
Abstract
Background
Due to a relatively high level of codominant inheritance and transferability within and among taxonomic groups, simple sequence repeat (SSR) markers are important elements in comparative mapping and delineation of genomic regions associated with traits of economic importance. Expressed sequence tags (ESTs) are a source of SSRs that can be used to develop markers to facilitate plant breeding and for more basic research across genera and higher plant orders.
Methods
Leaf and meristem tissue from ‘Heritage’ red raspberry (Rubus idaeus) and ‘Bristol’ black raspberry (R. occidentalis) were utilized for RNA extraction. After conversion to cDNA and library construction, ESTs were sequenced, quality verified, assembled and scanned for SSRs. Primers flanking the SSRs were designed and a subset tested for amplification, polymorphism and transferability across species. ESTs containing SSRs were functionally annotated using the GenBank non-redundant (nr) database and further classified using the gene ontology database.
Results
To accelerate development of EST-SSRs in the genus Rubus (Rosaceae), 1149 and 2358 cDNA sequences were generated from red raspberry and black raspberry, respectively. The cDNA sequences were screened using rigorous filtering criteria which resulted in the identification of 121 and 257 SSR loci for red and black raspberry, respectively. Primers were designed from the surrounding sequences resulting in 131 and 288 primer pairs, respectively, as some sequences contained more than one SSR locus. Sequence analysis revealed that the SSR-containing genes span a diversity of functions and share more sequence identity with strawberry genes than with other Rosaceous species.
Conclusion
This resource of Rubus-specific, gene-derived markers will facilitate the construction of linkage maps composed of transferable markers for studying and manipulating important traits in this economically important genus.
Similar content being viewed by others
Background
Red raspberry (Rubus idaeus L.) is an important fruit crop grown world-wide in the Northern and Southern hemispheres; black raspberry (R. occidentalis L.) is a specialty crop grown mainly in the Pacific Northwest of the United States. Interest in improvement of these crops is increasing in light of studies on their nutritional and nutraceutical value [1–4]. Development of new cultivars can benefit from reliable markers linked to important traits, including disease resistance, flowering traits, fruit quality characteristics, and plant architecture. Because interspecific hybridization was widely used by caneberry breeders [5, 6], markers that are transferrable between black and red raspberry and even between raspberry and blackberry would be especially useful. In addition, transferable Rubus markers could further illuminate mechanisms of sub-genomic organization in hybrids between disomic and polysomic species [7, 8]. Very few molecular markers exist for Rubus in general [9–12] and fewer are transferable between species [10, 13–15]. Several genetic linkage maps composed of various types of molecular markers are available for raspberry [14, 16–19], and one is available for blackberry [12], however, not all marker types used to construct these maps are transferable between taxa. Many more Rubus molecular markers and other genomic tools are needed to map important traits, facilitate cultivar development, maintain cultivar identity, and study basic genetic and genomic mechanisms.
Molecular markers designed from simple sequence repeats (SSR), tandem repeats of 1–6 nucleotides that frequently show co-dominant inheritance, are known to be highly variable even within species, and are transferable across taxa to a varying extent [20]. Gene-based SSR loci derived from expressed sequence tag (EST-SSR) are significantly more transferable across large taxonomic distances compared with genomic SSRs [21]. This feature makes EST-SSRs superior for comparative linkage mapping and interspecific cross-verification and manipulation of genomic regions associated with phenotypic traits [11, 18, 22–30]. However, EST resources available for the genus Rubus at the National Center for Biotechnology Information’s (NCBI) GenBank are scarce with only 3184 and 50 cDNA sequences for R. idaeus and R. occidentalis, respectively (accessed on January 24, 2015). A main impetus for this sequencing project was to generate a useful set of EST-SSR markers to enable further genetic research into the raspberry genome, and to increase the number of DNA sequences available for the Rosaceae research community and raspberry breeders. EST-SSRs reported here can significantly advance comparative linkage analysis among Rubus species.
Results and discussion
Red raspberry cDNA library construction and SSR discovery
A red raspberry cDNA library of 18,432 clones (48 plates in a 384-well format) was produced from Rubus idaeus cv. Heritage [31]. ‘Heritage’ is a widely grown, everbearing cultivar with resistance to most common raspberry diseases, and medium to large sized fruit with good color, flavor, firmness and freezing quality [32]. The cDNA library was prepared from the newly emerging leaves of a single plant. A cDNA library subset consisting of 1824 clones was sequenced with Sanger technology [33] (Clemson University Genomics & Computational Biology Laboratory, Clemson, SC, USA) yielding 1149 high quality sequences after removal of sequence shorter than 100 base pairs (bp) reported as accession numbers JZ840520 through JZ841668 in GenBank. The resulting sequences had an average length of 429 bp and an average Phred quality score [34] of 48. Transcripts derived from the same expressed gene sequence were assembled into 136 contiguous sequences (contigs) and 732 singletons, yielding a unique gene sequence or “unigene” of 868 sequences, thus reducing locus redundancy and inflation of marker numbers derived from a single locus.
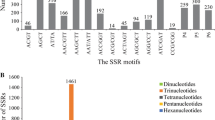
A search for SSR loci within the unigenes using the SSR mining script tool found in the Toolbox on the Genome Database for Rosaceae [35, 36] identified 121 short, perfect repeats in the unigene sequences, which are candidate regions for high polymorphism. Trimers, 3 bp repeats, are more common repeat lengths for gene coding regions, likely because their increase or decrease in repeat number does not cause a reading frame shift [37]. This dataset did demonstrate this tendency with 30 % dimers (2 bp repeat motif), 44 % trimers (3 bp repeat motif), 20 % tetramers (4 bp repeat motif) and 6 % pentamers (5 bp repeat motif). Primers were designed to facilitate the amplification of the SSR loci, yielding 131 primer pairs suitable for testing 98 individual unigenes (Additional file 1).
Black raspberry cDNA library construction and SSR discovery
Rubus occidentalis cv. Bristol [38] was chosen for construction of the black raspberry transcript library. ‘Bristol’ fruit ripen early, are medium sized and firm with excellent flavor; plants are susceptible to anthracnose and tolerant to powdery mildew [39]. The cDNA library was prepared from the newly emerging leaves of a single plant. The same number of cDNA clones was produced as for ‘Heritage’, 18,432. Because of expected low polymorphism rate in black raspberry [40–42], 4032 clones were sequenced with a final yield of 2358 high quality sequences after quality control analysis, reported as accession numbers JZ841669 through JZ844026 in GenBank. These sequences averaged 523 bp with an average Phred score of 50. The assembly consisted of 1422 unigenes (273 contigs, 1149 singletons).
A total of 257 SSR sequences were identified and showed a very similar composition to the red raspberry motif lengths: 35 % dimers, 40 % trimers, 21 % tetramers and 5 % pentamers. The final set of 288 primer pairs covers 207 unigenes (Additional file 2).
The percentages of each motif are generally as expected in plants [43, 44], and a high percentage of tetramers is not uncommon in plants [35]. An elevated number of tetramer repeats is thought to be an indication that the majority of this motif length may be found in non-coding regions of the expressed genes [43].
Amplification using designed primer pairs
A random selection of SSR loci was tested for PCR amplification, amplification of a polymorphic PCR product, and transferability between species. A subset of 36 primer pairs from the 131 designed to test 98 individual unigenes identified in red raspberry, and 24 primer pairs from the 288 designed to test 207 unigenes identified in black raspberry were assessed using two genotypes each of R. idaeus (‘Heritage’ and ZIH-e1) and R. occidentalis (‘Bristol’ and Preston_2).
Table 1 summarizes the results of the amplification test. Of the 36 primer pairs tested that were designed from R. idaeus sequences, 25 pairs amplified a product, 19 of which produced a polymorphic product in R. idaeus. Of the 24 primer pairs designed from R. occidentalis sequences, 20 pairs amplified a product, 13 of which produced a polymorphic product in R. occidentalis. Of the 60 total primer pairs tested, 46 (76 %) produced amplification products that could be used to distinguish between the two species. In general, number and size range of alleles produced were similar between the two species. In terms of transferability, 22 of the 36 primer pairs (61 %) designed from R. idaeus sequence amplified a product in R. occidentalis, 18 (50 %) of which were polymorphic in R. occidentalis. Transferability from R. occidentalis to R. idaeus was demonstrated with 19 of the 24 primer pairs (79 %) amplifying a product of which 17 (71 %) detected polymorphisms in R. idaeus. These results indicate that markers that amplify a polymorphic product in highly-homozygous black raspberry are likely to amplify a polymorphic product in red raspberry, regardless of the sequence source.
Sequence functional characterization
The main reason for creating the Rubus libraries and sequence resources was for marker discovery; however, functional annotation of the sequences is a useful supplement for mapping efforts. Functional annotation allows investigators to target specific functional signatures of interest when testing molecular markers and allows the application of the sequences in a broader range of research questions. The functional information also provides a quality check for the library; we expect to see almost all sequences matching a model plant species and spanning a diversity of functions characteristic of leaf tissue. For this purpose, we chose to combine the transcripts from the two raspberry libraries into a single unigene set to provide the maximum amount of information about genes expressed in raspberry leaves and get the longest possible transcripts for searching and comparing to other genes. The combined raspberry unigene set has 418 contigs and 1671 singletons for a total of 2089 unigenes. The number of combined contigs was less than the sum of the contigs from the two datasets used for SSR identification, as identical contigs derived from both Rubus species were combined.
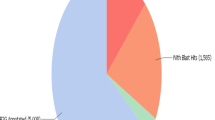
A basic local alignment search tool (BLAST) [45] comparison of the 2089 unigenes to the non-redundant (nr) protein database from the NCBI [46] yielded matches for 1664 unigenes (80 %). Only six of these (0.003 %) had a best match to an organism outside of green plants. The majority, 1570 (94 %) had a best match to a plant in the rosid clade (Fig. 1). This confirms that the library has little, if any, contamination with microbes from either the sampling or laboratory procedures.

A basic local alignment search tool (BLAST) comparison of the 2145 combined black and red raspberry unigene set to the non-redundant (nr) protein database from the National Center for Biotechnology Information (NCBI). Results indicate that the majority of the unigenes aligned to genera in the rosid clade
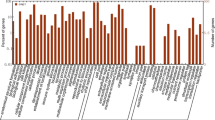
The unigene set was aligned to the Gene Ontology (GO) database [47] and classified according to the three basic categories: biological process, molecular function, and cellular component (Fig. 2). The most abundant sub-level two GO category was biological process with a total of 708 sequences associated with metabolic processes (211), cellular processes (187), and single organism processes (122). Other representative terms of biological process were response to stimulus (38), localization (38), and biological regulation (30) (Fig. 2a). GO assignments for the category molecular function totaled 366 sequences with functions for catalytic activity (148), binding (128), and structural molecule activity (47) (Fig. 2b). GO assignments for the category cellular component totaled 465 sequences assigned to cell part (164) and organelle (123) (Fig. 2c). A more detailed view of the GO sub-levels 3–5 reveals a significant fraction of genes related to metabolic processes such as macromolecule metabolism, organic substance metabolism, biosynthetic processes, and nitrogen/phosphorus metabolism (Additional file 3). Within the category molecular function, binding-related sub-categories such as cation binding, ion binding, and nucleoside binding were enriched. Finally, within the category cellular component, membrane, macromolecular complex, and symplast sub-categories were enriched (Additional file 3). Contig lengths ranged from 124 bp–1465 bp with an average length of 558 bp. To provide an example of functional diversity we aligned the ten longest unigenes to the GO database and identified a diversity of gene functions including heat shock, protease activity, and photosynthetic function (Additional file 4). All these annotations are reasonable for a set of genes from a plant leaf, and demonstrate the diversity of activities that were identified from a small set of ESTs.

The unigene set was aligned to the Gene Ontology (GO) database [47] and classified according to the three basic categories: biological process, molecular function, and cellular component. The most abundant level 2 GO category was biological process with a total of 708 sequences associated with metabolic processes (211), cellular processes (187), and single organism processes (122). Other representative terms of biological process were response to stimulus (38), localization (38), and biological regulation (30) (Fig. 2a). GO assignments for the category molecular function totaled 366 sequences with functions for catalytic activity (148), binding (128), and structural molecule activity (47) (Fig. 2b). GO assignments for the category cellular component totaled 465 sequences assigned to cell part (164) and organelle (123) (Fig. 2c)
Reference genomes have been published from members of the Rosaceae: diploid strawberry (Fragaria vesca L.) [48], which is in the same subfamily (Rosoideae) as raspberry [49], double haploid peach (Prunus persica L.) [50], apple (Malus × domestica Borkh.) [51], European pear (Pyrus communis L.) [52], and Asian pear (Pyrus bretschneideri Rehd.) [53]. If enough sequence conservation exists between these genomes and raspberry, some of these new raspberry-derived markers and primers designed from polymorphic regions may be transferable to the other genera. The gene space in particular should be well conserved; therefore the raspberry unigenes were aligned to the gene sets from strawberry, peach, and apple to evaluate the actual sequence conservation. The best match for each unigene was re-aligned with a Smith-Waterman search [54] to obtain the best possible alignment. Considering all of the best alignments between raspberry and strawberry genes, 56.1 % of the alignments had greater than 90 % identity; when aligned to the peach genome, 29.7 % of the matches had a greater than 90 % identity; and for apple genes, 15.7 % of the matches had greater than 90 % sequence identity. Figure 3 illustrates this trend for percent identity across all alignments, demonstrating that the raspberry unigenes have an overall higher percent identity to strawberry than to the other two gene sets, which is consistent with their closer phylogenetic relationship.

The distribution of percent sequence identities from alignments of raspberry unigenes to apple, peach, or strawberry genes. The greater similarity between raspberry and strawberry is a result of their close phylogenetic relationship relative to the other two crops
Conclusion
We have generated 121 and 257 EST-SSRs derived from leaf tissue of red raspberry (R. idaeus) and black raspberry (R. occidentalis) respectively. We have also designed 131 and 288 primer pairs for red and black raspberry, respectively. This resource constitutes a first step toward developing Rubus-specific, gene-derived markers that will facilitate the construction of linkage maps comprised of transferable markers for studying and manipulating important traits. The utility of some of these markers has been demonstrated already in the works of Dossett et al. 2010 [42] and Bushakra et al. 2012 [14], where some were used to evaluate genetic diversity among a wide selection of black raspberry genotypes and in genetic linkage map construction, respectively.
The advent of inexpensive next generation sequencing technologies has led to an increase in the use of SNP markers derived from high-throughput methods such as genotyping by sequencing (GBS) [55] and restriction site associated DNA (RAD) tags [56]. However, we argue that the long-utilized SSR is still the most effective and efficient marker type in certain circumstances. High-throughput sequencing costs are often reported as attractively low, but additional significant costs are associated with optimizing the restriction enzyme-based DNA preparations for a new species of interest, applying an appropriate informatics pipeline to manage the huge amount of sequence data, and finally to call the SNPs from an often “sparse” resulting data matrix [57, 58]. The same statistical power can be achieved with many fewer multiallelic SSRs than with biallelic SNPs derived from the complex GBS process. In the case of Rubus spp., where a reference genome is not yet available, the lack of key informatics poses an even more significant barrier to sequence-based SNP assays, such as the inability to align the SNPs to a reference, which requires additional work to assemble the sequencing reads. Also, specific to the Rubus spp. system, multiple species often are utilized and crossed in breeding programs. SSRs are significantly more likely than SNPs to transfer between species with little to no additional informatics investment. Considering the significant advantages, we selected SSRs as the best tool for straightforward yet effective genetic marker studies in Rubus species.
Methods
Plant material
Plants of ‘Heritage’ red raspberry and ‘Bristol’ black raspberry were purchased from Nourse Farms (Wately, Massachusetts, USA) and grown in pots in a greenhouse at Clemson University (Clemson, South Carolina, USA). Greenhouse conditions were 31.2 % relative humidity and 25 °C (76.7 °F). Approximately 5 g of young expanding leaf and meristem tissue from healthy plants was harvested from ‘Heritage’ and ‘Bristol’ on November 7, 2007 at approximately 10:00 a.m. EST, then immediately frozen in liquid nitrogen, and stored at −80 °C prior to RNA extraction. Leaf tissue from breeding selections ZIH-e1A, a red-fruited R. idaeus, and Preston_2, a yellow-fruited R. occidentalis, was kindly donated by Dr. Harry Swartz.
cDNA library construction and sequencing
Total RNA was extracted using modifications to the methodologies of Meisel et al. [59]. Polyadenylated RNA was enriched using the Ambion® PolyA+ purist kit (Life Technologies, Grand Island, NY, USA) and was the substrate for cDNA synthesis. First- and second-strand synthesis was performed with the BD biosystems SMART® PCR cDNA synthesis kit (Clontech Laboratories, Inc.) and directionally cloned into the sfiA/B site of the vector pDNR-LIB (Clontech Laboratories, Inc.). A survey of the size of the insert in a subset of 48 clones, as assessed by resolving a polymerase chain reaction (PCR) product on 1 % agarose gels, revealed an average insert size of 750 bp. DNA isolation was carried out in 96-well format using standard alkaline lysis conditions [60]. DNA sequencing was performed with BigDye v3.1 (Applied Biosystems, Inc.) and raw trace data collected on an ABI 3730xl DNA analyzer (Applied Biosystems, Inc.).
EST processing
The EST sequences were compared against the UniVec database from NCBI (ftp://ftp.ncbi.nih.gov/pub/UniVec/) to detect the presence of vector and adapter sequences. The program Cross_Match was implemented with the Consed package [61] and sequences quality trimmed of the vector and adapter sequences using the Lucy software [62]. Sequences with greater than 5 % ambiguous nucleotides (indicated by N) or fewer than 100 high quality bases (Phred score of ≥20) were discarded. The resulting high-quality cleaned ESTs were assembled into unigenes with the contig assembly program CAP3 [63] with empirically chosen parameters (−p 90 − d 60) to minimize assembly errors. The unigene set consists of the assembled contigs and the singletons output from CAP3.
A modified version (CUGISSR) of a Perl script SSRIT incorporated into the GDR tools [36, 64] was used to find perfect repeats meeting the following minimum requirements: 5 repeats of a 2 bp motif, 5 repeats of a 3 bp motif, 4 repeats of a 4 bp motif, or 3 repeats of a 5 bp motif. Primer sequences for the identified SSRs were generated using the Primer3 program [65]. To establish the SSR positions in relation to coding region, putative open reading frames (ORFs) were identified with the software FLIP [66]. All of these data are available in a Microsoft® Excel file through the Supplemental Materials.
The two sets of raspberry ESTs were combined into a single unigene with the CAP3 software program with empirically chosen parameters (−p 90 − d 60) prior to being functionally characterized. Homology searches using BLAST [45] were performed with an E-value cutoff of 1e-6 against the NCBI nr protein database. To assign GO terms, the software Blast2GO [67] was run utilizing the NCBI nr results. The GO results and discussion in this publication refer to the functional results from the combined unigene.
Further comparisons of the combined Rubus sequences to the wider Rosaceae taxa were completed by performing a BLAST search to the protein coding sequences (CDS features) associated with three recently published whole genome sequences: Fragaria vesca [48], Prunus persica [50], and Malus × domestica [51]. All three sets were downloaded from the Genome Database for Rosaceae (http://www.rosaceae.org/). The hybrid Rubus gene models were chosen for comparison to Fragaria vesca. To get the best possible contiguous alignment, each raspberry unigene was compared to its best CDS match in each of the three genomes with SSearch [68], a software program that performs a rigorous Smith-Waterman alignment.
PCR test of a subset of SSR primer pairs
A subset of 36 primer pairs from the 131 designed to test the 98 individual unigenes identified in red raspberry, and 24 primer pairs from the 288 designed to test the 207 unigenes identified in black raspberry were identified using random sorting of the source sequences in a Microsoft® Excel file and assessed in PCR. Primer pairs were evaluated for PCR amplification, production of polymorphic products and transferability between species. Amplification was tested with two genotypes each of R. idaeus (‘Heritage’ and ZIH-e1A) and R. occidentalis (‘Bristol’ and breeding selection Preston_2). DNA extraction, polymerase chain reactions (PCR) and sizing of PCR products followed Stafne et al. [69].
PCR products were visualized using an ABI 3730 Genetic Analyzer (Applied Biosystems, Inc.) and analyzed using ABI GeneMapper software v4.0.
References
Chen HS, Liu M, Shi LJ, Zhao JL, Zhang CP, Lin LQ, et al. Effects of raspberry phytochemical extract on cell proliferation, apoptosis, and serum proteomics in a rat model. J Food Sci. 2011;76(8):T192–8.
Jimenez-Garcia SN, Guevara-Gonzalez RG, Miranda-Lopez R, Feregrino-Perez AA, Torres-Pacheco I, Vazquez-Cruz MA. Functional properties and quality characteristics of bioactive compounds in berries: Biochemistry, biotechnology, and genomics. Food Res Int. 2012;54(1):1195–207.
Kafkas E, Özgen M, Özoğui Y, Türemiş N. Phytochemical and fatty acid profile of selected red raspberry cultivars: A comparative study. J Food Qual. 2008;31(1):67–78.
Olsson ME, Andersson CS, Oredsson S, Berglund RH, Gustavsson K-E. Antioxidant levels and inhibition of cancer cell proliferation in vitro by extracts from organically and conventionally cultivated strawberries. J Agric Food Chem. 2006;54(4):1248–55.
Dale A, Moore PP, McNicol RJ, Sjulin TM, Burmistrov LA. Genetic diversity of red raspberry varieties throughout the world. J Amer Soc Hortic Sci. 1993;118(1):119–29.
Darrow GM. Blackberry-raspberry hybrids. J Hered. 1955;46(2):67–71.
van Dijk T, Noordijk Y, Dubos T, Bink M, Meulenbroek B, Visser R, et al. Microsatellite allele dose and configuration establishment (MADCE): an integrated approach for genetic studies in allopolyploids. BMC Plant Biol. 2012;12(1):25.
van Dijk T, Pagliarani G, Pikunova A, Noordijk Y, Yilmaz-Temel H, Meulenbroek B, et al. Genomic rearrangements and signatures of breeding in the allo-octoploid strawberry as revealed through an allele dose based SSR linkage map. BMC Plant Biol. 2014;14(1):55.
Amsellem L, Dutech C, Billotte N. Isolation and characterization of polymorphic microsatellite loci in Rubus alceifolius Poir. (Rosaceae), an invasive weed in La Réunion island. Mol Ecol Notes. 2001;1(1–2):33–5.
Castillo NRF, Reed BM, Graham J, Fernández-Fernández F, Bassil NV. Microsatellite markers for raspberry and blackberry. J Amer Soc Hortic Sci. 2010;135(3):271–8.
Lewers K, Saski C, Cuthbertson B, Henry D, Staton M, Main D, et al. A blackberry (Rubus L.) expressed sequence tag library for the development of simple sequence repeat markers. BMC Plant Biol. 2008;8(1):69.
Castro P, Stafne ET, Clark JR, Lewers KS. Genetic map of the primocane-fruiting and thornless traits of tetraploid blackberry. Theor Appl Genet. 2013;126(10):2521–32.
Debnath SC. Inter simple sequence repeat (ISSR) markers and pedigree information to assess genetic diversity and relatedness within raspberry genotypes. Int J Fruit Sci. 2008;7(4):1–17.
Bushakra JM, Stephens MJ, Atmadjaja AN, Lewers KS, Symonds VV, Udall JA, et al. Construction of black (Rubus occidentalis) and red (R. idaeus) raspberry linkage maps and their comparison to the genomes of strawberry, apple, and peach. Theor Appl Genet. 2012;125(2):311–27.
Lewers KS, Styan SMN, Hokanson SC, Bassil NV. Strawberry GenBank-derived and genomic simple sequence repeat (SSR) markers and their utility with strawberry, blackberry, and red and black raspberry. J Amer Soc Hortic Sci. 2005;130(1):102–15.
Graham J, Smith K, MacKenzie K, Jorgenson L, Hackett C, Powell W. The construction of a genetic linkage map of red raspberry (Rubus idaeus subsp. idaeus) based on AFLPs, genomic-SSR and EST-SSR markers. Theor Appl Genet. 2004;109(4):740–9.
Sargent D, Fernández-Fernández F, Rys A, Knight V, Simpson D, Tobutt K. Mapping of A1 conferring resistance to the aphid Amphorophora idaei and dw (dwarfing habit) in red raspberry (Rubus idaeus L.) using AFLP and microsatellite markers. BMC Plant Biol. 2007;7(1):15.
Woodhead M, McCallum S, Smith K, Cardle L, Mazzitelli L, Graham J. Identification, characterisation and mapping of simple sequence repeat (SSR) markers from raspberry root and bud ESTs. Mol Breeding. 2008;22(4):555–63.
Ward J, Bhangoo J, Fernández-Fernández F, Moore P, Swanson J, Viola R, et al. Saturated linkage map construction in Rubus idaeus using genotyping by sequencing and genome-independent imputation. BMC Genomics. 2013;14(1):2.
Powell W, Machray GC, Provan J. Polymorphism revealed by simple sequence repeats. Trends Plant Sci. 1996;1(7):215–22.
Ellis JR, Burke JM. EST-SSRs as a resource for population genetic analyses. Heredity. 2007;99(2):125–32.
Cordeiro GM, Casu R, McIntyre CL, Manners JM, Henry RJ. Microsatellite markers from sugarcane (Saccharum spp.) ESTs cross transferable to erianthus and sorghum. Plant Sci. 2001;160(6):1115–23.
Eujayl I, Sorrells ME, Baum M, Wolters P, Powell W. Isolation of EST-derived microsatellite markers for genotyping the A and B genomes of wheat. Theor Appl Genet. 2002;104(2):399–407.
Decroocq V, Favé MG, Hagen L, Bordenave L, Decroocq S. Development and transferability of apricot and grape EST microsatellite markers across taxa. Theor Appl Genet. 2003;106(5):912–22.
Qureshi SN, Sukumar S, Kantety RV, Jenkins JN. EST-SSR: a new class of genetic markers in cotton. J Cotton Sci. 2004;8:112–23.
Bassil NV, Gunn M, Folta K, Lewers K. Microsatellite markers for Fragaria from ‘Strawberry Festival’ expressed sequence tags. Mol Ecol Notes. 2006;6(2):473–6.
Gil-Ariza DJ, Amaya I, Botella MA, Blanco JM, Caballero JL, López-Aranda JM, et al. EST-derived polymorphic microsatellites from cultivated strawberry (Fragaria × ananassa) are useful for diversity studies and varietal identification among Fragaria species. Mol Ecol Notes. 2006;6(4):1195–7.
Gasic K, Han Y, Kertbundit S, Shulaev V, Iezzoni A, Stover E, et al. Characteristics and transferability of new apple EST-derived SSRs to other Rosaceae species. Mol Breeding. 2009;23(3):397–411.
Zorrilla-Fontanesi Y, Cabeza A, Torres A, Botella M, Valpuesta V, Monfort A, et al. Development and bin mapping of strawberry genic-SSRs in diploid Fragaria and their transferability across the Rosoideae subfamily. Mol Breeding. 2011;27(2):137–56.
Varshney RK, Graner A, Sorrells ME. Genic microsatellite markers in plants: features and applications. Trends Biotechnol. 2005;23(1):48–55.
Ourecky DK, Slate GL. Heritage, a new fall bearing red raspberry. Fruit Varieties Hortic Digest. 1969;23(4):912–22.
Weber CA: Raspberry Variety Review. Cornell Cooperative Extension: Cornell University; 2012.
Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Nat Acad Sci. 1977;74(12):5463–7.
Ewing B, Green P. Base-calling of automated sequencer traces using Phred. II. Error probabilities. Genome Res. 1998;8(3):186–94.
Jung S, Jesudurai C, Staton M, Du Z, Ficklin S, Cho I, et al. GDR (Genome Database for Rosaceae): Integrated web resources for Rosaceae genomics and genetics research. BMC Bioinf. 2004;5(1):130.
Jung S, Staton M, Lee T, Blenda A, Svancara R, Abbott A, et al. GDR (Genome Database for Rosaceae): Integrated web-database for Rosaceae genomics and genetics data. Nucleic Acids Res. 2008;36 suppl 1:D1034–40.
Metzgar D, Bytof J, Wills C. Selection against frameshift mutations limits microsatellite expansion in coding DNA. Genome Res. 2000;10(1):72–80.
Slate GL. New or noteworthy fruits: XII. Small fruits. N Y State Agric Res Sta Bull. 1938;680:3–18.
Weber CA. Black raspberry performance and potential. N Y Fruit Q. 2007;15(4):19–22.
Weber CA. Genetic diversity in black raspberry detected by RAPD markers. Hortscience. 2003;38(2):269–72.
Dossett M, Bassil NV, Finn CE. Fingerprinting of black raspberry cultivars shows discrepancies in identification. Acta Hort (ISHS). 2012;946:49–53.
Dossett M, Bassil NV, Lewers KS, Finn CE. Genetic diversity in wild and cultivated black raspberry (Rubus occidentalis L.) evaluated by simple sequence repeat markers. Genet Resour Crop Evol. 2012;59(8):1849–65.
Ranade SS, Lin Y-C, Zuccolo A, Van de Peer Y, Garcia-Gil MR. Comparative in silico analysis of EST-SSRs in angiosperm and gynmosperm tree genera. BMC Plant Biol. 2014;14:220.
Vásquez A, López C. In silico genome comparison and distribution analysis of simple sequences repeats in cassava. Int J Genomics. 2014;2014:9. doi:10.1155/2014/471461.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–10.
Benson DA, Cavanaugh M, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, et al. GenBank. Nucleic Acids Res. 2013;41(D1):D36–42.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25(1):25–9.
Shulaev V, Sargent DJ, Crowhurst RN, Mockler TC, Folkerts O, Delcher AL, et al. The genome of woodland strawberry (Fragaria vesca). Nat Genet. 2011;43(2):109–16.
Potter D, Eriksson T, Evans RC, Oh S, Smedmark JEE, Morgan DR, et al. Phylogeny and classification of Rosaceae. Plant Syst Evol. 2007;266(1):5–43.
Verde I, Abbott AG, Scalabrin S, Jung S, Shu S, Marroni F, et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat Genet. 2013;45(5):487–94.
Velasco R, Zharkikh A, Affourtit J, Dhingra A, Cestaro A, Kalyanaraman A, et al. The genome of the domesticated apple (Malus x domestica Borkh.). Nat Genet. 2010;42(10):833–9.
Chagné D, Crowhurst RN, Pindo M, Thrimawithana A, Deng C, Ireland H, et al. The draft genome sequence of European pear (Pyrus communis L. ‘Bartlett’). PLoS One. 2014;9(4):e92644.
Wu J, Wang Z, Shi Z, Zhang S, Ming R, Zhu S, et al. The genome of the pear (Pyrus bretschneideri Rehd.). Genome Res. 2013;23(2):396–408.
Smith TF, Waterman MS. Identification of common molecular subsequences. J Mol Biol. 1981;147(1):195–7.
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One. 2011;6(5):e19379.
Miller MR, Dunham JP, Amores A, Cresko WA, Johnson EA. Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Res. 2007;17(2):240–8.
Yang X, Xu Y, Shah T, Li H, Han Z, Li J, et al. Comparison of SSRs and SNPs in assessment of genetic relatedness in maize. Genetica. 2011;139(8):1045–54.
Van Inghelandt D, Melchinger A, Lebreton C, Stich B. Population structure and genetic diversity in a commercial maize breeding program assessed with SSR and SNP markers. Theor Appl Genet. 2010;120(7):1289–99.
Meisel L, Fonseca B, González S, Baeza-Yates R, Cambiazo V, Campos R, et al. A rapid and efficient method for purifying high quality total RNA from peaches (Prunus persica) for functional genomics analyses. Biol Res. 2005;38:83–8.
Sambrook J, Fritsch E, Maniatis T. Molecular cloning: a laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Press; 1989.
Gordon D, Abajian C, Green P. Consed: a graphical tool for sequence finishing. Genome Res. 1998;8(3):195–202.
Chou H-H, Holmes MH. DNA sequence quality trimming and vector removal. Bioinformatics. 2001;17(12):1093–104.
Huang X, Madan A. CAP3: a DNA sequence assembly program. Genome Res. 1999;9(9):868–77.
Temnykh S, DeClerck G, Lukashova A, Lipovich L, Cartinhour S, McCouch S. Computational and experimental analysis of microsatellites in rice (Oryza sativa L.): Frequency, length variation, transposon associations, and genetic marker potential. Genome Res. 2001;11(8):1441–52.
Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. In: Misener S, Krawetz SA, editors. Bioinformatics methods and protocols: methods in molecular biology. Totowa, NJ: Humana Press Inc; 2000. p. 365–86.
Bossard N, Burger G. FLIP: A Unix program used to find/translate ORFs. Bionet Software. 1997. http://megasun.bch.umontreal.ca/ogmp/aboutflip.html.
Götz S, García-Gómez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008;36(10):3420–35.
Pearson WR, Lipman DJ. Improved tools for biological sequence comparison. Proc Nat Acad Sci. 1988;85(8):2444–8.
Stafne ET, Clark JR, Weber CA, Graham J, Lewers KS. Simple sequence repeat (SSR) markers for genetic mapping of raspberry and blackberry. J Amer Soc Hortic Sci. 2005;130(5):722–8.
Acknowledgements
The authors wish to thank Dr. Harry Swartz and the University of Maryland for donation of plant material for SSR testing. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture or Clemson University.
Funding
This project was funded by USDA-ARS Projects 8042-21220-254-00D and 2072-21220-002-00D, and by Clemson University.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interest
The authors declare that they have no competing interests.
Authors’ contributions
JMB analyzed PCR amplification data and led the drafting and revising of the manuscript. KSL conceived of the research idea, acquired all plant materials, oversaw all project activities including a contract with Clemson University for library construction, sequencing and SSR discovery, performed the PCR reactions and helped write the manuscript. MES performed bioinformatics analyses including read trimming, assembly, SSR identification and primer design. TZ participated in interpretation of results and revised a draft of the manuscript; CAS directed the library construction, sequencing, performed data analyses, and manuscript preparation. All authors read and approved the final manuscript.
Authors’ information
Not applicable.
Availability of data and materials
Not applicable.
Additional files
Additional file 1:
NCBI accession, locus name, and details of SSR, primer design and DNA sequence for red raspberry (R. idaeus). Highlight indicates those loci tested in R. idaeus and R. occidentalis genotypes with results shown in manuscript Table 1. (XLSX 48 kb)
Additional file 2:
NCBI accession, locus name, and details of SSR, primer design and DNA sequence for black raspberry (R. occidentalis). Highlight indicates those loci tested in R. idaeus and R. occidentalis genotypes with results shown in manuscript Table 1. (XLSX 93 kb)
Additional file 3:
Gene ontology term distribution for the categories Biological Process, Molecular Function, and Cellular Component. (XLSX 12 kb)
Additional file 4:
Top ten longest unigenes aligned to the Gene Ontology database with BLAST results. (XLSX 10 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Bushakra, J.M., Lewers, K.S., Staton, M.E. et al. Developing expressed sequence tag libraries and the discovery of simple sequence repeat markers for two species of raspberry (Rubus L.). BMC Plant Biol 15, 258 (2015). https://doi.org/10.1186/s12870-015-0629-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-015-0629-8