
Abstract
Background
To date, the contribution of BRCA1/2 mutations in Moroccan early onset breast cancer patients remains unknown. Here we assess these genetic alterations for the first time in a cohort from North of Morocco.
Methods
Thirty-three patients diagnosed with breast cancer at the age of ≤40 years were recruited irrespective of breast and/or ovarian cancer family history. Coding regions and intron-exon boundaries of BRCA1 and BRCA2 genes were sequenced from peripheral blood DNA using Ion Proton (Thermo Fisher Scientific) next generation sequencing platform.
Results
Overall, five BRCA germline mutations were identified (15.1%). The frequency of mutations among patients with family history of breast cancer was 16.7%. Three mutations were found in BRCA1 (9%) and two within the BRCA2 gene (6%). These are three frameshift mutations (c.798_799del, c.2125_2126insA, c.5116_5119delAATA), one missense (c.116G > A) and one nonsense mutation (c.289G > T). The mutation c.5116_5119delAATA has a founder effect in North Africa. Moreover, one variant of unknown significance was identified in BRCA2 (c.4090A > G). Most BRCA mutations carriers (80%) had no family history of breast cancer.
Conclusion
Our data do not support the hypothesis that BRCA mutations alone explain the higher frequency of breast cancer in Moroccan young women. The young age (≤40 years) for breast cancer diagnosis seems to be strongly predictive of BRCA mutation status in Moroccan patients. These results will help in decision making with regard to genetic counseling and testing in the national scale.
Similar content being viewed by others

Background
In Morocco, and other less developed countries, breast cancer occurs more frequently in young population. The young age for development of breast cancer often suggests a genetic predisposition especially germline mutations in BRCA1 and BRCA2 genes. These genetic alterations were shown to be involved in up to 12% of early-onset breast cancer (EOBC) cases in western countries [1]. It is well established that the frequency of these mutations differs according to inclusion criteria, screening methods, geographical location and ethnic origin. However, the prevalence of BRCA1/2 mutations is still unknown among Moroccan young breast cancer patients and it is still unclear whether these genetic factors may explain the higher rate of EOBC in Morocco.
The aim of the present study was to analyze BRCA1 and BRCA2 germline mutations in EOBC patients who were unselected for a family history. To the best of our knowledge, this is the first genetic study of its kind carried out in North of Morocco and the first study that used only young age as selection criteria irrespective of family history. This work is an important milestone in determining the prevalence of BRCA1/2 germline mutations in EOBC in the Moroccan population.
Methods
Study patients
From January 2010 to December 2015, a total of 82 young patients (≤ 40 years) with invasive breast carcinoma were referred to the Oncology Clinic AL AMAL in Tangier. Clinico-pathologic, prognostic and therapeutic features of the whole group were previously published [2]. Alive patients were invited to participate in the genetic study and were informed about potential implications of the test. All these patients were selected based on young age (≤ 40 years) irrespective of family history of breast and/or ovarian cancer. Thirty-four patients agreed to participate in the study (One patient withdrew consent for personal reasons). All recruited patients elected to receive their results.
Clinico-pathologic data was collected from medical records. Information about family history was completed during recruitment.
This study was approved by the Biomedical Research Ethics Committee in the Faculty of Medicine and Pharmacy in Rabat (CERB) and an informed written consent was obtained from all participants. Blood samples were obtained from each patient during the interview process. Results were treated with confidentiality and were not included in medical records.
Genetic testing
Genomic DNA was isolated from 200 μL peripheral-blood using QIAamp DNA isolation kit and stored at − 20 °C. Mutational analysis was performed at GENETRACER BIOTECH Laboratory, Cantabria in Spain or at the National Center of Scientific and Technical Research, Rabat (Units of Technical Support to Scientific Research) in Morocco. All coding sequences of BRCA1 and BRCA2 genes and exon–intron boundaries were analyzed using the Ion Proton (Thermo Fisher Scientific) next generation sequencing platform. The NGS library was constructed by a multiplex polymerase chain reaction (PCR)-based assay (Ampliseq technology) using Oncomine BRCA Research Assay according to the manufacturer’s protocol. Reads were aligned to the human genome reference sequence 19 (hg19) using Torrent Browser 5.2 software and Ion Reporter 5.2. Exonic sequence analysis was performed with an average coverage greater than 4500X.
The results obtained with the Ion Reporter 5.2 software were filtered by eliminating all the alterations considered as common polymorphisms by the University of California Santa Cruz (UCSC) genome browser. All recorded variants were germline heterozygous mutations. The mutations obtained by this filtering process were verified in the UCSC genome browser and in various databases including EXAC, dbSNP, COSMIC, UMD, LOVD, BIC and ClinVar and also in the scientific literature. Finally, the results obtained were verified by the algorithm Sophia DDM™-BRCA-Ampliseq™-GL-IonProton™ (Sophia Genetics).
Patients with pathogenic or likely pathogenic variants were considered to be mutation carriers. Patients with normal sequencing results and those with variants of unknown clinical significance (VUS), benign or probably benign variants were considered as non-carriers.
Results
The median age at diagnosis was 35 years (range: 28–40). More than half of patients (51.5%) were diagnosed before the age of 35 and 48.5% were aged 36–40 years old. Family history was unknown for one patient. A family history for breast cancer (1st or 2nd degree) was recorded for 18.8% of cases. Two patients had bilateral cancer. Almost one quarter of patients (24.2%) were presented at an advanced stage (Locally advanced: 21.2% and metastatic: 3%). Most patients had invasive carcinoma of no special type (NST) (93.9%), and 3% were diagnosed with lobular or mucinous carcinoma. 51.5 and 39.4% of patients had intermediate and high grade carcinomas, respectively and 30.3% of cases had triple negative tumors.
Among the 33 patients aged ≤40 years who were unselected for family history, five BRCA mutations were identified in five unrelated patients (15.1%). Three mutations were found in BRCA1 gene (9%) and two in BRCA2 (6%). Of the 17 patients who were aged ≤35 years, the frequency of BRCA1/2 mutations was 23.5%. Of the 16 patients aged 36–40 years, 6.2% were BRCA1/2 mutations carriers. When stratified by family history (1st or 2nd degree), 4 of 26 (15.4%) who were aged ≤40 with negative family history had BRCA mutations (3 BRCA1 and 1 BRCA2). In contrast, only 1 of 6 patients (16.7%) who were aged ≤40 with positive family history had BRCA mutations (1 BRCA2).
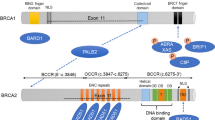
Of the three mutations identified on BRCA1 gene, two were located on exon 11 and one on exon 3. Of the two BRCA2 mutations, one was located on exon 11 and the other on exon 3. These include three frameshift mutations: BRCA1: c.798_799del (p.Ser267Lysfs), BRCA1: c.2125_2126insA (p.Phe709Tyrfs), BRCA1: c.5116_5119delAATA (p.Asn1706Leufs), one missense BRCA1: c.116G > A (p.Cys39Tyr) and one nonsense mutation BRCA2: c.289G > T (p.Glu97Ter). The mutation BRCA1: c.116G > A (p.Cys39Tyr) is a substitution of G with A at position 39 at exon 3 of the BRCA1 gene. The mutation BRCA1:c.2125_2126insA (p.Phe709Tyrfs) is an insertion of an A that produces an alteration in the reading frame and leads to a STOP codon in exon 10 of BRCA1. The mutation BRCA1: c.798_799del (p.Ser267Lysfs) is a deletion of 2 T bases that leads to the premature termination of protein synthesis at codon 285 of exon 10 of BRCA1. The mutation BRCA2: c.5116_5119delAATA (p.Asn1706Leufs) is a deletion of AATA that produces alteration in the reading frame and leads to a STOP codon in exon 11 of BRCA2. The mutation BRCA2: c.289G > T (p.Glu97Ter) is a G-base substitution by T leading to a premature STOP codon at exon 3 of the BRCA2 gene (Table 1).
All mutations have been identified only once. A single mutation was not previously reported in BIC, but has already been described in the literature BRCA2: c.289G > T (p.Glu97Ter). No patient presents simultaneously two mutations.
In addition to these mutations, one BRCA2 VUS was found BRCA2: c.4090A > G (p.Ile1364Val) and co-occurred with a likely pathogenic mutation BRCA1: c.116G > A (p.Cys39Tyr). Another variant BRCA2: c.6322C > T (p.Arg2108Cys) with conflicting interpretations of pathogenicity was also identified in BRCA2 (Table 1).
Most (80%) BRCA mutation carriers were aged ≤35 years, without 1st or 2nd degree breast cancer family history, had early-stage disease, and were diagnosed with unilateral breast cancer (4/5). All mutation carriers had NST carcinoma. Furthermore, 60% of our mutated patients had high-grade and triple negative tumors (3/5) (Table 2).
Discussion
Overall, only few genetic studies aiming to analyze BRCA1/2 germline mutations among unselected young breast cancer patients have been reported [3,4,5,6,7,8,9,10]. Historically, the prevalence among women aged under 40 varies from 4.8 to 11.6% [6, 8, 9, 11]. Our frequency (15.1%) is significantly higher than these western data. This large difference could be explained by the increased sensitivity of our screening method (NGS), unlike those older series which used mostly indirect techniques for mutations screening such as heteroduplex, Single Strand Conformation Polymorphism (SSCP), Denaturing Gradient Gel Electropheresis (DGGE)... More recently, a large multi-center study recruiting 2733 cases from 127 hospitals in the United Kingdom (POSH study) and using a NGS platform reported a frequency of 12% [1]. Our mutation rate did not differ greatly from these data suggesting thus similar contribution in western and Moroccan EOBC. Based on these data and in light of the limitations of our work, it seems that our findings do not support the hypothesis that BRCA mutations alone may explain the higher incidence of EOBC in our population. Larger multicenter studies are warranted to verify our results.
Studies among unselected EOBC (< 40 years) from Middle East and North Africa (MENA) region showed disparate results with a BRCA mutation rate ranging from 0 to 26% [12,13,14,15] (Table 3). Fortunately, all these reports have sequenced both BRCA genes and also analyzed large genomic rearrangements, unlike our work. However, the limited size of most of these studies makes it difficult to draw firm conclusions about the contribution of BRCA alterations to EOBC in this world region. In Morocco, to the best of our knowledge, the current study is the first study for BRCA testing in unselected EOBC patients. Earlier reports have been drawn from unusual cancer-rich or cancer-free families [16,17,18] that are not representative of the family history profiles of women in the general population who develop breast carcinoma in young age.
It has been suggested that BRCA1 and BRCA2 genes contribute equally to EOBC. We described a BRCA1/BRCA2 ratio of 1.5 (BRCA1 mutations: 9% and BRCA2 mutations: 6%), consistent with that reported in POSH study and other large population-based studies [1, 33, 34].
The BRCA1:c.116G > A (p.Cys39Tyr) mutation is classified as pathogenic in Clinvar with a pending classification in BIC. It is a recurrent mutation in Slovenia [35,36,37]. It was also found in North eastern Italy [38], in India [39], but never been reported in Morocco. This mutation affects the homologous recombination, leads to centrosome amplification, and may affect the formation of the BARD1/BRCA1 heterodimer [40]. This mutation was simultaneously present with a VUS, BRCA2:c.4090A > G (p.Ile1364Val), suggesting that this latter is a neutral variant. BRCA2:c.4090A > G has already been reported in the Moroccan population [16, 41]. In silico analysis with PolyPhen suggests lack of pathogenicity: PolyPhen-2 v2.2.2r398--HumDiv: Benign with a score of 0.005 (sensitivity: 0.97; specificity: 0.74); HumVar: Benign with a score of 0.003 (sensitivity: 0.98; specificity: 0.26). In silico analysis with NNSPLICE, MaxEntScan and Splice Site Finder show the creation of a donor site with a possible splicing effect.
The BRCA1:c.2125_2126insA (p.Phe709Tyrfs) mutation is a recurrent mutation in the French-Canadian population [42,43,44,45,46]. It has been also found in the United Kingdom [1]. More recently, it has been reported in Algeria in three young patients aged 33, 34 and 38 years respectively, all with triple negative cancer and a family history of breast and/or prostate cancer [47].
The BRCA1:c.798_799del (p.Ser267Lysfs) mutation has been detected in four Hispanic patients [48]. It was also identified in two Tunisian and Algerian families with a common haplotype to all carriers of this mutation [20], suggesting the presence of the first non-Jewish founder effect in North Africa. It was later identified in four Algerian patients [21, 47], three Moroccan women [16, 17] and five Tunisian women [22], all with a family history of breast or ovarian cancer and aged between 34 and 44 years.
The BRCA2:c.289G > T (p.Glu97Ter) mutation is not mentioned in BIC, but already reported in the literature. It has been identified in one patient in central Italy [49], in France [50] and in Korea [51].
The BRCA2:c.5116_5119delAATA (p.Asn1706Leufs) mutation is reported only once in BIC with a Caucasian ethnic origin. It was detected for the first time in northwestern Spain in Castilla y León [52] and has been described as a recurrent founder mutation in this region. Interestingly, it has been associated with EOBC (mean age 37.4 years; p = 0.033) [53]. Overall, this mutation has been identified in a group of eight independent Spanish families [53,54,55], and also in two Korean patients [56].
In summary, according to our data, it seems that there is a great heterogeneity in the spectrum of BRCA1/2 mutations in our population study. More specifically, this spectrum appears to be shared with other Mediterranean populations, which can be explained by the geographic location of our country.
The vast majority of BRCA1/2 mutations identified in our work occur in a non-family context (80%). This may be due to low penetrance of BRCA mutations due to genetic or environmental risk modifying factors. Another possible explanation is the limited size of families of mutation carriers, or a possible preponderance of males or normal alleles rather than segregated altered alleles. The presence of a memory bias is also not excluded since family history data were collected retrospectively after the cancer diagnosis and this information was not verified.
Young age of onset for breast cancer is a testing criterion in already published recommendations with a disparity in the age thresholds between guidelines (National comprehensive cancer network: ≤45 years, and Saint Gallen consensus: ≤35) [57, 58]. However, in Morocco the prevalence of BRCA1/2 mutations is yet to be well defined, which adds further challenge in tailoring international guidelines to the national context. We have shown that the frequency of mutations among young patients who were unselected for family history (15.1%) exceeds the probability threshold required for BRCA genetic testing (≥10%). Moreover, the presence of a family history seems to raise only slightly the mutations rate (16.7%) and a significant proportion of mutation carriers were sporadic cases (80%). Hence, limiting BRCA testing to high-risk women could lead to miss a significant number of mutations. In light of these data, we suggest that young age alone is a sufficient criterion to indicate a systematic genetic screening and that the age limit of ≤40 years is an adequate threshold for our population. This remains to be further investigated.
The present study shows some limitations. The sample size of our analyzed series (n = 33) and the number of detected BRCA1/2 mutations (n = 5) were small, thus we could not provide conclusive data about the prevalence and spectrum of BRCA1/2 mutations in young breast cancer patients from North of Morocco. The time interval between patient’s recruitment and molecular analysis may be another limitation for our study. Alive patients who were referred to our Clinic between January 2010 to December 2015 were recruited and molecular analysis was carried out in 2017 (33 patients from 82 initially identified). The patients “selection” by delayed molecular analysis can change some characteristics such as family history, metastatic cancer and triple negative breast cancers rates (26% in the initial group Vs. 18.8% in the analyzed group, 12.2% Vs. 3%, and 23% Vs. 30.3%, respectively) [2]. Lastly, our analysis was restricted to BRCA1/2 mutations and did not include large rearrangements nor copy number variations.
Conclusion
Our frequency of BRCA1/2 germline mutations does not differ greatly to Western data. These genetic alterations alone seem not to explain the higher incidence of breast cancer in young Moroccan women in contrast to what was expected. The implication of other gene mutations such as PALB2, TP53 should be investigated. Copy number variations were highly biologically relevant in breast cancer and their study would be of interest as some of them were suggested to contribute to the aggressive nature of tumors arising in younger patients.
Our data suggest that young age (≤ 40 years) irrespective of family history is a sufficient criterion for systematic genetic screening for Moroccan young breast cancer patients, following the western recommendations for BRCA1/2 genetic testing.
Finally, our study could not provide conclusive data due to some limitations including possible “patients selection” by delayed molecular analysis (retrospective analysis).Larger and prospective studies are warranted to confirm our findings.
Availability of data and materials
All the variants found in this study were deposited in the LOVD database: #0000643111(https://databases.lovd.nl/shared/variants/0000643111#00003478);
#0000643125 (https://databases.lovd.nl/shared/variants/0000643125#00003478);
#0000643126 (https://databases.lovd.nl/shared/variants/0000643126#00003478);
#0000643127 (https://databases.lovd.nl/shared/variants/0000643127#00003479);
#0000643128 (https://databases.lovd.nl/shared/variants/0000643128#00003479);
#0000643129 (https://databases.lovd.nl/shared/variants/0000643129#00003479);
#0000643130 (https://databases.lovd.nl/shared/variants/0000643130#00003479).
Abbreviations
- BIC:
-
Breast cancer Information Core
- DGGE:
-
Denaturing Gradient Electrophoresis
- EOBC:
-
Early Onset Breast Cancer
- MENA:
-
Middle East and North Africa
- MLPA:
-
Multiplex Ligation Probe Amplification
- NGS:
-
Next Generation Sequencing
- NST:
-
Invasive carcinoma of no special type
- SSCP:
-
Single-Strand Conformation Polymorphism
- UCSC:
-
University of California Santa Cruz
- VUS:
-
Variant of Unknown Significance
References
Copson ER, Maishman TC, Tapper WJ, Cutress RI, Greville-Heygate S, Altman DG, et al. Germline BRCA mutation and outcome in young-onset breast cancer (POSH): a prospective cohort study. Lancet Oncol. 2018;19:169–80.
Bakkach J, Mansouri M, Derkaoui T, Loudiyi A, Fihri M, Hassani S, et al. Clinicopathologic and prognostic features of breast cancer in young women: a series from north of Morocco. BMC Womens Health. 2017;17:106.
Langston AA, Malone KE, Thompson JD, Daling JR, Ostrander EA. BRCA1 mutations in a population-based sample of young women with breast Cancer. N Engl J Med. 1996;334:137–42.
Peto J, Collins N, Barfoot R, Seal S, Warren W, Rahman N, et al. Prevalence of BRCA1 and BRCA2 gene mutations in patients with early-onset breast Cancer. JNCI J Natl Cancer Inst. 1999;91:943–9.
Malone KE, Daling JR, Neal C, Suter NM, O’Brien C, Cushing-Haugen K, et al. Frequency ofBRCA1/BRCA2 mutations in a population-based sample of young breast carcinoma cases. Cancer. 2000;88:1393–402.
Loman N, Johannsson O, Kristoffersson U, Olsson H, Borg A. Family history of breast and ovarian cancers and BRCA1 and BRCA2 mutations in a population-based series of early-onset breast cancer. J Natl Cancer Inst. 2001;93:1215–23.
Tonin PN, Perret C, Lambert JA, Paradis A-J, Kantemiroff T, Benoît M-H, et al. FounderBRCA1 andBRCA2 mutations in early-onset French Canadian breast cancer cases unselected for family history. Int J Cancer. 2001;95:189–93.
de Sanjosé S, Léoné M, Bérez V, Izquierdo A, Font R, Brunet JM, et al. Prevalence of BRCA1 and BRCA2 germline mutations in young breast cancer patients: a population-based study: BRCA and breast Cancer in young women. Int J Cancer. 2003;106:588–93.
Martínez-Ferrandis JI, Vega A, Chirivella I, Marín-García P, Insa A, Lluch A, et al. Mutational analysis of BRCA1 and BRCA2 in Mediterranean Spanish women with early-onset breast cancer: identification of three novel pathogenic mutations. Hum Mutat. 2003;22:417–8.
Koumpis C, Dimitrakakis C, Antsaklis A, Royer R, Zhang S, Narod SA, et al. Prevalence of BRCA1 and BRCA2 mutations in unselected breast cancer patients from Greece. Hered Cancer Clin Pract. 2011;9:10.
Anton-Culver H, Cohen PF, Gildea ME, Ziogas A. Characteristics of BRCA1 mutations in a population-based case series of breast and ovarian cancer. Eur J Cancer. 2000;36:1200–8.
El Saghir NS, Zgheib NK, Assi HA, Khoury KE, Bidet Y, Jaber SM, et al. BRCA1 and BRCA2 mutations in ethnic Lebanese Arab women with high hereditary risk breast Cancer. Oncologist. 2015;20:357–64.
Henouda S, Bensalem A, Reggad R, Serrar N, Rouabah L, Pujol P. Contribution of BRCA1 and BRCA2 Germline mutations to early Algerian breast Cancer. Dis Markers. 2016;2016:1–7.
Abdel-Razeq H, Al-Omari A, Zahran F, Arun B. Germline BRCA1/BRCA2 mutations among high risk breast cancer patients in Jordan. BMC Cancer. 2018;18:152.
Ebrahimi E, Sellars E, Shirkoohi R, Harirchi I, Ghiasvand R, Mohebbi E et al. NGS-Based BRCA1, BRCA2, and PALB2 Mutation Testing in Iranian Population With Breast Cancer. J Glob Oncol. 2018; 4 suppl (2):208s-208s.
Tazzite A, Jouhadi H, Nadifi S, Aretini P, Falaschi E, Collavoli A, et al. BRCA1 and BRCA2 germline mutations in Moroccan breast/ovarian cancer families: novel mutations and unclassified variants. Gynecol Oncol. 2012;125:687–92.
Laraqui A, Uhrhammer N, Lahlou-Amine I, Rhaffouli HE, Baghdadi JE, Dehayni M, et al. Mutation screening of the BRCA1 gene in early onset and familial breast/ovarian Cancer in Moroccan population. Int J Med Sci. 2013;10:60–7.
Tazzite A, Nadiffi S, Kottwitz D, El Amrani M, Jouhadi H, Benider A, et al. Specific BRCA1 gene variations amongst young Moroccan breast cancer patients. Genet Mol Res. 2014;13:791–8.
Jouali F, Laarabi F-Z, Marchoudi N, Ratbi I, Elalaoui SC, Rhaissi H, et al. First application of next-generation sequencing in Moroccan breast/ovarian cancer families and report of a novel frameshift mutation of the BRCA1 gene. Oncol Lett. 2016;12:1192–6.
Uhrhammer N, Abdelouahab A, Lafarge L, Feillel V, Ben Dib A, Bignon Y-J. BRCA1 mutations in Algerian breast cancer patients: high frequency in young, sporadic cases. Int J Med Sci. 2008;5:197–202.
Cherbal F, Bakour R, Adane S, Boualga K, Benais-Pont G, Maillet P. BRCA1 and BRCA2 Germline mutations screening in Algerian breast/ovarian Cancer families. Dis Markers. 2010;28(6):377–84.
Mahfoudh W, Bouaouina N, Ahmed SB, Gabbouj S, Shan J, Mathew R, et al. Hereditary breast cancer in middle eastern and north African (MENA) populations: identification of novel, recurrent and founder BRCA1 mutations in the Tunisian population. Mol Biol Rep. 2012;39:1037–46.
Riahi A, Kharrat M, Ghourabi ME, Khomsi F, Gamoudi A, Lariani I, et al. Mutation spectrum and prevalence of BRCA 1 and BRCA 2 genes in patients with familial and early-onset breast/ovarian cancer from Tunisia: mutation spectrum and prevalence of BRCA 1 and BRCA 2 genes. Clin Genet. 2015;87:155–60.
Ibrahim SS, Hafez EE, Hashishe MM. Presymptomatic breast cancer in Egypt: role of BRCA1 and BRCA2 tumor suppressor genes mutations detection. J Exp Clin Cancer Res. 2010;29:82.
Yassaee VR, Zeinali S, Harirchi I, Jarvandi S, Mohagheghi MA, Hornby DP, et al. Novel mutations in the BRCA1 and BRCA2genes in Iranian women with early-onset breast cancer. Breast Cancer Res. 2002;4:R6.
Pietschmann A, Mehdipour P, Mehdipour P, Atri M, Hofmann W, Hosseini-Asl SS, et al. Mutation analysis of BRCA1 and BRCA2 genes in Iranian high risk breast cancer families. J Cancer Res Clin Oncol. 2005;131:552–8.
Keshavarzi F, Javadi GR, Zeinali S. BRCA1 and BRCA2 germline mutations in 85 Iranian breast cancer patients. Familial Cancer. 2012;11:57–67.
Yassaee VR, Ravesh Z, Soltani Z, Hashemi-Gorji F, Poorhosseini SM, Anbiaee R, et al. Mutation spectra of BRCA genes in Iranian women with early onset breast Cancer - 15 years experience. Asian Pac J Cancer Prev. 2016;17:149–53.
Khalil H, Monem F, Al-Quobaili F. Identification of three BRCA1/2 mutations and a study of the likelihood of an association with certain characteristics in Syrian familial breast Cancer patients. Middle East J Cancer. 2018;9:274–81.
Lolas Hamameh S, Renbaum P, Kamal L, Dweik D, Salahat M, Jaraysa T, et al. Genomic analysis of inherited breast cancer among Palestinian women: genetic heterogeneity and a founder mutation in TP53. Int J Cancer. 2017;141:750–6.
Alhuqail A-J, Alzahrani A, Almubarak H, Al-Qadheeb S, Alghofaili L, Almoghrabi N, et al. High prevalence of deleterious BRCA1 and BRCA2 germline mutations in Arab breast and ovarian cancer patients. Breast Cancer Res Treat. 2018;168:695–702.
Abulkhair O, Al Balwi M, Makram O, Alsubaie L, Faris M, Shehata H, et al. Prevalence of BRCA1 and BRCA2 mutations among high-risk Saudi patients with breast cancer. J Glob Oncol. 2018;4:JGO–18.
Malone KE, Daling JR, Doody DR, Hsu L, Bernstein L, Coates RJ, et al. Prevalence and predictors of BRCA1 and BRCA2 mutations in a population-based study of breast Cancer in white and black American women ages 35 to 64 years. Cancer Res. 2006;66:8297–308.
Goodwin PJ, Phillips K-A, West DW, Ennis M, Hopper JL, John EM, et al. Breast cancer prognosis in BRCA1 and BRCA2 mutation carriers: an international prospective breast Cancer family registry population-based cohort study. J Clin Oncol Off J Am Soc Clin Oncol. 2012;30:19–26.
Novaković S, Milatović M, Cerkovnik P, Stegel V, Krajc M, HočEvar M, et al. Novel BRCA1 and BRCA2 pathogenic mutations in Slovene hereditary breast and ovarian cancer families. Int J Oncol. 2012;41:1619–27.
Krajc M, Zadnik V, Novaković S, Stegel V, Teugels E, Bešič N, et al. Geographical distribution of Slovenian BRCA1/2 families according to family origin: implications for genetic screening: geographical distribution of Slovenian BRCA1/2 families. Clin Genet. 2014;85:59–63.
Stegel V, Krajc M, Žgajnar J, Teugels E, De Grève J, Hočevar M, et al. The occurrence of germline BRCA1 and BRCA2sequence alterations in Slovenian population. BMC Med Genet. 2011;12:9.
Cini G, Mezzavilla M, Della Puppa L, Cupelli E, Fornasin A, D’Elia AV, et al. Tracking of the origin of recurrent mutations of the BRCA1 and BRCA2 genes in the north-east of Italy and improved mutation analysis strategy. BMC Med Genet. 2016;17:11.
Sharma-Oates A, Shaaban AM, Tomlinson I, Wynne L, Cazier J-B, Sundar S. Heterogeneity of germline variants in high risk breast and ovarian cancer susceptibility genes in India. Precis Clin Med. 2018;1:75–87.
Kais Z, Chiba N, Ishioka C, Parvin JD. Functional differences among BRCA1 missense mutations in the control of centrosome duplication. Oncogene. 2012;31:799.
Cherbal F, Bakour R, Adane S, Boualga K. BRCA1 and BRCA2 germline mutation spectrum in hereditary breast/ovarian cancer families from Maghrebian countries. Breast Dis. 2013;34:1–8.
Laplace-Marieze V, Presneau N, Sylvain V, Kwiatkowski F, Lortholary A, Hardouin A, et al. Systematic sequencing of the BRCA-1 coding region for germ-line mutation detection in 70 French high-risk families. Int J Oncol. 1999;14:971–8.
Simard J, Dumont M, Moisan A-M, Gaborieau V, Vezina H, Durocher F, et al. Evaluation of BRCA1 and BRCA2 mutation prevalence, risk prediction models and a multistep testing approach in French-Canadian families with high risk of breast and ovarian cancer. J Med Genet. 2006;44:107–21.
Cavallone L, Arcand SL, Maugard CM, Nolet S, Gaboury LA, Mes-Masson A-M, et al. Comprehensive BRCA1 and BRCA2 mutation analyses and review of French Canadian families with at least three cases of breast cancer. Familial Cancer. 2010;9:507–17.
Castéra L, Krieger S, Rousselin A, Legros A, Baumann J-J, Bruet O, et al. Next-generation sequencing for the diagnosis of hereditary breast and ovarian cancer using genomic capture targeting multiple candidate genes. Eur J Hum Genet. 2014;22:1305–13.
Belanger MH, Dolman L, Arcand SL, Shen Z, Chong G, Mes-Masson A-M, et al. A targeted analysis identifies a high frequency of BRCA1 and BRCA2 mutation carriers in women with ovarian cancer from a founder population. J Ovarian Res. 2015;2015(8):1.
Gaceb H, Cherbal F, Bakour R, Ould-Rouis A, Mahfouf H. Clinicopathological and molecular study of triple-negative breast Cancer in Algerian patients. Pathol Oncol Res. 2018;24:297–308.
John EM, Miron A, Gong G, Phipps AI, Felberg A, Li FP, et al. Prevalence of pathogenic BRCA1 mutation carriers in 5 US racial/ethnic groups. Jama. 2007;298:2869–76.
Papi L, Putignano AL, Congregati C, Zanna I, Sera F, Morrone D, et al. Founder mutations account for the majority of BRCA1-attributable hereditary breast/ovarian cancer cases in a population from Tuscany, Central Italy. Breast Cancer Res Treat. 2009;117:497–504.
Caux-Moncoutier V, Castéra L, Tirapo C, Michaux D, Rémon M-A, Laugé A, et al. EMMA, a cost- and time-effective diagnostic method for simultaneous detection of point mutations and large-scale genomic rearrangements: application to BRCA1 and BRCA2 in 1,525 patients. Hum Mutat. 2011;32:325–34.
Korean Hereditary Breast Cancer Study Group, Kang E, Seong M-W, Park SK, Lee JW, Lee J, et al. The prevalence and spectrum of BRCA1 and BRCA2 mutations in Korean population: recent update of the Korean hereditary breast Cancer (KOHBRA) study. Breast Cancer Res Treat. 2015;151:157–68.
Infante M, Durán M, Esteban-Cardeñosa E, Miner C, Velasco E. High proportion of novel mutations of BRCA1 and BRCA2 in breast/ovarian cancer patients from Castilla-León (Central Spain). J Hum Genet. 2006;51:611–7.
Infante M, Durán M, Lasa A, Acedo A, de la Hoya M, Esteban-Cardeñosa E, et al. Two founder BRCA2 mutations predispose to breast cancer in young women. Breast Cancer Res Treat. 2010;122:567–71.
Blay P, Santamaría I, Pitiot AS, Luque M, Alvarado MG, Lastra A, et al. Mutational analysis of BRCA1 and BRCA2 in hereditary breast and ovarian cancer families from Asturias (northern Spain). BMC Cancer. 2013;13:243.
de Juan I, Palanca S, Domenech A, Feliubadaló L, Segura Á, Osorio A, et al. BRCA1 and BRCA2 mutations in males with familial breast and ovarian cancer syndrome. Results of a Spanish multicenter study. Familial Cancer. 2015;14:505–13.
Korean Breast Cancer Study Group, Kim H, Cho D-Y, Choi DH, Choi S-Y, Shin I, et al. Characteristics and spectrum of BRCA1 and BRCA2 mutations in 3,922 Korean patients with breast and ovarian cancer. Breast Cancer Res Treat. 2012;134:1315–26.
Burstein HJ, Curigliano G, Loibl S, Dubsky P, Gnant M, Poortmans P, et al. Estimating the benefits of therapy for early-stage breast cancer: the St. Gallen international consensus guidelines for the primary therapy of early breast cancer 2019. Ann Oncol. 2019;30(10):1541–57.
National Comprehensive Cancer Network (NCCN) Clinical Practices Guidelines in Oncology. Genetic/familial high-risk assessment: breast, ovarian and pancreatic. Version 1.2020-December 4, 2019. http://www.nccn.org. Accessed 12 June 2020.
Acknowledgments
A part of this work was published by the 11th Breast & gynecological international cancer conference (BGICC), Cairo-Egypt 17-18th January, 2019 (DOI: https://doi.org/10.1177/0300891619826184).
Funding
The authors have no support or funding to report.
Author information
Authors and Affiliations
Contributions
JB designed the study, drafted the manuscript, carried out the collection and assembly of data, performed data analysis and interpretation. MM participated in the design of the study, helped with data analysis, interpretation. TD and AL helped in the collection and assembly of data. EE was responsible for the genetics analysis. JMD, CCB were responsible also for the genetic analysis and performed genetic data analysis and interpretation. AB and NGG revised the paper. MBM supervised the study and revised critically the manuscript. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Ethics Committee for Biomedical Research of the Faculty of Medicine and Pharmacy of Rabat (CERB) registered under number IORG0006594. All participants gave a written informed consent to participate in the study.
Consent for publication
All participants provided written informed consent.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bakkach, J., Mansouri, M., Derkaoui, T. et al. Contribution of BRCA1 and BRCA2 germline mutations to early onset breast cancer: a series from north of Morocco. BMC Cancer 20, 859 (2020). https://doi.org/10.1186/s12885-020-07352-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-020-07352-9