
Abstract
Background
Inherited mutations in the breast cancer susceptibility genes BRCA1 and BRCA2 (BRCA1/2) confer high risks of breast and ovarian cancer. Because the contribution of BRCA1/2 germline mutations to BC in the Northeastern population of Morocco remains largely unknown, we conducted this first study to evaluate the prevalence and the phenotypic spectrum of two BRCA1/2 pathogenic mutations (the founder BRCA1 c.5309G>T and BRCA2 c.1310_1313delAAGA). This choice was also argued by the presence of an apparent specific geographical connection of these mutations and the Northeastern region of Morocco.
Methods
Screening for the germline mutations c.5309G>T and BRCA2 c.1310_1313delAAGA was performed by sequencing on a total of 184 breast cancer (BC) patients originated from the Northeastern region of Morocco.
The likelihood of identifying a BRCA mutation is calculated using the Eisinger scoring model. The clinical and pathologic features were compared between the BRCA-positive and BRCA-negative groups of patients. Difference in survival outcomes was compared between mutation carriers and non-carriers.
Results
BRCA1 c.5309G>T and BRCA2 c.1310_1313delAAGA are responsible for a significant proportion of all BC cases (12.5%) and at least 20% of familial BC. The screening of BRCA1/2 genes by NGS sequencing confirmed that there are no additional mutations detected among positive patients.
The clinicopathological features in positive patients were in accordance with typical characteristics of BRCA pathogenic mutations. The mean features in the carriers were the early onset of the disease, familial history, triple negative status (for BRCA1 c.5309G>T) and worse prognosis in terms of overall surviving.
Our study indicates that the Eisinger scoring model could be recommended to identify patients for referral to BRCA1/2 oncogenetic counseling.
Conclusion
Our findings suggest that BRCA1 c.5309G>T and BRCA2 c.1310_1313delAAGA mutations may have a strong founder and/or recurrent effect on breast cancer among the Northeastern Moroccan population. There contribution to breast cancer incidence is certainly substantial in this subgroup. Therefore, we believe that BRCA1 c.5309G>T and BRCA2 c.1310_1313delAAGA mutations have to be included in the array of tests aimed at revealing cancer syndrome carriers among subjects of Moroccan origin.
Similar content being viewed by others


Background
The vast majority of breast cancer (BC) cases are considered sporadic-appearing tumors for which environmental and life-style factors are the most important determinants of the risk, while 5% to10% of all cases are thought to develop because of a genetic predisposition [1].
Hereditary Breast and Ovarian Cancer (HBOC) is a genetic predisposing syndrome characterized by a young age of onset, a type of tumor as well as family history. HBOC like most of the other genetic predisposing syndromes to cancer is caused by germline mutations of oncogenes and tumor suppressor genes. Deleterious germline mutations of at last 15 genes (BRCA1/2, ATM, BARD1, CDH1, CDKN2A, CHEK2, MLH1, MSH2, MSH6, NF1, PALB2, PTEN, RAD51D, TP53, BRIP1) are associated with an increased risk (> 2.0 times) of breast cancer. Deleterious germline mutations of eleven genes are associated with an increased (> 2.0-fold) risk of ovarian cancer (ATM, BRCA1 / 2, BRIP1, MSH2, MSH6, NBN, PMS2, RAD51C, RAD51D, and TP53) [2].
Breast Cancer 1 gene (BRCA1) and Breast Cancer 2 gene (BRCA2) are included in the category of high penetrance genes. A plethora of germline pathogenic mutations of BRCA1/2 genes (over 3300) which are inherited in an autosomal dominant form are responsible for the major HBOC cases ( http://www.hgmd.cf.ac.uk/ac/index.php). These variants are closely related to high lifetime risk of developing HBOC; the reported cumulative lifetime risk of breast cancer is approximately 72% and 69%, up to the age of 80 years, for BRCA1 and BRCA2 respectively [3].
Both BRCA1 and BRCA2 are tumor suppressor genes, and are instrumental in a range of cellular regulating pathways, including regulating DNA double-strand breaks repair in the process of homologous recombination, genomic integrity, transcriptional regulation, apoptosis, chromosomal segregation and chromatin remodeling [4, 5]. There is also evidence that BRCA1 is an important link in the signal chain that starts with recognition of DNA damage (sensed by ATM) and leads to cell cycle arrest at the G2/M checkpoint [6].
BRCA1/2 mutations exhibit important differences in prevalence and spectrum across various racial/ethnic groups and geographical regions. In some ethnic communities or specific populations, mutations of BRCA1/2 genes are more frequent due to founder effects. This is particularly remarkable in Ashkenazi Jews population, Polish, Norwegian, Icelandic people and in several other area where isolated populations exists [1, 7, 8].
With the emergence of genetic testing, BRCA1/2 profiling was strongly recommended for women with a family history or early age onset of BC [9]. The assessment of BRCA1/2 mutation carriers in familial breast cancer has been proved to be valuable not only in the perspective of prevention and early detection of related cancers but also it has implications in implementation of personalized medicine and chemoprevention of recurrence. It has been suggested that breast cancer patients with BRCA1/2 mutations may benefit from precision treatments, such as platinum-based chemotherapy and poly ADP-ribose polymerase inhibitors [10]. On another side, the identification of the most prevalent or founder mutations in an ethnic population will facilitate earlier and rapid and especially cheaper molecular diagnosis of BC. Therefore, these have made it imperative for the recurrent and founder mutations of the BRCA1/2 genes within low income countries to be identified and included in breast cancer screening and diagnosis [11].
Although the highest rates of breast cancer incidence are observed in developed countries, the incidence of this disease has clearly risen in Arab countries including Morocco, a country of North-western Africa. BC is still a major cause of death by cancer among Moroccan women and accounted for 35.8% of all registered cancers. The latest statistics available reported an increasing incidence rate from 39.0 to 49.5 per 100.000 women in this population between 2008 and 2012 [12]. It remained the highest incidence among the countries of North-western Africa (Algeria and Tunisia) [13]. Data reported in Moroccan population, showed a higher proportion of BC among young women aged between 45 to 49 years, and a frequent clinical observation of family history. All this suggests a strong influence of high-penetrance genetic factors in BC etiology [14, 15].
To date, published studies on the contribution of BRCA1/2 mutations to BC in the Moroccan population are still limited. Only a partially characterized BRCA1 mutation landscape in BC Moroccans is available and includes the following deleterious mutations: c.68-69delAG, c.116G > A, c.181 T > G, c.798-799delTT, c.1016dupA, c.2126insA, c.2805delA, c.3279delC, c.3453delT, c.4942A > T, c.5062-5064delGTT and c.5095C > T [16,17,18,19]. The main pathogenic mutations detected in BRCA2 gene were c.289G > T, c.517-1G > A, c.1310_1313delAAGA, c.3381delT, c.3847_3848delGT, c.5073dupA, c.5116_5119delAATA, c.5576-5579delTTAA, c.6428C > A, c.7110delA and c.7234_7235insG [16, 17, 19,20,21]. For all of these mutations, there is no evidence provided on their founding effect in the Moroccan population. Furthermore, all of patients recruited in these previous studies have been diagnosed in few cancer institutions based primarily in the west of Morocco (mainly in the two large cities of Casablanca and Rabat), and therefore are not fully representative of the whole population. Interestingly and independently of the studies carried out in Morocco, another Spanish study, conducted by Quiles et al. [22], reported a new BRCA1 deleterious mutation with founder effect (c.5309G>T, G1770V) in five families of Moroccan origin but settled in Spain and Norway. Interestingly, all of the five independent families were originated from the same area of Morocco, mainly from the regions of Oujda and Nador located in Northeastern Morocco. Most importantly, this founder mutation has not been described in previous published Moroccan studies. Otherwise, another mutation, c.1310_1313delAAGA located on BRCA2 gene, was first described in Morocco in 2016 and 2017 [23, 24]. These studies which focused on a total of 122 patients originating from different regions of Morocco showed that 9 out of 14 positive cases (64.3%) shared the same geographic origin in the Northeast of Morocco essentially from the region of Oujda Angad; the other five patients (35.7%) were from neighboring central regions.
In the light of these observations, it seems essential to us to undertake the present study on a cohort of pathologically confirmed female breast cancer patients who originated from the Moroccan Northeastern region. Our intention was not only to ascertain the specificity and prevalence of the two BRCA1/2 mutations (BRCA1, c.5309G>T, and BRCA2, c.1310_1313delAAGA) in the Moroccan Northeastern region, but also to evaluate their role in tumor phenotypic spectrum and disease prognosis, and to establish an adapted and rapid procedure for BRCA1/2 mutations screening among the population of this region for better clinical management of BRCA mutation carriers.
Methods
Study participants
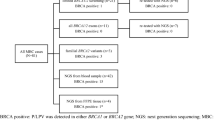
We established a cohort including 184 pathologically confirmed female breast cancer patients who originated from the Moroccan Northeastern region. Four BC male patients were also included. All patients were referred from the Hassan II Regional Oncology Center of Oujda. The study protocol was reviewed and approved by the ethics committee for Biomedical Research of the Faculty of Medicine and Pharmacy of Casablanca under the number 06/18. Written informed consent for research participation was obtained from all subjects prior to peripheral blood collection.
Clinical and pathological data were abstracted from patient’s medical files and pathology reports. The recorded information included age at diagnosis, family history of breast cancer, laterality, tumor histology type, Scarff-Bloom-Richardson (SBR) grade, tumor size, lymph node involvement, metastases, survival as well as hormone receptor status including: estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (Her-2).
Eisinger score carrier risk prediction
Germline mutations in BRCA1/2 genes have important implications for treatment of patients diagnosed with breast or ovarian cancers as well as unaffected carriers of these mutations. Various statistical models have been established to predict the likelihood of identifying a deleterious BRCA mutation based on an individual's personal and family history [25].
According to the criteria published by the national expertise INSERM-FNCLCC in 2004 based on personal and familial breast and ovarian cancer history, we used the Eisinger scoring system that has been used in Europe to calculate the likelihood of carrying a BRCA1/2 pathogenic variants for each patient [26]. The Eisinger score (seven factors, score from 0 to 5) is a simple family tree analysis risk assessment tool to validate the indication for an oncogenetic consultation and to consider a search for mutations. It also helps to gradate the risk of genetic predisposition to breast cancer in the absence of identified familial mutation (score = 5 or more: excellent indication; score = 3 or 4: possible indication; score = 1 or 2: no indication). Based on the calculation of Eisinger score for each patient in our cohort, 46.15% of the patients have criteria and personal and/or familial history suggesting an increased risk of breast cancer due to genetic predisposition (familial group; Eisinger score > 3). The other 53.85% of the patients have no such risk criteria and the cancers may be classified as sporadic (sporadic group; Eisinger score ≤ 3).
Molecular Analysis of BRCA1/2 genes
Peripheral blood samples from the patients were collected in EDTA coated tubes. Genomic DNA extraction was done using a standard salting-out method [27]. DNA concentration and quality were evaluated by NanoVue Plus™ spectrophotometer (biochrom, Harvard Bioscience Inc. Massachusetts, USA), and stored at − 20 °C until analysis.
The target screening of the c.5309G>T founder mutation was performed by PCR-based Sanger sequencingof BRCA1 exon 21. Amplification and sequencing of a 320 bp fragment were carried out with the following primers: forwardprimer 5’-cttgtccctgggaagtagca-3’and newly designed reverse primer 5’-gatgggggttcctcagattg-3’ (designed through Primer 3 software) [28]. To screen for c.1310_1313delAAGA mutation at exon 10 of BRCA2, a PCR product of 552 bp was amplified and sequenced with the following primers: forward primer 5’-tggaaccaaatgatactgatcc-3’and reverse primer 5’- cctctgaaagtggactggaaa-3’.
The PCR was performed in a volume of 25 μL. Cycling conditions were 94 °C for 30 s, 60 °C for 30 s, and 72 °C for 30 s for 30 cycles in the case of BRCA1 exon 21. Thirty five cycles of 94 °C for 30 s, 58 °C for 30 s, and 72 °C for 30 s were cycling conditions used for BRCA2 exon 10. Purifcation of the PCR products was done by ExoSAP-IT purifcation kit (Thermo Fisher Scientifc, Waltham, MA) according to the manufacture’s protocol. Purified products were sequenced bidirectionally using the Big Dye Terminator Cycle Sequencing kits ((Life technologies, Inc. Foster City, CA) and the same primers used for PCR. The sequence data were collected from an automated ABI Prism 3130XL capillary electrophoresis system (Life technologies, Inc. Foster City, CA). Alignment to the BRCA1 and BRCA2 reference genomic sequence (GenBank entries: NM_007294 and NM_000059 respectively) was done with SeqScanner v2 software (Applied Biosystems).
Next generation sequencing
Germline mutation profiling using NGS was performed in blood samples of 54 patients belonging to the present cohort and including all patients carrying the mutations BRCA1 c.5309G>T and BRCA2 c.1310_1313 DelAAGA.
BRCA1 and BRCA2 genes were screened by sequencing using Ion Proton next generation sequencing platform (Thermo Fisher Scientific). The NGS library was constructed using the Oncomine BRCA Research Assay, Chef-Ready Library Preparation (IonTorrent, Thermo Fisher Scientific, USA) according to the manufacturer’s instructions. Reads were aligned to the human genome reference sequence 19 (hg19) and variant identification were performed with the Torrent Suite v.5.12 software. The generated BAM files of each sample were imported to the Ion Reporter Software v5.18 and run using the Oncomine BRCA Research Germline workflow for variants annotations. Exonic sequence analysis was performed with average depth coverage of at least 200X.
Statistical analysis
The clinical and pathologic features were compared between the BRCA-positive and BRCA-negative groups of patients. Descriptive of clinical data were expressed in percentage or mean ± SD. The statistical significance of associations was evaluated for categorical variables based on Pearson’s chi-squared test or Fisher’s exact test. The Mann–Whitney test was used to compare the mean ages of the different groups. The p values were based on two sided tests and conducted at a 5% significance level. Statistical analyses were performed using SPSS software (version 21.0; IBM Corp. Armonk, NY, USA).
Overall survival
Overall survival (OS) was determined as the length of time from the date of diagnosis until either the date of death or the date of last follow-up. We estimated the survival function with the Kaplan–Meier estimator. Difference in survival outcomes between mutation carriers and non-carriers was compared with the Log-Rank test.
Results
Patient’s characteristics
The mean age of patients at the time of diagnosis was 42.3 ± 0.72 years (range from 25 to 72 years). Personal and family history informations of BC were used in the Eisinger score calculation to distinguish two groups of patients. The first group (47%) included female patients with personal and/or familial history suggesting an increased risk of breast cancer due to genetic predisposition (score > 3). We called this group “Familial”. The second group (53%) included patients who have no personal and/or familial cancer history that may suggest a genetic predisposition to breast cancer (score ≤ 3). We named this group “sporadic”. The Table 1 showed the clinical characteristics of BC in these two groups of patients. Clinicopathological data were not available for two positive female BC. The mean age at diagnosis was 38.6 ± 0.95 years and 45.6 ± 0.95 years for familial and sporadic groups respectively. There are more patients under 39 years of age in the familial group compared to the sporadic group (55.3% vs. 28.9%; p = 0.0003). Patients with bilateral breast cancer (9.4%) were found only in the familial group (8/85). There is no case of bilateral breast cancer in the sporadic group (p = 0.002). In addition, early stage tumor (T2) and histological grade III were slightly more frequent in patients with familial history compared to sporadic group (p = 0.03 and 0.04 respectively). Contrariwise, the proportion of patients developing metastasis and lymph node involvement was slightly higher in the latter group (0.026 and 0.001 respectively). Finally, there was no significant difference regarding histological type of the tumor and hormone receptor expression between the two groups of patients.
BRCA1 c.5309G>T and BRCA2 c.1310_1313 DelAAGA mutations
As indicated in Table 1, the overall prevalence of pathogenic BRCA1 c.5309G>T and BRCA2 c.1310_1313 DelAAGA mutations among east-Moroccan BC female patients was 22 (12.15%). The large and significant proportion was observed in familial group compared to sporadic one (16, (19%) and 4 (4.2%) respectively; p = 0.002). All positive cases were heterozygous for these mutations. The screening of BRCA1/2 genes by NGS sequencing confirmed that there are no additional mutations detected among the mutation carrier patients.
BRCA1 c.5309G>T mutation
The heterozygous BRCA1 c.5309G>T mutation was found in 10 patients (5.43%). the frequency of the mutation was significantly higher in familial BC group (9, (10.1%)) than in sporadic group (1, (1.1%) (p = 0.007).
BRCA2 c.1310_1313 DelAAGA mutation
In total, we identified 12 females (7.35%) carrying BRCA2 c.1310_1313 DelAAGA mutation. The highest prevalence of the mutation was observed in the in familial BC group (7, (10.1%)) than in sporadic group (3, (1.1%)). Data are missing for two patients; therefore these are not included in the calculations. Interestingly, the mutation was also encountered in two patients among four BC males.
Clinical features of BRCA1 c.5309G>T associated BC
Detailed clinicopathological features of 12 positive females BC and two males BC are listed in Table 2. The comparisons between female carriers and non carriers are summarized in Table 3. The average of age at diagnosis in patients with c.5309G>T mutation was 41 years (SD = 6.02, range: 33–50). Forty percent of carriers showed an age of onset below 39 years comparatively to the non carriers. There are also 50% who were triple negative. Most of patients (80%) shared similar histopathological features including unilateral BC, ductal invasive histological type and tumor grades I-II. Moreover, Table 3 showed that 55.6% of carriers had intermediate tumor lesion (T2) and positive lymph nodes. But only 20% developed metastasis. The distribution of these features in the two groups of patients was equivalent.
Clinical features of BRCA2 c.1310_1313 DelAAGA associated BC
Detailed clinicopathological features of positive patients are listed in Table 2. and the comparisons between carriers and non carriers are summarized in Table 3.
The mean age at diagnosis of BC in patients with BRCA2 c.1310_1313 DelAAGA mutation was 38.5 years (SD = 8.92, range: 28–54) with a large proportion (60%) diagnosed at 39 years of age or younger compared to noncarriers (37%). All BRCA2 carriers developed unilateral BC and were more likely to have a family history of BC (70%) when compared to the non carriers (41.3%). Invasive ductal carcinoma is the most common histopathologic type (80%). The occurrence of intermediate tumor lesion T2 (60%), positive lymph nodes (90%), early histological stage (I-II) and no metastasis (0%) was noted in carriers. Only 22.2% (2/12) of the mutation carrier patients are triple-negative.There was no statistically significant difference between positive patients and noncarriers.
Risk of BRCA1/2 mutations assessment: Eisinger Score
We aimed at addressing the practice of referral for genetic counseling and establishing whether the Eisinger prediction model could be helpful in identifying individual’s mutation risk and determining eligibility for BC genetic screening in our population.
All patients positive for the two tested mutations (except one case) fit the Eisinger guidelines for BRCA mutation screening (score > = 3; Table 2). The mean Eisinger Score in positive patients for the founder BRCA1 c.5309G>T mutation was 7.4 ± 1.18 (median = 7) and 5.63 ± 0.65 (median = 5.5) in patients positive BRCA2 c.1310_1313 DelAAGA mutation (Table 4). When considering the two mutations together, the mean Eisinger Score in positive patients was 6.61 ± 0.73, (mediane = 6) and that of negative patients ranged from 3.92 ± 0.21 to 4.02 ± 0.20 (mediane = 3) (Table 4).
A Mann–Whitney test was then carried out to examine whether or not the Eisinger prediction model was useful in the present population (Table 4). In order to compare the scores effectively, patients who tested positive for the founder mutation (group 1, n = 10), or those who tested positive for BRCA2 deletion mutation (group 2, n = 8), as well as the two groups together (group 3, n = 18) were compared with patients who tested negative for one or for both mutations. There was a significant difference in the Eisinger risk scores between carriers and non carriers in all three tested groups (p = 0.002; p = 0.02; p = 1.2 × 10–4, respectively).
Overall survival
In order to determine whether the two BRCA mutations (BRCA1 c.5309G>T and BRCA2 c.1310_1313 DelAAGA) affect survival, we compared the overall survival of patients with one BRCA mutation with the non carriers (Fig. 1). Association analysis showed that patients with the BRCA1 c.5309G > T mutation have worse OS than the negative group (p = 0.004). All positive patients are dead except one (data are missing for two patients). On the other side, survival was not significantly affected by the presence of c.1310_1313 DelAAGA mutation (p = 0.83).

Kaplan–Meier estimates of cummulative survival of patients with BRCA1 c.5309G>T or BRCA2 c.1310_1313 DelAAGA mutation vs. patients without mutation
Discussion
Genetic testing for pathogenic germline mutations in BRCA1/2 genes is strongly recommended for people with a BC family history. It may have important implications for clinical management of patients diagnosed with the disease as well as unaffected carriers of these mutations. It also influence prognosis of the current cancer and enable prevention of future cancers [29]. However, genetic testing is expensive and may be associated with adverse psychosocial effects. Identifying most prevalent or founder mutations in specific populations constitute a valuable opportunity for genetic screening since it facilitate earlier and rapid and especially cheaper molecular diagnosis of BC.
Insofar as only a small proportion of people with breast cancer in the general population are carriers of a mutation, it is not possible to propose testing for a mutation at all cases. To provide a cost-efficient and clinically appropriate genetic counseling service, genetic testing should be targeted at those individuals most likely to carry pathogenic mutations. Several algorithms that predict the likelihood of carrying a BRCA1 or a BRCA2 mutation are currently used in clinical practice to identify such individuals. Their widespread use would improve equity of access and the cost-effectiveness of genetic testing.
In the current study, we sought to present a first report on the prevalence and clinical significance of two particular BRCA1/2 mutations in the northeastern region of Morocco. We also evaluated the efficiency of a clinical prediction tool, the Eisinger scoring system, for use in clinical practice to select patients for mutation analysis in our cohort.
The first mutation was BRCA1 c.5309G>T (p.Gly1770Val; rs863224765) founder mutation that seems to be unique to this region of Morocco. Indeed, this mutation was first reported in two different families from Norway and three in Spain. Interestingly, all of these five families were of Moroccan origin, more precisely from the Moroccan northeast region [22, 30]. To date, only one study reported this mutation in two patients from Tanger (in the north of Morocco) [31]. In addition, the BRCA1 c.5309G>T mutation was reported as a Moroccan founder variant based on Microsatellite analysis in the five families studied by Quiles et al. [22]. The fact that this variant was never reported in other Moroccan studies nor worldwide, raises the probability of it being specific pathogenic variant for the north eastern Moroccan population.
BRCA1 c.5309G>T mutation is located in the functionally important BRCA1 carboxyl terminal (BRCT) domain, a domain known to harbor missense substitutions associated with increased risk of breast/ovarian cancer [32]. Using a functional complementation assay of BRCA1 sequence variants in a mouse-Brca1-null embryonic stem cells, Bouwman et al. [33] classified this variant deleterious because the gene was functionally impaired in the direct repeat (DR)-GFP and/or combined PARP inhibitor/cisplatin sensitivity assay. Quiles et al. [34] demonstrated that this variant significantly alters the BRCT structure and that it compromises the BRCA1 transcriptional activity. Further studies based on multifactorial likelihood analysis provided evidence that BRCA1 c.5309G>T should be treated as a disease-causing variant [32].
The second tested variant in our study was the BRCA2 c.1310_1313delAAGA frameshift mutation that has been reported as “pathogenic” in the ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/), the Breast Cancer Information Core (BIC) (http://www.research.nhgri.nih.gov/bic/), the HGMD (http://www.hgmd.cf.ac.uk/ac/index.php) databases. This sequence change deletes four nucleotides from exon 10 of the BRCA2 mRNA, causing a frameshift after codon 437 and the creation of a premature translational stop signal 22 amino acid residues.
BRCA2 c.1310_1313delAAGA was commonly circulated among north African patients with BC as recurrent mutation since it has been reported in some Moroccan [23, 24], Algerian [35] and Tunisian studies [36, 37]. Interestingly, the genealogic investigation in Moroccan patients revealed that all carriers of this mutation shared the same restricted geographic origin in the North-East of Morocco [24]. Its higher incidence in the north east of Morocco is suggestive of a founder effect which required confirmation by haplotype analysis. Moreover, this mutation was also found in other patients from European [38,39,40,41,42], Hispanic [43], Libanese [44] and Caribbean [45] origins.
In our cohort, all Moroccan BC patients were originating from the northeastern region of Morocco and were grouped into familial (47%) and sporadic (53%) groups using Eisinger scoring system. All of the patients were screened for the two mutations BRCA1 c.5309G>T and BRCA2 c.1310_1313delAAGA and the clinicopathological features of carriers were analyzed.
The first main finding was the high prevalence of these two mutations in our cohort (12.5%). The screening of BRCA1/2 genes by NGS sequencing confirmed that there are no additional mutations detected among positive patients for BRCA1 c.5309G>T or BRCA2 c.1310_1313delAAGA. The two mutations appeared to be the most frequent genetic cause of BC in our population preferentially in a strong familial context of the disease since it can explain at least 20% of familial cases. This result concur, as it has been documented by others, that the family history is an important criterion for the identification of BRCA1/2 mutation carriers [46, 47].
In addition, the cohort we screened for BRCA2 c.1310_1313delAAGA mutation included 4 BC males and among them, two are positive for this alteration. Interestingly, this mutation has also been previously detected in one Tunisian male [36]. This may suggest that this mutation has a relatively high penetrance in males. Male breast cancer (MBC) is a rare disease accounting for less than 1% of all breast cancer cases and it was previously shown that nearly 90% of MBC arising in BRCA mutation carriers are found to harbor a BRCA2 mutation [48].
Unexpectedly and interestingly, we found (work in progress in an ongoing study in our laboratory) that the screening of BRCA1/2 genes using NGS method, starting with 54 familial BC patients, revealed a reduced mutational landscape, characterized by the presence of 4 different mutations detected in 47% of the patients tested. Here, BRCA1 c.5309G>T and BRCA2 c.1310_1313delAAGA mutations accounted for the majority (92%) of these mutations. Taken together, these observations suggest either that a small number of mutations exist in this population, although increasing the number of patients is necessary to prove this hypothesis (this work is in progress using NGS to screen a larger number of patients), or that the diversity of the mutations in this population is far from being known.
The occurrence of several breast cancers in the same family is an example of a family history that may signal a genetic predisposition. When a breast cancer risk is suspected, it should lead to genetic counseling. The Moroccan population has limited access to clinical genetics and information regarding genetic counseling is usually delivered by other health-care professionals like oncologists.
The Eisinger’s score assessment [26] is a family tree analysis and is used here to decide whether an oncogenetic consultation is advisable. This scoring model has the advantage of being simpler and allowing the clinician to quickly identify patients for referral to oncogenetic counseling [49].
We have seen in our study that a threshold of 3 for the Eisinger score made it possible to diagnose 94.44% of patients with the disease carrying one of the two BRCA1/2 mutations tested here. Low score observed in one case was probably reflecting a lack of information rather than an absence of cancer occurring within these families. Nevertheless, these findings provide substantial evidence that the Eisinger scoring system is an efficient screening tool method to identify counselees BC patients at high risk for hereditary breast cancer for genetic counseling. Incorporating this carrier prediction algorithm into risk assessment may improve breast cancer management in our population.
On another side, we evaluated the phenotypic spectrum of BC according to the presence of BRCA1 c.5309G>T and BRCA2 c.1310_1313delAAGA mutations. It is noticeable that patients carrying these mutations are more prone to have early breast cancer onset. Although some patients were diagnosed at a relatively middle age (6 patients with age > 45 years), they have family members with BC diagnosed at younger age. Thereby, the relatively middle age of indexes could be due to delay in access to care services rather than late onset of disease.
The clinical features of most BRCA1 c.5309G>T related tumors were more frequently of the ductal invasive type, with intermediate grade and half of them were triple negative. This is in accordance with typical characteristics of BRCA1 pathogenic mutations [18, 22].
Although there was not statistically differences between BRCA1 c.5309G>T carriers group and BRCA2 c.1310_1313delAAGA carriers group in terms of tumor laterality, histopathological subtype, tumor grade, RE and RP expression, we detected proportional differences between patients groups. It was found that BRCA1 c.5309G>T mutation showed more aggressive features than BRCA2 c.1310_1313delAAGA in terms of triple negative status and overall survival. Indeed, we found that patients with BRCA1 c.5309G>T mutation had a significantly worse prognosis regarding OS than negative group. All positive patients are dead except one. On the other hand, BRCA2 c.1310_1313delAAGA mutation carriers were more likely to be diagnosed with breast cancer already spread to regional lymph nodes. Although the overall survival was not statistically affected by the presence of this deletion, there was a trend to reduce survival in many cases (death was reported in 7/12 patients (58%)).
These data indicated that BC patients with BRCA1 c.5309G>T and BRCA2 c.1310_1313delAAGA mutations had poor survival outcomes and hence screening patients with BC for BRCA mutations might help in strategizing their treatment and improving their survival. For persons carrying a constitutional genetic impairment related to HBOC syndrome, an appropriate follow-up strategy based on surveillance and/or preventive surgery should be provided. Performing prophylactic mastectomy to reduce the risk of contralateral breast cancer and prophylactic ovariectomy are two types of surgery proposed to women with BRCA1 and BCRA2 gene abnormalities [50]. Quiles et al. [22] who first described BRCA1 c.5309G>T mutation adopted the following protocol: In general, patients are offered prophylactic mastectomy and prophylactic salpingo-oophorectomy. Annual breast magnetic resonance imaging and mammography is an alternative if the patients do not wish to undergo prophylactic mastectomy. Breast surveillance is offered from the age of 25 and oophorectomy from the age of 35.
Conclusion
The knowledge about the contribution of BRCA1 and BRCA2 mutations in Moroccan BC will lead to better understanding of genetic risk factors of this disease. Altogether, the data in this study indicated that the BRCA1 c.5309G>T and BRCA2 c.1310_1313delAAGA mutations are currently the most significant BRCA genetic cause of BC in Northeastern Moroccan population. Systematic screening for this mutation in BC patients should be considered to facilitate early detection of subjects at high risk of BC among their relatives.
Our study indicates that the Eisinger scoring model could be recommended to decide whether an oncogenetic consultation is advisable to assess the probability of a BRCA1 or BRCA2 mutation.
To our best knowledge, this work represents the first study in North east of Morocco supporting the major contribution of the BRCA1 c.5309G>T and BRCA2 c.1310_1313delAAGA mutations to BC. However, we believe that the sample size is still small and larger cohorts are needed to trace a clear and complete BRCA1/2 mutational spectrum in this population.
Availability of data and materials
The datasets produced and analysed in the present study were deposited in the NCBI Sequence Read Archive. They can be accessed publicly using the BioProject accession number PRJNA949945 and the BioSample accession numbers SAMN33963498 to SAMN33963539. Additionally, the raw Sanger reads of all samples are available for retrieval on the Harvard Dataverse website through the following links: https://doi.org/10.7910/DVN/B5DG3Q and https://doi.org/10.7910/DVN/DAESLS.
Abbreviations
- BC:
-
Breast cancer
- BRCA1/2 :
-
Breast Cancer genes 1/2
- ER:
-
Estrogen receptor
- Her-2:
-
Human epidermal growth factor receptor 2
- NGS:
-
Next generation Sequencing
- OS:
-
Overall surviving
- PR:
-
Progesterone receptor
References
Mahdavi M, Nassiri M, Kooshyar MM, Vakili-Azghandi M, Avan A, Sandry R, et al. Hereditary breast cancer; genetic penetrance and current status with BRCA. J Cell Physiol. 2019;234(5):5741–50. https://doi.org/10.1002/jcp.27464.
Monteiro AN, Bouwman P, Kousholt AN, Eccles DM, Millot GA, Masson JY, et al. Variants of uncertain clinical significance in hereditary breast and ovarian cancer genes: best practices in functional analysis for clinical annotation. J Med Genet. 2020;57(8):509–18. https://doi.org/10.1136/jmedgenet-2019-106368.
Kuchenbaecker KB, Hopper JL, Barnes DR, Phillips KA, Mooij TM, Roos-Blom MJ, et al. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA. 2017;317(23):2402–16. https://doi.org/10.1001/jama.2017.7112.
Narod SA, Foulkes WD. BRCA1 and BRCA2: 1994 and beyond. Nat Rev Cancer. 2004;4(9):665–76. https://doi.org/10.1038/nrc1431.
Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 2002;108(2):171–82. https://doi.org/10.1016/s0092-8674(02)00615-3.
Gordon OK. Cancer of the Breast and Female Reproductive Tract. David Rimoin RPaBK, editor. Emery and Rimoin's Principles and Practice of Medical Genetics (Sixth Edition). Academic Press. Elsevier Science & Technology; 2013. p. 1–31. https://doi.org/10.1016/B978-0-12-383834-6.00093-8.
Laitman Y, Feng BJ, Zamir IM, Weitzel JN, Duncan P, Port D, et al. Haplotype analysis of the 185delAG BRCA1 mutation in ethnically diverse populations. Eur J Hum Genet. 2013;21(2):212–6. https://doi.org/10.1038/ejhg.2012.124.
Ferla R, Calo V, Cascio S, Rinaldi G, Badalamenti G, Carreca I, et al. Founder mutations in BRCA1 and BRCA2 genes. Ann Oncol. 2007;18(Suppl 6):vi93-8. https://doi.org/10.1093/annonc/mdm234.
Daly MB, Pilarski R, Axilbund JE, Berry M, Buys SS, Crawford B, et al. Genetic/Familial High-Risk Assessment: Breast and Ovarian, Version 2.2015. J Natl Compr Canc Netw. 2016;14(2):153–62. https://doi.org/10.6004/jnccn.2016.0018.
Lee A, Moon BI, Kim TH. BRCA1/BRCA2 pathogenic variant breast cancer: treatment and prevention strategies. Ann Lab Med. 2020;40(2):114–21. https://doi.org/10.3343/alm.2020.40.2.114.
Rotimi SO, Rotimi OA, Salhia B. A review of cancer genetics and genomics studies in Africa. Front Oncol. 2020;10:606400. https://doi.org/10.3389/fonc.2020.606400.
Fondation Lalla Salma Prévention et traitement des Cancers. Prévention et traitement des Cancers. Registre des cancers de la région du Grand Casablanca pour la période 2008–2012. 2016.
Hashim MJ, Al-Shamsi FA, Al-Marzooqi NA, Al-Qasemi SS, Mokdad AH, Khan G. Burden of breast cancer in the Arab World: findings from Global Burden of Disease, 2016. J Epidemiol Glob Health. 2018;8(1–2):54–8. https://doi.org/10.2991/j.jegh.2018.09.003.
Mrabti H, Sauvaget C, Benider A, Bendahhou K, Selmouni F, Muwonge R, et al. Patterns of care of breast cancer patients in Morocco - A study of variations in patient profile, tumour characteristics and standard of care over a decade. Breast. 2021;59:193–202. https://doi.org/10.1016/j.breast.2021.07.009.
Tazzite A, Jouhadi H, Saiss K, Benider A, Nadifi S. Relationship between family history of breast cancer and clinicopathological features in Moroccan patients. Ethiop J Health Sci. 2013;23(2):150–7.
Laarabi FZ, Jaouad IC, Ouldim K, Aboussair N, Jalil A, Gueddari BE, et al. Genetic testing and first presymptomatic diagnosis in Moroccan families at high risk for breast/ovarian cancer. Oncol Lett. 2011;2(2):389–93. https://doi.org/10.3892/ol.2011.248.
Tazzite A, Jouhadi H, Nadifi S, Aretini P, Falaschi E, Collavoli A, et al. BRCA1 and BRCA2 germline mutations in Moroccan breast/ovarian cancer families: novel mutations and unclassified variants. Gynecol Oncol. 2012;125(3):687–92. https://doi.org/10.1016/j.ygyno.2012.03.007.
Laraqui A, Uhrhammer N, Lahlou-Amine I, El Rhaffouli H, El Baghdadi J, Dehayni M, et al. Mutation screening of the BRCA1 gene in early onset and familial breast/ovarian cancer in Moroccan population. Int J Med Sci. 2013;10(1):60–7. https://doi.org/10.7150/ijms.5014.
Bakkach J, Mansouri M, Derkaoui T, Loudiyi A, El Fahime E, Barakat A, et al. Contribution of BRCA1 and BRCA2 germline mutations to early onset breast cancer: a series from north of Morocco. BMC Cancer. 2020;20(1):859. https://doi.org/10.1186/s12885-020-07352-9.
Guaoua S, Ratbi I, Lyahyai J, El Alaoui SC, Laarabi FZ, Sefiani A. Novel nonsense mutation of BRCA2 gene in a Moroccan man with familial breast cancer. Afr Health Sci. 2014;14(2):468–71. https://doi.org/10.4314/ahs.v14i2.25.
El Ansari FZ, Jouali F, Marchoudi N, Bennani MM, Ghailani NN, Barakat A, et al. Screening of BRCA1/2 genes mutations and copy number variations in patients with high risk for hereditary breast and ovarian cancer syndrome (HBOC). BMC Cancer. 2020;20(1):747. https://doi.org/10.1186/s12885-020-07250-0.
Quiles F, Teule A, Martinussen Tandstad N, Feliubadalo L, Tornero E, Del Valle J, et al. Identification of a founder BRCA1 mutation in the Moroccan population. Clin Genet. 2016;90(4):361–5. https://doi.org/10.1111/cge.12747.
Jouali F, Laarabi FZ, Marchoudi N, Ratbi I, Elalaoui SC, Rhaissi H, et al. First application of next-generation sequencing in Moroccan breast/ovarian cancer families and report of a novel frameshift mutation of the BRCA1 gene. Oncol Lett. 2016;12(2):1192–6. https://doi.org/10.3892/ol.2016.4739.
Laarabi FZ, Ratbi I, Elalaoui SC, Mezzouar L, Doubaj Y, Bouguenouch L, et al. High frequency of the recurrent c.1310_1313delAAGA BRCA2 mutation in the North-East of Morocco and implication for hereditary breast-ovarian cancer prevention and control. BMC Res Notes. 2017;10(1):188. https://doi.org/10.1186/s13104-017-2511-2.
Dutil J, Colon-Colon JL, Matta JL, Sutphen R, Echenique M. Identification of the prevalent BRCA1 and BRCA2 mutations in the female population of Puerto Rico. Cancer Genet. 2012;205(5):242–8. https://doi.org/10.1016/j.cancergen.2012.04.002.
Eisinger F, Bressac B, Castaigne D, Cottu PH, Lansac J, Lefranc JP, et al. Identification and management of hereditary breast/ovarian cancers (2004 update). La Lettre du Gynécologue. 2004;294:26–37.
Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16(3):1215. https://doi.org/10.1093/nar/16.3.1215.
Untergasser A, Cutcutache I, koressaar T, Ye J, Faircloth BC, Remm M, et al. Primer3 - new capabilities and interfaces. 2012.
Grindedal EM, Heramb C, Karsrud I, Ariansen SL, Maehle L, Undlien DE, et al. Current guidelines for BRCA testing of breast cancer patients are insufficient to detect all mutation carriers. BMC Cancer. 2017;17(1):438. https://doi.org/10.1186/s12885-017-3422-2.
Heramb C, Wangensteen T, Grindedal EM, Ariansen SL, Lothe S, Heimdal KR, et al. BRCA1 and BRCA2 mutation spectrum - an update on mutation distribution in a large cancer genetics clinic in Norway. Hered Cancer Clin Pract. 2018;16:3. https://doi.org/10.1186/s13053-017-0085-6.
Mansouri M, Derkaoui T, Bakkach J, Loudiyi A, Ghailani Nourouti N, Barakat A, et al. Screening of BRCA1 and BRCA2 germline mutations in unselected triple-negative breast cancer patients: A series from north of Morocco. Precis Med Sci. 2020;9:43–8. https://doi.org/10.1002/prm2.12009.
Tudini E, Moghadasi S, Parsons MT, van der Kolk L, van den Ouweland AMW, Niederacher D, et al. Substantial evidence for the clinical significance of missense variant BRCA1 c.5309G>T p.(Gly1770Val). Breast Cancer Res Treat. 2018;172(2):497–503. https://doi.org/10.1007/s10549-018-4903-y.
Bouwman P, van der Gulden H, van der Heijden I, Drost R, Klijn CN, Prasetyanti P, et al. A high-throughput functional complementation assay for classification of BRCA1 missense variants. Cancer Discov. 2013;3(10):1142–55. https://doi.org/10.1158/2159-8290.CD-13-0094.
Quiles F, Fernandez-Rodriguez J, Mosca R, Feliubadalo L, Tornero E, Brunet J, et al. Functional and structural analysis of C-terminal BRCA1 missense variants. PLoS One. 2013;8(4):e61302. https://doi.org/10.1371/journal.pone.0061302.
Cherbal F, Bakour R, Adane S, Boualga K, Benais-Pont G, Maillet P. BRCA1 and BRCA2 germline mutations screening in Algerian breast/ovarian cancer families. Dis Markers. 2010;28(6):377–84. https://doi.org/10.3233/DMA-2010-0718.
Fourati A, Louchez MM, Fournier J, Gamoudi A, Rahal K, El May MV, et al. Screening for common mutations in BRCA1 and BRCA2 genes: interest in genetic testing of Tunisian families with breast and/or ovarian cancer. Bull Cancer. 2014;101(11):E36-40. https://doi.org/10.1684/bdc.2014.2049.
Ben Ayed-Guerfali D, Ben Kridis-Rejab W, Ammous-Boukhris N, Ayadi W, Charfi S, Khanfir A, et al. Novel and recurrent BRCA1/BRCA2 germline mutations in patients with breast/ovarian cancer: a series from the south of Tunisia. J Transl Med. 2021;19(1):108. https://doi.org/10.1186/s12967-021-02772-y.
Caputo S, Benboudjema L, Sinilnikova O, Rouleau E, Beroud C, Lidereau R, et al. Description and analysis of genetic variants in French hereditary breast and ovarian cancer families recorded in the UMD-BRCA1/BRCA2 databases. Nucleic Acids Res. 2012;40(Database issue):D992-1002. https://doi.org/10.1093/nar/gkr1160.
Meisel C, Sadowski CE, Kohlstedt D, Keller K, Staritz F, Grubling N, et al. Spectrum of genetic variants of BRCA1 and BRCA2 in a German single center study. Arch Gynecol Obstet. 2017;295(5):1227–38. https://doi.org/10.1007/s00404-017-4330-z.
Tea MK, Kroiss R, Muhr D, Fuerhauser-Rappaport C, Oefner P, Wagner TM, et al. Central European BRCA2 mutation carriers: birth cohort status correlates with onset of breast cancer. Maturitas. 2014;77(1):68–72. https://doi.org/10.1016/j.maturitas.2013.09.012.
Vos JR, Teixeira N, van der Kolk DM, Mourits MJ, Rookus MA, van Leeuwen FE, et al. Variation in mutation spectrum partly explains regional differences in the breast cancer risk of female BRCA mutation carriers in the Netherlands. Cancer Epidemiol Biomarkers Prev. 2014;23(11):2482–91. https://doi.org/10.1158/1055-9965.EPI-13-1279.
Wojcik P, Jasiowka M, Strycharz E, Sobol M, Hodorowicz-Zaniewska D, Skotnicki P, et al. Recurrent mutations of BRCA1, BRCA2 and PALB2 in the population of breast and ovarian cancer patients in Southern Poland. Hered Cancer Clin Pract. 2016;14:5. https://doi.org/10.1186/s13053-016-0046-5.
Cruz-Correa M, Perez-Mayoral J, Dutil J, Echenique M, Mosquera R, Rivera-Roman K, et al. Hereditary cancer syndromes in Latino populations: genetic characterization and surveillance guidelines. Hered Cancer Clin Pract. 2017;15:3. https://doi.org/10.1186/s13053-017-0063-z.
El Saghir NS, Zgheib NK, Assi HA, Khoury KE, Bidet Y, Jaber SM, et al. BRCA1 and BRCA2 mutations in ethnic Lebanese Arab women with high hereditary risk breast cancer. Oncologist. 2015;20(4):357–64. https://doi.org/10.1634/theoncologist.2014-0364.
Donenberg T, Ahmed H, Royer R, Zhang S, Narod SA, George S, et al. A Survey of BRCA1, BRCA2, and PALB2 mutations in women with breast cancer in Trinidad and Tobago. Breast Cancer Res Treat. 2016;159(1):131–8. https://doi.org/10.1007/s10549-016-3870-4.
Gayther SA, Russell P, Harrington P, Antoniou AC, Easton DF, Ponder BA. The contribution of germline BRCA1 and BRCA2 mutations to familial ovarian cancer: no evidence for other ovarian cancer-susceptibility genes. Am J Hum Genet. 1999;65(4):1021–9. https://doi.org/10.1086/302583.
Maillet P, Chappuis PO, Khoshbeen-Boudal M, Sciretta V, Sappino AP, Testing SNfCP, et al. Twenty-three novel BRCA1 and BRCA2 sequence variations identified in a cohort of Swiss breast and ovarian cancer families. Cancer Genet Cytogenet. 2006;169(1):62–8. https://doi.org/10.1016/j.cancergencyto.2006.03.010.
Silvestri V, Barrowdale D, Mulligan AM, Neuhausen SL, Fox S, Karlan BY, et al. Male breast cancer in BRCA1 and BRCA2 mutation carriers: pathology data from the Consortium of Investigators of Modifiers of BRCA1/2. Breast Cancer Res. 2016;18(1):15. https://doi.org/10.1186/s13058-016-0671-y.
Bonaiti B, Alarcon F, Bonadona V, Pennec S, Andrieu N, Stoppa-Lyonnet D, et al. A new scoring system for the diagnosis of BRCA1/2 associated breast-ovarian cancer predisposition. Bull Cancer. 2011;98(7):779–95. https://doi.org/10.1684/bdc.2011.1397.
Deng M, Chen HH, Zhu X, Luo M, Zhang K, Xu CJ, et al. Prevalence and clinical outcomes of germline mutations in BRCA1/2 and PALB2 genes in 2769 unselected breast cancer patients in China. Int J Cancer. 2019;145(6):1517–28. https://doi.org/10.1002/ijc.32184.
Acknowledgements
We would like to thank all the patients for their participation in this study.
Funding
Internal support for this study was provided by the Mohammed First University-Oujda, Morocco.
Author information
Authors and Affiliations
Contributions
R.M. conceived the study, carried out mutation screening, performed the statistical analyses, interpreted the results and wrote the manuscript. N.B. conceived and coordinated the study and drafted manuscript. M.M. carried out NGS analysis. T.E. was involved in the recruitment of the patients and provided the clinical data. S.A. contributed to clinical data management. All authors reviewed the manuscript. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethic approval and consent to participate
The study protocol was reviewed and approved by the ethics committee for Biomedical Research of the Faculty of Medicine and Pharmacy of Casablanca under the number 06/18.
Informed and written consent was obtained from all individual participants included in the study.
All methods were carried out in accordance with relevant guidelines and regulations.
Consent for publication
Not Applicale.
Competing interests
The authors declare that they have no competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Melki, R., Melloul, M., Aissaoui, S. et al. Increased prevalence of the founder BRCA1 c.5309G>T and recurrent BRCA2 c.1310_1313delAAGA mutations in breast cancer families from Northerstern region of Morocco: evidence of geographical specificity and high relevance for genetic counseling. BMC Cancer 23, 339 (2023). https://doi.org/10.1186/s12885-023-10822-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-023-10822-5