological as well as pathological conditions. Based on such data, new antagonistic approaches have been suggested (including the treatment with RNase or DNase, antagomirs and others) to combat certain neurological disorders (summarized in Table 2). Whether such regimen will find their way into the broad medical field remains to be seen, at least some miRNA-based biomarkers appear to be used in diagnostic analysis.
Nevertheless, the topic addressed in this review is often underappreciated and dismissed as a normal consequence of cell stress, tissue injury, infection, or the healing process in the brain, but has potent translational impact for medical applications in neuroinflammatory and neurodegenerative diseases. A discussion on these disorders, exacerbated by extracellular nucleic acids, is virtually non-existent in the literature, and the here presented approaches may find their way into possible new treatments. It is hoped that this review would stimulate basic researchers as well as clinical scientists in their research fields devoted to neurological pathologies.
Availability of data and materials
Not applicable.
Abbreviations
- Aβ:
-
Amyloid-β
- AD:
-
Alzheimer's disease
- AIM2:
-
Absent in melanoma
- ALS:
-
Amyotrophic lateral sclerosis
- AMPK:
-
AMP-activated kinase
- ANXA2:
-
Annexin A2
- AP2B1:
-
Adaptor-related protein complex 2 subunit β1
- APC:
-
Antigen-presenting cell
- APP:
-
Amyloid precursor protein
- AUF1:
-
AU-rich element/poly(U)-binding/degradation factor 1
- AXIN1:
-
Axis inhibition protein 1
- BACE1:
-
β-Site APP cleaving enzyme-1
- BBB:
-
Blood–brain barrier
- BMI1:
-
B lymphoma Mo-MLV insertion region 1 homolog
- CaMKKβ:
-
Calcium/calmodulin-dependent protein kinase kinase beta
- CARD:
-
Caspase activation and recruitment domain
- cfDNA:
-
Cell-free DNA
- cGAS:
-
Cyclic GMP/AMP (cGAMP) synthase
- circRNA:
-
Circular RNA
- CNS:
-
Central nervous system
- CSF:
-
Cerebrospinal fluid
- CREB:
-
CAMP response element-binding protein
- DAMPs:
-
Danger-associated molecular patterns
- dsDNA:
-
Double-stranded DNA
- dsRNA:
-
Double-stranded RNA
- EAE:
-
Experimental autoimmune encephalomyelitis
- ET:
-
Extracellular trap
- EV:
-
Extracellular vesicle
- FIRRE:
-
Functional intergenic RNA repeat element
- FUS:
-
Fused in sarcoma
- GAS5:
-
Growth arrest-specific 5
- GDF-15:
-
Growth differentiation factor 15
- HAGLROS:
-
HAGLR opposite strand
- HD:
-
Huntington's disease
- HDAC1:
-
Histone deacetylase 1
- HLA-DR:
-
Human leukocyte antigen-DR isotype
- HMGB1:
-
High mobility group box protein 1
- hnRNP K:
-
Heterogeneous nuclear ribonucleoprotein K
- HOTAIR:
-
HOX transcript antisense RNA
- IFN:
-
Interferon
- IKZF3:
-
Ikaros family zinc finger protein 3
- IL:
-
Interleukin
- iNOS:
-
Inducible nitric oxide synthase
- I/R:
-
Ischemia/reperfusion
- IRAK1:
-
IL-1 receptor-associated kinase 1
- LCP1:
-
Lymphocyte cytosolic protein 1
- LGP2:
-
Laboratory of genetics and physiology 2
- lincRNA-p21:
-
Long intergenic noncoding RNA-p21
- lncRNA:
-
Long non-coding RNA
- LPS:
-
Lipopolysaccharide
- LRRK2:
-
Leucine rich repeat kinase 2
- Maclpil:
-
Macrophage contained LCP1 related pro-inflammatory lncRNA
- MALAT1:
-
Metastasis-associated lung adenocarcinoma transcript 1
- MAF:
-
Musculoaponeurotic fibrosarcoma
- MAVS:
-
Mitochondrial antiviral-signaling protein
- MDA5:
-
Melanoma differentiation-associated protein 5
- mexDNA:
-
Mitochondrial exDNA
- MIB1:
-
E3 Ubiquitin ligase Mindbomb1
- miRNA:
-
MicroRNA
- MPP+:
-
1-Methyl-4-phenylpyridinium
- MPTP:
-
1-Methyl-1-4-phenyl-1,2,3,6-tetrahydripyridine hydrochloride
- mRNA:
-
Messenger RNA
- MS:
-
Multiple sclerosis
- mtDNA:
-
Mitochondrial DNA
- mTOR:
-
Mechanistic target of rapamycin
- MyD88:
-
Myeloid differentiation factor 88
- NBR1:
-
Next to BRCA1 gene 1
- ncRNA:
-
Non-coding RNA
- NEAT1:
-
Nuclear paraspeckle assembly transcript 1
- Nespas:
-
Nesp-antisense
- NETs:
-
Neutrophil extracellular traps
- NF-κB:
-
Nuclear factor-kappa B
- NFT:
-
Intraneuronal neurofibrillary tangle
- NLR:
-
Nucleotide-binding oligomerization domain (NOD)-like receptor
- NLRP3:
-
NLR family pyrin domain containing protein 3
- nucDNA:
-
Nuclear DNA
- OGD/R:
-
Oxygen–glucose deprivation/reoxygenation
- PAD4:
-
Peptidyl-arginine-deiminase-4
- PAMPs:
-
Pathogen-associated molecular patterns
- PBMC:
-
Peripheral blood mononuclear cell
- PD:
-
Parkinson's disease
- PINK1:
-
PTEN-induced kinase 1
- PPARγ:
-
Peroxisome proliferator-activated receptor gamma
- PPMS:
-
Primary progressive MS
- PRC2:
-
Polycomb repressive complex 2
- PRR:
-
Pattern recognition receptor
- RAGE:
-
Receptor for advanced glycation end-products
- rexRNA:
-
Extracellular rRNA
- RLR:
-
Retinoic acid-inducible gene-1 (RIG-1)-like receptor
- RMST:
-
Rhabdomyosarcoma 2-associated transcript
- RN7SK:
-
RNA component of 7SK nuclear ribonucleoprotein
- ROS:
-
Reactive oxygen species
- RRMS:
-
Relapsing–remitting MS
- rRNA:
-
Ribosomal RNA
- rt-PA:
-
Recombinant tissue plasminogen activator
- SAH:
-
Subarachnoid hemorrhage
- SFPQ:
-
Splicing factor proline- and glutamine-rich
- SHIP1:
-
SH2 domain-containing inositol 5′-phosphatase 1
- SIGMAR1:
-
Sigma non-opioid intracellular receptor 1
- SIRT1:
-
Sirtuin 1
- SLE:
-
Systemic lupus erythematosus
- SNCA:
-
α-Synuclein
- SNHG:
-
Small nucleolar RNA host gene
- SOCS:
-
Suppressor of cytokine signaling
- SPMS:
-
Secondary progressive MS
- ssRNA:
-
Single-stranded RNA
- STAT6:
-
Signal transducer and activator of transcription 6
- STING:
-
Stimulator of interferon genes
- TAK1:
-
Transforming growth factor-β-activated kinase 1
- tau:
-
Tubulin associated unit
- TBI:
-
Traumatic brain injury
- TBK1:
-
TANK-binding kinase
- TFAM:
-
Mitochondrial transcription factor A
- TIR:
-
Toll/IL-1 receptor
- TLR:
-
Toll-like receptor
- TNF-α:
-
Tumor necrosis factor-α
- TRAF6:
-
TNF receptor-associated factor 6
- TREM2:
-
Triggering receptor expressed on myeloid cells 2
- tRNA:
-
Transfer RNA
- TUG1:
-
Taurine upregulated 1
- UCHL1:
-
Ubiquitin C-terminal hydrolase L1
- ULK1:
-
UNC51-like kinase-1
- VEGF:
-
Vascular endothelial growth factor
- ZFAS1:
-
ZNFX1 antisense RNA 1
References
Abplanalp WT, Fischer A, John D, Zeiher AM, Gosgnach W, Darville H, Montgomery R, Pestano L, Allee G, Paty I, Fougerousse F, Dimmeler S. Efficiency and target derepression of anti-miR-92a: results of a first in human study. Nucleic Acid Ther. 2020;30(6):335–45.
Abuelezz NZ, Nasr FE, Abdel Aal WM, Molokhia T, Zaky A. Sera miR-34a, miR-29b and miR-181c as potential novel diagnostic biomarker panel for Alzheimers in the Egyptian population. Exp Gerontol. 2022;169: 111961.
Adamczak S, Dale G, de Rivero Vaccari JP, Bullock MR, Dietrich WD, Keane RW. Inflammasome proteins in cerebrospinal fluid of brain-injured patients as biomarkers of functional outcome: clinical article. J Neurosurg. 2012;117(6):1119–25.
Aharon A, Spector P, Ahmad RS, Horrany N, Sabbach A, Brenner B, Aharon-Peretz J. Extracellular vesicles of Alzheimer’s disease patients as a biomarker for disease progression. Mol Neurobiol. 2020;57(10):4156–69.
Ahlbrecht J, Martino F, Pul R, Skripuletz T, Suhs KW, Schauerte C, Yildiz O, Trebst C, Tasto L, Thum S, Pfanne A, Roesler R, Lauda F, Hecker M, Zettl UK, Tumani H, Thum T, Stangel M. Deregulation of microRNA-181c in cerebrospinal fluid of patients with clinically isolated syndrome is associated with early conversion to relapsing-remitting multiple sclerosis. Mult Scler. 2016;22(9):1202–14.
Al-Kafaji G, Bakheit HF, Alharbi MA, Farahat AA, Jailani M, Ebrahin BH, Bakhiet M. Mitochondrial DNA copy number in peripheral blood as a potential non-invasive biomarker for multiple sclerosis. Neuromol Med. 2020;22(2):304–13.
Alborelli I, Generali D, Jermann P, Cappelletti MR, Ferrero G, Scaggiante B, Bortul M, Zanconati F, Nicolet S, Haegele J, Bubendorf L, Aceto N, Scaltriti M, Mucci G, Quagliata L, Novelli G. Cell-free DNA analysis in healthy individuals by next-generation sequencing: a proof of concept and technical validation study. Cell Death Dis. 2019;10(7):534.
Albornoz EA, Woodruff TM, Gordon R. Inflammasomes in CNS diseases. Exp Suppl. 2018;108:41–60.
Ali MA, Shaker OG, Khalifa AA, Ezzat EM, Elghobary HA, Abdel Mawla TS, Elkhateeb AF, Elebiary AMA, Elamir AM. LncRNAs NEAT1, HOTAIR, and GAS5 expression in hypertensive and non-hypertensive associated cerebrovascular stroke patients, and its link to clinical characteristics and severity score of the disease. Noncoding RNA Res. 2023;8(1):96–108.
Allen C, Thornton P, Denes A, McColl BW, Pierozynski A, Monestier M, Pinteaux E, Rothwell NJ, Allan SM. Neutrophil cerebrovascular transmigration triggers rapid neurotoxicity through release of proteases associated with decondensed DNA. J Immunol. 2012;189(1):381–92.
Aloi MS, Prater KE, Sopher B, Davidson S, Jayadev S, Garden GA. The pro-inflammatory microRNA miR-155 influences fibrillar beta-Amyloid1 -42 catabolism by microglia. Glia. 2021;69(7):1736–48.
Altincicek B, Stotzel S, Wygrecka M, Preissner KT, Vilcinskas A. Host-derived extracellular nucleic acids enhance innate immune responses, induce coagulation, and prolong survival upon infection in insects. J Immunol. 2008;181(4):2705–12.
Andersen JV, Schousboe A, Verkhratsky A. Astrocyte energy and neurotransmitter metabolism in Alzheimer’s disease: integration of the glutamate/GABA-glutamine cycle. Prog Neurobiol. 2022;217: 102331.
Angelopoulou E, Paudel YN, Piperi C. miR-124 and Parkinson’s disease: a biomarker with therapeutic potential. Pharmacol Res. 2019;150: 104515.
Ansari A, Maffioletti E, Milanesi E, Marizzoni M, Frisoni GB, Blin O, Richardson JC, Bordet R, Forloni G, Gennarelli M, Bocchio-Chiavetto L, PharmaCog C. miR-146a and miR-181a are involved in the progression of mild cognitive impairment to Alzheimer’s disease. Neurobiol Aging. 2019;82:102–9.
Antonyova V, Kejik Z, Brogyanyi T, Kaplanek R, Pajkova M, Talianova V, Hromadka R, Masarik M, Sykora D, Miksatkova L, Martasek P, Jakubek M. Role of mtDNA disturbances in the pathogenesis of Alzheimer’s and Parkinson’s disease. DNA Repair. 2020;91–92: 102871.
Baghi M, Delavar DM, Yadegari E, Peymani M, Pozo D, Hossein Nasr-Esfahani M, Ghaedi K. Modified level of miR-376a is associated with Parkinson’s disease. J Cell Mol Med. 2020;24(4):2622–34.
Bai X, Tang Y, Yu M, Wu L, Liu F, Ni J, Wang Z, Wang J, Fei J, Wang W, Huang F, Wang J. Downregulation of blood serum microRNA 29 family in patients with Parkinson’s disease. Sci Rep. 2017;7(1):5411.
Balint E, Ashkar AA. Remote hyperinflammation drives neurological disease via T-cell-mediated innate-like cytotoxicity. Cell Mol Immunol. 2021;18(7):1638–40.
Bao MH, Szeto V, Yang BB, Zhu SZ, Sun HS, Feng ZP. Long non-coding RNAs in ischemic stroke. Cell Death Dis. 2018;9(3):281.
Barrat FJ, Meeker T, Gregorio J, Chan JH, Uematsu S, Akira S, Chang B, Duramad O, Coffman RL. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J Exp Med. 2005;202(8):1131–9.
Batistela MS, Josviak ND, Sulzbach CD, de Souza RL. An overview of circulating cell-free microRNAs as putative biomarkers in Alzheimer’s and Parkinson’s diseases. Int J Neurosci. 2017;127(6):547–58.
Bauernfried S, Scherr MJ, Pichlmair A, Duderstadt KE, Hornung V. Human NLRP1 is a sensor for double-stranded RNA. Science. 2021;371(6528): eabd0811.
Behbahanipour M, Peymani M, Salari M, Hashemi MS, Nasr-Esfahani MH, Ghaedi K. Expression profiling of blood microRNAs 885, 361, and 17 in the patients with the Parkinson’s disease: integrating interaction data to uncover the possible triggering age-related mechanisms. Sci Rep. 2019;9(1):13759.
Bertheloot D, Naumovski AL, Langhoff P, Horvath GL, Jin T, Xiao TS, Garbi N, Agrawal S, Kolbeck R, Latz E. RAGE enhances TLR responses through binding and internalization of RNA. J Immunol. 2016;197(10):4118–26.
Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81(1):1–5.
Bierhaus A, Humpert PM, Morcos M, Wendt T, Chavakis T, Arnold B, Stern DM, Nawroth PP. Understanding RAGE, the receptor for advanced glycation end products. J Mol Med. 2005;83(11):876–86.
Bonasio R, Shiekhattar R. Regulation of transcription by long noncoding RNAs. Annu Rev Genet. 2014;48:433–55.
Borsche M, Konig IR, Delcambre S, Petrucci S, Balck A, Bruggemann N, Zimprich A, Wasner K, Pereira SL, Avenali M, Deuschle C, Badanjak K, Ghelfi J, Gasser T, Kasten M, Rosenstiel P, Lohmann K, Brockmann K, Valente EM, Youle RJ, Grunewald A, Klein C. Mitochondrial damage-associated inflammation highlights biomarkers in PRKN/PINK1 parkinsonism. Brain. 2020;143(10):3041–51.
Bortolotti P, Faure E, Kipnis E. Inflammasomes in tissue damages and immune disorders after trauma. Front Immunol. 2018;9:1900.
Bracko O, Njiru BN, Swallow M, Ali M, Haft-Javaherian M, Schaffer CB. Increasing cerebral blood flow improves cognition into late stages in Alzheimer’s disease mice. J Cereb Blood Flow Metab. 2020;40(7):1441–52.
Brea D, Sobrino T, Rodriguez-Yanez M, Ramos-Cabrer P, Agulla J, Rodriguez-Gonzalez R, Campos F, Blanco M, Castillo J. Toll-like receptors 7 and 8 expression is associated with poor outcome and greater inflammatory response in acute ischemic stroke. Clin Immunol. 2011;139(2):193–8.
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–5.
Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16(7):407–20.
Burge DJ, Eisenman J, Byrnes-Blake K, Smolak P, Lau K, Cohen SB, Kivitz AJ, Levin R, Martin RW, Sherrer Y, Posada JA. Safety, pharmacokinetics, and pharmacodynamics of RSLV-132, an RNase-Fc fusion protein in systemic lupus erythematosus: a randomized, double-blind, placebo-controlled study. Lupus. 2017;26(8):825–34.
Cabrera-Fuentes HA, Lopez ML, McCurdy S, Fischer S, Meiler S, Baumer Y, Galuska SP, Preissner KT, Boisvert WA. Regulation of monocyte/macrophage polarisation by extracellular RNA. Thromb Haemost. 2015;113(3):473–81.
Cabrera-Fuentes HA, Ruiz-Meana M, Simsekyilmaz S, Kostin S, Inserte J, Saffarzadeh M, Galuska SP, Vijayan V, Barba I, Barreto G, Fischer S, Lochnit G, Ilinskaya ON, Baumgart-Vogt E, Boning A, Lecour S, Hausenloy DJ, Liehn EA, Garcia-Dorado D, Schluter KD, Preissner KT. RNase1 prevents the damaging interplay between extracellular RNA and tumour necrosis factor-alpha in cardiac ischaemia/reperfusion injury. Thromb Haemost. 2014;112(6):1110–9.
Caggiu E, Paulus K, Mameli G, Arru G, Sechi GP, Sechi LA. Differential expression of miRNA 155 and miRNA 146a in Parkinson’s disease patients. eNeurologicalSci. 2018;13:1–4.
Cao B, Wang T, Qu Q, Kang T, Yang Q. Long noncoding RNA SNHG1 promotes neuroinflammation in Parkinson’s disease via regulating miR-7/NLRP3 pathway. Neuroscience. 2018;388:118–27.
Cao DW, Liu MM, Duan R, Tao YF, Zhou JS, Fang WR, Zhu JR, Niu L, Sun JG. The lncRNA Malat1 functions as a ceRNA to contribute to berberine-mediated inhibition of HMGB1 by sponging miR-181c-5p in poststroke inflammation. Acta Pharmacol Sin. 2020;41(1):22–33.
Cao F, Liu Z, Sun G. Diagnostic value of miR-193a-3p in Alzheimer’s disease and miR-193a-3p attenuates amyloid-beta induced neurotoxicity by targeting PTEN. Exp Gerontol. 2020;130: 110814.
Cavassani KA, Ishii M, Wen H, Schaller MA, Lincoln PM, Lukacs NW, Hogaboam CM, Kunkel SL. TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J Exp Med. 2008;205(11):2609–21.
Cervera-Carles L, Alcolea D, Estanga A, Ecay-Torres M, Izagirre A, Clerigue M, Garcia-Sebastian M, Villanua J, Escalas C, Blesa R, Martinez-Lage P, Lleo A, Fortea J, Clarimon J. Cerebrospinal fluid mitochondrial DNA in the Alzheimer’s disease continuum. Neurobiol Aging. 2017;53(192):e191-192.
Chavakis T, Bierhaus A, Nawroth PP. RAGE (receptor for advanced glycation end products): a central player in the inflammatory response. Microbes Infect. 2004;6(13):1219–25.
Chen C, Cai J, Zhang S, Gan L, Dong Y, Zhu T, Ma G, Li T, Zhang X, Li Q, Cheng X, Wu C, Yang J, Zuo Y, Liu J. Protective effect of RNase on unilateral nephrectomy-induced postoperative cognitive dysfunction in aged mice. PLoS ONE. 2015;10(7): e0134307.
Chen C, Feng Y, Zou L, Wang L, Chen HH, Cai JY, Xu JM, Sosnovik DE, Chao W. Role of extracellular RNA and TLR3-Trif signaling in myocardial ischemia-reperfusion injury. J Am Heart Assoc. 2014;3(1): e000683.
Chen C, Zhang S, Wei Y, Sun X. LncRNA RMST regulates neuronal apoptosis and inflammatory response via sponging miR-150-5p in Parkinson’s disease. NeuroImmunoModulation. 2022;29(1):55–62.
Chen J, Liu P, Dong X, Jin J, Xu Y. The role of lncRNAs in ischemic stroke. Neurochem Int. 2021;147: 105019.
Chen X, Ren G, Li Y, Chao W, Chen S, Li X, Xue S. Level of LncRNA GAS5 and hippocampal volume are associated with the progression of Alzheimer’s disease. Clin Interv Aging. 2022;17:745–53.
Chen X, Zhang X, Su C, Huang S. Long noncoding RNA HULC in acute ischemic stroke: association with disease risk, severity, and recurrence-free survival and relation with IL-6, ICAM1, miR-9, and miR-195. J Clin Lab Anal. 2020;34(11): e23500.
Chen Y, Song Y, Huang J, Qu M, Zhang Y, Geng J, Zhang Z, Liu J, Yang GY. Increased circulating exosomal miRNA-223 is associated with acute ischemic stroke. Front Neurol. 2017;8:57.
Chen Y, Zhang H, Hu X, Cai W, Ni W, Zhou K. Role of NETosis in central nervous system injury. Oxid Med Cell Longev. 2022;2022:3235524.
Chen Z, Wang K, Huang J, Zheng G, Lv Y, Luo N, Liang M, Huang L. Upregulated serum MiR-146b serves as a biomarker for acute ischemic stroke. Cell Physiol Biochem. 2018;45(1):397–405.
Cheng J, Duan Y, Zhang F, Shi J, Li H, Wang F, Li H. The role of lncRNA TUG1 in the Parkinson disease and its effect on microglial inflammatory response. Neuromol Med. 2021;23(2):327–34.
Cieslak M, Kukulski F, Komoszynski M. Emerging role of extracellular nucleotides and adenosine in multiple sclerosis. Purinergic Signal. 2011;7(4):393–402.
Conte C. Possible link between SARS-CoV-2 infection and Parkinson’s disease: the role of toll-like receptor 4. Int J Mol Sci. 2021;22(13):7135.
Costanza M, Poliani PL, Portararo P, Cappetti B, Musio S, Pagani F, Steinman L, Colombo MP, Pedotti R, Sangaletti S. DNA threads released by activated CD4(+) T lymphocytes provide autocrine costimulation. Proc Natl Acad Sci U S A. 2019;116(18):8985–94.
Creemers EE, Tijsen AJ, Pinto YM. Circulating microRNAs: novel biomarkers and extracellular communicators in cardiovascular disease? Circ Res. 2012;110(3):483–95.
Crescitelli R, Lasser C, Szabo TG, Kittel A, Eldh M, Dianzani I, Buzas EI, Lotvall J. Distinct RNA profiles in subpopulations of extracellular vesicles: apoptotic bodies, microvesicles and exosomes. J Extracell Vesicles. 2013;2:20677.
Crivello M, O’Riordan SL, Woods I, Cannon S, Halang L, Coughlan KS, Hogg MC, Lewandowski SA, Prehn JHM. Pleiotropic activity of systemically delivered angiogenin in the SOD1(G93A) mouse model. Neuropharmacology. 2018;133:503–11.
Cruz Hernandez JC, Bracko O, Kersbergen CJ, Muse V, Haft-Javaherian M, Berg M, Park L, Vinarcsik LK, Ivasyk I, Rivera DA, Kang Y, Cortes-Canteli M, Peyrounette M, Doyeux V, Smith A, Zhou J, Otte G, Beverly JD, Davenport E, Davit Y, Lin CP, Strickland S, Iadecola C, Lorthois S, Nishimura N, Schaffer CB. Neutrophil adhesion in brain capillaries reduces cortical blood flow and impairs memory function in Alzheimer’s disease mouse models. Nat Neurosci. 2019;22(3):413–20.
Cui G, Ye X, Zuo T, Zhao H, Zhao Q, Chen W, Hua F. Chloroquine pretreatment inhibits toll-like receptor 3 signaling after stroke. Neurosci Lett. 2013;548:101–4.
da Silva DJ, Borges AF, Souza PO, de Souza PR, Cardoso CR, Dorta ML, de Oliveira MA, Teixeira AL, Ribeiro-Dias F. Decreased toll-like receptor 2 and toll-like receptor 7/8-induced cytokines in parkinson’s disease patients. NeuroImmunoModulation. 2016;23(1):58–66.
Dahl S, Anders E, Gidlof O, Svensson D, Nilsson BO. The host defense peptide LL-37 triggers release of nucleic acids from human mast cells. Peptides. 2018;109:39–45.
Dakterzada F, David BI, Targa A, Llado A, Torres G, Romero L, de Gonzalo-Calvo D, Moncusi-Moix A, Tort-Merino A, Huerto R, Sanchez-de-la-Torre M, Barbe F, Pinol-Ripoll G. Reduced levels of miR-342-5p in plasma are associated with worse cognitive evolution in patients with mild Alzheimer’s disease. Front Aging Neurosci. 2021;13: 705989.
Danielson KM, Rubio R, Abderazzaq F, Das S, Wang YE. High throughput sequencing of extracellular RNA from human plasma. PLoS ONE. 2017;12(1): e0164644.
Davis CH, Kim KY, Bushong EA, Mills EA, Boassa D, Shih T, Kinebuchi M, Phan S, Zhou Y, Bihlmeyer NA, Nguyen JV, Jin Y, Ellisman MH, Marsh-Armstrong N. Transcellular degradation of axonal mitochondria. Proc Natl Acad Sci USA. 2014;111(26):9633–8.
De Bondt M, Hellings N, Opdenakker G, Struyf S. Neutrophils: underestimated players in the pathogenesis of multiple sclerosis (MS). Int J Mol Sci. 2020;21(12):4558.
de Buhr N, von Kockritz-Blickwede M. Detection, visualization, and quantification of neutrophil extracellular traps (NETs) and NET markers. Methods Mol Biol. 2020;2087:425–42.
De Meyer SF, Suidan GL, Fuchs TA, Monestier M, Wagner DD. Extracellular chromatin is an important mediator of ischemic stroke in mice. Arterioscler Thromb Vasc Biol. 2012;32(8):1884–91.
de Oliveira LG, Angelo YS, Iglesias AH, Peron JPS. Unraveling the link between mitochondrial dynamics and neuroinflammation. Front Immunol. 2021;12: 624919.
De Strooper B, Karran E. The cellular phase of Alzheimer’s disease. Cell. 2016;164(4):603–15.
De Vito F, Musella A, Fresegna D, Rizzo FR, Gentile A, Stampanoni Bassi M, Gilio L, Buttari F, Procaccini C, Colamatteo A, Bullitta S, Guadalupi L, Caioli S, Vanni V, Balletta S, Sanna K, Bruno A, Dolcetti E, Furlan R, Finardi A, Licursi V, Drulovic J, Pekmezovic T, Fusco C, Bruzzaniti S, Hornstein E, Uccelli A, Salvetti M, Matarese G, Centonze D, Mandolesi G. MiR-142–3p regulates synaptopathy-driven disease progression in multiple sclerosis. Neuropathol Appl Neurobiol. 2022;48(2): e12765.
Decout A, Katz JD, Venkatraman S, Ablasser A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat Rev Immunol. 2021;21(9):548–69.
Deerhake ME, Biswas DD, Barclay WE, Shinohara ML. Pattern recognition receptors in multiple sclerosis and its animal models. Front Immunol. 2019;10:2644.
Deleidi M, Hallett PJ, Koprich JB, Chung CY, Isacson O. The Toll-like receptor-3 agonist polyinosinic:polycytidylic acid triggers nigrostriatal dopaminergic degeneration. J Neurosci. 2010;30(48):16091–101.
Deng L, Guo Y, Liu J, Wang X, Chen S, Wang Q, Rao J, Wang Y, Zuo T, Hu Q, Zhao X, Dong Z. miR-671-5p attenuates neuroinflammation via suppressing NF-kappaB expression in an acute ischemic stroke model. Neurochem Res. 2021;46(7):1801–13.
Deng L, Pan J, Peng Q, Dong Z, Wang Y. Toll-like receptor 3 and interferon beta mRNA expressions were increased in peripheral blood of ischemic stroke patients with good outcome. J Stroke Cerebrovasc Dis. 2017;26(3):559–66.
Deng Y, Chen D, Wang L, Gao F, Jin B, Lv H, Zhang G, Sun X, Liu L, Mo D, Ma N, Song L, Huo X, Yan T, Miao Z. Silencing of long noncoding RNA nespas aggravates microglial cell death and neuroinflammation in ischemic stroke. Stroke. 2019;50(7):1850–8.
Denorme F, Portier I, Rustad JL, Cody MJ, de Araujo CV, Hoki C, Alexander MD, Grandhi R, Dyer MR, Neal MD, Majersik JJ, Yost CC, Campbell RA. Neutrophil extracellular traps regulate ischemic stroke brain injury. J Clin Invest. 2022;132(10): e154225.
Dhawan A. Extracellular miRNA biomarkers in neurologic disease: is cerebrospinal fluid helpful? Biomark Med. 2021;15(15):1377–88.
Dhir A, Dhir S, Borowski LS, Jimenez L, Teitell M, Rotig A, Crow YJ, Rice GI, Duffy D, Tamby C, Nojima T, Munnich A, Schiff M, de Almeida CR, Rehwinkel J, Dziembowski A, Szczesny RJ, Proudfoot NJ. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature. 2018;560(7717):238–42.
Diling C, Yinrui G, Longkai Q, Xiaocui T, Yadi L, Xin Y, Guoyan H, Ou S, Tianqiao Y, Dongdong W, Yizhen X, Yang BB, Qingping W. Circular RNA NF1-419 enhances autophagy to ameliorate senile dementia by binding Dynamin-1 and Adaptor protein 2 B1 in AD-like mice. Aging. 2019;11(24):12002–31.
Ding Y, Luan W, Shen X, Wang Z, Cao Y. LncRNA BDNF-AS as ceRNA regulates the miR-9-5p/BACE1 pathway affecting neurotoxicity in Alzheimer’s disease. Arch Gerontol Geriatr. 2022;99: 104614.
Dolle C, Flones I, Nido GS, Miletic H, Osuagwu N, Kristoffersen S, Lilleng PK, Larsen JP, Tysnes OB, Haugarvoll K, Bindoff LA, Tzoulis C. Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nat Commun. 2016;7:13548.
Dong LH, Sun L, Zhang WJ, Wang XY, Li JM. Reduced serum miR-202 may promote the progression of Alzheimer’s disease patients via targeting amyloid precursor protein. Kaohsiung J Med Sci. 2021;37(8):730–8.
Doring Y, Libby P, Soehnlein O. Neutrophil extracellular traps participate in cardiovascular diseases: recent experimental and clinical insights. Circ Res. 2020;126(9):1228–41.
Dorostgou Z, Yadegar N, Dorostgou Z, Khorvash F, Vakili O. Novel insights into the role of circular RNAs in Parkinson disease: an emerging renaissance in the management of neurodegenerative diseases. J Neurosci Res. 2022;100:1775.
Doxakis E. Cell-free microRNAs in Parkinson’s disease: potential biomarkers that provide new insights into disease pathogenesis. Ageing Res Rev. 2020;58: 101023.
Doxakis E. Insights into the multifaceted role of circular RNAs: implications for Parkinson’s disease pathogenesis and diagnosis. NPJ Parkinsons Dis. 2022;8(1):7.
Du C, Liu C, Kang J, Zhao G, Ye Z, Huang S, Li Z, Wu Z, Pei G. MicroRNA miR-326 regulates TH-17 differentiation and is associated with the pathogenesis of multiple sclerosis. Nat Immunol. 2009;10(12):1252–9.
Duan Y, Wang Y, Liu Y, Jin Z, Liu C, Yu X, Chen K, Meng D, Xi J, Fang B. Circular RNAs in Parkinson’s disease: reliable biological markers and targets for rehabilitation. Mol Neurobiol. 2023;60(6):3261–76.
Ducroux C, Di Meglio L, Loyau S, Delbosc S, Boisseau W, Deschildre C, BenMaacha M, Blanc R, Redjem H, Ciccio G, Smajda S, Fahed R, Michel JB, Piotin M, Salomon L, Mazighi M, Ho-Tin-Noe B, Desilles JP. Thrombus neutrophil extracellular traps content impair tPA-induced thrombolysis in acute ischemic stroke. Stroke. 2018;49(3):754–7.
Dykes IM, Emanueli C. Transcriptional and post-transcriptional gene regulation by long non-coding RNA. Genomics Proteomics Bioinform. 2017;15(3):177–86.
Edwardson MA, Zhong X, Fiandaca MS, Federoff HJ, Cheema AK, Dromerick AW. Plasma microRNA markers of upper limb recovery following human stroke. Sci Rep. 2018;8(1):12558.
Ekstrom K, Valadi H, Sjostrand M, Malmhall C, Bossios A, Eldh M, Lotvall J. Characterization of mRNA and microRNA in human mast cell-derived exosomes and their transfer to other mast cells and blood CD34 progenitor cells. J Extracell Vesicles. 2012;1:18389.
Elkhodiry AA, Zamzam DA, El Tayebi HM. miR-155 and functional proteins of CD8+ T cells as potential prognostic biomarkers for relapsing-remitting multiple sclerosis. Mult Scler Relat Disord. 2021;53: 103078.
Ellwardt E, Zipp F. Molecular mechanisms linking neuroinflammation and neurodegeneration in MS. Exp Neurol. 2014;262(Pt A):8–17.
Elsemuller AK, Tomalla V, Gartner U, Troidl K, Jeratsch S, Graumann J, Baal N, Hackstein H, Lasch M, Deindl E, Preissner KT, Fischer S. Characterization of mast cell-derived rRNA-containing microvesicles and their inflammatory impact on endothelial cells. FASEB J. 2019;33(4):5457–67.
Estfanous S, Daily KP, Eltobgy M, Deems NP, Anne MNK, Krause K, Badr A, Hamilton K, Carafice C, Hegazi A, Abu Khweek A, Kelani H, Nimjee S, Awad H, Zhang X, Cormet-Boyaka E, Haffez H, Soror S, Mikhail A, Nuovo G, Barrientos RM, Gavrilin MA, Amer AO. Elevated expression of MiR-17 in microglia of Alzheimer’s disease patients abrogates autophagy-mediated amyloid-beta degradation. Front Immunol. 2021;12: 705581.
Fallarino F, Gargaro M, Mondanell G, Downer EJ, Hossain MJ, Gran B. Delineating the role of toll-like receptors in the neuro-inflammation model EAE. Methods Mol Biol. 2016;1390:383–411.
Fang P, Wu Y, Zhang Z, Cui C, Dong X, Hu K, Jia J, Duan X, Zhang Y, Huo H. The clinical value of long noncoding RNA GAS5 in acute ischemic stroke: correlation with disease risk, inflammation, severity, and risk of recurrence. J Clin Lab Anal. 2022;36(1): e24171.
Fann DY, Lee SY, Manzanero S, Tang SC, Gelderblom M, Chunduri P, Bernreuther C, Glatzel M, Cheng YL, Thundyil J, Widiapradja A, Lok KZ, Foo SL, Wang YC, Li YI, Drummond GR, Basta M, Magnus T, Jo DG, Mattson MP, Sobey CG, Arumugam TV. Intravenous immunoglobulin suppresses NLRP1 and NLRP3 inflammasome-mediated neuronal death in ischemic stroke. Cell Death Dis. 2013;4(9): e790.
Fazeli S, Motovali-Bashi M, Peymani M, Hashemi MS, Etemadifar M, Nasr-Esfahani MH, Ghaedi K. A compound downregulation of SRRM2 and miR-27a-3p with upregulation of miR-27b-3p in PBMCs of Parkinson’s patients is associated with the early stage onset of disease. PLoS ONE. 2020;15(11): e0240855.
Feng F, Jiao P, Wang J, Li Y, Bao B, Luoreng Z, Wang X. Role of long noncoding RNAs in the regulation of cellular immune response and inflammatory diseases. Cells. 2022;11(22):3642.
Feng L, Guo J, Ai F. Circulating long noncoding RNA ANRIL downregulation correlates with increased risk, higher disease severity and elevated pro-inflammatory cytokines in patients with acute ischemic stroke. J Clin Lab Anal. 2019;33(1): e22629.
Feng X, Zhan F, Luo D, Hu J, Wei G, Hua F, Xu G. LncRNA 4344 promotes NLRP3-related neuroinflammation and cognitive impairment by targeting miR-138-5p. Brain Behav Immun. 2021;98:283–98.
Ferreira SA, Romero-Ramos M. Microglia response during Parkinson’s disease: alpha-synuclein intervention. Front Cell Neurosci. 2018;12:247.
Fischer S, Cabrera-Fuentes HA, Noll T, Preissner KT. Impact of extracellular RNA on endothelial barrier function. Cell Tissue Res. 2014;355(3):635–45.
Fischer S, Gerriets T, Wessels C, Walberer M, Kostin S, Stolz E, Zheleva K, Hocke A, Hippenstiel S, Preissner KT. Extracellular RNA mediates endothelial-cell permeability via vascular endothelial growth factor. Blood. 2007;110(7):2457–65.
Fischer S, Gesierich S, Griemert B, Schanzer A, Acker T, Augustin HG, Olsson AK, Preissner KT. Extracellular RNA liberates tumor necrosis factor-alpha to promote tumor cell trafficking and progression. Cancer Res. 2013;73(16):5080–9.
Fischer S, Nasyrov E, Brosien M, Preissner KT, Marti HH, Kunze R. Self-extracellular RNA promotes pro-inflammatory response of astrocytes to exogenous and endogenous danger signals. J Neuroinflamm. 2021;18(1):252.
Fischer S, Nishio M, Peters SC, Tschernatsch M, Walberer M, Weidemann S, Heidenreich R, Couraud PO, Weksler BB, Romero IA, Gerriets T, Preissner KT. Signaling mechanism of extracellular RNA in endothelial cells. FASEB J. 2009;23(7):2100–9.
Fischer S, Preissner KT. Extracellular nucleic acids as novel alarm signals in the vascular system. Mediators of defence and disease. Hamostaseologie. 2013;33(1):37–42.
Fissolo N, Cervera-Carles L, Villar Guimerans LM, Lleo A, Clarimon J, Drulovic J, Dujmovic I, Voortman M, Khalil M, Gil E, Navarro L, Alvarez-Cermeno JC, Montalban X, Comabella M. Cerebrospinal fluid mitochondrial DNA levels in patients with multiple sclerosis. Mult Scler. 2019;25(11):1535–8.
Fotuhi SN, Khalaj-Kondori M, Hoseinpour Feizi MA, Talebi M. Long non-coding RNA BACE1-AS may serve as an Alzheimer’s disease blood-based biomarker. J Mol Neurosci. 2019;69(3):351–9.
Frank S, Copanaki E, Burbach GJ, Muller UC, Deller T. Differential regulation of toll-like receptor mRNAs in amyloid plaque-associated brain tissue of aged APP23 transgenic mice. Neurosci Lett. 2009;453(1):41–4.
Freund I, Eigenbrod T, Helm M, Dalpke AH. RNA modifications modulate activation of innate toll-like receptors. Genes. 2019;10(2):92.
Fruh A, Tielking K, Schoknecht F, Liu S, Schneider UC, Fischer S, Vajkoczy P, Xu R. RNase A inhibits formation of neutrophil extracellular traps in subarachnoid hemorrhage. Front Physiol. 2021;12: 724611.
Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176(2):231–41.
Fuchs TA, Kremer Hovinga JA, Schatzberg D, Wagner DD, Lammle B. Circulating DNA and myeloperoxidase indicate disease activity in patients with thrombotic microangiopathies. Blood. 2012;120(6):1157–64.
Galloway DA, Carew SJ, Blandford SN, Benoit RY, Fudge NJ, Berry T, Moore GRW, Barron J, Moore CS. Investigating the NLRP3 inflammasome and its regulator miR-223-3p in multiple sclerosis and experimental demyelination. J Neurochem. 2022;163(2):94–112.
Gambardella S, Limanaqi F, Ferese R, Biagioni F, Campopiano R, Centonze D, Fornai F. ccf-mtDNA as a potential link between the brain and immune system in neuro-immunological disorders. Front Immunol. 2019;10:1064.
Gamdzyk M, Doycheva DM, Araujo C, Ocak U, Luo Y, Tang J, Zhang JH. cGAS/STING pathway activation contributes to delayed neurodegeneration in neonatal hypoxia-ischemia rat model: possible involvement of LINE-1. Mol Neurobiol. 2020;57(6):2600–19.
Gandhi R, Healy B, Gholipour T, Egorova S, Musallam A, Hussain MS, Nejad P, Patel B, Hei H, Khoury S, Quintana F, Kivisakk P, Chitnis T, Weiner HL. Circulating microRNAs as biomarkers for disease staging in multiple sclerosis. Ann Neurol. 2013;73(6):729–40.
Ganguly D, Chamilos G, Lande R, Gregorio J, Meller S, Facchinetti V, Homey B, Barrat FJ, Zal T, Gilliet M. Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J Exp Med. 2009;206(9):1983–94.
Ge L, Zhou X, Ji WJ, Lu RY, Zhang Y, Zhang YD, Ma YQ, Zhao JH, Li YM. Neutrophil extracellular traps in ischemia-reperfusion injury-induced myocardial no-reflow: therapeutic potential of DNase-based reperfusion strategy. Am J Physiol Heart Circ Physiol. 2015;308(5):H500-509.
Ghafouri-Fard S, Taheri M. A comprehensive review of non-coding RNAs functions in multiple sclerosis. Eur J Pharmacol. 2020;879: 173127.
Giuliani A, Gaetani S, Sorgentoni G, Agarbati S, Laggetta M, Matacchione G, Gobbi M, Rossi T, Galeazzi R, Piccinini G, Pelliccioni G, Bonfigli AR, Procopio AD, Albertini MC, Sabbatinelli J, Olivieri F, Fazioli F. Circulating inflamma-miRs as potential biomarkers of cognitive impairment in patients affected by Alzheimer’s disease. Front Aging Neurosci. 2021;13: 647015.
Giuliani A, Lattanzi S, Ramini D, Graciotti L, Danni MC, Procopio AD, Silvestrini M, Olivieri F, Sabbatinelli J. Potential prognostic value of circulating inflamma-miR-146a-5p and miR-125a-5p in relapsing-remitting multiple sclerosis. Mult Scler Relat Disord. 2021;54: 103126.
Gong C, Maquat LE. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3’ UTRs via Alu elements. Nature. 2011;470(7333):284–8.
Gong T, Liu L, Jiang W, Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol. 2020;20(2):95–112.
Goubau D, Deddouche S, Reis e Sousa C. Cytosolic sensing of viruses. Immunity. 2013;38(5):855–69.
Grazioli S, Pugin J. Mitochondrial damage-associated molecular patterns: from inflammatory signaling to human diseases. Front Immunol. 2018;9:832.
Grosse GM, Blume N, Abu-Fares O, Gotz F, Ernst J, Leotescu A, Gabriel MM, van Gemmeren T, Worthmann H, Lichtinghagen R, Imker R, Falk CS, Weissenborn K, Schuppner R, de Buhr N. Endogenous deoxyribonuclease activity and cell-free deoxyribonucleic acid in acute ischemic stroke: a cohort study. Stroke. 2022;53(4):1235–44.
Grossi I, Radeghieri A, Paolini L, Porrini V, Pilotto A, Padovani A, Marengoni A, Barbon A, Bellucci A, Pizzi M, Salvi A, De Petro G. MicroRNA-34a-5p expression in the plasma and in its extracellular vesicle fractions in subjects with Parkinson’s disease: an exploratory study. Int J Mol Med. 2021;47(2):533–46.
Gu Y, Zhou H, Yu H, Yang W, Wang B, Qian F, Cheng Y, He S, Zhao X, Zhu L, Zhang Y, Jin M, Lu E. miR-99a regulates CD4(+) T cell differentiation and attenuates experimental autoimmune encephalomyelitis by mTOR-mediated glycolysis. Mol Ther Nucleic Acids. 2021;26:1173–85.
Guedes JR, Custodia CM, Silva RJ, de Almeida LP, Pedroso de Lima MC, Cardoso AL. Early miR-155 upregulation contributes to neuroinflammation in Alzheimer’s disease triple transgenic mouse model. Hum Mol Genet. 2014;23(23):6286–301.
Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21(7):677–87.
Gupta M, Martens K, Metz LM, de Koning AJ, Pfeffer G. Long noncoding RNAs associated with phenotypic severity in multiple sclerosis. Mult Scler Relat Disord. 2019;36: 101407.
Haider L, Fischer MT, Frischer JM, Bauer J, Hoftberger R, Botond G, Esterbauer H, Binder CJ, Witztum JL, Lassmann H. Oxidative damage in multiple sclerosis lesions. Brain. 2011;134(Pt 7):1914–24.
Han C, Guo L, Yang Y, Guan Q, Shen H, Sheng Y, Jiao Q. Mechanism of microRNA-22 in regulating neuroinflammation in Alzheimer’s disease. Brain Behav. 2020;10(6): e01627.
Han D, Zhou Y. YY1-induced upregulation of lncRNA NEAT1 contributes to OGD/R injury-induced inflammatory response in cerebral microglial cells via Wnt/beta-catenin signaling pathway. In Vitro Cell Dev Biol Anim. 2019;55(7):501–11.
Han J, Zhuang W, Feng W, Dong F, Hua F, Yao R, Qu X. The circular RNA circINPP4B acts as a sponge of miR-30a to regulate Th17 cell differentiation during progression of experimental autoimmune encephalomyelitis. Cell Mol Immunol. 2021;18(9):2177–87.
Hanan M, Simchovitz A, Yayon N, Vaknine S, Cohen-Fultheim R, Karmon M, Madrer N, Rohrlich TM, Maman M, Bennett ER, Greenberg DS, Meshorer E, Levanon EY, Soreq H, Kadener S. A Parkinson’s disease CircRNAs resource reveals a link between circSLC8A1 and oxidative stress. EMBO Mol Med. 2020;12(11): e13551.
Hanke ML, Kielian T. Toll-like receptors in health and disease in the brain: mechanisms and therapeutic potential. Clin Sci. 2011;121(9):367–87.
Hara N, Kikuchi M, Miyashita A, Hatsuta H, Saito Y, Kasuga K, Murayama S, Ikeuchi T, Kuwano R. Serum microRNA miR-501-3p as a potential biomarker related to the progression of Alzheimer’s disease. Acta Neuropathol Commun. 2017;5(1):10.
Haridy SFA, Shahin NN, Shabayek MI, Selim MM, Abdelhafez MA, Motawi TK. Diagnostic and prognostic value of the RUNXOR/RUNX1 axis in multiple sclerosis. Neurobiol Dis. 2023;178: 106032.
Hartmann G. Nucleic acid immunity. Adv Immunol. 2017;133:121–69.
Havla J, Hohlfeld R. Antibody therapies for progressive multiple sclerosis and for promoting repair. Neurotherapeutics. 2022;19:774.
Hawez A, Al-Haidari A, Madhi R, Rahman M, Thorlacius H. MiR-155 regulates PAD4-dependent formation of neutrophil extracellular traps. Front Immunol. 2019;10:2462.
Hayakawa K, Esposito E, Wang X, Terasaki Y, Liu Y, Xing C, Ji X, Lo EH. Transfer of mitochondria from astrocytes to neurons after stroke. Nature. 2016;535(7613):551–5.
He L, Chen Z, Wang J, Feng H. Expression relationship and significance of NEAT1 and miR-27a-3p in serum and cerebrospinal fluid of patients with Alzheimer’s disease. BMC Neurol. 2022;22(1):203.
He XW, Shi YH, Liu YS, Li GF, Zhao R, Hu Y, Lin CC, Zhuang MT, Su JJ, Liu JR. Increased plasma levels of miR-124-3p, miR-125b-5p and miR-192-5p are associated with outcomes in acute ischaemic stroke patients receiving thrombolysis. Atherosclerosis. 2019;289:36–43.
He XW, Shi YH, Zhao R, Liu YS, Li GF, Hu Y, Chen W, Cui GH, Su JJ, Liu JR. Plasma levels of miR-125b-5p and miR-206 in acute ischemic stroke patients after recanalization treatment: a prospective observational study. J Stroke Cerebrovasc Dis. 2019;28(6):1654–61.
Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci. 2015;16(6):358–72.
Honardoost MA, Kiani-Esfahani A, Ghaedi K, Etemadifar M, Salehi M. miR-326 and miR-26a, two potential markers for diagnosis of relapse and remission phases in patient with relapsing-remitting multiple sclerosis. Gene. 2014;544(2):128–33.
Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458(7237):514–8.
Hornung V, Latz E. Intracellular DNA recognition. Nat Rev Immunol. 2010;10(2):123–30.
Huang L, Ma Q, Li Y, Li B, Zhang L. Inhibition of microRNA-210 suppresses pro-inflammatory response and reduces acute brain injury of ischemic stroke in mice. Exp Neurol. 2018;300:41–50.
Huang R, Zhang W, Li W, Gao Y, Zheng D, Bi G. Overexpressing circ_0000831 is sufficient to inhibit neuroinflammation and vertigo in cerebral ischemia through a miR-16-5p-dependent mechanism. Exp Neurol. 2022;353: 114047.
Huang S, Zhao J, Huang D, Zhuo L, Liao S, Jiang Z. Serum miR-132 is a risk marker of post-stroke cognitive impairment. Neurosci Lett. 2016;615:102–6.
Huttunen HJ, Kuja-Panula J, Sorci G, Agneletti AL, Donato R, Rauvala H. Coregulation of neurite outgrowth and cell survival by amphoterin and S100 proteins through receptor for advanced glycation end products (RAGE) activation. J Biol Chem. 2000;275(51):40096–105.
Hyakkoku K, Hamanaka J, Tsuruma K, Shimazawa M, Tanaka H, Uematsu S, Akira S, Inagaki N, Nagai H, Hara H. Toll-like receptor 4 (TLR4), but not TLR3 or TLR9, knock-out mice have neuroprotective effects against focal cerebral ischemia. Neuroscience. 2010;171(1):258–67.
Iparraguirre L, Munoz-Culla M, Prada-Luengo I, Castillo-Trivino T, Olascoaga J, Otaegui D. Circular RNA profiling reveals that circular RNAs from ANXA2 can be used as new biomarkers for multiple sclerosis. Hum Mol Genet. 2017;26(18):3564–72.
Isobe C, Abe T, Terayama Y. Levels of reduced and oxidized coenzyme Q-10 and 8-hydroxy-2’-deoxyguanosine in the CSF of patients with Alzheimer’s disease demonstrate that mitochondrial oxidative damage and/or oxidative DNA damage contributes to the neurodegenerative process. J Neurol. 2010;257(3):399–404.
Jaax ME, Krauel K, Marschall T, Brandt S, Gansler J, Furll B, Appel B, Fischer S, Block S, Helm CA, Muller S, Preissner KT, Greinacher A. Complex formation with nucleic acids and aptamers alters the antigenic properties of platelet factor 4. Blood. 2013;122(2):272–81.
Janeway CA Jr. The immune system evolved to discriminate infectious nonself from noninfectious self. Immunol Today. 1992;13(1):11–6.
Jayaraj RL, Azimullah S, Beiram R, Jalal FY, Rosenberg GA. Neuroinflammation: friend and foe for ischemic stroke. J Neuroinflamm. 2019;16(1):142.
Ji Q, Ji Y, Peng J, Zhou X, Chen X, Zhao H, Xu T, Chen L, Xu Y. Increased brain-specific MiR-9 and MiR-124 in the serum exosomes of acute ischemic stroke patients. PLoS ONE. 2016;11(9): e0163645.
Ji Y, Zhou Y, Pan J, Li X, Wang H, Wang Y. Temporal pattern of Toll-like receptor 9 upregulation in neurons and glial cells following cerebral ischemia reperfusion in mice. Int J Neurosci. 2016;126(3):269–77.
Jia LH, Liu YN. Downregulated serum miR-223 servers as biomarker in Alzheimer’s disease. Cell Biochem Funct. 2016;34(4):233–7.
Jiang F, Liu X, Cui X, Hu J, Wang L, Xue F, Guo S, Wang X. Circ_0000518 promotes macrophage/microglia M1 polarization via the FUS/CaMKKbeta/AMPK pathway to aggravate multiple sclerosis. Neuroscience. 2022;490:131–43.
Jiang GL, Yang XL, Zhou HJ, Long J, Liu B, Zhang LM, Lu D. cGAS knockdown promotes microglial M2 polarization to alleviate neuroinflammation by inhibiting cGAS-STING signaling pathway in cerebral ischemic stroke. Brain Res Bull. 2021;171:183–95.
Jiang X, Wang X, Tuo M, Ma J, Xie A. RAGE and its emerging role in the pathogenesis of Parkinson’s disease. Neurosci Lett. 2018;672:65–9.
Jiang X, Xiao L, Jiang X, Li G, Lu Z. Screening of Parkinson’s differential MicroRNA Based on GEO database and its clinical verification. Biomed Res Int. 2021;2021:8171236.
Jin F, Xing J. Circulating pro-angiogenic and anti-angiogenic microRNA expressions in patients with acute ischemic stroke and their association with disease severity. Neurol Sci. 2017;38(11):2015–23.
Jin JJ, Kim HD, Maxwell JA, Li L, Fukuchi K. Toll-like receptor 4-dependent upregulation of cytokines in a transgenic mouse model of Alzheimer’s disease. J Neuroinflamm. 2008;5:23.
Johnson TP, Tyagi R, Patel K, Schiess N, Calabresi PA, Nath A. Impaired toll-like receptor 8 signaling in multiple sclerosis. J Neuroinflamm. 2013;10:74.
Junker A, Krumbholz M, Eisele S, Mohan H, Augstein F, Bittner R, Lassmann H, Wekerle H, Hohlfeld R, Meinl E. MicroRNA profiling of multiple sclerosis lesions identifies modulators of the regulatory protein CD47. Brain. 2009;132(Pt 12):3342–52.
Jurcau A, Simion A. Neuroinflammation in cerebral ischemia and ischemia/reperfusion injuries: from pathophysiology to therapeutic strategies. Int J Mol Sci. 2021;23(1):14.
Kabelitz D, Medzhitov R. Innate immunity–cross-talk with adaptive immunity through pattern recognition receptors and cytokines. Curr Opin Immunol. 2007;19(1):1–3.
Kadir RRA, Alwjwaj M, Bayraktutan U. MicroRNA: an emerging predictive, diagnostic, prognostic and therapeutic strategy in ischaemic stroke. Cell Mol Neurobiol. 2022;42(5):1301–19.
Kang L, Yu H, Yang X, Zhu Y, Bai X, Wang R, Cao Y, Xu H, Luo H, Lu L, Shi MJ, Tian Y, Fan W, Zhao BQ. Neutrophil extracellular traps released by neutrophils impair revascularization and vascular remodeling after stroke. Nat Commun. 2020;11(1):2488.
Kariko K, Ni H, Capodici J, Lamphier M, Weissman D. mRNA is an endogenous ligand for Toll-like receptor 3. J Biol Chem. 2004;279(13):12542–50.
Kawada J, Kimura H, Ito Y, Hara S, Iriyama M, Yoshikawa T, Morishima T. Systemic cytokine responses in patients with influenza-associated encephalopathy. J Infect Dis. 2003;188(5):690–8.
Kawai T, Akira S. Pathogen recognition with Toll-like receptors. Curr Opin Immunol. 2005;17(4):338–44.
Kenny A, McArdle H, Calero M, Rabano A, Madden SF, Adamson K, Forster R, Spain E, Prehn JHM, Henshall DC, Medina M, Jimenez-Mateos EM, Engel T. Elevated plasma microRNA-206 levels predict cognitive decline and progression to dementia from mild cognitive impairment. Biomolecules. 2019;9(11):734.
Kieran NW, Suresh R, Dorion MF, MacDonald A, Blain M, Wen D, Fuh SC, Ryan F, Diaz RJ, Stratton JA, Ludwin SK, Sonnen JA, Antel J, Healy LM. MicroRNA-210 regulates the metabolic and inflammatory status of primary human astrocytes. J Neuroinflamm. 2022;19(1):10.
Kierdorf K, Fritz G. RAGE regulation and signaling in inflammation and beyond. J Leukoc Biol. 2013;94(1):55–68.
Kigerl KA, de Rivero Vaccari JP, Dietrich WD, Popovich PG, Keane RW. Pattern recognition receptors and central nervous system repair. Exp Neurol. 2014;258:5–16.
Kim KM, Abdelmohsen K, Mustapic M, Kapogiannis D, Gorospe M. RNA in extracellular vesicles. Wiley Interdiscip Rev RNA. 2017;8(4): e1413.
Kong L, Li W, Chang E, Wang W, Shen N, Xu X, Wang X, Zhang Y, Sun W, Hu W, Xu P, Liu X. mtDNA-STING axis mediates microglial polarization via IRF3/NF-kappaB signaling after ischemic stroke. Front Immunol. 2022;13: 860977.
Kortam MA, Elfar N, Shaker OG, El-Boghdady NA, Abd-Elmawla MA. MAGI2-AS3 and miR-374b-5p as putative regulators of multiple sclerosis via modulating the PTEN/AKT/IRF-3/IFN-beta axis: new clinical insights. ACS Chem Neurosci. 2023;14(6):1107–18.
Kozin MS, Kulakova OG, Favorova OO. Involvement of mitochondria in neurodegeneration in multiple sclerosis. Biochemistry. 2018;83(7):813–30.
Kramer TJ, Hubener P, Pottker B, Golz C, Neulen A, Pantel T, Goetz H, Ritter K, Schafer MKE, Thal SC. Ribonuclease-1 treatment after traumatic brain injury preserves blood-brain barrier integrity and delays secondary brain damage in mice. Sci Rep. 2022;12(1):5731.
Kristensen LS, Andersen MS, Stagsted LVW, Ebbesen KK, Hansen TB, Kjems J. The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet. 2019;20(11):675–91.
Krug A. Nucleic acid recognition receptors in autoimmunity. Handb Exp Pharmacol. 2008;183:129–51.
Kumar V. Toll-like receptors in the pathogenesis of neuroinflammation. J Neuroimmunol. 2019;332:16–30.
Lampron A, Elali A, Rivest S. Innate immunity in the CNS: redefining the relationship between the CNS and Its environment. Neuron. 2013;78(2):214–32.
Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang YH, Homey B, Cao W, Wang YH, Su B, Nestle FO, Zal T, Mellman I, Schroder JM, Liu YJ, Gilliet M. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature. 2007;449(7162):564–9.
Laridan E, Denorme F, Desender L, Francois O, Andersson T, Deckmyn H, Vanhoorelbeke K, De Meyer SF. Neutrophil extracellular traps in ischemic stroke thrombi. Ann Neurol. 2017;82(2):223–32.
Lasch M, Kleinert EC, Meister S, Kumaraswami K, Buchheim JI, Grantzow T, Lautz T, Salpisti S, Fischer S, Troidl K, Fleming I, Randi AM, Sperandio M, Preissner KT, Deindl E. Extracellular RNA released due to shear stress controls natural bypass growth by mediating mechanotransduction in mice. Blood. 2019;134(17):1469–79.
Lauretti E, Dabrowski K, Pratico D. The neurobiology of non-coding RNAs and Alzheimer’s disease pathogenesis: pathways, mechanisms and translational opportunities. Ageing Res Rev. 2021;71: 101425.
Leal NS, Dentoni G, Schreiner B, Naia L, Piras A, Graff C, Cattaneo A, Meli G, Hamasaki M, Nilsson P, Ankarcrona M. Amyloid beta-peptide increases mitochondria-endoplasmic reticulum contact altering mitochondrial function and autophagosome formation in Alzheimer’s disease-related models. Cells. 2020;9(12):2552.
Lehmann SM, Kruger C, Park B, Derkow K, Rosenberger K, Baumgart J, Trimbuch T, Eom G, Hinz M, Kaul D, Habbel P, Kalin R, Franzoni E, Rybak A, Nguyen D, Veh R, Ninnemann O, Peters O, Nitsch R, Heppner FL, Golenbock D, Schott E, Ploegh HL, Wulczyn FG, Lehnardt S. An unconventional role for miRNA: let-7 activates Toll-like receptor 7 and causes neurodegeneration. Nat Neurosci. 2012;15(6):827–35.
Lehmann SM, Rosenberger K, Kruger C, Habbel P, Derkow K, Kaul D, Rybak A, Brandt C, Schott E, Wulczyn FG, Lehnardt S. Extracellularly delivered single-stranded viral RNA causes neurodegeneration dependent on TLR7. J Immunol. 2012;189(3):1448–58.
Lehnardt S, Lehmann S, Kaul D, Tschimmel K, Hoffmann O, Cho S, Krueger C, Nitsch R, Meisel A, Weber JR. Toll-like receptor 2 mediates CNS injury in focal cerebral ischemia. J Neuroimmunol. 2007;190(1–2):28–33.
Leurs CE, Podlesniy P, Trullas R, Balk L, Steenwijk MD, Malekzadeh A, Piehl F, Uitdehaag BM, Killestein J, van Horssen J, Teunissen CE. Cerebrospinal fluid mtDNA concentration is elevated in multiple sclerosis disease and responds to treatment. Mult Scler. 2018;24(4):472–80.
Li C, Xing Y, Zhang Y, Hua Y, Hu J, Bai Y. Neutrophil extracellular traps exacerbate ischemic brain damage. Mol Neurobiol. 2022;59(1):643–56.
Li G, Morris-Blanco KC, Lopez MS, Yang T, Zhao H, Vemuganti R, Luo Y. Impact of microRNAs on ischemic stroke: from pre- to post-disease. Prog Neurobiol. 2018;163–164:59–78.
Li H, Yu L, Li M, Chen X, Tian Q, Jiang Y, Li N. MicroRNA-150 serves as a diagnostic biomarker and is involved in the inflammatory pathogenesis of Parkinson’s disease. Mol Genet Genomic Med. 2020;8(4): e1189.
Li L, Acioglu C, Heary RF, Elkabes S. Role of astroglial toll-like receptors (TLRs) in central nervous system infections, injury and neurodegenerative diseases. Brain Behav Immun. 2021;91:740–55.
Li P, Duan S, Fu A. Long noncoding RNA NEAT1 correlates with higher disease risk, worse disease condition, decreased miR-124 and miR-125a and predicts poor recurrence-free survival of acute ischemic stroke. J Clin Lab Anal. 2020;34(2): e23056.
Li Q, Cao Y, Dang C, Han B, Han R, Ma H, Hao J, Wang L. Inhibition of double-strand DNA-sensing cGAS ameliorates brain injury after ischemic stroke. EMBO Mol Med. 2020;12(4): e11002.
Li S, Sheng J, Hu JK, Yu W, Kishikawa H, Hu MG, Shima K, Wu D, Xu Z, Xin W, Sims KB, Landers JE, Brown RH Jr, Hu GF. Ribonuclease 4 protects neuron degeneration by promoting angiogenesis, neurogenesis, and neuronal survival under stress. Angiogenesis. 2013;16(2):387–404.
Li Y, Fan H, Sun J, Ni M, Zhang L, Chen C, Hong X, Fang F, Zhang W, Ma P. Circular RNA expression profile of Alzheimer’s disease and its clinical significance as biomarkers for the disease risk and progression. Int J Biochem Cell Biol. 2020;123: 105747.
Lian L, Zhang Y, Liu L, Yang L, Cai Y, Zhang J, Xu S. Neuroinflammation in ischemic stroke: focus on MicroRNA-mediated polarization of microglia. Front Mol Neurosci. 2020;13: 612439.
Liang C, Zou T, Zhang M, Fan W, Zhang T, Jiang Y, Cai Y, Chen F, Chen X, Sun Y, Zhao B, Wang Y, Cui L. MicroRNA-146a switches microglial phenotypes to resist the pathological processes and cognitive degradation of Alzheimer’s disease. Theranostics. 2021;11(9):4103–21.
Liang Y, Wang L. Inflamma-microRNAs in Alzheimer’s disease: from disease pathogenesis to therapeutic potentials. Front Cell Neurosci. 2021;15: 785433.
Liao Y, Cheng J, Kong X, Li S, Li X, Zhang M, Zhang H, Yang T, Dong Y, Li J, Xu Y, Yuan Z. HDAC3 inhibition ameliorates ischemia/reperfusion-induced brain injury by regulating the microglial cGAS-STING pathway. Theranostics. 2020;10(21):9644–62.
Lim KH, Staudt LM. Toll-like receptor signaling. Cold Spring Harb Perspect Biol. 2013;5(1): a011247.
Lin X, Wang R, Li R, Tao T, Zhang D, Qi Y. Diagnostic performance of miR-485-3p in patients with Parkinson’s disease and its relationship with neuroinflammation. Neuromolecular Med. 2022;24(2):195–201.
Lind NA, Rael VE, Pestal K, Liu B, Barton GM. Regulation of the nucleic acid-sensing Toll-like receptors. Nat Rev Immunol. 2022;22(4):224–35.
Liu B, Ye X, Zhao G, Jin L, Shi J. Association of RAGE with acute ischemic stroke prognosis in type 2 diabetes. Ir J Med Sci. 2021;190(2):625–30.
Liu D, Li L, Xu J, Luo W, Pan L, Niu Q, Wei D. Upregulated lncRNA NORAD can diagnose acute cerebral ischemic stroke patients and predict poor prognosis. Folia Neuropathol. 2023;61(1):105–10.
Liu D, Zhao D, Zhao Y, Wang Y, Zhao Y, Wen C. Inhibition of microRNA-155 alleviates cognitive impairment in Alzheimer’s disease and involvement of neuroinflammation. Curr Alzheimer Res. 2019;16(6):473–82.
Liu G, Gack MU. Distinct and orchestrated functions of RNA sensors in innate immunity. Immunity. 2020;53(1):26–42.
Liu H, Zheng W, Song Z. circDlgap4 alleviates cerebral ischaemic injury by binding to AUF1 to suppress oxidative stress and neuroinflammation. Mol Neurobiol. 2022;59(5):3218–32.
Liu L, Chen X, Chen YH, Zhang K. Identification of circular RNA hsa_Circ_0003391 in peripheral blood is potentially associated with Alzheimer’s disease. Front Aging Neurosci. 2020;12: 601965.
Liu M, Liu X, Zhou M, Guo S, Sun K. Impact of CircRNAs on ischemic stroke. Aging Dis. 2022;13(2):329–39.
Liu Q, Lei C. Neuroprotective effects of miR-331-3p through improved cell viability and inflammatory marker expression: correlation of serum miR-331-3p levels with diagnosis and severity of Alzheimer’s disease. Exp Gerontol. 2021;144: 111187.
Liu X, Zhou F, Yang Y, Wang W, Niu L, Zuo D, Li X, Hua H, Zhang B, Kou Y, Guo J, Kong F, Pan W, Gao D, Meves JM, Sun H, Xue M, Zhang Q, Wang Y, Tang R. MiR-409-3p and MiR-1896 co-operatively participate in IL-17-induced inflammatory cytokine production in astrocytes and pathogenesis of EAE mice via targeting SOCS3/STAT3 signaling. Glia. 2019;67(1):101–12.
Liu Y, Cheng X, Li H, Hui S, Zhang Z, Xiao Y, Peng W. Non-coding RNAs as novel regulators of neuroinflammation in Alzheimer’s disease. Front Immunol. 2022;13: 908076.
Liu Y, Xu Y, Yu M. MicroRNA-4722-5p and microRNA-615-3p serve as potential biomarkers for Alzheimer’s disease. Exp Ther Med. 2022;23(3):241.
Lowes H, Pyle A, Duddy M, Hudson G. Cell-free mitochondrial DNA in progressive multiple sclerosis. Mitochondrion. 2019;46:307–12.
Lowes H, Pyle A, Santibanez-Koref M, Hudson G. Circulating cell-free mitochondrial DNA levels in Parkinson’s disease are influenced by treatment. Mol Neurodegener. 2020;15(1):10.
Lu D, Ho ES, Mai H, Zang J, Liu Y, Li Y, Yang B, Ding Y, Tsang CK, Xu A. Identification of blood circular RNAs as potential biomarkers for acute ischemic stroke. Front Neurosci. 2020;14:81.
Lu J, Liu L, Chen J, Zhi J, Li J, Li L, Jiang Z. LncRNA HOTAIR in exercise-induced neuro-protective function in Alzheimer’s disease. Folia Neuropathol. 2022;60(4):414–20.
Lu Y, Tan L, Wang X. Circular HDAC9/microRNA-138/sirtuin-1 pathway mediates synaptic and amyloid precursor protein processing deficits in Alzheimer’s disease. Neurosci Bull. 2019;35(5):877–88.
Lundberg AM, Drexler SK, Monaco C, Williams LM, Sacre SM, Feldmann M, Foxwell BM. Key differences in TLR3/poly I: C signaling and cytokine induction by human primary cells: a phenomenon absent from murine cell systems. Blood. 2007;110(9):3245–52.
Lv Q, Wang Z, Zhong Z, Huang W. Role of long noncoding RNAs in Parkinson’s disease: putative biomarkers and therapeutic targets. Parkinsons Dis. 2020;2020:5374307.
Lv Q, Zhong Z, Hu B, Yan S, Yan Y, Zhang J, Shi T, Jiang L, Li W, Huang W. MicroRNA-3473b regulates the expression of TREM2/ULK1 and inhibits autophagy in inflammatory pathogenesis of Parkinson disease. J Neurochem. 2021;157(3):599–610.
Lyman M, Lloyd DG, Ji X, Vizcaychipi MP, Ma D. Neuroinflammation: the role and consequences. Neurosci Res. 2014;79:1–12.
Lyu Y, Bai L, Qin C. Long noncoding RNAs in neurodevelopment and Parkinson’s disease. Animal Model Exp Med. 2019;2(4):239–51.
Ma P, Li Y, Zhang W, Fang F, Sun J, Liu M, Li K, Dong L. Long non-coding RNA MALAT1 inhibits neuron apoptosis and neuroinflammation while stimulates neurite outgrowth and its correlation with MiR-125b mediates PTGS2, CDK5 and FOXQ1 in Alzheimer’s disease. Curr Alzheimer Res. 2019;16(7):596–612.
Ma W, Li Y, Wang C, Xu F, Wang M, Liu Y. Serum miR-221 serves as a biomarker for Parkinson’s disease. Cell Biochem Funct. 2016;34(7):511–5.
Macdonald RL, Diringer MN, Citerio G. Understanding the disease: aneurysmal subarachnoid hemorrhage. Intensive Care Med. 2014;40(12):1940–3.
Maffioletti E, Milanesi E, Ansari A, Zanetti O, Galluzzi S, Geroldi C, Gennarelli M, Bocchio-Chiavetto L. miR-146a plasma levels are not altered in Alzheimer’s disease but correlate with age and illness severity. Front Aging Neurosci. 2019;11:366.
Mancuso R, Hernis A, Agostini S, Rovaris M, Caputo D, Clerici M. MicroRNA-572 expression in multiple sclerosis patients with different patterns of clinical progression. J Transl Med. 2015;13:148.
Manda-Handzlik A, Demkow U. The brain entangled: the contribution of neutrophil extracellular traps to the diseases of the central nervous system. Cells. 2019;8(12):1477.
Mandolesi G, Rizzo FR, Balletta S, Stampanoni Bassi M, Gilio L, Guadalupi L, Nencini M, Moscatelli A, Ryan CP, Licursi V, Dolcetti E, Musella A, Gentile A, Fresegna D, Bullitta S, Caioli S, Vanni V, Sanna K, Bruno A, Buttari F, Castelli C, Presutti C, De Santa F, Finardi A, Furlan R, Centonze D, De Vito F. The microRNA let-7b-5p is negatively associated with inflammation and disease severity in multiple sclerosis. Cells. 2021;10(2):330.
Mangold A, Alias S, Scherz T, Hofbauer M, Jakowitsch J, Panzenbock A, Simon D, Laimer D, Bangert C, Kammerlander A, Mascherbauer J, Winter MP, Distelmaier K, Adlbrecht C, Preissner KT, Lang IM. Coronary neutrophil extracellular trap burden and deoxyribonuclease activity in ST-elevation acute coronary syndrome are predictors of ST-segment resolution and infarct size. Circ Res. 2015;116(7):1182–92.
Marek LR, Kagan JC. Deciphering the function of nucleic acid sensing TLRs one regulatory step at a time. Front Biosci. 2011;16(6):2060–8.
Marta M, Andersson A, Isaksson M, Kampe O, Lobell A. Unexpected regulatory roles of TLR4 and TLR9 in experimental autoimmune encephalomyelitis. Eur J Immunol. 2008;38(2):565–75.
MartInez-Coria H, Arrieta-Cruz I, Cruz ME, Lopez-Valdes HE. Physiopathology of ischemic stroke and its modulation using memantine: evidence from preclinical stroke. Neural Regen Res. 2021;16(3):433–9.
Marttinen M, Takalo M, Natunen T, Wittrahm R, Gabbouj S, Kemppainen S, Leinonen V, Tanila H, Haapasalo A, Hiltunen M. Molecular mechanisms of synaptotoxicity and neuroinflammation in Alzheimer’s disease. Front Neurosci. 2018;12:963.
Masoumi F, Ghorbani S, Talebi F, Branton WG, Rajaei S, Power C, Noorbakhsh F. Malat1 long noncoding RNA regulates inflammation and leukocyte differentiation in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2019;328:50–9.
Massberg S, Grahl L, von Bruehl ML, Manukyan D, Pfeiler S, Goosmann C, Brinkmann V, Lorenz M, Bidzhekov K, Khandagale AB, Konrad I, Kennerknecht E, Reges K, Holdenrieder S, Braun S, Reinhardt C, Spannagl M, Preissner KT, Engelmann B. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med. 2010;16(8):887–96.
Mathew A, Lindsley TA, Sheridan A, Bhoiwala DL, Hushmendy SF, Yager EJ, Ruggiero EA, Crawford DR. Degraded mitochondrial DNA is a newly identified subtype of the damage associated molecular pattern (DAMP) family and possible trigger of neurodegeneration. J Alzheimers Dis. 2012;30(3):617–27.
Matute C, Torre I, Perez-Cerda F, Perez-Samartin A, Alberdi E, Etxebarria E, Arranz AM, Ravid R, Rodriguez-Antiguedad A, Sanchez-Gomez M, Domercq M. P2X(7) receptor blockade prevents ATP excitotoxicity in oligodendrocytes and ameliorates experimental autoimmune encephalomyelitis. J Neurosci. 2007;27(35):9525–33.
Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045.
McKenzie BA, Mamik MK, Saito LB, Boghozian R, Monaco MC, Major EO, Lu JQ, Branton WG, Power C. Caspase-1 inhibition prevents glial inflammasome activation and pyroptosis in models of multiple sclerosis. Proc Natl Acad Sci U S A. 2018;115(26):E6065–74.
McKenzie JA, Spielman LJ, Pointer CB, Lowry JR, Bajwa E, Lee CW, Klegeris A. Neuroinflammation as a common mechanism associated with the modifiable risk factors for Alzheimer’s and Parkinson’s diseases. Curr Aging Sci. 2017;10(3):158–76.
Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, Loewer A, Ziebold U, Landthaler M, Kocks C, le Noble F, Rajewsky N. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495(7441):333–8.
Millan MJ. Linking deregulation of non-coding RNA to the core pathophysiology of Alzheimer’s disease: an integrative review. Prog Neurobiol. 2017;156:1–68.
Mills J, Capece M, Cocucci E, Tessari A, Palmieri D. Cancer-derived extracellular vesicle-associated MicroRNAs in intercellular communication: one cell’s trash is another cell’s treasure. Int J Mol Sci. 2019;20(24):6109.
Miyake K, Saitoh SI, Fukui R, Shibata T, Sato R, Murakami Y. Dynamic control of nucleic-acid-sensing Toll-like receptors by the endosomal compartment. Int Immunol. 2021;33(12):835–40.
Miyake K, Shibata T, Ohto U, Shimizu T. Emerging roles of the processing of nucleic acids and Toll-like receptors in innate immune responses to nucleic acids. J Leukoc Biol. 2017;101(1):135–42.
Mo M, Xiao Y, Huang S, Cen L, Chen X, Zhang L, Luo Q, Li S, Yang X, Lin X, Xu P. MicroRNA expressing profiles in A53T mutant alpha-synuclein transgenic mice and Parkinsonian. Oncotarget. 2017;8(1):15–28.
Moradi A, Rahimi Naiini M, Yazdanpanahi N, Tabatabaeian H, Nabatchian F, Baghi M, Azadeh M, Ghaedi K. Evaluation of the expression levels of three long non-coding rnas in multiple sclerosis. Cell J. 2020;22(2):165–70.
Moreno-Garcia L, Lopez-Royo T, Calvo AC, Toivonen JM, de la Torre M, Moreno-Martinez L, Molina N, Aparicio P, Zaragoza P, Manzano R, Osta R. Competing endogenous RNA networks as biomarkers in neurodegenerative diseases. Int J Mol Sci. 2020;21(24):9582.
Muller T, Hamm S, Bauer S. TLR9-mediated recognition of DNA. Handb Exp Pharmacol. 2008;183:51–70.
Mycko MP, Zurawska AE, Selmaj I, Selmaj KW. Impact of diminished expression of circRNA on multiple sclerosis pathomechanisms. Front Immunol. 2022;13: 875994.
Naegele M, Tillack K, Reinhardt S, Schippling S, Martin R, Sospedra M. Neutrophils in multiple sclerosis are characterized by a primed phenotype. J Neuroimmunol. 2012;242(1–2):60–71.
Nakamura K, Shichita T. Cellular and molecular mechanisms of sterile inflammation in ischaemic stroke. J Biochem. 2019;165(6):459–64.
Nasi M, Bianchini E, De Biasi S, Gibellini L, Neroni A, Mattioli M, Pinti M, Iannone A, Mattioli AV, Simone AM, Ferraro D, Vitetta F, Sola P, Cossarizza A. Increased plasma levels of mitochondrial DNA and pro-inflammatory cytokines in patients with progressive multiple sclerosis. J Neuroimmunol. 2020;338: 577107.
Nasi M, De Gaetano A, Bianchini E, De Biasi S, Gibellini L, Neroni A, Mattioli M, Pinti M, Lo TD, Borella R, Mattioli AV, Chester J, Melegari A, Simone AM, Ferraro D, Vitetta F, Sola P, Cossarizza A. Mitochondrial damage-associated molecular patterns stimulate reactive oxygen species production in human microglia. Mol Cell Neurosci. 2020;108: 103538.
Nemutlu SD, Akcay G, Yildirim S, Ozkan A, Ceker T, Derin N, Tanriover G, Aslan M, Agar A, Ozbey G. Vortioxetine ameliorates motor and cognitive impairments in the rotenone-induced Parkinson’s disease via targeting TLR-2 mediated neuroinflammation. Neuropharmacology. 2022;208: 108977.
Neumann A, Brogden G, von Kockritz-Blickwede M. Extracellular traps: an ancient weapon of multiple kingdoms. Biology. 2020;9(2):34.
Newman LE, Shadel GS. Pink1/Parkin link inflammation, mitochondrial stress, and neurodegeneration. J Cell Biol. 2018;217(10):3327–9.
Ni J, Wang X, Chen S, Liu H, Wang Y, Xu X, Cheng J, Jia J, Zhen X. MicroRNA let-7c-5p protects against cerebral ischemia injury via mechanisms involving the inhibition of microglia activation. Brain Behav Immun. 2015;49:75–85.
Ni Y, Huang H, Chen Y, Cao M, Zhou H, Zhang Y. Investigation of long non-coding RNA expression profiles in the substantia nigra of Parkinson’s disease. Cell Mol Neurobiol. 2017;37(2):329–38.
Nies YH, MohamadNajib NH, Lim WL, Kamaruzzaman MA, Yahaya MF, Teoh SL. MicroRNA dysregulation in Parkinson’s disease: a narrative review. Front Neurosci. 2021;15: 660379.
Nociti V, Santoro M. What do we know about the role of lncRNAs in multiple sclerosis? Neural Regen Res. 2021;16(9):1715–22.
Noll F, Behnke J, Leiting S, Troidl K, Alves GT, Muller-Redetzky H, Preissner KT, Fischer S. Self-extracellular RNA acts in synergy with exogenous danger signals to promote inflammation. PLoS ONE. 2017;12(12): e0190002.
Oliveira SR, Dionisio PA, CorreiaGuedes L, Goncalves N, Coelho M, Rosa MM, Amaral JD, Ferreira JJ, Rodrigues CMP. Circulating inflammatory miRNAs associated with Parkinson’s disease pathophysiology. Biomolecules. 2020;10(6):945.
Oliveira SR, Dionisio PA, Gaspar MM, Correia Guedes L, Coelho M, Rosa MM, Ferreira JJ, Amaral JD, Rodrigues CMP. miR-335 targets LRRK2 and mitigates inflammation in Parkinson’s disease. Front Cell Dev Biol. 2021;9: 661461.
Ozinsky A, Underhill DM, Fontenot JD, Hajjar AM, Smith KD, Wilson CB, Schroeder L, Aderem A. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc Natl Acad Sci U S A. 2000;97(25):13766–71.
Pai MC, Kuo YM, Wang IF, Chiang PM, Tsai KJ. The role of methylated circulating nucleic acids as a potential biomarker in Alzheimer’s disease. Mol Neurobiol. 2019;56(4):2440–9.
Palm NW, Medzhitov R. Pattern recognition receptors and control of adaptive immunity. Immunol Rev. 2009;227(1):221–33.
Pan Y, Jiao Q, Wei W, Zheng T, Yang X, Xin W. Emerging role of LncRNAs in ischemic stroke-novel insights into the regulation of inflammation. J Inflamm Res. 2021;14:4467–83.
Patergnani S, Morciano G, Carinci M, Leo S, Pinton P, Rimessi A. The, “mitochondrial stress responses”: the “Dr. Jekyll and Mr. Hyde” of neuronal disorders. Neural Regen Res. 2022;17(12):2563–75.
Paudel YN, Angelopoulou E, Piperi C, Othman I, Aamir K, Shaikh MF. Impact of HMGB1, RAGE, and TLR4 in Alzheimer’s Disease (AD): from risk factors to therapeutic targeting. Cells. 2020;9(2):383.
Pazmandi K, Agod Z, Kumar BV, Szabo A, Fekete T, Sogor V, Veres A, Boldogh I, Rajnavolgyi E, Lanyi A, Bacsi A. Oxidative modification enhances the immunostimulatory effects of extracellular mitochondrial DNA on plasmacytoid dendritic cells. Free Radic Biol Med. 2014;77:281–90.
Pena-Martinez C, Duran-Laforet V, Garcia-Culebras A, Cuartero MI, Moro MA, Lizasoain I. Neutrophil extracellular trap targeting protects against ischemic damage after fibrin-rich thrombotic stroke despite non-reperfusion. Front Immunol. 2022;13: 790002.
Pena-Martinez C, Duran-Laforet V, Garcia-Culebras A, Ostos F, Hernandez-Jimenez M, Bravo-Ferrer I, Perez-Ruiz A, Ballenilla F, Diaz-Guzman J, Pradillo JM, Lizasoain I, Moro MA. Pharmacological modulation of neutrophil extracellular traps reverses thrombotic stroke tPA (tissue-type plasminogen activator) resistance. Stroke. 2019;50(11):3228–37.
Peng J, Wang H, Gong Z, Li X, He L, Shen Q, Pan J, Peng Y. Idebenone attenuates cerebral inflammatory injury in ischemia and reperfusion via dampening NLRP3 inflammasome activity. Mol Immunol. 2020;123:74–87.
Peng X, Jing P, Chen J, Xu L. The role of circular RNA HECTD1 expression in disease risk, disease severity, inflammation, and recurrence of acute ischemic stroke. J Clin Lab Anal. 2019;33(7): e22954.
Picca A, Calvani R, Coelho-Junior HJ, Landi F, Bernabei R, Marzetti E. Mitochondrial dysfunction, oxidative stress, and neuroinflammation: intertwined roads to neurodegeneration. Antioxidants. 2020;9(8):647.
Pickrell AM, Huang CH, Kennedy SR, Ordureau A, Sideris DP, Hoekstra JG, Harper JW, Youle RJ. Endogenous Parkin preserves dopaminergic substantia nigral neurons following mitochondrial DNA mutagenic stress. Neuron. 2015;87(2):371–81.
Pietronigro EC, Della BV, Zenaro E, Constantin G. NETosis in Alzheimer’s disease. Front Immunol. 2017;8:211.
Podlesniy P, Llorens F, Puigros M, Serra N, Sepulveda-Falla D, Schmidt C, Hermann P, Zerr I, Trullas R. Cerebrospinal fluid mitochondrial DNA in rapid and slow progressive forms of Alzheimer’s disease. Int J Mol Sci. 2020;21(17):6298.
Posada J, Valadkhan S, Burge D, Davies K, Tarn J, Casement J, Jobling K, Gallagher P, Wilson D, Barone F, Fisher BA, Ng WF. Improvement of severe fatigue following nuclease therapy in patients with primary Sjogren’s syndrome: a randomized clinical trial. Arthr Rheumatol. 2021;73(1):143–50.
Preissner KT, Fischer S, Deindl E. Extracellular RNA as a versatile DAMP and alarm signal that influences leukocyte recruitment in inflammation and infection. Front Cell Dev Biol. 2020;8: 619221.
Preissner KT, Herwald H. Extracellular nucleic acids in immunity and cardiovascular responses: between alert and disease. Thromb Haemost. 2017;117(7):1272–82.
Prinz M, Garbe F, Schmidt H, Mildner A, Gutcher I, Wolter K, Piesche M, Schroers R, Weiss E, Kirschning CJ, Rochford CD, Bruck W, Becher B. Innate immunity mediated by TLR9 modulates pathogenicity in an animal model of multiple sclerosis. J Clin Invest. 2006;116(2):456–64.
Puigros M, Calderon A, Perez-Soriano A, de Dios C, Fernandez M, Colell A, Marti MJ, Tolosa E, Trullas R. Cell-free mitochondrial DNA deletions in idiopathic, but not LRRK2, Parkinson’s disease. Neurobiol Dis. 2022;174: 105885.
Pyle A, Anugrha H, Kurzawa-Akanbi M, Yarnall A, Burn D, Hudson G. Reduced mitochondrial DNA copy number is a biomarker of Parkinson’s disease. Neurobiol Aging. 2016;38(216):e217.
Pyle A, Brennan R, Kurzawa-Akanbi M, Yarnall A, Thouin A, Mollenhauer B, Burn D, Chinnery PF, Hudson G. Reduced cerebrospinal fluid mitochondrial DNA is a biomarker for early-stage Parkinson’s disease. Ann Neurol. 2015;78(6):1000–4.
Qi X, Shao M, Sun H, Shen Y, Meng D, Huo W. Long non-coding RNA SNHG14 promotes microglia activation by regulating miR-145-5p/PLA2G4A in cerebral infarction. Neuroscience. 2017;348:98–106.
Qian C, Cao X. Regulation of Toll-like receptor signaling pathways in innate immune responses. Ann N Y Acad Sci. 2013;1283:67–74.
Qu S, Yang X, Li X, Wang J, Gao Y, Shang R, Sun W, Dou K, Li H. Circular RNA: a new star of noncoding RNAs. Cancer Lett. 2015;365(2):141–8.
Quan Y, Wang J, Wang S, Zhao J. Association of the plasma long non-coding RNA MEG3 With Parkinson’s disease. Front Neurol. 2020;11: 532891.
Rainer TH, Leung LY, Chan CPY, Leung YK, Abrigo JM, Wang D, Graham CA. Plasma miR-124-3p and miR-16 concentrations as prognostic markers in acute stroke. Clin Biochem. 2016;49(9):663–8.
Reddy PH, Yin X, Manczak M, Kumar S, Pradeepkiran JA, Vijayan M, Reddy AP. Mutant APP and amyloid beta-induced defective autophagy, mitophagy, mitochondrial structural and functional changes and synaptic damage in hippocampal neurons from Alzheimer’s disease. Hum Mol Genet. 2018;27(14):2502–16.
Regnault A, Lankar D, Lacabanne V, Rodriguez A, Thery C, Rescigno M, Saito T, Verbeek S, Bonnerot C, Ricciardi-Castagnoli P, Amigorena S. Fcgamma receptor-mediated induction of dendritic cell maturation and major histocompatibility complex class I-restricted antigen presentation after immune complex internalization. J Exp Med. 1999;189(2):371–80.
Rehwinkel J, Gack MU. RIG-I-like receptors: their regulation and roles in RNA sensing. Nat Rev Immunol. 2020;20(9):537–51.
Ren B, Song Z, Chen L, Niu X, Feng Q. Long non-coding RNA UCA1 correlates with elevated disease severity, Th17 cell proportion, inflammatory cytokines, and worse prognosis in acute ischemic stroke patients. J Clin Lab Anal. 2021;35(3): e23697.
Ren H, Wu F, Liu B, Song Z, Qu D. Association of circulating long non-coding RNA MALAT1 in diagnosis, disease surveillance, and prognosis of acute ischemic stroke. Braz J Med Biol Res. 2020;53(12): e9174.
Richter D, Weber R, Eyding J, Bartig D, Misselwitz B, Grau A, Hacke W, Krogias C. Acute ischemic stroke care in Germany - further progress from 2016 to 2019. Neurol Res Pract. 2021;3(1):14.
Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–66.
Roers A, Hiller B, Hornung V. Recognition of endogenous nucleic acids by the innate immune system. Immunity. 2016;44(4):739–54.
Rykova EY, Morozkin ES, Ponomaryova AA, Loseva EM, Zaporozhchenko IA, Cherdyntseva NV, Vlassov VV, Laktionov PP. Cell-free and cell-bound circulating nucleic acid complexes: mechanisms of generation, concentration and content. Expert Opin Biol Ther. 2012;12(Suppl 1):S141-153.
Sabry R, El Sharkawy RE, Gad NM. MiRNA -483-5p as a potential noninvasive biomarker for early detection of Alzheimer’s disease. Egypt J Immunol. 2020;27(2):59–72.
Saeidi N, Goudarzvand H, Mohammadi H, Mardi A, Ghoreishizadeh S, Shomali N, Goudarzvand M. Dysregulation of miR-193a serves as a potential contributor to MS pathogenesis via affecting RhoA and Rock1. Mult Scler Relat Disord. 2023;69: 104468.
Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, Lohmeyer J, Preissner KT. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS ONE. 2012;7(2): e32366.
Saffarzadeh M, Preissner KT. Fighting against the dark side of neutrophil extracellular traps in disease: manoeuvres for host protection. Curr Opin Hematol. 2013;20(1):3–9.
Santoro M, Nociti V, Lucchini M, De Fino C, Losavio FA, Mirabella M. Expression profile of long non-coding RNAs in serum of patients with multiple sclerosis. J Mol Neurosci. 2016;59(1):18–23.
Saresella M, Gatti A, Tortorella P, Marventano I, Piancone F, La Rosa F, Caputo D, Rovaris M, Biasin M, Clerici M. Toll-like receptor 3 differently modulates inflammation in progressive or benign multiple sclerosis. Clin Immunol. 2014;150(1):109–20.
Saridas F, Tezcan UH, Cecener G, Egeli U, Sabour TM, Sabour TL, Tunca B, Zarifoglu M, Turan OF, Taskapilioglu O. The expression and prognostic value of miR-146a and miR-155 in Turkish patients with multiple sclerosis. Neurol Res. 2022;44(3):217–23.
Scherrer N, Fays F, Mueller B, Luft A, Fluri F, Christ-Crain M, Devaux Y, Katan M. MicroRNA 150–5p improves risk classification for mortality within 90 days after acute ischemic stroke. J Stroke. 2017;19(3):323–32.
Schmidt AM, Yan SD, Yan SF, Stern DM. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest. 2001;108(7):949–55.
Scholtzova H, Kascsak RJ, Bates KA, Boutajangout A, Kerr DJ, Meeker HC, Mehta PD, Spinner DS, Wisniewski T. Induction of toll-like receptor 9 signaling as a method for ameliorating Alzheimer’s disease-related pathology. J Neurosci. 2009;29(6):1846–54.
Schorn C, Janko C, Latzko M, Chaurio R, Schett G, Herrmann M. Monosodium urate crystals induce extracellular DNA traps in neutrophils, eosinophils, and basophils but not in mononuclear cells. Front Immunol. 2012;3:277.
Senousy MA, Shaker OG, Sayed NH, Fathy N, Kortam MA. LncRNA GAS5 and miR-137 polymorphisms and expression are associated with multiple sclerosis risk: mechanistic insights and potential clinical impact. ACS Chem Neurosci. 2020;11(11):1651–60.
Shademan B, Nourazarian A, Nikanfar M, Biray Avci C, Hasanpour M, Isazadeh A. Investigation of the miRNA146a and miRNA155 gene expression levels in patients with multiple sclerosis. J Clin Neurosci. 2020;78:189–93.
Shao P. MiR-216a-5p ameliorates learning-memory deficits and neuroinflammatory response of Alzheimer’s disease mice via regulation of HMGB1/NF-kappaB signaling. Brain Res. 2021;1766: 147511.
Sharp AJ, Polak PE, Simonini V, Lin SX, Richardson JC, Bongarzone ER, Feinstein DL. P2x7 deficiency suppresses development of experimental autoimmune encephalomyelitis. J Neuroinflamm. 2008;5:33.
Shi Z, Chen T, Yao Q, Zheng L, Zhang Z, Wang J, Hu Z, Cui H, Han Y, Han X, Zhang K, Hong W. The circular RNA ciRS-7 promotes APP and BACE1 degradation in an NF-kappaB-dependent manner. FEBS J. 2017;284(7):1096–109.
Shu Y, Qian J, Wang C. Aberrant expression of microRNA-132-3p and microRNA-146a-5p in Parkinson’s disease patients. Open Life Sci. 2020;15(1):647–53.
Simsekyilmaz S, Cabrera-Fuentes HA, Meiler S, Kostin S, Baumer Y, Liehn EA, Weber C, Boisvert WA, Preissner KT, Zernecke A. Role of extracellular RNA in atherosclerotic plaque formation in mice. Circulation. 2014;129(5):598–606.
Singh D. Astrocytic and microglial cells as the modulators of neuroinflammation in Alzheimer’s disease. J Neuroinflamm. 2022;19(1):206.
Sirois CM, Jin T, Miller AL, Bertheloot D, Nakamura H, Horvath GL, Mian A, Jiang J, Schrum J, Bossaller L, Pelka K, Garbi N, Brewah Y, Tian J, Chang C, Chowdhury PS, Sims GP, Kolbeck R, Coyle AJ, Humbles AA, Xiao TS, Latz E. RAGE is a nucleic acid receptor that promotes inflammatory responses to DNA. J Exp Med. 2013;210(11):2447–63.
Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, Burman JL, Li Y, Zhang Z, Narendra DP, Cai H, Borsche M, Klein C, Youle RJ. Parkin and PINK1 mitigate STING-induced inflammation. Nature. 2018;561(7722):258–62.
Smalheiser NR. Mining clinical case reports to identify new lines of investigation in Alzheimer’s disease: the curious case of DNase I. J Alzheimers Dis Rep. 2019;3(1):71–6.
Smyth LCD, Murray HC, Hill M, van Leeuwen E, Highet B, Magon NJ, Osanlouy M, Mathiesen SN, Mockett B, Singh-Bains MK, Morris VK, Clarkson AN, Curtis MA, Abraham WC, Hughes SM, Faull RLM, Kettle AJ, Dragunow M, Hampton MB. Neutrophil-vascular interactions drive myeloperoxidase accumulation in the brain in Alzheimer’s disease. Acta Neuropathol Commun. 2022;10(1):38.
Soltanmoradi S, Tavakolpour V, Moghadasi AN, Kouhkan F. Expression analysis of NF-kappaB-associated long noncoding RNAs in peripheral blood mononuclear cells from relapsing-remitting multiple sclerosis patients. J Neuroimmunol. 2021;356: 577602.
Soni C, Reizis B. Self-DNA at the epicenter of SLE: immunogenic forms, regulation, and effects. Front Immunol. 2019;10:1601.
Sorensen SS, Nygaard AB, Carlsen AL, Heegaard NHH, Bak M, Christensen T. Elevation of brain-enriched miRNAs in cerebrospinal fluid of patients with acute ischemic stroke. Biomark Res. 2017;5:24.
Soreq L, Salomonis N, Guffanti A, Bergman H, Israel Z, Soreq H. Whole transcriptome RNA sequencing data from blood leukocytes derived from Parkinson’s disease patients prior to and following deep brain stimulation treatment. Genom Data. 2015;3:57–60.
Stephenson J, Nutma E, van der Valk P, Amor S. Inflammation in CNS neurodegenerative diseases. Immunology. 2018;154(2):204–19.
Sternberg Z, Ostrow P, Vaughan M, Chichelli T, Munschauer F. AGE-RAGE in multiple sclerosis brain. Immunol Invest. 2011;40(2):197–205.
Stetson DB, Medzhitov R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity. 2006;24(1):93–103.
Sun D, Yu Z, Fang X, Liu M, Pu Y, Shao Q, Wang D, Zhao X, Huang A, Xiang Z, Zhao C, Franklin RJ, Cao L, He C. LncRNA GAS5 inhibits microglial M2 polarization and exacerbates demyelination. EMBO Rep. 2017;18(10):1801–16.
Sun Q, Wang S, Chen J, Cai H, Huang W, Zhang Y, Wang L, Xing Y. MicroRNA-190 alleviates neuronal damage and inhibits neuroinflammation via Nlrp3 in MPTP-induced Parkinson’s disease mouse model. J Cell Physiol. 2019;234(12):23379–87.
Sun XL, Wang ZL, Wu Q, Jin SQ, Yao J, Cheng H. LncRNA RMST activates TAK1-mediated NF-kappaB signaling and promotes activation of microglial cells via competitively binding with hnRNPK. IUBMB Life. 2019;71(11):1785–93.
Suzuki H, Tsukahara T. A view of pre-mRNA splicing from RNase R resistant RNAs. Int J Mol Sci. 2014;15(6):9331–42.
Taghizadeh E, Gheibihayat SM, Taheri F, Afshani SM, Farahani N, Saberi A. LncRNAs as putative biomarkers and therapeutic targets for Parkinson’s disease. Neurol Sci. 2021;42(10):4007–15.
Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805–20.
Tan Q, Guo P, Zhou J, Zhang J, Zhang B, Lan C, Xian J, Ge M, Feng H, Chen Z. Targeting neutrophil extracellular traps enhanced tPA fibrinolysis for experimental intracerebral hemorrhage. Transl Res. 2019;211:139–46.
Tawil SE, Muir KW. Thrombolysis and thrombectomy for acute ischaemic stroke. Clin Med. 2017;17(2):161–5.
Tetz V, Tetz G. Effect of deoxyribonuclease I treatment for dementia in end-stage Alzheimer’s disease: a case report. J Med Case Rep. 2016;10(1):131.
Thome AD, Harms AS, Volpicelli-Daley LA, Standaert DG. microRNA-155 regulates alpha-synuclein-induced inflammatory responses in models of Parkinson disease. J Neurosci. 2016;36(8):2383–90.
Thompson JW, Hu R, Huffaker TB, Ramstead AG, Ekiz HA, Bauer KM, Tang WW, Ghazaryan A, Round JL, Fujinami RS, O’Connell RM. MicroRNA-155 plays selective cell-intrinsic roles in brain-infiltrating immune cell populations during neuroinflammation. J Immunol. 2023;210(7):926–34.
Tian C, Li Z, Yang Z, Huang Q, Liu J, Hong B. Plasma microRNA-16 is a biomarker for diagnosis, stratification, and prognosis of hyperacute cerebral infarction. PLoS ONE. 2016;11(11): e0166688.
Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, Parroche P, Drabic S, Golenbock D, Sirois C, Hua J, An LL, Audoly L, La Rosa G, Bierhaus A, Naworth P, Marshak-Rothstein A, Crow MK, Fitzgerald KA, Latz E, Kiener PA, Coyle AJ. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007;8(5):487–96.
Tian J, Liu Y, Wang Z, Zhang S, Yang Y, Zhu Y, Yang C. LncRNA Snhg8 attenuates microglial inflammation response and blood-brain barrier damage in ischemic stroke through regulating miR-425-5p mediated SIRT1/NF-kappaB signaling. J Biochem Mol Toxicol. 2021;35(5): e22724.
Tielking K, Fischer S, Preissner KT, Vajkoczy P, Xu R. Extracellular RNA in central nervous system pathologies. Front Mol Neurosci. 2019;12:254.
Tillack K, Naegele M, Haueis C, Schippling S, Wandinger KP, Martin R, Sospedra M. Gender differences in circulating levels of neutrophil extracellular traps in serum of multiple sclerosis patients. J Neuroimmunol. 2013;261(1–2):108–19.
Tripathi S, Shree B, Mohapatra S, Swati, Basu A, Sharma V. The expanding regulatory mechanisms and cellular functions of long non-coding RNAs (lncRNAs) in neuroinflammation. Mol Neurobiol. 2021;58(6):2916–39.
Tsai NW, Lin TK, Chen SD, Chang WN, Wang HC, Yang TM, Lin YJ, Jan CR, Huang CR, Liou CW, Lu CH. The value of serial plasma nuclear and mitochondrial DNA levels in patients with acute ischemic stroke. Clin Chim Acta. 2011;412(5–6):476–9.
Tsang JC, Lo YM. Circulating nucleic acids in plasma/serum. Pathology. 2007;39(2):197–207.
Tsao CW, Aday AW, Almarzooq ZI, Alonso A, Beaton AZ, Bittencourt MS, Boehme AK, Buxton AE, Carson AP, Commodore-Mensah Y, Elkind MSV, Evenson KR, Eze-Nliam C, Ferguson JF, Generoso G, Ho JE, Kalani R, Khan SS, Kissela BM, Knutson KL, Levine DA, Lewis TT, Liu J, Loop MS, Ma J, Mussolino ME, Navaneethan SD, Perak AM, Poudel R, Rezk-Hanna M, Roth GA, Schroeder EB, Shah SH, Thacker EL, VanWagner LB, Virani SS, Voecks JH, Wang NY, Yaffe K, Martin SS. Heart disease and stroke statistics-2022 update: a report from the American Heart Association. Circulation. 2022;145(8):e153–639.
Tumburu L, Ghosh-Choudhary S, Seifuddin FT, Barbu EA, Yang S, Ahmad MM, Wilkins LHW, Tunc I, Sivakumar I, Nichols JS, Dagur PK, Yang S, Almeida LEF, Quezado ZMN, Combs CA, Lindberg E, Bleck CKE, Zhu J, Shet AS, Chung JH, Pirooznia M, Thein SL. Circulating mitochondrial DNA is a proinflammatory DAMP in sickle cell disease. Blood. 2021;137(22):3116–26.
Vaibhav K, Braun M, Alverson K, Khodadadi H, Kutiyanawalla A, Ward A, Banerjee C, Sparks T, Malik A, Rashid MH, Khan MB, Waters MF, Hess DC, Arbab AS, Vender JR, Hoda N, Baban B, Dhandapani KM. Neutrophil extracellular traps exacerbate neurological deficits after traumatic brain injury. Sci Adv. 2020;6(22): eaax8847.
Vaillant-Beuchot L, Mary A, Pardossi-Piquard R, Bourgeois A, Lauritzen I, Eysert F, Kinoshita PF, Cazareth J, Badot C, Fragaki K, Bussiere R, Martin C, Mary R, Bauer C, Pagnotta S, Paquis-Flucklinger V, Buee-Scherrer V, Buee L, Lacas-Gervais S, Checler F, Chami M. Accumulation of amyloid precursor protein C-terminal fragments triggers mitochondrial structure, function, and mitophagy defects in Alzheimer’s disease models and human brains. Acta Neuropathol. 2021;141(1):39–65.
Vajpeyee A, Wijatmiko T, Vajpeyee M, Taywade O. Cell free DNA: a novel predictor of neurological outcome after intravenous thrombolysis and/or mechanical thrombectomy in acute ischemic stroke patients. Neurointervention. 2018;13(1):13–9.
Vajpeyee A, Wijatmiko T, Vajpeyee M, Taywade O, Pandey S, Chauhan PS. Clinical usefulness of cell-free DNA as a prognostic marker in acute ischemic stroke. Neurologist. 2020;25(1):11–3.
Varhaug KN, Vedeler CA, Myhr KM, Aarseth JH, Tzoulis C, Bindoff LA. Increased levels of cell-free mitochondrial DNA in the cerebrospinal fluid of patients with multiple sclerosis. Mitochondrion. 2017;34:32–5.
Vasudeva K, Dutta A, Munshi A. Role of lncRNAs in the development of ischemic stroke and their therapeutic potential. Mol Neurobiol. 2021;58(8):3712–28.
Vickers KC, Palmisano BT, Shoucri BM, Shamburek RD, Remaley AT. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat Cell Biol. 2011;13(4):423–33.
Vlassov VV, Laktionov PP, Rykova EY. Extracellular nucleic acids. BioEssays. 2007;29(7):654–67.
Voet S, Srinivasan S, Lamkanfi M, van Loo G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol Med. 2019;11(6): e10248.
Volkman R, Ben-Zur T, Kahana A, Garty BZ, Offen D. Myeloperoxidase deficiency inhibits cognitive decline in the 5xfad mouse model of Alzheimer’s disease. Front Neurosci. 2019;13:990.
von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, Khandoga A, Tirniceriu A, Coletti R, Kollnberger M, Byrne RA, Laitinen I, Walch A, Brill A, Pfeiler S, Manukyan D, Braun S, Lange P, Riegger J, Ware J, Eckart A, Haidari S, Rudelius M, Schulz C, Echtler K, Brinkmann V, Schwaiger M, Preissner KT, Wagner DD, Mackman N, Engelmann B, Massberg S. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209(4):819–35.
Walberer M, Tschernatsch M, Fischer S, Ritschel N, Volk K, Friedrich C, Bachmann G, Mueller C, Kaps M, Nedelmann M, Blaes F, Preissner KT, Gerriets T. RNase therapy assessed by magnetic resonance imaging reduces cerebral edema and infarction size in acute stroke. Curr Neurovasc Res. 2009;6(1):12–9.
Walker DG, Tang TM, Lue LF. Increased expression of toll-like receptor 3, an anti-viral signaling molecule, and related genes in Alzheimer’s disease brains. Exp Neurol. 2018;309:91–106.
Walko TD 3rd, Bola RA, Hong JD, Au AK, Bell MJ, Kochanek PM, Clark RS, Aneja RK. Cerebrospinal fluid mitochondrial DNA: a novel DAMP in pediatric traumatic brain injury. Shock. 2014;41(6):499–503.
Wang G, Zhou Y, Zhong T, Song A, Xue Q. The role of blood lnc-ZFAS1 in acute ischemic stroke: correlation with neurological impairment, inflammation, and survival profiles. J Clin Lab Anal. 2022;36(2): e24219.
Wang H, Liao S, Li H, Chen Y, Yu J. Long Non-coding RNA TUG1 sponges Mir-145a-5p to regulate microglial polarization after oxygen-glucose deprivation. Front Mol Neurosci. 2019;12:215.
Wang J, Chen C, Zhang Y. An investigation of microRNA-103 and microRNA-107 as potential blood-based biomarkers for disease risk and progression of Alzheimer’s disease. J Clin Lab Anal. 2020;34(1): e23006.
Wang J, Zhao H, Fan Z, Li G, Ma Q, Tao Z, Wang R, Feng J, Luo Y. Long Noncoding RNA H19 promotes neuroinflammation in ischemic stroke by driving histone deacetylase 1-dependent M1 microglial polarization. Stroke. 2017;48(8):2211–21.
Wang M, Chen W, Geng Y, Xu C, Tao X, Zhang Y. Long non-coding RNA MEG3 promotes apoptosis of vascular cells and is associated with poor prognosis in ischemic stroke. J Atheroscler Thromb. 2020;27(7):718–26.
Wang Q, Han CL, Wang KL, Sui YP, Li ZB, Chen N, Fan SY, Shimabukuro M, Wang F, Meng FG. Integrated analysis of exosomal lncRNA and mRNA expression profiles reveals the involvement of lnc-MKRN2-42:1 in the pathogenesis of Parkinson’s disease. CNS Neurosci Ther. 2020;26(5):527–37.
Wang R, Zhang J. Clinical significance of miR-433 in the diagnosis of Alzheimer’s disease and its effect on Abeta-induced neurotoxicity by regulating JAK2. Exp Gerontol. 2020;141: 111080.
Wang R, Zhu Y, Liu Z, Chang L, Bai X, Kang L, Cao Y, Yang X, Yu H, Shi MJ, Hu Y, Fan W, Zhao BQ. Neutrophil extracellular traps promote tPA-induced brain hemorrhage via cGAS in mice with stroke. Blood. 2021;138(1):91–103.
Wang X, Chen S, Ni J, Cheng J, Jia J, Zhen X. miRNA-3473b contributes to neuroinflammation following cerebral ischemia. Cell Death Dis. 2018;9(1):11.
Wang Y, Huang J, Ma Y, Tang G, Liu Y, Chen X, Zhang Z, Zeng L, Wang Y, Ouyang YB, Yang GY. MicroRNA-29b is a therapeutic target in cerebral ischemia associated with aquaporin 4. J Cereb Blood Flow Metab. 2015;35(12):1977–84.
Wang Y, Liu C, Chen Y, Chen T, Han T, Xue L, Xu B. Systemically silencing long non-coding RNAs maclpil with short interfering RNA nanoparticles alleviates experimental ischemic stroke by promoting macrophage apoptosis and anti-inflammatory activation. Front Cardiovasc Med. 2022;9: 876087.
Wang Y, Luo Y, Yao Y, Ji Y, Feng L, Du F, Zheng X, Tao T, Zhai X, Li Y, Han P, Xu B, Zhao H. Silencing the lncRNA Maclpil in pro-inflammatory macrophages attenuates acute experimental ischemic stroke via LCP1 in mice. J Cereb Blood Flow Metab. 2020;40(4):747–59.
Wasner K, Smajic S, Ghelfi J, Delcambre S, Prada-Medina CA, Knappe E, Arena G, Mulica P, Agyeah G, Rakovic A, Boussaad I, Badanjak K, Ohnmacht J, Gerardy JJ, Takanashi M, Trinh J, Mittelbronn M, Hattori N, Klein C, Antony P, Seibler P, Spielmann M, Pereira SL, Grunewald A. Parkin deficiency impairs mitochondrial DNA dynamics and propagates inflammation. Mov Disord. 2022;37:1405.
Watanabe T, Takada S, Mizuta R. Cell-free DNA in blood circulation is generated by DNase1L3 and caspase-activated DNase. Biochem Biophys Res Commun. 2019;516(3):790–5.
Wen Y, Yu Y, Fu X. LncRNA Gm4419 contributes to OGD/R injury of cerebral microglial cells via IkappaB phosphorylation and NF-kappaB activation. Biochem Biophys Res Commun. 2017;487(4):923–9.
Wen Y, Zhang X, Dong L, Zhao J, Zhang C, Zhu C. Acetylbritannilactone modulates microRNA-155-mediated inflammatory response in ischemic cerebral tissues. Mol Med. 2015;21:197–209.
West AP, Shadel GS. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol. 2017;17(6):363–75.
Wieczorek AJ, Rhyner C, Block LH. Isolation and characterization of an RNA-proteolipid complex associated with the malignant state in humans. Proc Natl Acad Sci U S A. 1985;82(10):3455–9.
Wilhelm I, Nyul-Toth A, Kozma M, Farkas AE, Krizbai IA. Role of pattern recognition receptors of the neurovascular unit in inflamm-aging. Am J Physiol Heart Circ Physiol. 2017;313(5):H1000–12.
Wilkins HM, Carl SM, Weber SG, Ramanujan SA, Festoff BW, Linseman DA, Swerdlow RH. Mitochondrial lysates induce inflammation and Alzheimer’s disease-relevant changes in microglial and neuronal cells. J Alzheimers Dis. 2015;45(1):305–18.
Wilkins HM, Koppel SJ, Weidling IW, Roy N, Ryan LN, Stanford JA, Swerdlow RH. Extracellular mitochondria and mitochondrial components act as damage-associated molecular pattern molecules in the mouse brain. J Neuroimmune Pharmacol. 2016;11(4):622–8.
Wolska M, Jarosz-Popek J, Junger E, Wicik Z, Porshoor T, Sharif L, Czajka P, Postula M, Mirowska-Guzel D, Czlonkowska A, Eyileten C. Long non-coding RNAs as promising therapeutic approach in ischemic stroke: a comprehensive review. Mol Neurobiol. 2021;58(4):1664–82.
Wong SL, Wagner DD. Peptidylarginine deiminase 4: a nuclear button triggering neutrophil extracellular traps in inflammatory diseases and aging. FASEB J. 2018;32:6358.
Wu H, Zheng J, Xu S, Fang Y, Wu Y, Zeng J, Shao A, Shi L, Lu J, Mei S, Wang X, Guo X, Wang Y, Zhao Z, Zhang J. Mer regulates microglial/macrophage M1/M2 polarization and alleviates neuroinflammation following traumatic brain injury. J Neuroinflamm. 2021;18(1):2.
Wu J, Fan CL, Ma LJ, Liu T, Wang C, Song JX, Lv QS, Pan H, Zhang CN, Wang JJ. Distinctive expression signatures of serum microRNAs in ischaemic stroke and transient ischaemic attack patients. Thromb Haemost. 2017;117(5):992–1001.
Wu L, Xu Q, Zhou M, Chen Y, Jiang C, Jiang Y, Lin Y, He Q, Zhao L, Dong Y, Liu J, Chen W. Plasma miR-153 and miR-223 levels as potential biomarkers in Parkinson’s disease. Front Neurosci. 2022;16: 865139.
Wu X, Zeng H, Cai L, Chen G. Role of the extracellular traps in central nervous system. Front Immunol. 2021;12: 783882.
Xiao Y, Chen H, Liao J, Zhang Q, He H, Lei J, Huang J, Ouyang Q, Shen Y, Wang J. The potential circular RNAs biomarker panel and regulatory networks of Parkinson’s disease. Front Neurosci. 2022;16: 893713.
Xie C, Zhu B, Gu J, Sun M. The correlation of lncRNA SNHG16 with inflammatory cytokines, adhesion molecules, disease severity, and prognosis in acute ischemic stroke patients. J Clin Lab Anal. 2022;36(6): e24439.
Xie F, Tan Q, Yu A, Guo P, Wang L, Zeng Z, Liang L, Xian J, Feng H, Chen Z. The role of cell-free DNA in fibrinolysis for intraventricular hemorrhage. J Neurosurg. 2021;135:1105.
Xin C, Liu J. Long non-coding RNAs in Parkinson’s disease. Neurochem Res. 2021;46(5):1031–42.
Xing YH, Chen LL. Processing and roles of snoRNA-ended long noncoding RNAs. Crit Rev Biochem Mol Biol. 2018;53(6):596–606.
Xiong W, Li D, Feng Y, Jia C, Zhang X, Liu Z. CircLPAR1 promotes neuroinflammation and oxidative stress in APP/PS1 mice by inhibiting SIRT1/Nrf-2/HO-1 axis through destabilizing GDF-15 mRNA. Mol Neurobiol. 2023;60(4):2236–51.
Xu W, Zhang L, Geng Y, Liu Y, Zhang N. Long noncoding RNA GAS5 promotes microglial inflammatory response in Parkinson’s disease by regulating NLRP3 pathway through sponging miR-223-3p. Int Immunopharmacol. 2020;85: 106614.
Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301(5633):640–3.
Yamamoto M, Sato S, Mori K, Hoshino K, Takeuchi O, Takeda K, Akira S. Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the Toll-like receptor signaling. J Immunol. 2002;169(12):6668–72.
Yan JH, Hua P, Chen Y, Li LT, Yu CY, Yan L, Zhang H, He Y, Zheng H, Chen H, Zhang ZJ, Yao QH, Dong H, Liu WG. Identification of microRNAs for the early diagnosis of Parkinson’s disease and multiple system atrophy. J Integr Neurosci. 2020;19(3):429–36.
Yanai H, Ban T, Wang Z, Choi MK, Kawamura T, Negishi H, Nakasato M, Lu Y, Hangai S, Koshiba R, Savitsky D, Ronfani L, Akira S, Bianchi ME, Honda K, Tamura T, Kodama T, Taniguchi T. HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature. 2009;462(7269):99–103.
Yang H. LncRNA MALAT1 potentiates inflammation disorder in Parkinson’s disease. Int J Immunogenet. 2021;48(5):419–28.
Yang P, Lin G, Wang M, Chen X, Huang J. Long non-coding RNA ANRIL interacts with microRNA-34a and microRNA-125a, and they all correlate with disease risk and severity of Parkinson’s disease. J Clin Lab Anal. 2022;36(1): e24037.
Yang Q, Zhao Q, Yin Y. miR-133b is a potential diagnostic biomarker for Alzheimer’s disease and has a neuroprotective role. Exp Ther Med. 2019;18(4):2711–8.
Yang R, Xu B, Yang B, Fu J, Chen H, Wang X. Non-coding RNAs: the extensive and interactive regulators of the blood-brain barrier permeability. RNA Biol. 2021;18(sup1):108–16.
Yang Y, Li Y, Yang H, Guo J, Li N. Circulating microRNAs and long non-coding RNAs as potential diagnostic biomarkers for Parkinson’s disease. Front Mol Neurosci. 2021;14: 631553.
Yang Z, Li T, Li S, Wei M, Qi H, Shen B, Chang RC, Le W, Piao F. Altered expression levels of microRNA-132 and Nurr1 in peripheral blood of Parkinson’s disease: potential disease biomarkers. ACS Chem Neurosci. 2019;10(5):2243–9.
Ye Y, He X, Lu F, Mao H, Zhu Z, Yao L, Luo W, Sun X, Wang B, Qian C, Zhang Y, Lu G, Zhang S. A lincRNA-p21/miR-181 family feedback loop regulates microglial activation during systemic LPS- and MPTP- induced neuroinflammation. Cell Death Dis. 2018;9(8):803.
Yi J, Chen B, Yao X, Lei Y, Ou F, Huang F. Upregulation of the lncRNA MEG3 improves cognitive impairment, alleviates neuronal damage, and inhibits activation of astrocytes in hippocampus tissues in Alzheimer’s disease through inactivating the PI3K/Akt signaling pathway. J Cell Biochem. 2019;120(10):18053–65.
Yi X, Wang Y, Yu FS. Corneal epithelial tight junctions and their response to lipopolysaccharide challenge. Invest Ophthalmol Vis Sci. 2000;41(13):4093–100.
Yoon JH, Abdelmohsen K, Gorospe M. Posttranscriptional gene regulation by long noncoding RNA. J Mol Biol. 2013;425(19):3723–30.
Yost CC, Schwertz H, Cody MJ, Wallace JA, Campbell RA, Vieira-de-Abreu A, Araujo CV, Schubert S, Harris ES, Rowley JW, Rondina MT, Fulcher JM, Koening CL, Weyrich AS, Zimmerman GA. Neonatal NET-inhibitory factor and related peptides inhibit neutrophil extracellular trap formation. J Clin Invest. 2016;126(10):3783–98.
Yousuf A, Qurashi A. Non-coding RNAs in the pathogenesis of multiple sclerosis. Front Genet. 2021;12: 717922.
Yu G, Zheng S, Zhang H. Inhibition of myeloperoxidase by N-acetyl lysyltyrosylcysteine amide reduces experimental autoimmune encephalomyelitis-induced injury and promotes oligodendrocyte regeneration and neurogenesis in a murine model of progressive multiple sclerosis. NeuroReport. 2018;29(3):208–13.
Yu L, Wang L, Chen S. Endogenous toll-like receptor ligands and their biological significance. J Cell Mol Med. 2010;14(11):2592–603.
Zang Y, Zhou X, Wang Q, Li X, Huang H. LncRNA FIRRE/NF-kB feedback loop contributes to OGD/R injury of cerebral microglial cells. Biochem Biophys Res Commun. 2018;501(1):131–8.
Zenaro E, Pietronigro E, Della BV, Piacentino G, Marongiu L, Budui S, Turano E, Rossi B, Angiari S, Dusi S, Montresor A, Carlucci T, Nani S, Tosadori G, Calciano L, Catalucci D, Berton G, Bonetti B, Constantin G. Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat Med. 2015;21(8):880–6.
Zeng H, Fu X, Cai J, Sun C, Yu M, Peng Y, Zhuang J, Chen J, Chen H, Yu Q, Xu C, Zhou H, Cao Y, Hu L, Li J, Cao S, Gu C, Yan F, Chen G. Neutrophil extracellular traps may be a potential target for treating early brain injury in subarachnoid hemorrhage. Transl Stroke Res. 2022;13(1):112–31.
Zeng L, Liu J, Wang Y, Wang L, Weng S, Tang Y, Zheng C, Cheng Q, Chen S, Yang GY. MicroRNA-210 as a novel blood biomarker in acute cerebral ischemia. Front Biosci. 2011;3(4):1265–72.
Zernecke A, Preissner KT. Extracellular ribonucleic acids (RNA) enter the stage in cardiovascular disease. Circ Res. 2016;118(3):469–79.
Zha C, Meng X, Li L, Mi S, Qian D, Li Z, Wu P, Hu S, Zhao S, Cai J, Liu Y. Neutrophil extracellular traps mediate the crosstalk between glioma progression and the tumor microenvironment via the HMGB1/RAGE/IL-8 axis. Cancer Biol Med. 2020;17(1):154–68.
Zhang C, Li B. The correlation between LncRNA-17A expression in peripheral blood mononuclear cells and Wnt/beta-catenin signaling pathway and cognitive function in patients with Alzheimer disease. Am J Transl Res. 2021;13(10):11981–6.
Zhang G, Wang Q, Su D, Xie Y. Long non-coding RNAMALAT1 knockdown alleviates cerebral ischemia/reperfusion injury of rats through regulating the miR-375/PDE4D axis. Front Neurol. 2020;11: 578765.
Zhang H, Ray A, Miller NM, Hartwig D, Pritchard KA, Dittel BN. Inhibition of myeloperoxidase at the peak of experimental autoimmune encephalomyelitis restores blood-brain barrier integrity and ameliorates disease severity. J Neurochem. 2016;136(4):826–36.
Zhang J, Dongwei Z, Zhang Z, Xinhui Q, Kunwang B, Guohui L, Jian D. miR-let-7a suppresses alpha-Synuclein-induced microglia inflammation through targeting STAT3 in Parkinson’s disease. Biochem Biophys Res Commun. 2019;519(4):740–6.
Zhang J, Wang R. Deregulated lncRNA MAGI2-AS3 in Alzheimer’s disease attenuates amyloid-beta induced neurotoxicity and neuroinflammation by sponging miR-374b-5p. Exp Gerontol. 2021;144: 111180.
Zhang JS, Hou PP, Shao S, Manaenko A, Xiao ZP, Chen Y, Zhao B, Jia F, Zhang XH, Mei QY, Hu Q. microRNA-455-5p alleviates neuroinflammation in cerebral ischemia/reperfusion injury. Neural Regen Res. 2022;17(8):1769–75.
Zhang L-W, Zhang J, Wang K, Wang R-B. Serum microRNA-30c-5p and microRNA-373 expressions as potential biomarkers for Parkinson’s disease. All Life. 2020;13(1):194–200.
Zhang LJ, Sen GL, Ward NL, Johnston A, Chun K, Chen Y, Adase C, Sanford JA, Gao N, Chensee M, Sato E, Fritz Y, Baliwag J, Williams MR, Hata T, Gallo RL. Antimicrobial peptide LL37 and MAVS signaling drive interferon-beta production by epidermal keratinocytes during skin injury. Immunity. 2016;45(1):119–30.
Zhang M, Bian Z. The emerging role of circular RNAs in Alzheimer’s disease and Parkinson’s disease. Front Aging Neurosci. 2021;13: 691512.
Zhang M, Han W, Xu Y, Li D, Xue Q. Serum miR-128 serves as a potential diagnostic biomarker for Alzheimer’s disease. Neuropsychiatr Dis Treat. 2021;17:269–75.
Zhang N, Gao Y, Yu S, Sun X, Shen K. Berberine attenuates Abeta42-induced neuronal damage through regulating circHDAC9/miR-142-5p axis in human neuronal cells. Life Sci. 2020;252: 117637.
Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464(7285):104–7.
Zhang S, Cao Y, Du J, Liu H, Chen X, Li M, Xiang M, Wang C, Wu X, Liu L, Wang C, Wu Y, Li Z, Fang S, Shi J, Wang L. Neutrophil extracellular traps contribute to tissue plasminogen activator resistance in acute ischemic stroke. FASEB J. 2021;35(9): e21835.
Zhang S, Sun WC, Liang ZD, Yin XR, Ji ZR, Chen XH, Wei MJ, Pei L. LncRNA SNHG4 attenuates inflammatory responses by sponging miR-449c-5p and up-regulating STAT6 in microglial during cerebral ischemia-reperfusion injury. Drug Des Devel Ther. 2020;14:3683–95.
Zhang W, Li G, Luo R, Lei J, Song Y, Wang B, Ma L, Liao Z, Ke W, Liu H, Hua W, Zhao K, Feng X, Wu X, Zhang Y, Wang K, Yang C. Cytosolic escape of mitochondrial DNA triggers cGAS-STING-NLRP3 axis-dependent nucleus pulposus cell pyroptosis. Exp Mol Med. 2022;54(2):129–42.
Zhang X, Zhu XL, Ji BY, Cao X, Yu LJ, Zhang Y, Bao XY, Xu Y, Jin JL. LncRNA-1810034E14Rik reduces microglia activation in experimental ischemic stroke. J Neuroinflamm. 2019;16(1):75.
Zhang Y, Han JJ, Liang XY, Zhao L, Zhang F, Rasouli J, Wang ZZ, Zhang GX, Li X. miR-23b suppresses leukocyte migration and pathogenesis of experimental autoimmune encephalomyelitis by targeting CCL7. Mol Ther. 2018;26(2):582–92.
Zhang Y, Niu C. The correlation of long non-coding RNA intersectin 1–2 with disease risk, disease severity, inflammation, and prognosis of acute ischemic stroke. J Clin Lab Anal. 2020;34(2): e23053.
Zhang Y, Zhao Y, Ao X, Yu W, Zhang L, Wang Y, Chang W. The role of non-coding RNAs in Alzheimer’s disease: from regulated mechanism to therapeutic targets and diagnostic biomarkers. Front Aging Neurosci. 2021;13: 654978.
Zhang Z, Xue Z, Liu Y, Liu H, Guo X, Li Y, Yang H, Zhang L, Da Y, Yao Z, Zhang R. MicroRNA-181c promotes Th17 cell differentiation and mediates experimental autoimmune encephalomyelitis. Brain Behav Immun. 2018;70:305–14.
Zhao H, Peng R, Liu Q, Liu D, Du P, Yuan J, Peng G, Liao Y. The lncRNA H19 interacts with miR-140 to modulate glioma growth by targeting iASPP. Arch Biochem Biophys. 2016;610:1–7.
Zhao Y, Alexandrov PN, Jaber V, Lukiw WJ. Deficiency in the ubiquitin conjugating enzyme UBE2A in Alzheimer’s Disease (AD) is linked to deficits in a natural circular miRNA-7 Sponge (circRNA; ciRS-7). Genes. 2016;7(12):116.
Zhao Y, Yang G. Potential of extracellular vesicles in the Parkinson’s disease - pathological mediators and biomarkers. Neurochem Int. 2021;144: 104974.
Zheng L, Xiong Y, Liu J, Yang X, Wang L, Zhang S, Liu M, Wang D. MMP-9-related microRNAs as prognostic markers for hemorrhagic transformation in cardioembolic stroke patients. Front Neurol. 2019;10:945.
Zhong L, Ju K, Chen A, Cao H. Circulating circRNAs panel acts as a biomarker for the early diagnosis and severity of Parkinson’s disease. Front Aging Neurosci. 2021;13: 684289.
Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, Wong J, Ding S, Seki E, Schnabl B, Hevener AL, Greenberg HB, Kisseleva T, Karin M. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature. 2018;560(7717):198–203.
Zhou J, Guo P, Hao X, Sun X, Feng H, Chen Z. Neutrophil extracellular traps (NETs): a new therapeutic target for neuroinflammation and microthrombosis after subarachnoid hemorrhage? Transl Stroke Res. 2022;14:443.
Zhou S, Yu X, Wang M, Meng Y, Song D, Yang H, Wang D, Bi J, Xu S. Long non-coding RNAs in pathogenesis of neurodegenerative diseases. Front Cell Dev Biol. 2021;9: 719247.
Zhou Y, Liu Y, Kang Z, Yao H, Song N, Wang M, Song C, Zhang K, Ding J, Tang J, Hu G, Lu M. CircEPS15, as a sponge of MIR24–3p ameliorates neuronal damage in Parkinson disease through boosting PINK1-PRKN-mediated mitophagy. Autophagy. 2023. https://doi.org/10.1080/15548627.2023.2196889.
Zhou Z, Wu Q, Yan Z, Zheng H, Chen CJ, Liu Y, Qi Z, Calandrelli R, Chen Z, Chien S, Su HI, Zhong S. Extracellular RNA in a single droplet of human serum reflects physiologic and disease states. Proc Natl Acad Sci U S A. 2019;116(38):19200–8.
Zhu M, Li N, Luo P, Jing W, Wen X, Liang C, Tu J. Peripheral blood leukocyte expression of lncRNA MIAT and its diagnostic and prognostic value in ischemic stroke. J Stroke Cerebrovasc Dis. 2018;27(2):326–37.
Zhu Z, Huang P, Sun R, Li X, Li W, Gong W. A novel long-noncoding RNA LncZFAS1 prevents MPP(+)-induced neuroinflammation through MIB1 activation. Mol Neurobiol. 2022;59(2):778–99.
Zhuang J, Cai P, Chen Z, Yang Q, Chen X, Wang X, Zhuang X. Long noncoding RNA MALAT1 and its target microRNA-125b are potential biomarkers for Alzheimer’s disease management via interactions with FOXQ1, PTGS2 and CDK5. Am J Transl Res. 2020;12(9):5940–54.
Ziegler G, Harhausen D, Schepers C, Hoffmann O, Rohr C, Prinz V, Konig J, Lehrach H, Nietfeld W, Trendelenburg G. TLR2 has a detrimental role in mouse transient focal cerebral ischemia. Biochem Biophys Res Commun. 2007;359(3):574–9.
Zindel J, Kubes P. DAMPs, PAMPs, and LAMPs in immunity and sterile inflammation. Annu Rev Pathol. 2020;15:493–518.
Zingale VD, Gugliandolo A, Mazzon E. MiR-155: an important regulator of neuroinflammation. Int J Mol Sci. 2021;23(1):90.
Zou J, Guo Y, Wei L, Yu F, Yu B, Xu A. Long noncoding RNA POU3F3 and alpha-synuclein in plasma L1CAM exosomes combined with beta-glucocerebrosidase activity: potential predictors of Parkinson’s disease. Neurotherapeutics. 2020;17(3):1104–19.
Zuo L, Xie J, Liu Y, Leng S, Zhang Z, Yan F. Down-regulation of circular RNA CDC14A peripherally ameliorates brain injury in acute phase of ischemic stroke. J Neuroinflamm. 2021;18(1):283.
Zuo L, Zhang L, Zu J, Wang Z, Han B, Chen B, Cheng M, Ju M, Li M, Shu G, Yuan M, Jiang W, Chen X, Yan F, Zhang Z, Yao H. Circulating circular RNAs as biomarkers for the diagnosis and prediction of outcomes in acute ischemic stroke. Stroke. 2020;51(1):319–23.
Zurawska A, Mycko MP, Selmaj KW. Circular RNAs as a novel layer of regulatory mechanism in multiple sclerosis. J Neuroimmunol. 2019;334: 576971.
Zurawska AE, Mycko MP, Selmaj I, Raine CS, Selmaj KW. Multiple sclerosis: circRNA profile defined reveals links to B-cell function. Neurol Neuroimmunol Neuroinflamm. 2021;8(5): e1041.
Acknowledgements
Not applicable.
Funding
Work in authors' laboratories related to this topic has been supported by Grants from the German Research Council/Deutsche Forschungsgemeinschaft (DFG, Bonn, Germany; Grant No. KU 3791/4-1 to RK and Grant No. FI 1543/4-1 to SF) and the von-Behring-Röntgen Foundation (Marburg, Germany; Grant No. 65-0021 to SF).
Author information
Authors and Affiliations
Contributions
RK, SF, HHM, and KTP conceptualized, wrote and edited the manuscript. RK and SF prepared the figure and all tables. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kunze, R., Fischer, S., Marti, H.H. et al. Brain alarm by self-extracellular nucleic acids: from neuroinflammation to neurodegeneration. J Biomed Sci 30, 64 (2023). https://doi.org/10.1186/s12929-023-00954-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12929-023-00954-y