
Abstract
Background
The incidence of breast cancer (BC) and/or ovarian cancer (OC) is increasing in Tunisia especially in young women and mostly those with family history. However, the spectrum of BRCA mutations remains little explored in Tunisian patients in particular in the southern region.
Methods
We sequenced the entire coding regions of BRCA1and BRCA2 genes using next generation sequencing (NGS) in 134 selected patients with BC and/or OC.
Results
Among the 134 patients, 19 (14.17%) carried pathogenic mutations (10 are BRCA1 mutation carriers and 9 are BRCA2 mutation carriers) that are mainly frameshift index (76.9%). Interestingly, 5 out of the 13 variants (38.46%) were found at least twice in unrelated patients, as the c.1310-1313 delAAGA in BRCA2 and the c.5030_5033 delCTAA that has been identified in 4/98 BC patients and in 3/15 OC patients from unrelated families with strong history of cancer. Besides recurrent mutations, 6 variant (4 in BRCA1 and 2 in BRCA2) were not reported previously. Furthermore, 3 unrelated patients carried the VUS c.9976A > T, (K3326*) in BRCA2 exon 27. BRCA carriers correlated significantly with tumor site (p = 0.029) and TNBC cases (p = 0.008). In the groups of patients aged between 31 and 40, and 41–50 years, BRCA1 mutations occurred more frequently in patients with OC than those with BC, and conversely BRCA2 carriers are mostly affected with BC (p = 0.001, and p = 0.044 respectively).
Conclusions
The overall frequency of the BRCA germline mutations was 14.17% in patients with high risk of breast/ovarian cancer. We identified recurrent mutations as the c.1310_1313 delAAGA in BRCA2 gene and the c.5030_5033 delCTAA in BRCA1 gene that were found in 4% and 20% of familial BC and OC respectively. Our data will contribute in the implementation of genetic counseling and testing for families with high-risk of BC and/or OC.
Similar content being viewed by others


Background
Breast Cancer (BC) is the most prevalent cancer worldwide and the second leading cause of death by cancer in women [1, 2]. In Tunisia, the incidence of BC is 27.1/100.000 per year affecting more often young women (< 35 years), and with more aggressive clinical behavior [3, 4]. Approximately 5 to 10% of BC patients harbor germline mutations that predispose to this malignancy at earlier onset compared to the general population [5, 6].
Evidently, the BC susceptibility genes BRCA1 (MIM# 113705) and BRCA2 (MIM# 600185) are tumor suppressor genes that play a key role in DNA repair through the homologous recombination pathway [7, 8]. Therefore, loss of the BRCA function results to an inefficient DNA repair process that increases the mutation rates and thus contributes to tumor development [9, 10]. Indeed, women carrying pathogenic germline mutations in BRCA1/BRCA2 genes have an increased risk of BOC compared to those with wild type BRCA genes [11,12,13]. Screening of BRCA1/BRCA2 genes for the identification of pathogenic mutations is essential to provide genetic counseling to the members of affected families, for medical follow-up, and for the targeted therapy selection which is based on the inhibitor of poly-ADP-ribose polymerase inhibitor namely the Olaparib [14]. Despite the importance of the mutational spectrum of BRCA1/BRCA2 genes, only few studies using Sanger DNA sequencing, investigated Tunisian patients with HBOC. In a recent study, Laitamn et al., reported all the pathogenic variants in both BRCA1 and BRCA2 genes in the Middle East, North Africa, and Southern Europe [15]. The authors identified 232 and 239 pathogenic sequence variants in BRCA1 and BRCA2, respectively that include only few variants that were found in Tunisian patients [15].
Besides, Riahi et al., reported twelve pathogenic mutations (25%); among them, three were found in BRCA1 (c.211dupA, c.5266dupC and c.1504_1508delTTAAA) and two were novel mutations detected in BRCA2 (c.1313dupT and c.7654dupT). The study was carried out on 92 families and was performed by direct sequencing [16]. Furthermore, two other studies reported deleterious mutations of the BRCA1/BRCA2 genes in 19% and 18% of familial patients from the north region of Tunisia respectively [17, 18]. Moreover, Fourati et al. by targeting the exons 5, 11, and 20 of the BRCA1 gene and exons 10 and 11 of the BRCA2 gene, showed that among 66 patients, only 12 patients (18%) had deleterious mutations in the BRCA1 or BRCA2 genes [19].
The study of Mahfoudh et al., included 16 Tunisian high-risk BC families and they were screened for only BRCA1 gene. The authors showed that the prevalence of BRCA1 carriers was 37.5% and identified 3 truncating mutations (c.916 delTT, c.3450 delCAAG, c.5382 insC) and one splice site mutation c.212 + 2insG [20].
Technological advances in DNA sequencing have improved and facilitated the screening procedure of BRCA1/BRCA2 genes since the large size of these genes and the absence of hotspots regions make this procedure expensive and time-consuming. In fact, NGS has resolved these problems and allows, henceforth the detection of mutations with higher sensibility [21,22,23]. Our study is the first using NGS to screen the whole exons of BRCA1/BRCA2 genes in 134 selected patients with high risk of BOC in the south region of Tunisia.
Methods
Patients
A total of 134 patients were selected (between 2016 and 2019) among them 113 patients with HBOC (98 cases) and/or OC (15 cases) that meet one of the following criteria: (1) Presence of at least three related first or second-degree BC cases; (2) BC in young patients aged less than 35 years, (3) Presence of male BC among the first or second-degree relatives, (4) Presence of at least two cases of BC or OC, regardless of age, and at least one case of prostate cancer in a related first or second degree patient.
In addition, 21 cases without evidence of family history were included in this study, 12 were diagnosed with BC at the age ≤ 40, and 9 patients diagnosed with OC at the age ≤ 60 years. All patients were recruited from the department of Medical Oncology of the CHU Habib Bourguiba of Sfax (Tunisia), and had provided written informed consent for participation in the research study and for the genetic testing.
Genetic testing
Genomic DNA was isolated from 0.2 mL of peripheral blood of the proband from each selected family using the “QIAamp DNA Blood Mini kit” (Qiagen), following the manufacturer’s instructions. Isolated DNA was quantified by Qubit 3.0 Fluorometric quantitation (Thermo Fisher Scientific). The BRCA/BRCA2 germline mutations were detected using the next-generation sequencing approach (NGS). Briefly, 50 ng of genomic DNA from each sample was used to prepare library using the “AmpliSeq™BRCA Panel”, “AmpliSeq™Library Plus” and “AmpliSeq™CD Indexes” according to the protocol provided by Illumina. The adaptors and sample unique DNA barcodes were incorporated into the amplified amplicons with a second PCR. Libraries were quantified with the Qubit® dsDNA HS Assay Kit (Life Technologies).The DNA library was pooled and prepared for sequencing using the Illumina MiSeq sequencer with Miseq Reagent Kit v2 (300-cycles) (Illumina, San Diego, CA) according to the manufacturer’s instructions to generate paired-end reads with a 151-bp read length. Reads were trimmed to remove low-quality sequences and then aligned to the human reference genome (GRCh37/hg19) using the Burrows-Wheeler alignment (BWA) package. The total PF Reads is 16,755,084 and the Q30 is 93.56%.
The BRCA1 (NM_007300.3) and BRCA2 (NM_000059.3) sequences from the National Center for Biotechnology Information (NCBI) database (http://www.ncbi.nlm.nih.gov) were used as reference. The NGS data was analyzed using the BaseSpace Variant Interpreter (https://basespace.illumina.com).
Sanger sequencing was performed to confirm the BRCA1/BRCA2 pathogenic mutation identified by NGS (see Additional file 1: Fig.1 in Supplementary data). Sequencing primers were designed using Primer 5.0 software and provided on request. PCR products were purified using the QIAquick PCR Purification Kit (Qiagen) and labeled using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems), and the sequence analysis was performed by BioEdit.
Variant annotation and classification
We annotated the mutations using in silico prediction tools to evaluate the functional effects of the candidate variants: PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), which predicts the effects of an amino acid substitution on the structure and function of a protein. The PolyPhen score represents the probability that a substitution is damaging, so values between 0.9 to 1 are more confidently predicted to be deleterious. The program sorting intolerant from tolerant (SIFT) predicts whether an amino acid substitution is likely to affect protein function based on sequence homology and the physicochemical similarity between the alternate amino acid. The score is the normalized probability that the amino acid change is deleterious if the score is < 0.05 and tolerated if the score is > 0.05. Mutation Taster (http://www.Mutationtaster.org/) was used to assess the impact of mutations on protein function.
Statistical analysis
Associations between patient characteristics and presence of pathogenic BRCA1/BRCA2 mutations were tested by Chi-square test. A significance level of p ≤ 0.05 was used in the analysis. All analyses were performed using SPSS (version 20.0).
Results
Patients characteristics
A total of 134 breast and/or ovarian cancer subjects were included in this study. The characteristics of the study group are presented in Table 1. Unilateral BC was diagnosed in 104 (77.6%) patients, 6 (4.5%) women had bilateral BC, 22 (16.4%) had OC, and 2 (1.5%) had both BC and OC. Age at diagnosis of BC ranged from 27 to 65 years (median: 42.78 years), and of OC ranged from 38 to 64 years (median: 50.37 years). For patients with BC only 9 (6.7%) were diagnosed before the age of 30 years and 55/134 (41%) before 40 years of age. In the group of patients with OC only 4 patients were diagnosed at an age ≤ 40 years. Family history of BC and/or OC was found in 113 (84.3%) patients. Among the 21 patients without family history of BC/OC, 12 were diagnosed with TNBC and 9 with OC (Table 1).
BRCA1/BRCA2 germline pathogenic mutations
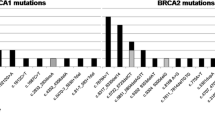
Pathogenic BRCA1/BRCA2 germline mutations were identified in 14.17% (19/134) of the patients (Fig. 1, Table 2). Among 113 patients with strong family history for breast/ovarian cancer (HBOC), 18 (15.9%) were positive for heterozygous BRCA mutations (9 in BRCA1 and 9 in BRCA2). In the group of patients without evidence of HBOC, including 12 patients with TNBC and 9 patients with OC, only one patient 4.8% (1/21) with OC carried the c.2338C > T (Gln780X) pathogenic BRCA1 mutation and 2 cases (1 TNBC and 1 OC) carried the VUS c.9976A > T (Lys3326X) in BRCA2 exon 27 (Fig. 1, Tables 2,4). Most of the BRCA mutations were pathogenic (class 2 to 6) and 76.9% of the mutation (10/13) were frameshift deletion, only 1 was frameshift duplication, 2 nonsens mutations and one splice site mutation (Table 2, Fig. 1). Among the 13 mutations detected, 5 were identified at least twice in unrelated patients. Notably, the frameshift mutation c.1310_1313delAAGA of the BRCA2 gene was detected in 4 unrelated BC patients, and the c.17-20 delAAGA in 2 unrelated young patients with strong family history of BC. In BRCA1, the c.5030_5033delCTAA was found in 3 unrelated young patients (≤ 40 years) diagnosed with OC (2 without and 1 with BC), and the c.2338C > T mutation was detected in 2 unrelated patients with OC only. Interestingly, our data indicated that the c.1310_1313delAAGA of the BRCA2 gene, and the c.5030_5033delCTAA of the BRCA1 gene were found in 4% (4/98) and in 20% (3/15) of familial BC and of early onset familial OC respectively. We also detected the c.632-1G > A mutation in the splice acceptor site in 2 patients with BC (Table 2, Fig. 2). It is interesting to note that the 2 unrelated patients with the c.632-1G > A variant, carried also the pathogenic mutation c.1310_1313delAAGA in BRCA2. Our study outlined 6 out of 13 (46%) novel pathogenic BRCA mutations, according to the BIC and ClinVar databases.

Schematic diagram of BRCA1 and BRCA2 proteins with the position of all identified pathogenic variants. Novel variants are marked with arrow

BRCA mutation rate (%) in all BC (a), in all OC (b), and in TNBC (c) patients divided according to age at diagnosis (≤ 30, 31–40, 41–50, and ≥ 51 years old)
Association of BRCA carriers and clinico-pathological parameters
In BC, there is a significant association between Triple Negative status and BRCA mutations. Among the 26 TNBC cases, 5 (19.23%) carried BRCA mutations and 4 (80%) were BRCA1 mutation carriers (p = 0.008, Table 1). Additionally, patients with OC (with or without BC) carried only mutations in BRCA1 gene as opposed to BRCA2 gene (5/24 vs 0/24), whereas BRCA2 mutations were more frequently detected in patients with only BC (9/110 vs 5/110, p = 0.029, Table 1). No other associations were observed with clinico-pathological parameters (Table 1).
On the other hand, for patients diagnosed with BC at an age ≤ 30 years, the BRCA mutation rate was 22.2% (2/9), and none of the patients was diagnosed with OC at an age ≤ 30 years in our series. In the group of patients between 31 and 40 years, BRCA mutation was detected in 13.72% (7/51) and in 75% (3/4) of women diagnosed with BC and OC respectively (p = 0.001, Fig. 2a, b; Table 3). Concerning women aged from 41 to ≤ 50 years old diagnosed with BC or OC, 8.33% (2/24) and 22.2% (2/9) carried a BRCA mutation respectively (p = 0.044, Fig. 2a, b; Table 3). None of OC patients over 51 years carried BRCA mutation while 11.5% (3/26) of BC patients were BRCA2 carriers (Fig. 2a, b; Table 3). The mutation rate for the BRCA gene in patients with only OC was 18.2% (4/22), all were BRCA1 carriers and only one OC patient carried the variant K3326X in the BRCA2 gene (Tables 1, 3). Furthermore, in young patients (≤ 30 years), 40% (2/5) of TNBC carried BRCA mutation while in the group of TNBC patients aged from 31 to 40 years old, BRCA mutations occurred more frequently in BRCA1 than in BRCA2 gene (3/13, (23%) vs 0/13 (0%), (p = 0.041, Fig. 2c, Table 3).
Variants of uncertain significance in BRCA1/BRCA2 genes
In addition to deleterious BRCA1/BRCA2 mutations, 10 VUS (6 in BRCA1 and 4 in BRCA2) were predicted to be pathogenic by PolyPhen, SIFT or Mutation Taster (Table 4). Each VUS was identified in more than one patient and the c.9976A > T in exon 27 of the BRCA2 gene was detected in 3 unrelated patients, 2 with BC diagnosed at an age ≤ 40 years and 1 patient with OC (Table 4). All VUS have been reported in the BIC database except the variant N550H in BRCA1 exon 11.
Discussion
BRCA1 and BRCA2 have key roles in the development of breast/ovarian cancer [9, 10]. The prevalence of BRCA1/BRCA2 mutations varies in different populations due to founder mutation effect [15, 24,25,26,27,28]. Genetic testing of patients with family history for breast/ovarian cancer have become standard clinical management in Western countries, however, in Tunisia studies of BRCA-associated breast/ovarian cancer remain less investigated. Clearly, the majority of studies that reported the spectrum of BRCA mutations in Tunisia included a small number of patients and used Sanger method for DNA sequencing [16,17,18,19,20]. Riahi et al. reported that the rate of BRCA mutations in patients from the north of Tunisia was 25% (12/48) [16]. Furthermore, in another study, it was showed that deleterious BRCA1 mutations were detected in 37% (6/16) of patients selected from high-risk breast cancer families [20]. In this study, we used NGS to determine the BRCA mutation rates in high risk breast and/or ovarian cancer in patients from the south of Tunisia. Thirteen truncating mutations and one splice acceptor mutation were identified in 19 among 134 patients (14.17%). The rate of BRCA1 mutations found in this study (7.46%, 10/134) was lower than those reported in previous studies. In fact, in a review of the literature, Cherbal et al., reported that in breast/ovarian cancer families from Algeria, Morocco and Tunisia, BRCA1 was mutated in 17.43% of cases (34/195) and that the c.798_799delTT was a recurrent mutation [29]. In our study none of the previously reported mutations in North African patients was identified except for the c.1310_1313delAAGA in BRCA2. Indeed, this mutation was detected in 4% (4/98) of patients from unrelated families with high risk of BC and among 9 BRCA2 carriers, 4 (44.4%) had the c.1310_1313delAAGA mutation suggesting that it might be recurrent in our population. Our finding is in line with a previous study showing that the c.1310_1313delAAGA mutation was detected in 6% (4/66) of patients from the north of Tunisia [19]. Furthermore, it was reported that the c.1310_1313delAAGA mutation was detected in 11.4% (14/122) of patients from the North-East region of Morocco, and in one Algerian family among 10 carrying BRCA mutation [30, 31]. According to the Breast Cancer Information Core database (BIC; http://research.nhgri.nih.gov/bic/), this mutation was found in different European patients and was recorded several times in the French UMD-BRCA2 database and classified as founder mutation [32]. Furthermore, we found that 2 BC patients are double heterozygous carrying both the c.1310_1313delAAGA frameshift mutation and the splice site acceptor variant c.-632G < A. This splicing mutation has been reported two times in the ClinVar database therefore, and in the best of our knowledge this is the first report describing the association of the splice site and a frameshift mutation in the BRCA2 gene in 2 unrelated patients with strong family history of BC.
The pathogenic mutation c.17-20 delAAGA (Lys6Xfs) in BRCA2 exon 2 was detected in 2 unrelated young patients (39 year-old) with strong family history of BC. This mutation has not been reported previously in the ClinVar and BIC databases.
We also detected in the BRCA2 exon 27, the c.9976A > T (K3326X) in 3 unrelated patients, 2 were diagnosed with family BC at 28 and 36 years old and one patient with OC at 56 years old and without family history. The BRCA2 variant (K3326X), was firstly interpreted as pathogenic, but its identification in control populations led to its classification as a benign polymorphism [33]. However, recent studies showed the association of K3326X variant with the risk of developing melanoma, pancreatic, breast and ovarian cancers [34,35,36]. Moreover, the large study of Meeks et al., provided evidence that the K3326X variant is associated with the risk of developing BC and OC independently of other pathogenic variants in BRCA2 [37]. Altogether, it was suggested that the K3326X variant is not neutral and that it may be included in SNP panels for evaluating BC risk.
In BRCA1gene, we identified the c.5030_5033delCTAA mutation in 3 out of 15 (20%) unrelated patients with early onset HBOC. This mutation has not been reported in North Africa but was found in patients from Greece, Italy, Jordan, Lebanon, Kuwait, and Saudia Arabia [15]. In addition, the nonsense mutation c.2338C > T was shared by 2 young patients (< 50 years-old) diagnosed with OC. The c. 2338C > T has been reported in Caucasian patients according to the BIC and ClinVar databases. Altogether, these findings may suggest ethnic and genetic associations between unrelated populations, or that these mutations occur in mutational hotspots region.
On the other hand, it is interesting to note that the BRCA mutation rate in young patients diagnosed with BC (age ≤ 30 years) was higher (22.2%) compared to older patients (13.7% (31–40), 8.3% (41–50), and 11.5% (≥ 51 years). The recent study of Bakkach et al., reported that the frequency of mutations among young patients (≤ 40 years) with family history of BC was 16.7% (5 out of 33 patients) suggesting that the young age for BC diagnosis seems to be strongly predictive of BRCA mutation status in Moroccan patients [38].
Of patients with OC (with and without BC), 20.83% (5/24) carried pathogenic variants. Mutation detection rates were 75% (3/4) for patients diagnosed between 31 and 40 years compared to only 22.2% (2/9) for women between 41 and 50 years. All patients over 51 years, carried no mutations in BRCA1/BRCA2 genes.
It is well established that TNBC exhibit an aggressive behavior and has a worse prognosis [39]. In our cohort, among 110 patients with only BC, 26 (23.6%) were TN including 14 patients with family history of BC and 12 without evidence of family history of BOC. Five patients (5/26, 19.23%) carried BRCA mutations and 4 among them (80%) were BRCA1 carriers. Consistent with our findings, in a previous study, the BRCA1 mutation carriers had a higher incidence in TNBC than BRCA2 mutation carriers [40,41,42,43]. Moreover, in a recent work, Toss et al., showed that in TNBC Italian patients, BRCA mutation prevalence was 22.6% (21.4% BRCA1) and that 64.2% are ≤ 30 years old, which is in line with our findings [44]. Furthermore, Mahfoudh et al., reported that the 5382insC mutation in BRCA1 was detected in 25% (2/8) of Tunisian patients with TNBC [45]. In our study, none of the patients carried the BRCA1 5382insC mutation, however, in TNBC we detected 3 mutations in BRCA1 (c.3254delG, c.3364_3370dupACAGATT, c.4067_4071delAAGAA), and no BRCA2 pathogenic mutations were detected in TNBC cases except the VUS c.9976A > T identified in 2 unrelated patients (Additional file 2: Table 1 in Supplementary data).
It is important to note that previous studies used Sanger sequencing to identify BRCA mutations in Tunisian families with a history of BOC unlike the present study where the whole BRCA genes were screened by the NSG which allowed us to identify new mutations in Tunisian patients. However, some recent studies have performed the whole exome sequencing to identify candidate genes in few BRCA negative families. Hamdi et al., performed whole exome sequencing on seven Tunisian families with HBOC and identified four novel BC candidate genes (MMS19, DNAH3, POLK and KATB6) [46]. Furthermore, other studies identified by exome sequencing, RCC1 and RAD50 as BC candidate susceptibility genes in Tunisian BRCA negative families [47, 48].
Conclusions
This study assessed the prevalence of germline mutations and identified novel and recurrent mutations for BRCA1/BRCA2 genes in patients with high risk of BOC from the south region of Tunisia. Giving the high prevalence of pathogenic mutations in BRCA1/BRCA2 genes (14.17%), our data will contribute to the establishment of a service dedicated to the genetic screening and to the counseling of the families with high risk of HBOC in Tunisia.
Availability of data and materials
All data generated are included in this article. Raw data are not publically available due to protect the confidentiality of patients, but are available from the corresponding author on request.
Abbreviations
- BRCA1:
-
BReast CAncer susceptibility gene 1
- BRCA2:
-
BReast CAncer susceptibility gene 2
- BIC:
-
Breast cancer Information Core
- BC:
-
Breast cancer
- OC:
-
Ovarian cancer
- ER:
-
Estrogen receptor
- PR:
-
Progesteron receptor
- HER2:
-
Human epidermal growth factor receptor 2
- TNBC:
-
Triple negative breast cancer
- NGS:
-
Next generation sequencing
- VUS:
-
Variant of uncertain significance
References
Coleman MP, Quaresma M, Berrino F, Lutz J, de Angelis R, Capocaccia R, et al. Cancer survival in five continents: a worldwide population-based study (CONCORD). Lancet Oncol. 2008;9:730–56.
GLOBOCAN Breast cancer incidence and mortality worldwide, 2012. International agency for research on cancer. http://globocan.iarc.fr/Pages/fact_sheets_population.aspx. (2012).
Missaoui N, Jaidene L, Abdelkrim SB, Abdelkader AB, Beizig N, Yaacoub LB, et al. Breast cancer in Tunisia: clinical and pathological findings. Asian Pac J Cancer Prev. 2011;121:69–72.
Ben Abdallah M, Zehani S, Maalej M, Hsairi M, Hechiche M, Ben Romdhane K, et al. Cancer du sein en Tunisie: caractéristiques épidémiologiques et tendance évolutive de l’incidence. Tunis Med. 2009;87:417–25.
Foulkes WD. Inherited susceptibility to common cancers. N Engl J Med. 2008;359:2143–53.
Peto J, Collins N, Batfoot R, Seal S, Warren W, Rahman N, et al. Prevalence of BRCA1 and BRCA2 gene mutations in patients with early onset breast cancer. J Natl Cancer Inst. 1999;91:943–9.
Venkitaraman AR. Functions of BRCA1 and BRCA2 in the biological response to DNA damage. J Cell Sci. 2001;114:3591–8.
Deng CX, Wang RH. Roles of BRCA1 in DNA damage repair: a link between development and cancer. Hum Mol Genet. 2003;12:113–23.
Yoshida K, Miki Y. Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Sci. 2004;95:866–71.
Jasin M. Homologous repair of DNA damage and tumorigenesis: the BRCA connection. Oncogene. 2002;2:8981–93.
Antoniou A, Pharoah PD, Narod S, Risch HA, Eyfjord JE, Hopper JL, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet. 2003;72:1117–30.
Brose MS, Rebbeck TR, Calzone KA, Stopfer JE, Nathanson KL, Weber BL. Cancer risk estimates for BRCA1 mutation carriers identified in a risk evaluation program. J Natl Cancer Inst. 2002;94:1365–72.
Mavaddat N, Peock S, Frost D, Ellis S, Platte R, Fineberg E, et al. Cancer risks for BRCA1 and BRCA2 mutation carriers: results from prospective analysis of EMBRACE. J Natl Cancer Inst. 2013;105:812–22.
Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, et al. Oral poly(ADP-ribose) polymerase inhib-itor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376:235–44.
Laitman Y, Friebel TM, Yannoukakos D, Fostira F, Konstantopoulou I, Figlioli G, et al. The spectrum of BRCA1 and BRCA2 pathogenic sequence variants in Middle Eastern, North African, and South European countries. Hum Mutat. 2019;40:e1–23.
Riahi A, Ghourabi ME, Fourati A, Chaabouni-Bouhamed H. Family history predictors of BRCA1/BRCA2 mutation status among Tunisian breast/ovarian cancer families. Breast Cancer. 2017;24:238–44.
Riahi A, Kharrat M, Ghourabi ME, Khomsi F, Gamoudi A, Lariani I, et al. Mutation spectrum and prevalence of BRCA1 and BRCA2 genes in patients with familial and early-onset breast/ovarian cancer from Tunisia. Clin Genet. 2015;87:155–60.
Troudi W, Uhrhammer N, Sibille C, Dahan C, Mahfoudh W, Bouchlaka Souissi C, et al. Contribution of the BRCA1 and BRCA2 mutations to breast cancer in Tunisia. J Hum Genet. 2007;52:915–20.
Fourati A, Louchez MM, Fournier J, Gamoudi A, Rahal K, El May MV, et al. Screening for common mutations in BRCA1 and BRCA2 genes: interest in genetic testing of Tunisian families with breast and/or ovarian cancer. Bull Cancer. 2014;101:e36–40.
Mahfoudh W, Bouaouina N, Ben Ahmed S, Gabbouj S, Shan J, Mathew R, et al. Hereditary breast cancer in Middle Eastern and North African (MENA) populations: identification of novel, recurrent and founder BRCA1 mutations in the Tunisian population. Mol Biol Rep. 2012;39:1037–46.
Chan M, Ji SM, Yeo ZX, Gan L, Yap E, Yap YS, et al. Development of a next-generation sequenc-ing method for BRCA mutation screening: a comparison between a high-throughput and a benchtop platform. J Mol Diag. 2012;14:602–12.
Park K, Kim MK, Lee T, Hong J, Kim HK, Ahn S, et al. Performance evaluation of an amplicon-based next-generation sequencing panel for BRCA1 and BRCA2 variant detection. Clin Lab Anal. 2020;19:e23524.
Sim WC, Lee CY, Richards R, Bettens K, Mottier V, Goh LL. Validation of a next generation sequencing assay for BRCA1, BRCA2, CHEK2 and PALB2 genetic testing. Exp Mol Pathol. 2020;116:104483. https://doi.org/10.1016/j.yexmp.2020.104483.
Díez O, Osorio A, Durán M, Martinez-Ferrandis JI, de la Hoya M, Salazar R, et al. Analysis of BRCA1 and BRCA2 genes in Spanish breast/ovarian cancer patients: a high proportion of mutations unique to Spain and evidence of founder effects. Hum Mutat. 2003;22:301–12.
Lieberman S, Tomer A, Ben-Chetrit A, Olsha O, Strano S, Beeri R, et al. Population screening for BRCA1/BRCA2 founder mutations in Ashkenazi Jews: proactive recruitment compared with self-referral. Genet Med. 2017;19:754–62.
Alvarez C, Tapia T, Perez-Moreno E, Gajardo-Meneses P, Ruiz C, Rios M, et al. BRCA1 and BRCA2 founder mutations account for 78% of germline carriers among hereditary breast cancer families in Chile. Oncotarget. 2017;8:74233–43.
Bu R, Siraj AK, Al-Obaisi KA, Beg S, Al Hazmi M, Ajarim D, et al. Identification of novel BRCA founder mutations in Middle Eastern breast cancer patients using capture and Sanger sequencing analysis. Int J Cancer. 2016;139:1091–7.
Laraqui A, Uhrhammer N, Rhaffouli HE, Sekhsokh Y, Lahlou-Amine I, Bajjou T, et al. BRCA genetic screening in Middle Eastern and North African: mutational spectrum and founder BRCA1 mutation (c.798_799delTT) in North African. Dis Markers. 2015. https://doi.org/10.1155/2015/194293.
Cherbal F, Bakour R, Adane S, Boualga K. BRCA1 and BRCA2 germline mutation spectrum in hereditary breast/ovarian cancer families from Maghrebian countries. Breast Dis. 2012;34:1–8.
Laarabi FZ, Ratbi I, Elalaoui SC, Mezzouar L, Doubaj Y, Bouguenouch L, et al. High frequency of the recurrent c.1310_1313delAAGA BRCA2 mutation in the North-East of Morocco and implication for hereditary breast-ovarian cancer prevention and control. BMC Res Notes. 2017;10:188.
Cherbal F, Bakour R, Adane S, Boualga K, Benais-Pont G, Maillet P. BRCA1 and BRCA2 germline mutations screening in Algerian breast/ovarian cancer families. Dis Markers. 2010;28:377–84.
Caputo S, Benboudjema L, Sinilnikova O, Rouleau E, Béroud C, Lidereau R, et al. Description and analysis of genetic variants in French hereditary breast and ovarian cancer families recorded in the UMD-BRCA1/BRCA2 databases. Nucleic Acids Res. 2012;40:992–1002.
Thompson ER, Gorringe KL, Rowley SM, Li N, McInerny S, Wong-Brown MW, et al. Reevaluation of the BRCA2 truncating allele c.9976A > T (pLys3326Ter) in a familial breast cancer context. Sci Rep. 2015;5:14800. https://doi.org/10.1038/srep14800.
Tuominen R, Engstrom PG, Helgadottir H, Eriksson H, Unneberg P, Kjellqvist S, et al. The role of germline alterations in the DNA damage response genes BRIP1 and BRCA2 in melanoma susceptibility. Genes Chromosom Cancer. 2016;55:601–11.
Martin ST, Matsubayashi H, Rogers CD, et al. Increased prevalence of the BRCA2 polymorphic stop codon K3326X among individuals with familial pancreatic cancer. Oncogene. 2005;24(22):3652–6.
Palmirotta R, Lovero D, Stucci LS, Silvestris E, Quaresmini D, Cardascia A, Silvestris F. Double Heterozygosity for BRCA1 Pathogenic Variant and BRCA2 Polymorphic Stop Codon K3326X: a Case Report in a Southern Italian Family. J Mol Sci. 2018;19(1):285. https://doi.org/10.3390/ijms19010285.
Meeks HD, Song H, Michailidou K, Bolla MK, Dennis J, Wang Q, et al. BRCA2 Polymorphic Stop Codon K3326X and the Risk of Breast, Prostate, and Ovarian Cancers. J. Natl. Cancer. 2016. https://doi.org/10.1093/jnci/djv315.
Bakkach J, Mansouri M, Derkaoui T, Loudiyi A, El Fahime E, Barakat A, et al. Contribution of BRCA1 and BRCA2 germline mutations to early onset breast cancer: a series from north of Morocco. BMC Cancer. 2020;20:859. https://doi.org/10.1186/s12885-020-07352-9.
Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. 2016;13:674–90.
Greenup R, Buchanan A, Lorizio W, Rhoads K, Chan S, Leedom T, et al. Prevalence of BRCA mutations among women with triple-negative breast cancer (TNBC) in a genetic counseling cohort. Ann Surg Oncol. 2013;20:3254–8.
Sharma P, Klemp JR, Kimler BF, Mahnken JD, Geier LJ, Khan QJ, et al. Germline BRCA mutation evaluation in a prospective triple-negative breast cancer registry: implications for hereditary breast and/or ovarian cancer syndrome testing. Breast Cancer Res Treat. 2014;145:707.
Muendlein A, Rohde BH, Gasser K, Haid A, Rauch S, Kinz E, et al. Evaluation of BRCA1/2 mutational status among German and Austrian women with triple-negative breast cancer. J Cancer Res Clin Oncol. 2015;141:2005–12.
Engel C, Rhiem K, Hahnen E, Loibl S, Weber KE, Seiler S, et al. Prevalence of pathogenic BRCA1/2 germline mutations among 802 women with unilateral triple-negative breast cancer without family cancer history. BMC Cancer. 2018;18:265. https://doi.org/10.1186/s12885-018-4029-y.
Toss A, Molinaro E, Venturelli M, Domati F, Marcheselli L, Piana S, et al. BRCA detection rate in an Italian cohort of luminal early-onset and triple-negative breast cancer patients without family history: when biology overcomes genealogy. Cancers. 2020. https://doi.org/10.3390/cancers12051252.
Mahfoudh W, Bettaieb I, Ghedira R, Snoussi K, Bouzid N, Klayech Z, et al. Contribution of BRCA1 5382insC mutation in triple negative breast cancer in Tunisia. J Transl Med. 2019;17:123. https://doi.org/10.1186/s12967-019-1873-8.
Hamdi Y, Boujemaa M, Ben Rekaya M, Ben Hamda C, Mighri N, El Benna H, et al. Family specific genetic predisposition to breast cancer: results from Tunisian whole exome sequenced breast cancer cases. J Trans Med. 2018;16(1):158. https://doi.org/10.1186/s12967-018-1504-9.
Riahi A, Radmanesh H, Schurmann P, Bogdanova N, Geffers R, et al. Exome sequencing and case-control analyses identify RCC1 as a candidate breast cancer susceptibility gene. Int J Cancer. 2018;142(12):2512–7. https://doi.org/10.1002/ijc.31273.
Mighri N, Hamdi Y, Boujemaa M, Othman H, Ben Nasr S, El Benna H, et al. Identification of novel BRCA1 and RAD50 mutations associated with breast cancer predisposition in Tunisian patients. Front Genet. 2020;11:552971. https://doi.org/10.3389/fgene.2020.552971.
Acknowledgements
The authors thank the patients involved in the study.
Funding
This work was supported by a grant of the CIC (Centre d’Investigation Clinique of the Habib Bourguiba Hospital-Sfax) and The Ministry of Higher Education and Scientific Research of Tunisia.
Author information
Authors and Affiliations
Contributions
WBK did the selection of families and patient recruitment. DBAG, NAB, WA performed experiments (DNA extraction, NGS and Sanger sequencing) and data compilation. SC, TSB performed the IHC of ER, PR and Her-2. RMG was involved in writing the manuscript, data analysis and bioinformatics analysis. AK, MF, JD contributed in the interpretation of results, and funding acquisition. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Institutional Review Board of the CHU Habib Bourguiba of Sfax-Tunisia. All patients provided written informed consent prior to their participation.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure 1.
Chromatograms showing the wild-type and the mutant DNA sequence together with the IVG or Genome Browser for the following BRCA mutations: a) c.4067_4071 delAAGAA in BRCA1 gene; b) c. 2338C > T in BRCA1 gene; c) c.17_20delAAGA in BRCA2 gene; d)c.1310_1313 delAAGA in BRCA2 gene and e) c.1796_1800 delCTTAT in BRCA2 gene.
Additional file 2: Table 1.
List of Benign/Likely Benign variants identified in the 134 selected patients with breast/ovarian cancer.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ben Ayed-Guerfali, D., Ben Kridis-Rejab, W., Ammous-Boukhris, N. et al. Novel and recurrent BRCA1/BRCA2 germline mutations in patients with breast/ovarian cancer: a series from the south of Tunisia. J Transl Med 19, 108 (2021). https://doi.org/10.1186/s12967-021-02772-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-021-02772-y