
Abstract
Background
Hearing loss (HL) is the most frequent sensory deficit in humans, HL has strong genetic heterogeneity. The genetic diagnosis of HL is very important to aid treatment decisions and to provide prognostic information and genetic counseling for the patient’s family.
Methods
We undertook pedigree analysis in 92 Chinese non-syndromic HL patients by targeted next-generation sequencing and Sanger sequencing.
Results
Among the 92 HL patients, 18 were assigned a molecular diagnosis with 33 different variants in 14 deafness genes. Eighteen of the variants in 12 deafness genes were novel. Variants in TMC1, CDH23, LOXHD1 and USH2A were each detected in two probands, and variants in POU3F4, OTOA, GPR98, GJB6, TRIOBP, SLC26A4, MYO15A, TNC, STRC and TMPRSS3 were each detected in one proband.
Conclusion
Our findings expand the spectrum of deafness gene variation, which will inform genetic diagnosis of deafness and add to the theoretical basis for the prevention of deafness.
Similar content being viewed by others


Background
Hearing loss (HL) is the most frequent sensory deficit in humans, with a prevalence of approximately 1/1000 in newborns [1, 2]. Hearing loss in approximately 50% to 60% of individuals is caused by genetic factors [3]. Among these, approximately 70% are non-syndromic HL (NSHL), in which the hearing impairment is the only distinctive clinical feature, while 30% of HL patients are syndromic with other abnormalities [4]. NSHL also has strong genetic heterogeneity.
The genetic diagnosis of NSHL is very important to aid treatment decisions and to provide prognostic information and genetic counseling for the patient’s family [5, 6]. The genetic mode of NSHL inheritance can be autosomal recessive, autosomal dominant, mitochondrial, or X/Y-linked. The development of molecular diagnostic technology has greatly reduced the cost of testing, and next-generation sequencing (NGS) has become an effective way of providing comprehensive and efficient diagnosis for NSHL [7]. To date, 224 genes have been reported to be associated with hearing loss (https://morl.lab.uiowa.edu/genes-included-otoscope-v9). Sixty-six are autosomal dominant, 117 are autosomal recessive, 21 are autosomal dominant/autosomal recessive, 9 are mitochondrial, and 5 are X-linked. However, most of the variations in these genes are rare and have only been reported in one or a few families [8].
Molecular epidemiological studies have found that the three common deafness genes GJB2, SLC26A4, and mtDNA 12S rRNA accounted for 30–50% of congenital HL [9]. In China, nine variants in four genes are the most common causes of NSHL, including c.235delC (18.3%), c.299_300delAT (5.6%), c.176del16 (1.8%) and c.35delG (0.14%) of GJB2; c.919-2A>G (15.4%) and c.2168A>G (1.08%) of SLC26A4; m.1555A>G (1.76%) and m.1494C>T (0.16%) of mtDNA 12S rRNA; c.538C>T (0.41%) of GJB3 [10,11,12]. A large neonatal cohort study in Beijing, China, showed that the heterozygous carrier rate of GJB2 gene was 2.3%, the SLC26A4 was 1.6%, the mtDNA 12S rRNA was 0.2% and the GJB3 was 0.3% [12].
Here, we recruited 92 Chinese Han NSHL families, who were confirmed not to carry the common HL variants in GJB2, SLC26A4 and MT-RNR1. Targeted NGS for known deafness genes was performed on the probands of each family to search for the genetic etiology of HL.
Methods
Recruitment of patients
92 patients with non-syndromic deafness were clinically diagnosed with bilateral sensorineural hearing loss at the Chinese People's Liberation Army (PLA) General Hospital (Beijing). Audiological tests were performed in the hearing center of the Chinese PLA General Hospital. Tests included pure-tone audiometry (or behavioral audiometry) for patients > 4 years old and multiple-frequency auditory steady-state evoked response (ASSR) tests for patients ≤ 4 years old [13]. All the probands were from non-consanguineous families. They were aged from 6 months to 54 years, and the age of onset ranged from birth to 22 years (Table 2).
Genomic DNA preparation
Blood samples (1–2 mL) were collected from the probands and their parents. Genomic DNA was extracted using a Tiangen DNA extraction kit (Tiangen Biotech, Beijing, China) according to the manufacturer’s instructions and quantified spectrophotometrically by NanoDrop 2000 manufacturer (ThermoScientific, USA).
Targeted -NGS and Sanger sequencing
Targeted capture of candidate disease genes (Table 1) was performed using the GenCap™ Custom Enrichment kit (MyGenostics, Beijing, China). Data analysis and bioinformatics analysis were performed according to method described by previous study [6]. Candidate variants were confirmed in the proband’s parents in each family by Sanger sequencing. The PCR products were bi-directionally sequenced using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, USA) on an ABI 3500DX Genetic Analyzer (Applied Biosystems, USA) after purification of the products in 2% agarose gels by using a Tiangen Midi Purification kit (Tiangen Biotech, Beijing, China).
Bioinformatics analysis
Variants are described according to the nomenclature recommended by the Human Genome Variation Society (www.hgvs.org/). Variants were annotated using ANNOVAR (https://annovar.openbioinformatics.org/en/) and filtered according to their predicted effects and allele frequencies in the public database, gnomAD (http://gnomad.broadinstitute.org/). Novel variants were checked in the Human Gene Variant Database (HGMD; www.hgmd.cf.ac.uk/), ClinVar database (www.ncbi. nlm.nih.gov/clinvar/) and gnomAD database. We use PolyPhen2(Polymorphism Phenotyping, http://genetics.bwh.harvard.edu/pph2) and PROVEAN (http://provean.jcvi.org/index.php) tools to assess the possible functional role of the novel variant. The conservativeness of the novel site is evaluated on the UCSC website (https://genome.ucsc.edu/). InterVar (http://wintervar.wglab.org/) was used to evaluate the pathogenicity of all variants according to the standards and guidelines of the American College of Medical Genetics and Genomics (ACMG) [14].
Results
Variant analysis
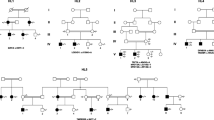
Among 92 probands analyzed, we determined a genetic diagnosis in 18, and all the 92 probands were from non-consanguineous families. Three modes of inheritance were observed, including 15 autosomal recessive cases, 2 autosomal dominant cases, and 1 X-linked recessive case (Table 2). Fourteen deafness gene variants were detected. Those in TMC1, CDH23, LOXHD1 and USH2A were each detected in two probands, and those in 10 other deafness genes were each detected in one proband (Table 2). The 18 probands carried 33 different variants (Table 2), of which 18 were novel, accounting for 54.5% of the total variants (18/33). These 33 variants included six different variant types, including 11 missense variants (33.3%, 11/33), 9 nonsense variants (27.3%, 9/33), 8 frameshift variants (24.2%, 8/33), 1 non-frameshift variant (3.0%, 1/33), 3 splice site variants (9.1%, 3/33), and 1 copy number variation (CNV) variant (3.0%, 1/33).
According to the ACMG guidelines and InterVar sofware, 10 of the novel variants were categorized as “pathogenic”, and 8 were “likely pathogenic” (Table 3).
The copy number variation verification of STRC
The target NGS showed that there was a heterozygous deletion of STRC in proband 12932. Quantitative RT-PCR was performed to estimate STRC copy number in members of proband 12932’s family and in healthy people. Each sample was assayed in triplicate for each gene using SYBR Green PCR Master Mix and a StepOnePlus Real Time PCR System. The primers used to amplify STRC and the internal reference gene GAPDH were showed in Table 4. The STRC copy number was calculated by dividing the yield of the STRC gene by that of the reference gene. The amplification conditions were: 95 °C for 3 min, then 40 cycles of 95 °C for 15 s, 60 °C for 30 s, then 50 °C for 15 s. The relative quantitative analysis Cq value was determined using the 2−△△Ct method to calculate the relative STRC copy number in the family members and healthy people. The results showed that the relative copy number of STRC in proband 12932 and his mother (12932-2) was only approximately 50% of that of a normal person (Fig. 1).

The real time PCR verification of the STRC copy number variant
Discussion
In this study, we performed a variant analysis of 92 unrelated Chinese NSHL patients. We determined a molecular diagnosis in 18 probands, with 33 different variants in 14 deafness genes (Table 2). We identified 18 novel variants in 12 deafness genes, which were not previously reported in ClinVar or HGMD. According to the ACMG guidelines and InterVar software, 10 variants of them were categorized as “pathogenic” variant, and 8 were categorized as “likely pathogenic” variants (Table 3).
Among the 18 probands who received a genetic diagnosis, 15 were autosomal recessive, two were autosomal dominant, and one was X/Y-linked. Yang et al. [23] recruited 190 NSHL patients, and after excluding the common GJB2, SLC26A4 and MT-RNR1 variants, 33 probands were determined to have rare HL variants, 28 were autosomal recessive, four were autosomal dominant, and one was mitochondrial. The number of autosomal recessive patients was much lower than in our study, which might be caused by regional differences. In our study, variants in TMC1, CDH23, LOXHD1 and USH2A were each detected in two probands, while variants in POU3F4, OTOA, GPR98, GJB6, TRIOBP, SLC26A4, MYO15A, TNC, STRC and TMPRSS3 were each detected in one proband. Among the 33 rare HL cases reported by Yang et al., the most frequently detected variant was in MYO15A (four times), then in TMC1, USH2A, PCDH15, and GPR98 (three times each) [23]. Although the detection rates of the TMC1 and USH2A variants were high in both this study and that of Yang et al., we detected an MYO15A variant in only one case, while Yang et al. did not detect any LOXHD1 variants, which we detected twice in our patients. These differences may be caused by regional differences between north and south China. Of course, this may also be caused by the sample size not being large enough.
Some deafness gene screening techniques can screen for hot-spot variants in SLC26A4. However, targeted screening tests might miss rare variants of SLC26A4. In patient 12751, we detected a compound heterozygous variant, c.589G>A/ c.1238A>G, which was not in the variant hot-spots of SLC26A4. Therefore, for patients with deafness, it is best not to use deafness gene screening technology. Targeted sequencing technology or whole exome sequencing technology should be used for diagnosis.
CNV is one of the main forms of structural genome variation, and is a cause of many genetic diseases. NGS is increasingly used to test for CNVs in many diseases. In patient 12932, we detected a CNV (a heterozygous deletion) in STRC, which has been previously reported [27, 28]. STRC CNV is common in HL patients [31] and 72 types of deletion and 35 duplications of STRC are included in the ClinVar database. Targeted-NGS methods to detect CNVs in HL patients can still be improved, for example specificity and sensitivity can be enhanced; however, whole exome sequencing (WES) or whole-genome sequencing (WGS) are recommended to detect CNV in HL patients.
In our study, we identified 18 novel variants in 12 deafness genes. These variants included eight missense variants, four nonsense variants, five frameshift variants and 1 splice site variants (Table 3). The nonsense variants and frameshift variants caused the peptide chain to terminate prematurely, which shortened the length of the peptide chain, and then affected the function of the gene. We use PolyPhen2 and PROVEAN tools to assess the possible functional role of the eight novel missense variant. The missense variants c.805C>T, c.5957T>C and c.6830C>A of CDH23, c.774A>C of OTOA were assessed as probably damaging by PolyPhen2, however, these four missense variants were assessed as neutral by PROVEAN. Then we checked the conservative of these four missense variants, all the four variant were highly conserved in different species. Combined with ACMG guidelines and InterVar software, we speculated that these four missense variants were “likely pathogenic” variants.
Conclusions
We used targeted-NGS for genetic diagnosis of 18 NSHL probands. We identified 18 novel variants in 12 deafness genes, which enlarged the variant spectrum of deafness genes in the Han Chinese population. These findings help inform the genetic diagnosis of deafness and add to the theoretical basis for the prevention of deafness. However, 74 patients in our cohort did not receive a clear genetic diagnosis; therefore, further WES or WGS testing is needed to identify mutations in other HL-causing genes or to discover new disease-causing genes for these patients.
Availability of data and materials
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2021) in National Genomics Data Center (Nucleic Acids Res 2021), China National Center for Bioinformation / Beijing Institute of Genomics, Chinese Academy of Sciences (GSA-Human:HRA001546) that are publicly accessible at https://bigd.big.ac.cn/gsa-human/browse/HRA001546.
Abbreviations
- ACMG:
-
American College of Medical Genetics and Genomics
- ASSR:
-
Auditory steady-state evoked response
- CNV:
-
Copy number variation
- HGMD:
-
Human Gene Variant Database
- HL:
-
Hearing loss
- NSHL:
-
Non-syndromic hearing loss
- NGS:
-
Next-generation sequencing
- WES:
-
Whole exome sequencing
- WGS:
-
Whole-genome sequencing
References
Fortnum HM, Summerfield AQ, Marshall DH, et al. Prevalence of permanent childhood hearing impairment in the United Kingdom and implications for universal neonatal hearing screening: questionnaire based ascertainment study. BMJ. 2001;323:536–40.
Du W, Wang Q, Zhu Y, et al. Associations between GJB2, mitochondrial 12S rRNA, SLC26A4 variants, and hearing loss among three ethnicities. Biomed Res Int. 2014;2014: 746838.
Nance WE, Lim BG, Dodson KM. Importance of congenital cytomegalovirus infections as a cause for pre-lingual hearing loss. J Clin Virol. 2006;35:221–5.
Zhou Y, Li C, Li M, et al. Mutation analysis of common deafness genes among 1,201 patients with non-syndromic hearing loss in Shanxi Province. Mol Genet Genomic Med. 2019;7(3): e537.
Sloan-Heggen CM, Bierer AO, Shearer AE, et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum Genet. 2016;135:441–50.
Zhang C, Hao S, Liu Y, et al. A novel LOXHD1 variant in a Chinese couple with hearing loss. J Int Med Res. 2019;47(12):6082–90.
Yang T, Wei X, Chai Y, et al. Genetic etiology study of the non-syndromic deafness in Chinese Hans by targeted next-generation sequencing. Orphanet J Rare Dis. 2013;14(8):85.
Wesdorp M, Schreur V, Beynon AJ, et al. Further audiovestibular characterization of DFNB77, caused by deleterious variants in LOXHD1, and investigation into the involvement of Fuchs corneal dystrophy. Clin Genet. 2018;94:221–31.
Yuan Y, Li Q, Su Y, et al. Comprehensive genetic testing of Chinese SNHL patients and variants interpretation using ACMG guidelines and ethnically matched normal controls. Eur J Hum Genet. 2020;28(2):231–43.
Dai P, Yu F, Han B, et al. GJB2 mutation spectrum in 2,063 Chinese patients with nonsyndromic hearing impairment. J Transl Med. 2009;7:26.
Yuan Y, You Y, Huang D, et al. Comprehensive molecular etiology analysis of nonsyndromic hearing impairment from typical areas in China. J Transl Med. 2009;7:79.
Dai P, Huang LH, Wang GJ, et al. Concurrent hearing and genetic screening of 180,469 neonates with follow-up in Beijing. China Am J Hum Genet. 2019;105(4):803–12.
Guo C, Huang SS, Yuan YY, et al. Hearing phenotypes of patients with hearing loss homozygous for the GJB2 c.235delc mutation. Neural Plast. 2020;2020:8841522.
Li Q, Wang K. InterVar: clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am J Hum Genet. 2017;100:267–80.
García-García G, Aller E, Jaijo T, et al. Novel deletions involving the USH2A gene in patients with Usher syndrome and retinitis pigmentosa. Mol Vis. 2014;25(20):1398–410.
Dai H, Zhang X, Zhao X, et al. Identification of five novel mutations in the long isoform of the USH2A gene in Chinese families with Usher syndrome type II. Mol Vis. 2008;14:2067–75.
Park JH, Kim NK, Kim AR, et al. Exploration of molecular genetic etiology for Korean COCHlear implantees with severe to profound hearing loss and its implication. Orphanet J Rare Dis. 2014;9:167.
Le Quesne SP, Saihan Z, Rangesh N, et al. Comprehensive sequence analysis of nine Usher syndrome genes in the UK National Collaborative Usher Study. J Med Genet. 2012;49(1):27–36.
Wang QJ, Zhao YL, Rao SQ, et al. A distinct spectrum of SLC26A4 mutations in patients with enlarged vestibular aqueduct in China. Clin Genet. 2007;72(3):245–54.
Yuan Y, Guo W, Tang J, et al. Molecular epidemiology and functional assessment of novel allelic variants of SLC26A4 in non-syndromic hearing loss patients with enlarged vestibular aqueduct in China. PLoS ONE. 2012;7(11): e49984.
Ji YB, Han DY, Wang DY, et al. Evaluation of deaf-mute patients with sensitive deafness gene screening in Shandong province. Zhonghua Yi Xue Za Zhi. 2009;89(36):2531–5.
Brownstein Z, Friedman LM, Shahin H, et al. Targeted genomic capture and massively parallel sequencing to identify genes for hereditary hearing loss in Middle Eastern families. Genome Biol. 2011;12:R89.
Yang T, Wei X, Chai Y, et al. Genetic etiology study of the non-syndromic deafness in Chinese Hans by targeted next-generation sequencing. Orphanet J Rare Dis. 2013;8:85.
BenSaïd M, Hmani-Aifa M, Amar I, et al. High frequency of the p.R34X mutation in the TMC1 gene associated with nonsyndromic hearing loss is due to founder effects. Genet Test Mol Biomarkers. 2010;14(3):307–11.
Sirmaci A, Duman D, Oztürkmen-Akay H, et al. Mutations in TMC1 contribute significantly to nonsyndromic autosomal recessive sensorineural hearing loss: a report of five novel mutations. Int J Pediatr Otorhinolaryngol. 2009;73(5):699–705.
Hilgert N, Alasti F, Dieltjens N, et al. Mutation analysis of TMC1 identifies four new mutations and suggests an additional deafness gene at loci DFNA36 and DFNB7/11. Clin Genet. 2008;74(3):223–32.
Bademci G, Foster J, Mahdieh N, et al. Comprehensive analysis via exome sequencing uncovers genetic etiology in autosomal recessive nonsyndromic deafness in a large multiethnic cohort. Genet Med. 2016;18(4):364–71.
Bademci G, Diaz-Horta O, Guo S, et al. Identification of copy number variants through whole-exome sequencing in autosomal recessive nonsyndromic hearing loss. Genet Test Mol Biomarkers. 2014;18(9):658–61.
Elbracht M, Senderek J, Eggermann T, et al. Autosomal recessive postlingual hearing loss (DFNB8): compound heterozygosity for two novel TMPRSS3 mutations in German siblings. J Med Genet. 2007;44(6): e81.
Lee J, Baek JI, Choi JY, et al. Genetic analysis of TMPRSS3 gene in the Korean population with autosomal recessive nonsyndromic hearing loss. Gene. 2013;532(2):276–80.
Shearer AE, Kolbe DL, Azaiez H, et al. Copy number variants are a common cause of non-syndromic hearing loss. Genome Med. 2014;6(5):37.
Acknowledgements
The authors are grateful to the patients and the pedigree members who participated in this study.
Funding
This work was supported by the National Key Research and Development Program of China (2016YFC1000307). The funding plays an important role in establishing the lines of research of its scientific team. The funding covered the costs of materials required for DNA extraction and expenses of Targeted -NGS and Sanger sequencing. In addition, it provides full infrastructure for the ethical and legal collection. In the course of the present study, the scientific team was responsible for performing the analysis and interpretation of the data.
Author information
Authors and Affiliations
Contributions
XHJ, PD, HFG and XM designed the study and write the manuscript. XHJ, SSH, LSA and CZ performed the molecular tests. All authors contributed to the editing of the manuscript and the scientific discussions. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was undertaken according with the tenets of the Declaration of Helsinki 1975 and its later amendments. This study was approved by the Ethics Committee of the Chinese People's Liberation Army (PLA) General Hospital (Beijing) (reference number S2016-120-02). Written informed consent was obtained from all of the adult participants and written informed consent of patients younger than 16 years old was obtained from their parents.
Consent for publication
Written consent was obtained from all the participants and for those younger than 18 years old, obtained from their parents.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Jin, X., Huang, S., An, L. et al. Variant analysis of 92 Chinese Han families with hearing loss. BMC Med Genomics 15, 12 (2022). https://doi.org/10.1186/s12920-022-01158-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-022-01158-3