
Abstract
Background
SQUAMOSA promoter-binding protein-like (SPL) transcription factors are widely present in plants and are involved in signal transduction, the stress response and development. The SPL gene family has been characterized in several model species, such as A. thaliana and G. max. However, there is no in-depth analysis of the SPL gene family in forage, especially alfalfa (Medicago sativa L.), one of the most important forage crops worldwide.
Result
In total, 76 putative MsSPL genes were identified in the alfalfa genome with an uneven distribution. Based on their identity and gene structure, these MsSPLs were divided into eight phylogenetic groups. Seventy-three MsSPL gene pairs arose from segmental duplication events, and the MsSPLs on the four subgenomes of individual chromosomes displayed high collinearity with the corresponding M. truncatula genome. The prediction of the cis-elements in the promoter regions of the MsSPLs detected two copies of ABA (abscisic acid)-responsive elements (ABREs) on average, implying their potential involvement in alfalfa adaptation to adverse environments. The transcriptome sequencing of MsSPLs in roots and leaves revealed that 54 MsSPLs were expressed in both tissues. Upon salt treatment, three MsSPLs (MsSPL17, MsSPL23 and MsSPL36) were significantly regulated, and the transcription level of MsSPL36 in leaves was repressed to 46.6% of the control level.
Conclusion
In this study, based on sequence homology, we identified 76 SPL genes in the alfalfa. The SPLs with high identity shared similar gene structures and motifs. In total, 71.1% (54 of 76) of the MsSPLs were expressed in both roots and leaves, and the majority (74.1%) preferred underground tissues to aerial tissues. MsSPL36 in leaves was significantly repressed under salt stress. These findings provide comprehensive information regarding the SPB-box gene family for improve alfalfa tolerance to high salinity.
Similar content being viewed by others

Background
Alfalfa (Medicago sativa L.) is the most widely grown forage legume crop worldwide [1]. Alfalfa has been widely used in animal feed because of its high biomass yield, good palatability, and strong adaptability. Currently, almost half of the world’s irrigated land and approximately 20.0% of cultivated land are affected by salinity [2]. However, many plant growth areas, such as China, are on saline-alkali soil, which seriously affects the growth and development of alfalfa. Transcription factors (TFs) play extremely important roles in controlling the growth and development of plants. TFs greatly affect plant development, secondary metabolism, and abiotic stress tolerance by binding cis-acting elements in the promoter regions of target genes [3, 4]. Although the SPL gene can regulate inflorescence formation and fruit development and enhance stress resistance, knowledge regarding the MsSPL gene in alfalfa is limited. Therefore, it is important to explore the possible functions of MsSPL genes to understand the regulation of growth, development, and abiotic stress in alfalfa.
SPLs constitute a plant-specific family and are widely distributed in green plants. SPL is a general term for a type of transcription factor, and its structure is similar to the SBP box [5]. The SQUAMOSA promoter-binding protein (SBP) domain is highly conserved, with a length of approximately 76 amino acids [6]. The SBP domain contains two tandem zinc fingers (Cys-Cys-His-Cys and Cys-Cys-Cys-His) and one nuclear localization signal (NLS) motif [6, 7]. The members of this transcription factor family share a highly conserved DNA binding domain, the SBP. The SBP box was first isolated from the A. majus cDNA library, and because of its ability to recognize and bind the SQUAMOSA (SQUA) promoter, it was named SQUA [8]. An increasing amount of evidence suggests that TFs play an important role in the regulatory network of plant growth and development [9]. Various gene families, such as TEOSINTE BRANCHED 1, CYCLOIDEA, PCF1 (TCP) [10], and IQ67-Domain (IQD) [11], have been found in eukaryotes.
With the publication of many plant genomes, SPL gene family members has been identified and characterized in A. thaliana [12], O. sativa [13], and G. max [14]. According to the sequence homology and hylogenetic analyses of SPL genes, this family is usually divided into 6–9 subgroups. In P. trichocarpa, there are 28 PtSPL genes, and these genes are divided into 8 subgroups [15]. In A. thaliana, a total of 16 members have been identified as SPL proteins and named AtSPL1 to AtSPL16. They were divided into eight groups according to the amino acid sequence [16]. The functions of these SPL genes in A. thaliana have also been identified, and these genes play an important role in leaf, flower, and shoot development [17, 18].
MicroRNAs are small RNAs of approximately 16–26 nucleotides in length that regulate gene expression at the posttranscriptional level in a sequence-specific manner [19]. As a key regulatory factor in most biological processes, the miR156/SPL module participates in the transformation from the vegetative stage to the reproductive stage, fruit ripening, and yield improvement [20]. However, the miR156/SPL module also responds to abiotic stresses in many plant species [21, 22]. When A. thaliana is under heat stress, the SPL gene is posttranscriptionally downregulated by miR156, which is essential for adapting to repeated heat stress [23]. The overexpression of the SPL gene in B. platyphylla is also very obvious and can improve the scavenging of reactive oxygen species to enhance tolerance to salt and drought stress [24]. Some related studies have also been carried out in alfalfa. In alfalfa, the transcript abundance of the miR156-targeted SPL8 and SPL13 genes was related to salt and drought tolerance [25, 26]. Studies have shown that drought stress increases the expression of miR156 by increasing leaf gas exchange and abscisic acid (ABA) while reducing water loss, thereby increasing the resistance of alfalfa to this stress [25]. To date, SPL genes have been isolated and identified in many plants, such as A. thaliana and O. sativa, but few studies investigated SPL genes in alfalfa, and the function of these proteins is unclear.
Despite the support of physiological, biochemical, and molecular data, the biological function of SPL transcription factor genes is still unclear. In this study, the gene structure, motif composition, chromosome location, and gene duplication of 76 recently completed alfalfa genome sequences were analyzed, and the evolutionary relationship of M. sativa was compared with those of A. thaliana, M. truncatula, and G. max. A quantitative real-time PCR (qRT–PCR) analysis was performed to examine the gene expression patterns in different tissues and their responses to salt stress. Through an overall expression analysis in alfalfa, the role of the members of the specific SPL gene family in the different biological processes of alfalfa was determined. This study not only provided valuable information for screening SPL genes important for the growth and development of alfalfa but also provided a method for mining SPL gene families in other plants.
Results
Sequence identification of the SPL genes in M. sativa
As a plant-specific transcription factor, SQUAMOSA promoter-binding protein-like (SPL) genes are involved in the plant response to adverse environmental conditions. To obtain SPL genes in the forage legume alfalfa, the SBP domain, a canonical feature of SPL, was screened from a Chinese landrace (Medicago sativa L. XinJiangDaYe) genome using HMM and BLASTP [27]. The hits were then confirmed by Pfam (http://pfam.xfam.org/) and the Conserved Domain Database (CDD) (https://www.ncbi.nlm.nih.gov/cdd). Ultimately, 76 genes were designated MsSPL and named MsSPL1—MsSPL76 (Table S1).
The prediction of the subcellular location showed that MsSPLs were localized in the nucleus and that 16 (21.1%) SPLs were also distributed in the cytoplasm, suggesting that the putative MsSPL transcription factors function mainly in the nucleus. The predicted MsSPLs vary in terms of the protein length and isoelectric point (pI), ranging from 100 (MsSPL42) to 1,026 (MsSPL76) amino acids (aa) and 5.24 (MsSPL42) to 9.65 (MsSPL49), respectively (Table S1).
Sequence alignment and phylogenetic analysis of MsSPLs
The multiple alignment showed that in addition to the conserved SBP domain (approximately 78 amino acid residues in length), most MsSPLs shared a highly conserved nuclear localization signal (NLS) and two zinc finger-like structures, namely, Zn-1 and Zn-2 (Fig. S1). In Zn-1, the CCCH is at positions 214, 219, 236, and 239, while in Zn-2, the CCHC is at positions 255, 258, 262, and 274 amino acids. Consistent with a previous report [28], the predicted NLS possesses conserved residues, such as lysine (K) and arginine (R), at positions 271–287.
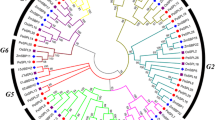
To analyze the phylogenetic relationship, a neighbor-joining tree of SPLs in M. sativa (76), M. truncatula (23) and A. thaliana (16) was constructed using MEGA (Fig. 1). The MsSPLs similar to their counterparts in M. truncatula and A. thaliana were clustered into eight groups (Groups I ~ VIII) with varying numbers of members. The largest group (Group II) contained 18 members, accounting for 23.7% of the MsSPLs, whereas the smallest groups (Group I and Group III) possessed four members. Relative to A. thaliana SPLs, the homologs in the two legumes (M. sativa and M. truncatula) are closer. Interestingly, compared with MtSPL, multiple (2–6) MsSPL counterparts were grouped into the same cluster, indicating the expansion of MsSPL, probably due to genome duplication of the tetraploid forage.

Phylogenetic analysis of SPLs from M. sativa, M. truncatula and A. thaliana. Proteins with SBP domains from M. sativa (Ms), A. thaliana (At) and M. truncatula (Mt) were searched and designated as SPL. Neighbor-joining (NJ) was used for the unrooted phylogenetic tree with the maximum likelihood method (1000 bootstraps)
Gene structure and motif composition of MsSPLs
A phylogenetic tree was constructed based on the predicted full-length MsSPL protein sequences and these proteins were also roughly divided into eight subgroups (Fig. 2A). An analysis of the gene structure of the SPL family in alfalfa revealed that the number of exons varied from 1–11. It seems that the MsSPL members in one group share a similar number of exons, with difference in intron sizes (Fig. 2B). Approximately half (52.6%) of the MsSPLs consist of 3–4 exons, of which 34.2% (26 out of 76) with 3 exons and 18.4% (14 out of 76) with 4 exons (Fig. 2B). The members of Groups I and II, excluding MsSPL30, contain relatively more exons (10–12) than the average number of exons in MsSPLs. The MsSPLs in Group VI comprise no more than three (1–3) exons. In addition to the conserved SBP domain at the N-terminus of MsSPLs, there is a conserved ankyrin (ANK) domain at the C-terminus of some Group II members (70.6% = 12/17), which is involved in protein–protein interactions in G. max [14]. The findings were consistent with a previous report in S. miltiorrhiza [29]. Therefore, MsSPLs from the same group share a similar gene structure, and the length of the exons is conserved correspondingly.

Analysis of the gene structure and conserved motifs of MsSPL based on phylogenetic relationships. A A phylogenetic tree was constructed based on the full-length sequences of M. sativa SPL proteins. B Exon/intron structure of MsSPLs. The black lines indicate introns. C Motif composition of putative MsSPL proteins. Motifs are displayed by colored boxes numbered 1–20. CDS stands for coding
We examined the motif composition of the MsSPLs using MEME. In total, 20 motifs (motifs 1–20) were identified in the 76 putative MsSPLs ranging from 2 (MsSPL42) to 18 (MsSPL1, MsSPL2, MsSPL24, MsSPL38, MsSPL39 and MsSPL61) for individual proteins (Fig. 2C). On average, Groups II and VI contained the most and the fewest motifs, respectively. Among them, motifs 1, 2 and 8 were the top three motifs present in 97.4%, 89.5% and 96.1% of the MsSPLs, respectively, suggesting that these motifs are the most important components of MsSPL proteins. Similar to the gene structure, the MsSPL members from the same group, particularly Group I, shared similar motif compositions, including the motif type and number. Some motifs were present in certain groups. For example, motifs 3, 5 and 7 were present exclusively in the SPL members in Groups I and II (Fig. 2C). Among the Group II members, 13 contain a conserved motif, and 6 contain 18 of the 20 motifs, except for Motif 13 and Motif 20. Therefore, MsSPL members of the same group share a similar gene structure and motif composition, while SPLs from different groups are likely to have specific structures, implying that the functional conservation and diversity of the MsSPLs evolved during evolution.
Analysis of the distribution, gene duplication and synteny of the MsSPLs
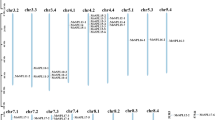
The predicted MsSPLs were mapped based on the genome database of M. sativa (XinJiangDaYe) [27]. Seventy-four of the 76 MsSPLs were unevenly distributed on 26 chromosomes, and the remaining two (MsSPL38 and MsSPL49) have not yet been assembled (Fig. 3). On average, there are approximately 2.8 SPL genes on each chromosome (Chr). Among them, no SPL was identified on Chr4.3, Chr5.2 or Chr6, while Chr4.2 possessed seven SPLs, probably due to gene duplication events.

Distribution of SPL genes in the M. sativa genome. A Location of genes on chr1.1 to 4.1. B Location of genes on chr4.2 to 8.4. Chromosomes are indicated in different colors and numbered on the top. The scale (Mb) represents the lengths of the chromosomes
The analysis of the duplication event in the MsSPL family showed that there were 73 pairs of segmental duplicates but no tandem duplications (Fig. 4). MsSPL homologs (such as MsSPL8 and MsSPL68) from different chromosomes share higher identity. This result suggests that segment duplication may contribute to MsSPL expansion.

Schematic representation of the interchromosomal relationship among the MsSPLs. Syntenic blocks in the alfalfa genome are indicated by lines in orange
Evolutionary analysis of the MsSPLs and orthologs from three model species
To explore the evolutionary origin of MsSPLs, we performed a syntenic analysis of SPLs from three model species A. thaliana, M. truncatula and G. max. In total, 16, 23, and 41 SPLs have been identified in A. thaliana (125 Mb) [30], M. truncatula (500 Mb) [31] and G. max (1.025 Gb) [32]. In total, 57 MsSPLs displayed syntenic relationships with M. truncatula, 56 MsSPLs displayed syntenic relationships with G. max and 33 MsSPLs displayed syntenic relationships with A. thaliana (Fig. 5). Among these MsSPLs, 131 pairs of orthologous genes were found with G. max, 79 pairs of orthologous genes were found with M. truncatula, and 40 pairs of orthologous genes were found with A. thaliana. Consistent with the MsSPL distribution on the chromosome, chromosomes 1, 2 and 7 of alfalfa accounted for the top three SPL homologous gene pairs with the three model plants (Table S2). Syntenic blocks between the two Medicago species showed that the four subgenomes of alfalfa had high collinearity with the corresponding M. truncatula genome. Approximately 63.3% (50 of 79) of the MsSPLs paired with MtSPLs on the same chromosome, suggesting the relatively conserved distribution of SPLs between the two legumes. The pairing of 36.7% of the SPLs across chromosomes implies the occurrence of interchromosomal rearrangements, particularly between chromosome 4 and chromosome 8, as reported by Li et al. [33].

Synteny analysis of SPL genes between M. sativa and three representative plant species. Gray lines in the background indicate collinear blocks within M. sativa and the indicated plant, whereas the red lines highlight syntenic SPL gene pairs
Prediction of the cis-acting elements in the promoter of MsSPLs
The cis-elements 2 kb upstream of the start codon (ATG) of MsSPLs were analyzed using the PlantCARE database (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/). According to the classification of wheat [34], 25 cis-elements were found for MsSPLs, with 11 (44.0%) related to hormone and stress responses, nine (36.0%) related to light responsiveness, and five (20.0%) related to plant growth and development (Fig. 6 and Table S3). Regarding the cis-acting elements predicted to be associated with hormone and stress responses, abscisic acid (ABA)-responsive elements (ABREs) and AU-rich elements (AREs) were predominant in MsSPLs, accounting for 89.4% and 86.8%, respectively. Both cis-elements are present in individual MsSPLs with an average of two copies. The presence of ABRE, the major cis-element in ABA-responsive genes, implies the potential involvement of MsSPLs in alfalfa resistance to osmotic stresses, including drought and salinity, as previously reported in O. sativa and A. thaliana [23, 35].

Analysis of cis-acting elements in the putative promoter of MsSPLs. The number in color represents the copy of the cis-acting element (at the bottom) of individual MsSPLs (on the right)
Comparison of the expression patterns of MsSPLs between roots and leaves
To compare the expression pattern of the MsSPLs between roots and leaves, 14-day-old alfalfa seedlings were used for RNA sequencing. Based on fragments per kilobase of transcript per million fragments mapped (FPKM), 68 MsSPLs were expressed in either tissue tested. Among them, 54 MsSPLs were detected simultaneously in both tissues, with 74.1% (40 of 54) showing higher expression in roots than in leaves (Fig. 7A). Nine MsSPLs were detected in roots rather than leaves, with four (SPL15, SPL55, SPL60 and SPL62) from Group V and two (SPL5 and SPL68) from Group VIII (Fig. 7B). In contrast, five MsSPLs exhibited the opposite expression pattern, showing expression in leaves but not roots (Fig. 7C). Our results show that approximately 72.1% (49 of 68) of MsSPLs had higher expression in roots relative to aerial tissues, indicating that these MsSPLs are preferentially expressed in underground tissues at this stage.

FPKM of MsSPLs in roots and leaves using RNA sequencing. A FPKM of MsSPLs expressed in both roots and leaves. The R/L ratio represents the ratio of FPKM in roots to that in leaves. B FPKM of the MsSPLs detected in roots but not leaves. C FPKM of the MsSPLs detected in leaves rather than roots. The color represents the FPKM normalized value. The blue and orange colors represent higher and lower expression, respectively
To investigate the response of alfalfa to salt stress at the transcription level, we measured the expression of MsSPLs in roots and leaves from seedlings treated with NaCl (300 mM) for 2 weeks. The data were arbitrarily filtered by the absolute value of Log2(FoldChange) ≥ 1 and Padj < 0.05 (Fig. S2). Three MsSPLs (MsSPL17, MsSPL23 and MsSPL36) were differentially expressed under the salt treatment, with the former MsSPL upregulated in the roots by 17.3% and the latter two downregulated in the leaves by 48.0% and 46.6%, respectively. Although both MsSPL17 and MsSPL23 showed leaf-preference under normal conditions, upon salt stress, the expression of the two genes contrasted each other, suggesting that these genes likely play opposite roles in the alfalfa response to long-term high salinity. The results were confirmed by qRT–PCR verification (Fig. 8 and Table S4).

Expression levels analyses of three MsSPL genes under salt treatment by qRT-PCR. A Expression levels of three MsSPL genes in root. B Expression levels of three MsSPL genes in leaf. “CK” represents normal growth condition, “Salt” represents salt treatment. The levels in root and leaf of the CK were arbitrarily set to 1. Error bars represent the standard deviations of three technical replicates
Discussion
SQUAMOSA-promoter Binding Protein Like (SPLs) encode a family of plant-specific transcription factors containing a conserved SQUAMOSA-promoter Binding Protein (SBP) domain and are involved in the regulation of the flowering time [36], plant development [37] and the stress response [38]. The SPL family has been mostly identified in model plants, such as A. thaliana [39], O. sativa [13] and M. truncatula [40]. The release of the alfalfa genome sequence in recent years [27, 41] has facilitated the identification of MsSPLs from the most valuable forage worldwide. The findings of this study could benefit alfalfa production and breeding, especially the generation of varieties with improved tolerance to environmental stresses, such as alkalinity and salinity.
M. sativa SPLs (MsSPLs) are canonical and highly conserved with SPLs from the model plant species. One line of evidence shows that similar to the orthologs from M. truncatula and A. thaliana, the 76 MsSPLs we identified here were phylogenetically clustered into eight groups based on their sequence identity, gene structure and motif composition. In individual groups, the number of MsSPLs was greater than that of MtSPLs or AtSPLs, with MsSPL members much closer to M. truncatula than A. thaliana. Moreover, the MsSPLs from the four subgenomes of alfalfa showed high collinearity with M. truncatula orthologs from the corresponding chromosome, and neither species had an SPL gene on chromosome 6 [40]. Multiple MsSPLs are probably attributed to the genome duplication of the tetraploid legume in comparison with MtSPLs. Interestingly, although there were 73 pairs of segment duplications among the MsSPLs, no tandemly duplicated MsSPL pairs were found (Fig. 4), indicating that the two legumes diverged prior to the occurrence of genome recombination for SPLs. Notably, in addition to the conserved motifs, such as Motif 1, Motif 2 or Motif 8, several unique motifs were present in certain groups of MsSPLs, suggesting a potential contribution to the specific biological functions of the dedicated MsSPL groups.
It appears that MsSPL36 is a crucial candidate for improving alfalfa tolerance to salt stress. Plant SPLs are well known as targets of microRNA156 in regulating phase change (from the vegetative to reproductive stage) and the stress response [3, 42]. For example, miR156-mediated downregulation of three SPLs, i.e., SPL2, SPL9 and SPL11, enhanced A. thaliana tolerance to heat stress [23]. Thirty-one maize SPLs displayed variations in their expression behavior upon exposure to one or more stresses, such as dehydration, salinity, cold and ABA [38], and transgenic tobacco expressing ZmmiR156 exhibited improved performance against drought and salt [42]. In addition, the SPLs in T. chinensis and D. glomerata have been shown to respond to salt, drought and heat stresses via the negative control of miR156 [21, 43]. In China, most alfalfa plantations are located in regions with saline-alkali soil due to the lack of farmland [44]. An urgent and promising task for alfalfa production in the nation is to breed varieties with tolerance to high salinity [45]. Our prediction of the cis-elements present in the putative promoter region of the MsSPLs revealed that 89.4% of MsSPLs possessed ABA-responsive elements (ABREs) [46], suggesting a possible contribution to the alfalfa abiotic stress response. Similar results have been documented in wheat and Betula luminifera [34, 47]. The transcriptomic analysis revealed that although 10.5% of the MsSPLs were undetected in our experiments, 89.5% of the detected MsSPLs were expressed in either the roots or leaves of 2-week-old plants, with 74.1% preferring roots to leaves. Under long-term high salinity (0.3 M NaCl) pressure, three MsSPLs were differentially expressed with MsSPL17 up-regulated in roots, while MsSPL23 and MsSPL36 down-regulated in leaves of two-week-old plants. Our verification by quantitative RT–PCR showed that MsSPL36 in leaves was repressed by the salt treatment to 46.6% of the control level. Experimental evidence is needed to support the notion that MsSPL36 is involved in the alfalfa response to environmental stresses, especially salt. The generation of transgenic alfalfa with MsSPL36 knockout via miR156 or overexpression could facilitate the elucidation of its functions in forage under adverse environmental conditions. A comprehensive analysis with multiple time points of salt treatment could be helpful for a dynamic illustration of MsSPL expression profiles in response to stress.
Conclusion
The phylogeny and diversification of SPL genes in alfalfa were investigated at different levels, including gene structures, evolutionary relationships, synteny analysis and expression patterns. All 76 MsSPL genes were divided into 8 groups, and genes in the same group shared similar evolutionary features and expression patterns, implying potentially similar functions for MsSPL genes. SPLs with a high identity shared similar gene structures and motifs. In total, 71.1% of the MsSPLs were expressed in both roots and leaves, and the majority (74.1%) preferred underground tissues to aerial tissues. The expression of MsSPL36 in leaves was significantly repressed by salt stress. Our findings provide comprehensive information regarding the SPB-box gene family in alfalfa and have a certain value for alfalfa to improve salt tolerance.
Methods
Plant materials and growth conditions
Alfalfa seeds (Cultivar Zhongmu No. 1) from the Institute of Animal Science of the Chinese Academy of Agricultural Sciences were germinated in a petri dish, treated at 4 °C for 3 days and then grown in a greenhouse at 24 °C (day)/20 °C (night) under a 16 h light/8 h dark photoperiod at a relative humidity of 70 to 80% for 4 days. The germinated seedlings were transferred to flowerpots placed in the greenhouse and developed for 7 days. The two-week-old seedlings were irrigated either with 20 ml 300 mM NaCl solution every two days or water as a control group. After 14 days of treatment, the roots and leaves of the alfalfa seedlings were sampled, immediately placed in liquid nitrogen, and stored at -80 °C until further use.
Identification of SPL genes in alfalfa
The alfalfa genome was downloaded from the alfalfa Genome Project (https://figshare.com/projects/whole_genome_sequencing_and_assembly_of_Medicago_sativa/66380). The A. thaliana protein sequences were obtained from A. thaliana Information Resource (TAIR) (https://www.arabidopsis.org/), and the M. truncatula genome was searched on a website (http://www.medicagogenome.org/). The largest number of SPL genes was screened from the alfalfa genome by two BLASTp methods, and the hidden MarKov model (HMM) profiles corresponding to the SBP domain (PF03110) were downloaded from the Pfam protein family database (https://pfam.xfam.org/). In total, 76 MsSPL genes were identified in the M. sativa genome using BLAST with a cutoff E-value > 1e−9. We collected the amino acid sequence of A. thaliana SPL proteins from the TAIR library, which ranges from 131 to 1035 aa. Similar SPL genes from the alfalfa genome were identified by using the SPL gene sequence of A. thaliana as a target. Subsequently, we analyzed the conserved domain of the MsSPL genes and removed the gene that did not contain the SBP conserved domain. Finally, 76 genes containing the SBP domain were screened from the alfalfa genome. The ExPASy website (https://web.expasy.org/compute_pi/) was used to analyze the MsSPL gene sequences to obtain the theoretical isoelectric points (pIs) and molecular weights (MWs).
Phylogenetic analysis and intron–exon structure determination
The SPL protein sequences for the phylogenetic tree were obtained from the UniProt database (https://www.UniProt.org). The multiple amino acid sequences of identified MsSPL genes were aligned using Clustalx2.0 software with the default parameters. Phylogenetic trees comparing M. sativa, A. thaliana and M. truncatula were constructed with the NJ method, and the specific parameters were Poisson model and 1000 bootstrap replications by using the MEGA software. The SPL protein sequences from M. sativa, A. thaliana and M. truncatula were also aligned using the Clustalx2.0 program before the phylogenetic tree was constructed. Then, the MsSPL gene structure was predicted by an online gene structure editor (http://gsds.cbi.pku.edu.cn/) website to align the coding and genome sequences. The determination of the conserved motifs in the MsSPL proteins was conducted by the MEME online program (http:/meme.nbcr.net/meme/intro.html), and the parameters were set to the optimum mode width of 6 to 200 and the maximum number of motifs of 20.
Chromosome location, gene duplication and synteny analysis
Information concerning the chromosomal location of MsSPL genes, including the chromosome length, gene direction, and gene start and stop positions, was obtained from the alfalfa genome database. MCScanX software was used to analyze the MsSPL replication events and detect collinear regions between MsSPLs and collinear blocks of MsSPL genes with A. thaliana, M. truncatula, and G. max. All function and chromosomal location information was obtained by TBtools software [48].
Cis-element analysis
The upstream 2 kb sequence was extracted as the promoter region for the prediction of cis-acting elements. The homeopathy components of the promoter sequence were predicted by the online tool PlantCARE, and the predicted results were drawn by GSDS online software.
Gene expression pattern of MsSPL gene families with RNA-seq data
The Illumina HiSeq 2500 platform was used to sequence the cDNA library based on synthetic sequencing technology, and a large amount of high-read data was obtained. Two replicates were prepared for the construction of a sequencing library per sample. We used RNA-seq data to analyze the gene expression patterns of MsSPL genes. The data were filtered and compared to the reference genome of XinJiangDaYe alfalfa. In addition, we applied FPKM (fragments per kilobase of transcript per million fragments mapped) to calculate the gene expression level according to the number of reads mapped to the reference sequence. The heatmap of the MsSPL gene expression profile was constructed by R software.
Gene pattern analyses of MsSPL genes by real-time quantitative RT-PCR
Total RNA was extracted from the roots and leaves of normally growing and salt-treated alfalfa seedlings with TRIzol reagent according to the manufacturer's instructions. Then, the cDNA library was constructed for the subsequent reactions using the Genesand Kit (UnionScript First-strand cDNA Synthesis Mix for qPCR). SYBR Premix Ex Taq II (TaKaRa) with a CFX96 real-time PCR system (Bio-Rad) was used to conduct the RT–PCR experiments. The qRT–PCR primers were designed on the NCBI website (https://blast.ncbi.nlm.nih.gov/) (Table S4). MsActin was used as the internal reference gene for data normalization. A total of four samples (CK_root, CK_leaf, Salt_root and Salt_leaf) were used in this study. Three independent biological replicates and three technical repeats were taken. Roots and leaves under control conditions were selected as the control samples for measuring gene expression under salt treatment. The data were quantified by the 2−△△CT method [49].
Availability of data and materials
RNA sequence data from roots and leaves after 14 days of salt treatment in a greenhouse has been submitted to The NCBI Sequence Read Archive (BioProject: PRJNA777963).
References
He F, Kang JM, Zhang F, Long RC, Yang QC. Genetic mapping of leaf-related traits in autotetraploid alfalfa (Medicago sativa L.). Mol Breed. 2019;39(10):147.
Paul D, Lade H. Plant-growth-promoting rhizobacteria to improve crop growth in saline soils: a review. Agron Sustain Dev. 2014;34(4):737–52.
Gao J, Cui L, et al. The miR156-SPL9-DFR pathway coordinates the relationship between development and abiotic stress tolerance in plants. Plant J. 2014;80(6):1108–17.
Hai W, Wang H. The miR156/SPL module, a regulatory hub and versatile toolbox, gears up crops for enhanced agronomic traits. Mol Plant. 2015;8(005):677–88.
Liu M, Sun W, Ma Z, Huang L, Chen H. Genome-wide identification of the SPL gene family in Tartary Buckwheat (Fagopyrum tataricum) and expression analysis during fruit development stages. BMC Plant Biol. 2019;19(1):1–14.
Yamasaki K, Kigawa T, Inoue M, Tateno M, Yamasaki T, Yabuki T, Aoki M, Seki E, Matsuda T, Nunokawa E. A novel zinc-binding motif revealed by solution structures of DNA-binding domains of Arabidopsis SBP-family transcription factors. J Mol Biol. 2004;337(1):49–63.
Klein J, Saedler H, Huijser P. A new family of DNA binding proteins includes putative transcriptional regulators of the Antirrhinum majus floral meristem identity gene SQUAMOSA. Mol Gen Genet. 1996;250(1):7–16.
Huijser P, Klein J, Lönnig WE, Meijer H, Saedler H, Sommer H. Bracteomania, an inflorescence anomaly, is caused by the loss of function of the MADS-box gene squamosa in Antirrhinum majus. Embo J. 1992;11(4):1239–49.
Riechmann JL, Heard J, Martin G, Reuber L, Yu GL. Arabidopsis Transcription Factors: Genome-Wide Comparative Analysis Among Eukaryotes. Science. 2001;290(5499):2105–10.
Huo Y, Xiong W, Su K, Li Y, Sun Z. Genome-wide analysis of the TCP gene family in switchgrass ( Panicum virgatum L.). Int J Genomics. 2019;2019(1):1–13.
Yuan J, Liu T, Yu Z, Li Y, Ren H, Hou X, Li Y. Genome-wide analysis of the Chinese cabbage IQD gene family and the response of Br IQD5 in drought resistance. Plant Mol Biol. 2019;99(6):603–20.
Xu M, Hu T, Zhao J, et al. Developmental Functions of miR156-Regulated SQUAMOSA PROMOTER BINDING PROTEIN-LIKE (SPL) Genes in Arabidopsis thaliana. PLoS Genet. 2016;12(8):e1006263.
Xie K, Wu C, Xiong L. Genomic organization, differential expression, and interaction of SQUAMOSA promoter-binding-like transcription factors and microRNA156 in rice. Plant Physiol. 2006;142(1):280–93.
Tripathi RK, Goel R, Kumari S, Dahuja A. Genomic organization, phylogenetic comparison, and expression profiles of the SPL family genes and their regulation in soybean. Dev Genes Evol. 2017;227(2):101–19.
Li C, Lu S. Molecular characterization of the SPL gene family in Populus trichocarpa. BMC Plant Biol. 2014;14(1):1–15.
Birkenbihl RP, Jach G, Saedler H, Huijser P. Functional dissection of the plant-specific SBP-domain: overlap of the DNA-binding and nuclear localization domains. J Mol Biol. 2005;352(3):585–96.
Gandikota M, Birkenbihl RP, Hmann SH, Cardon GH, Saedler H. The miRNA156/157 recognition element in the 3’ UTR of the Arabidopsis SBP box gene SPL3 prevents early flowering by translational inhibition in seedlings. Plant J. 2010;49(4):683–93.
Wu G. Temporal regulation of shoot development in Arabidopsis thaliana by miR156 and its target SPL3. Development. 2006;133(18):3539–47.
Sun G. MicroRNAs and their diverse functions in plants. Plant Mol Biol. 2012;80(1):17–36.
Wu Z, Cao Y, Yang R, Qi T, Hang Y, Lin H, Zhou G, Wang ZY, Fu C. Switchgrass SBP-box transcription factors PvSPL1 and 2 function redundantly to initiate side tillers and affect biomass yield of energy crop. Biotechnol Biofuels. 2016;9(1):1–15.
Wang J, Ye Y, Xu M, Feng L, Xu L. Roles of the SPL gene family and miR156 in the salt stress responses of tamarisk (Tamarix chinensis). BMC Plant Biol. 2019;19(1):1–11.
Aung B, Gruber MY, Hannoufa A. The MicroRNA156 system: a tool in plant biotechnology. Biocatal Agric Biotechnol. 2015;4(4):432–42.
Stief A, Altmann S, Hoffmann K, Pant BD, Scheible WR, Bäurle I. Arabidopsis miR156 regulates tolerance to recurring environmental stress through SPL transcription factors. Plant Cell. 2014;26(4):1792–807.
Ning K, Chen S, Huang H, Jiang J, Yuan H, Li H. Molecular characterization and expression analysis of the SPL gene family with BpSPL9 transgenic lines found to confer tolerance to abiotic stress in Betula platyphylla Suk. Plant Cell. 2017;130(3):469–81.
Arshad M, Feyissa BA, Amyot L, Aung B, Hannoufa A. MicroRNA156 improves drought stress tolerance in alfalfa (Medicago sativa) by silencing SPL13. Plant Sci. 2017;258:122–36.
Gou J, Debnath S, Sun L, Flanagan A, Tang Y, Jiang Q, Wen J, Wang ZY. From model to crop: functional characterization of SPL 8 in M. truncatula led to genetic improvement of biomass yield and abiotic stress tolerance in alfalfa. Plant Biotechnol J. 2018;16(4):951–62.
Chen H, Zeng Y, Yang Y, Huang L, Qiu Q. Allele-aware chromosome-level genome assembly and efficient transgene-free genome editing for the autotetraploid cultivated alfalfa. Nat Commun. 2020;11(1):1–11.
Li S, Li L, Jiang Y, Wu J, Sun H, Zhao M, Jiang Y, Zhu L, Wang Y, Su Y. SQUAMOSA Promoter Binding Protein-Like (SPL) Gene Family: TRANSCRIPTOME-Wide Identification, Phylogenetic Relationship, Expression Patterns and Network Interaction Analysis in Panax ginseng CA Meyer. Plants. 2020;9(3):354.
Zhang L, Wu B, Zhao D, Li C, Shao F, Lu S. Genome-wide analysis and molecular dissection of the SPL gene family in Salvia miltiorrhiza. J Integr Plant Biol. 2014;56(1):38–50.
Schneeberger K, Ossowski S, Ott F, Klein JD, Wang X, Lanz C, Smith LM, Cao J, Fitz J, Warthmann N. Reference-guided assembly of four diverse Arabidopsis thaliana genomes. Proc Natl Acad Sci Belarus-Agrar Ser. 2011;108(25):10249–54.
Young ND, Udvardi M. Translating Medicago truncatula genomics to crop legumes. Curr Opin Plant Biol. 2009;12(2):193–201.
Shen Y, Liu J, Geng H, Zhang J, Liu Y, Zhang H, Xing S, Du J, Ma S, Tian Z. De novo assembly of a Chinese soybean genome. Sci China-Life Sci. 2018;61(8):871–84.
Li A, Liu A, Du X, Chen JY, Yin M, Hu HY, Shrestha N, Wu SD, Wang HQ, Dou QW. A chromosome-scale genome assembly of a diploid alfalfa, the progenitor of autotetraploid alfalfa. Hortic Res. 2020;7(1):1–12.
Zhu T, Liu Y, Ma L, Wang X, Zhang D, Han Y, Ding Q, Ma L. Genome-wide identification, phylogeny and expression analysis of the SPL gene family in wheat. BMC Plant Biol. 2020;20(1):1–14.
Mukherjee K, Choudhury AR, Gupta B, Gupta S, Sengupta DN. An ABRE-binding factor, OSBZ8, is highly expressed in salt tolerant cultivars than in salt sensitive cultivars of indica rice. BMC Plant Biol. 2006;6(1):1–14.
Wang JW, Czech B, Weigel D. miR156-regulated SPL transcription factors define an endogenous flowering pathway in Arabidopsis thaliana. Cell. 2009;138(4):738–49.
Cardon GH, Höhmann S, Nettesheim K, Saedler H, Huijser P. Functional analysis of the Arabidopsis thaliana SBP-box gene SPL3: a novel gene involved in the floral transition. Plant J. 1997;12(2):367–77.
Mao HD, Yu LJ, Li ZJ, Yan Y, Han R, Liu H, Ma M. Genome-wide analysis of the SPL family transcription factors and their responses to abiotic stresses in maize. Plant Gene. 2016;6:1–12.
Yang Z, Wang X, Gu S, Hu Z, Xu H, Xu C. Comparative study of SBP-box gene family in Arabidopsis and rice. Gene. 2008;407(1):1–11.
Wang H, Lu Z, Xu Y, Kong L, Han L. Genome-wide characterization of SPL family in Medicago truncatula reveals the novel roles of miR156/SPL module in spiky pod development. BMC Genomics. 2019;20(1):1–14.
Shen C, Du HL, Chen Z, Lu HW, Zhu FG, Chen H, Meng XZ, Liu QQ, Liu P, Zheng LH, Li XX, Dong JL, Liang CZ, Wang T. The chromosome-level genome sequence of the autotetraploid alfalfa and resequencing of core germplasms provide genomic resources for alfalfa research. Mol Plant. 2020;13(9):1250–61.
Kang T, Yu CY, Liu Y, Song WM, Bao Y, Guo XT, Li B, Zhang HX. Subtly manipulated expression of ZmmiR156 in tobacco improves drought and salt tolerance without changing the architecture of transgenic plants. Front Plant Sci. 2020;10:1664.
Feng G, Han J, Yang Z, Liu Q, Shuai Y, Xu X, Nie G, Huang L, Liu W, Zhang X. Genome-wide identification, phylogenetic analysis, and expression analysis of the SPL gene family in orchardgrass (Dactylis glomerata L.). Genomics. 2021;113(4):2413–25.
Wang Y, Wang J, Guo D, Zhang H, Che Y, Li Y, Tian B, Wang Z, Sun G, Zhang H. Physiological and comparative transcriptome analysis of leaf response and physiological adaption to saline alkali stress across pH values in alfalfa (Medicago sativa). Plant Physiol Biochem. 2021;167:140–52.
Wei TJ, Jiang CJ, Jin YY, Zhang GH, Wang MM, Liang ZW. Ca2+/Na+ ratio as a critical marker for field evaluation of saline-alkaline tolerance in alfalfa (Medicago sativa L.). Agronomy. 2020;10(2):191.
Nakashima K, Yamaguchi-Shinozaki K. ABA signaling in stress-response and seed development. Plant Cell Rep. 2013;32(7):959–70.
Li XY, Lin EP, Huang HH, et al. Molecular characterization of SQUAMOSA PROMOTER BINDING PROTEIN-LIKE (SPL) gene family in Betula luminifera. Front Plant Sci. 2018;9:608.
Chen C, Chen H, Zhang Y, Thomas HR, Xia R. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol Plant. 2020;13(8):1194–202.
Kenneth JL, Thomas DS. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. 2001;25(4):402–8.
Acknowledgements
We thank all colleagues in our laboratory for providing useful discussions and technical assistance. We are very grateful to the editor and reviewers for critically evaluating the manuscript and providing constructive comments for its improvement.
Funding
This work was supported by the National Natural Science Foundation of China (31971758), the Agricultural Science and Technology Innovation Program (ASTIP No. CAAS-ZDRW202201 and ASTIP-IAS14) and Central Public-interest Scientific Institution Basal Research Fund (No.2022-YWF-ZYSQ-04).
Author information
Authors and Affiliations
Contributions
Fei He, Ruicai Long, and Zhen Wang planned and designed the research and analyzed data. Zhen Wang, Fei He, Ruicai Long wrote the manuscript. Chunxue Wei, Mingna Li, Yunxiu Zhang, Junmei Kang, Qingchuan Yang performed the experiments. Zhen Wang, Lin Chen supervised the research. Fei He, Ruicai Long contributed equally. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The cultivar Zhongmu No.1 used in the experiment is supplied by Institute of Animal Science of Chinese Academy of Agricultural Sciences. These plant materials are widely used in China. This article does not contain any studies with human participants or animals and did not involve any endangered or protected species. The collection of plant material and use comply with relevant institutional, national, and international guidelines and legislation.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
List of the 76 MsSPL genes identified in this study.
Additional file 2: Table S2.
One-to-one orthologous relationships between Medicago sativa L. and other plants.
Additional file 3: Table S3.
Cis_elements contained in the MsSPL genes promoter region.
Additional file 4: Table S4.
The primer sequences of qRT-PCR.
Additional file 5: Fig. S1.
Alignment of the conserved SBP domain in MsSPL proteins.
Additional file 6: Fig. S2.
Expression profile MsSPL genes across different tissues.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
{kind=link}
Cite this article
He, F., Long, R., Wei, C. et al. Genome-wide identification, phylogeny and expression analysis of the SPL gene family and its important role in salt stress in Medicago sativa L.. BMC Plant Biol 22, 295 (2022). https://doi.org/10.1186/s12870-022-03678-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-022-03678-7