
Abstract
Information about the genetic diversity and population structure in elite breeding material is of fundamental importance for the improvement of crops. The objectives of our study were to (a) examine the population structure and the genetic diversity in elite maize germplasm based on simple sequence repeat (SSR) markers, (b) compare these results with those obtained from single nucleotide polymorphism (SNP) markers, and (c) compare the coancestry coefficient calculated from pedigree records with genetic distance estimates calculated from SSR and SNP markers. Our study was based on 1,537 elite maize inbred lines genotyped with 359 SSR and 8,244 SNP markers. The average number of alleles per locus, of group specific alleles, and the gene diversity (D) were higher for SSRs than for SNPs. Modified Roger’s distance (MRD) estimates and membership probabilities of the STRUCTURE matrices were higher for SSR than for SNP markers but the germplasm organization in four heterotic pools was consistent with STRUCTURE results based on SSRs and SNPs. MRD estimates calculated for the two marker systems were highly correlated (0.87). Our results suggested that the same conclusions regarding the structure and the diversity of heterotic pools could be drawn from both markers types. Furthermore, although our results suggested that the ratio of the number of SSRs and SNPs required to obtain MRD or D estimates with similar precision is not constant across the various precision levels, we propose that between 7 and 11 times more SNPs than SSRs should be used for analyzing population structure and genetic diversity.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In hybrid breeding of maize, knowledge of genetic relationships among inbreds is useful for germplasm organization and cultivar protection (Melchinger et al. 1991; Bernardo 2002). In the context of germplasm organization, inbreds can be grouped according to their estimates of genetic similarity and assigned to heterotic pools. For plant variety protection, information on genetic distances among inbreds is important for the identification of essential derivation as well as legal protection of germplasm (Smith et al. 1995). Therefore, information about the genetic diversity and population structure in elite breeding material is of fundamental importance for the improvement of crops (Hallauer and Miranda 1988). Various avenues have been suggested in the literature to achieve this goal.
A widely used measure in this context is the coancestry coefficient f calculated from pedigree records, which is defined as the probability that two homologous genes drawn at random from two individuals are identical by descent (Malécot 1948). This approach has been often used in autogamous crops, such as wheat, oat, barley, or soybean, where keeping pedigree records has a long tradition. In maize breeding too, pedigree information is commonly employed to assign newly developed inbreds to heterotic pools (Messmer et al. 1993). Nevertheless, pedigree records tracing back to more than two generations are rare. A further shortcoming is that some founder inbreds of heterotic pools were derived from open pollinated populations. Hence, calculation of f is often not feasible or dubious in maize (Lübberstedt et al. 2000).
Alternatively, the genetic similarity between genotypes can be assessed with DNA markers (Melchinger and Gumber 1998). Until now, simple sequence repeat (SSR) markers have been the most widely used DNA marker type to characterize germplasm collections of crops because of their easy use, relatively low price, and high degree of polymorphism provided by the large number of alleles per locus (Vignal et al. 2002). More recently, single nucleotide polymorphism (SNP) markers received high attention because they occur at much higher frequency in the genome than SSRs. Furthermore, their genotyping can be easily automated. However, most SNPs are biallelic, and, thus, have a lower information content. Given the advantages and disadvantages of both marker systems, their usefulness in different fields of application must be compared.
When assessing the repeatability of genotyping results and proportion of missing data for SSR and SNP markers, Jones et al. (2007) found a clear advantage for SNPs. In contrast, Hamblin et al. (2007) investigated the usefulness of 89 SSRs versus 847 SNPs for assessing relatedness and evaluating genetic diversity in a set of public maize inbreds and found that SSRs performed better with respect to the assignment of inbreds to sub-populations. These authors suggested that compared with their study a considerable higher number of SNP markers might be required in order to have an equivalent discriminating power as with SSRs. Nevertheless, to our knowledge, no earlier study examined this issue, especially in elite maize germplasm, nor considered the differences in costs for genotyping SSRs and SNPs.
The objectives of our study were to (a) examine the population structure and the genetic diversity in elite maize germplasm based on SSR markers, (b) compare these results with those obtained from SNP markers, and (c) compare the coancestry coefficient calculated from pedigree records with genetic distance estimates calculated from SSR and SNP markers.
Materials and methods
Plant materials and molecular markers
A set of 1,537 maize inbred lines obtained by the plant breeding company Limagrain (France) representing founder (6%) as well as elite (94%) inbred lines of Europe and North-America was used in this study. Pedigree information of these genotypes was available up to six generations back. In addition, all inbreds were classified into four heterotic pools, namely Flint (396 inbreds), Lancaster (399 inbreds), Stiff Stalk (SSS; 377 inbreds) and Iodent (365 inbreds).
The 1,537 inbred lines were examined with 359 SSR and 8,244 SNP markers. The SSRs (80% public and 20% proprietary) were selected over years with respect to their polymorphism information content (PIC) value (Botstein et al. 1980) in various sets of maize inbreds. The SNPs (100% proprietary) of our study were discovered by sequencing 2,973 amplicons in a set of 30 diverse maize inbreds (development set). From the identified SNPs, those were selected for genotyping the entire germplasm set which showed an Illumina designability score >0.4 and were not in complete linkage disequilibrium (LD) in the development set. Each of the 359 SSRs and 8,244 SNPs, which were designated as loci, showed less than 20% missing data and the average proportion of missing data was 5.1 and 2.7% for SSRs and SNPs, respectively.
All markers were mapped in the IBM population (Lee et al. 2002), where 59, 42, 41, 34, 36, 31, 36, 31, 27, and 22 of the SSR markers were located on chromosomes 1–10, with average marker distances of 12.86, 9.41, 12.76, 11.12, 11.31, 10.42, 11.56, 12.65, 13.48, and 11.86 cM. In addition, 1,456, 858, 902, 898, 1,002, 633, 578, 632, 699, and 586 of the SNPs were mapped to chromosomes 1–10, with average marker distances of 0.42, 0.81, 0.58, 0.44, 0.41, 0.53, 0.76, 0.61, 0.46, and 0.45 cM. The total length of the SSR map was 4,265 cM, whereas that of the SNPs was 4,378 cM.
Genotyping of the SSRs was performed by Limagrain Verneuil Holding (Riom, France) using standard protocols. Genotyping of the SNPs was performed by using an Illumina Infinium iSelect chip developed by Biogemma (Clermont-Ferrand, France, unpublished data). In our study, the full cost pricing for genotyping of the 359 SSRs and 8,244 SNPs was comparable. The fact that in the near future also for most other plant species a high number of SNP markers will be publicly available makes our assumption of neglecting the costs for marker development in the economic considerations realistic for other plant species than maize.
Statistical analyses
All analyses described below were performed for SSRs as well as SNPs. The average and range of the number of alleles per locus, the number of group specific alleles, and the gene diversity D (Nei 1987), identical to PIC, were determined for each heterotic pool and for all 1,537 genotypes. Furthermore, the average and the range of the modified Roger’s distance (MRD) (Wright 1978) within and between heterotic pools and across all genotypes were calculated. An FST analysis according to Wright (1965) was performed. Associations among genotypes were revealed with principal coordinate analysis (PCoA) (Gower 1966) based on MRD estimates between pairs of inbred lines. The most important founder lines of each heterotic pool were accentuated in the PCoA plot.
To determine the sampling variance of MRD and D estimates calculated from SSRs and SNPs, a bootstrap analysis was performed. In each of the 100 repetitions, a subset of the markers (1, 2, 3, 5, 10, 15, 20, 25, 50, and 75% of the total set of markers) was either randomly selected (random sampling) or sampled in such a way that the selected markers were equally distributed across the genome (stratified sampling). Based on the selected markers, the MRD was calculated for each pair of inbreds and D was estimated for the four heterotic pools as well as the entire germplasm set. Finally, the coefficient of variation (CV) across all repetitions was determined.
The CV enables a direct comparison of the two marker types, because it is independent from the ratio ω of the number of polymorphic markers between two individuals and the total number of markers, which is not true for the sampling variance (Melchinger, unpublished data). For all calculations, R (R Development Core Team 2006) routines were used.
The model-based approach implemented in software package STRUCTURE (Pritchard et al. 2000) was used to reveal population structure. For the SSR markers, we first run STRUCTURE assuming one sub-group (K = 1), to infer the allele frequency parameter λ. The burn-in time and the number of iterations were 100,000, respectively. The mean value of λ across five replications was used in a second step to run STRUCTURE for K = 1–20. For each value of K, five replications were performed, where the genetic map information was neglected to reduce the computational burden. To determine the most probable value of K, the ad hoc criterion described by Evanno et al. (2005) was used.
For the SNPs, the computational burden was reduced by running STRUCTURE only for the most probable K value n, which was identified based on the SSR markers. The genetic map information was used but not all other setups of the program were changed. For both marker types, the replication of K = n showing the maximum likelihood was used to assign genotypes with membership probability surpassing a certain threshold (0.0, 0.5, 0.7, or 0.9) to a sub-group. Inbreds that showed for none of the sub-groups a membership probability surpassing the threshold were non-assigned.
The coancestry coefficient f (Malécot 1948) between all pairs of inbreds was calculated from the available pedigree records using SAS (SAS Institute 2004) under the following assumptions: (a) all ancestors without pedigree information were regarded as completely unrelated, (b) all parents that were inbreds were assigned an inbreeding coefficient F = 1 and all parents that were hybrids were assigned F = 0, and (c) each parent of a biparental cross contributed equally to the progeny derived from the cross.
Results
The average number of alleles per locus was 14.57 for the SSRs and 2.00 for the SNPs (Table 1). When regarding each heterotic pool separately, the average number of alleles per locus ranged from 8.45 to 10.93 for the SSRs and from 1.96 to 1.99 for the SNPs. The number of group specific alleles varied from 142 (Iodent) to 634 (Flint) for the SSRs and from four (Iodent and SSS) to 25 (Flint) for the SNPs. The total gene diversity D was 0.69 for the SSRs and half as much (0.32) for the SNPs. D estimates of the heterotic pools ranged from 0.50 (SSS) to 0.65 (Lancaster) for the SSRs and from 0.23 (Iodent and SSS) to 0.30 (Lancaster) for the SNPs. The overall fixation index FST was 0.16 (0.06–0.27) and 0.19 (0.06–0.29) for the SSR and SNP markers, respectively.
For the SSRs, the average MRD between pairs of inbreds of one heterotic pool ranged from 0.71 (SSS) to 0.80 (Flint and Lancaster) (Fig. 1) and the average MRD between pairs of inbreds of different heterotic pools varied between 0.81 (SSS/Iodent) and 0.88 (Flint/SSS) (Table 2). By comparison, for SNPs the average MRD between inbreds ranged from 0.48 (Iodent) to 0.55 (Lancaster) within heterotic pools and from 0.55 (SSS/Iodent) to 0.61 (Flint/SSS) between heterotic pools. The average distance between inbreds of one heterotic pool calculated from pedigree records (1 − f) varied from 0.86 (SSS) to 0.96 (Lancaster) (Fig. 1).

Distribution of a modified Roger’s distance estimates (MRD) calculated from simple sequence repeat (SSR) markers, b MRD calculated from single nucleotide polymorphism (SNP) markers and c distance calculated from coancestry coefficient 1 − f, for all genotypes and the four heterotic pools. Means were plotted in black on the histograms. Y axis scale is different for each plot
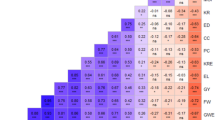
Spearman’s rank correlation coefficient between MRD estimates based on SSRs and SNPs was 0.87*** across all pairs of genotypes and ranged from 0.86*** (Flint) to 0.96*** (SSS) for pairs of inbreds from the same heterotic pool (Table 3). For both marker types, the correlation coefficient between MRD estimates and 1 − f was much lower (0.45*** for SSR and 0.42*** for SNP) (Table 3, Supplementary material S1).
In PCoA based on MRD estimates of all 1,537 maize genotypes, the first and second principle coordinate (PC) explained 9.1 and 6.9% of the molecular variance for SSRs and 10.8 and 7.9%, respectively, of the molecular variance for SNPs (Fig. 2). For both marker types, PC1 and PC2 clearly separated four clusters, which were for SSRs and SNPs in good harmony with the heterotic pool information.

Principal coordinate analysis of 1,537 maize inbred lines based on modified Roger’s distance calculated from 359 SSR (a) or from 8,244 SNP (b) marker loci. Genotypes were assigned to sub-group according to maximum membership probability. PC 1 and PC 2 are the first and second principal coordinate, respectively, and number in parentheses refer to the proportion of variance explained by the principal coordinates. Symbols identify the heterotic pools and colors the STRUCTURE groups
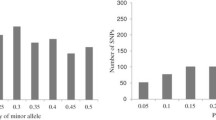
For the SSRs, the model-based approach of STRUCTURE indicated K = 4 as the most probable number of sub-groups (Supplementary material S3). For K = 4, the assignment of individuals to STRUCTURE sub-groups based on the maximum membership probability criterion was for 97% of the inbreds identical for SSRs and SNPs (Fig. 2). Introducing different assignment thresholds (0.5, 0.7, or 0.9) resulted in a much sharper increase of unassigned inbreds for SNPs than for the SSRs (Fig. 3, Supplementary material S2).

Percentage of unassigned genotypes using STRUCTURE based on different thresholds of the membership probability P with simple sequence repeat (SSR) and single nucleotide polymorphism (SNP) markers
The percentage of inbreds that were assigned, based on the maximum membership probability criterion, to the STRUCTURE sub-group corresponding to the heterotic pool defined by breeders were similar for SSRs (90.6) and SNPs (89.2) (Table 4). For both marker types, the percentage of inbreds for which the STRUCTURE sub-group and the heterotic pool were in accordance was highest for the Flint and lowest for the Lancaster pool. Furthermore, for both marker types, there were discrepancies between the size of the STRUCTURE sub-groups and the size of the corresponding heterotic pools (Table 4). For the Iodent pool, the STRUCTURE sub-group sizes were overestimated whereas the opposite was true for the Lancaster pool. This difference was higher for the SNP than for the SSR markers.
The CV of the MRD estimates increased exponentially with decreasing number of SSR and SNP markers (Fig. 4). Across the two sampling strategies, the CV was higher for the SSRs than for the SNPs. For both marker types, the CV of the stratified sampling strategy was slightly lower than that of the random sampling strategy. For both marker types as well as for all heterotic pools, the average D across all repetitions showed the same trend independently of the number markers (Supplementary material S4). The CV of the D estimates increased exponentially with decreasing number of SSR and SNP markers.

Mean coefficient of variation (CV) of the modified Roger’s distance (MRD) estimates between 1,537 maize genotypes assessed by random and stratified sampling of different numbers of simple sequence repeat (No SSR) and single nucleotide polymorphism (No SNP) markers in percent of the total number of markers (% Total). For details, see “Materials and methods”
Discussion
Population structure and genetic diversity assessed with SSRs
The concordance of the STRUCTURE analysis results, revealing four sub-groups (Supplementary material S3), with the PCoA clusters and the heterotic pools (Fig. 2a) were in accordance with the results of Maurer et al. (2006). Furthermore, for 90.6% of the inbreds, the assignment to a sub-group was in accordance with the heterotic pool information (Table 4). This indicates, not surprisingly, that in maize the heterotic pools describe very reliably the population structure. Thus, these heterotic pools were the basis for the genetic diversity analyses in our study.
Nevertheless, for about 150 inbreds, the heterotic pool information was not in accordance with their clustering in the PCoA and/or with the assignment to sub-groups using STRUCTURE (Fig. 2a). This finding can be partially explained by wrong or incomplete pedigree records (20%) especially for inbreds licensed from foundation seed companies but moreover by mixed pedigree information (80%) for inbreds selected from inter-pool crosses for which the assignment to heterotic pools is often uncertain. These results suggest that heterotic pools might be established in silico, corroborating the conclusions of Melchinger et al. (1991) and Smith et al. (1997) that molecular markers allow a better classification of genotypes than do pedigree records. However, for genotypes with mixed origin, the assignment to heterotic pools based on molecular marker information should be confirmed by field data examining the combining ability with testers from different heterotic pools (Melchinger 1999). In addition to the assignment of inbreds to sub-groups, the relationship between the different sub-groups is interesting for plant breeders and, thus, was examined in this study.
The MRD estimates between the Flint pool and the three other pools were higher than those among the three Non-Flint pools (Table 2). This can be explained by the breeding history of maize (Schnell 1992) separating Flint and Dent germplasm. In particular, the average distance between the Iodent and the Stiff Stalk pool was small in comparison with the distances between the other pools. This observation can be explained by one common ancestor of these two heterotic pools which is Reid Yellow Dent (Troyer 1999). Furthermore, this result can be explained by the origin of Limagrain’s Iodent pool which was developed from crosses between Stiff Stalk and original Iodent genotypes.
Long-term selection gain requires genetic variability and, thus, it is important to examine not only population structure but also the genetic diversity within the heterotic pools. Since estimates of D are not affected by differences in sample size, direct comparisons between different studies but also different heterotic pools are possible. Across the 1,537 elite maize inbred lines examined, we observed a total gene diversity D of 0.69 (Table 1). Our findings were in accordance with the results of an earlier study on European maize germplasm (Stich et al. 2005). However, Liu et al. (2003) detected with 0.82 a considerably higher estimate of D. This difference can be explained by the high proportion of diverse inbreds with tropical genetic background in their survey. Although our population size and that of other studies were not comparable, our observations on D were supported by the results on the average number alleles per locus. We observed a considerably higher number of alleles per locus (14.6) than previously reported by Jones et al. (2007) (5.1) and Stich et al. (2005) (9.8), but fewer than the number (21.7) reported by Liu et al. (2003).
Since the four heterotic pools of our study have similar size, direct comparisons between pools were possible for all genetic measures. We observed a higher number of (a) alleles per locus and (b) group-specific alleles as well as higher D estimates for the Flint and Lancaster pools than for the Iodent and Stiff Stalk pools (Table 1). Furthermore, the genetic distances between inbreds of the Flint as well as the Lancaster pools were on average higher than those observed for the Iodent and Stiff Stalk pools (Fig. 1). These findings can be explained by the fact that the Iodent and Stiff Stalk pools have a narrow genetic base (Hallauer and Miranda 1988; Troyer 1999). Furthermore, the selection pressure applied to adapt these heterotic pools originating from the US to the cooler climatic conditions prevailing in Western Europe might have decreased the genetic diversity.
In contrast, the high genetic diversity of the Flint pool can be explained by its very broad base. It includes progenies from almost all original European landraces, such as Lacaune from France (F2), Lizargarote from Spain (EP1), Gelber Badischer Landmais from Germany (DK105), and Italian Orange Flint (ILO904) (Messmer et al. 1992; Rebourg et al. 2001) but also Canadian germplasm (CO255) (Fig. 2). The same holds true for the Lancaster pool of Limagrain, which is a Flint-Dent mixed pool comprising not only true Lancaster Sure Crop inbreds such as Mo17 and Oh 43 but also inbreds from several diverse origins like Danube, Wisconsin (W117), and also exotic germplasm from China and Central America.
Comparison between SSRs and SNPs
Description of population structure and genetic diversity
Using the model-based approach of Pritchard et al. (2000), we found that, independently of the membership threshold we used for the assignment of inbreds to the sub-groups, far more genotypes could not be assigned to a sub-group for the SNPs than for the SSRs (Fig. 3). This observation could be due to the fact that STRUCTURE was run, for computational reasons, only for K = 4 for the SNP markers. However, the number of clusters revealed by the PCoA for SNPs and the very high correlation between MRD estimates based on SSR and SNP markers indicated that this simplification could not explain the difference in the proportion of unassigned inbreds between the two marker types.
Nevertheless, this difference is in accordance with the results of Hamblin et al. (2007). The lower gene diversity D of the SNPs compared with the SSRs might explain the above-described finding. The combination of SNP alleles at different loci to haplotypes has the potential to make the results more comparable to those of the SSRs (Hamblin et al. 2007). However, because no information about the extent and distribution of LD, which determines the number of SNPs to be combined into haplotypes, was available for our germplasm set, we did not examine SNP haplotypes in our study. Finally, we found in our study a larger proportion of SNP markers that were not discriminating with respect to heterotic pools compared with SSR markers (data not shown).
Nevertheless, the assignment to a sub-group based on SSRs and SNPs was for 97% of the inbreds identical, when using the highest membership probability criterion. Furthermore, the sub-groups identified in this scenario were in accordance with the heterotic pools as well as with the clusters revealed by PCoA (Fig. 2) and the percentage of genotypes assigned to the correct STRUCTURE groups was similar for SSR and SNP markers (Table 4). These observations suggested that for SNPs the assignment of inbreds to a sub-group, for which the highest membership probability was observed, is more promising than using other thresholds. However, if the number of sub-populations is very high, this criterion might be inappropriate for individuals with mixed origin, as the absolute membership probability for a sub-group can be very low, despite it is the highest one. Furthermore, for most association mapping methods, genotypes are not assigned to sub-groups but the matrices from STRUCTURE comprising the membership probabilities are used as cofactors (Yu et al. 2006). Consequently, one would expect that the differences in the absolute membership probabilities between SSRs and SNPs might have an influence on the results of association mapping approaches. However, this needs further research.
The above mentioned observation that for both marker types the sub-groups identified by STRUCTURE were in good accordance with the clusters revealed by the PCoA indicated that for the assignment of genotypes to sub-groups both clustering methods are equally appropriate. However, the high computational requirements of STRUCTURE analyses, especially when thousands of SNP markers are examined as in our study, suggest that the use of PCoA should be preferred.
Estimation of genetic diversity within sub-populations
The average number of alleles per SSR locus was considerably higher than that for the SNPs (Table 1). This is due to the fact that the SNPs are usually biallelic (Vignal et al. 2002). This property of SNPs explains together with the definition of gene diversity D that D values found for SNPs are lower than those for SSRs (cf. Jones et al. 2007). Theoretical considerations show that the maximum gene diversity D observable with biallelic markers is 0.5, whereas for multi-allelic markers such as SSRs the maximum can approach 1. Another factor which contributes to the observed difference in the D estimates of SSRs and SNPs is the selection history of the two marker types. The SSRs were selected over years with respect to their PIC value in various sets of maize inbred lines, whereas the SNPs have not undergone such a selection procedure. Therefore, it is expected that in the future the D estimates of the SNPs increase towards the above mentioned theoretical maximum of 0.5.
Despite this difference in the average of D estimates calculated for SSRs and SNPs, we observed for the SNPs the same trends in gene diversity D across the heterotic pools as found for the SSRs. The same is true for the number of group-specific alleles. Those results indicate that both marker types are equally appropriate to estimate genetic diversity in elite germplasm. Furthermore, the results of the bootstrap analysis for D (Supplementary material S4) suggested that this statement is not only true for the 359 SSRs and 8,244 SNPs examined in our study but even when examining only 2% thereof. However, in the context of genetic diversity analysis not only the absolute D estimates are an important criterion for marker applications but also the variance associated with these estimates. Based on our results, about 90 SSRs or 650 SNPs, which corresponds to a 1:7 SSR:SNP ratio, are required to reach for the examined germplasm set a stabilized plateau in the CV of D (Supplementary material S4).
The overall fixation index FST, useful as an overall measure of population differentiation, is low for both marker types, indicating that the majority of variation is found within heterotic pools rather than between heterotic pools. However, FST calculated from SNPs indicated a slightly higher differentiation (0.19) between the heterotic pools versus total differentiation than SSRs did (0.16) (Wright 1978). This is in accordance with results of Hamblin et al. (2007).
In analogy to D, our results revealed that the range of the MRD estimates was considerably lower for the SNPs than for the SSRs (Fig. 1). This finding is in accordance with the results of Hamblin et al. (2007) and Jones et al. (2007) and can be explained by the difference between the two marker systems with respect to the number of alleles per locus. A decreasing number of alleles per locus, which in turn increases the average allele frequency, decreases the proportion of polymorphic markers between two individuals ω. Because MRD can be expressed as a function of the ratio ω (data not shown), this leads to a decrease of the genetic distance estimates. This indicates that new thresholds have to be defined, if essentially derived varieties will be, in future, identified based on SNP markers instead of SSR or restriction fragment length polymorphism markers (cf. International Seed Federation 2008).
Despite this difference in the range of MRD estimates calculated from SSRs and SNPs, the estimates were correlated (Table 3). The imperfect correlation between the MRD estimates is most probably due to the fact that the mutation rate of SSRs is considerably higher than that of SNPs so that on the level of germplasm collections SNP-based distances will be almost entirely due to drift, while SSR-based distances will also be in part due to mutation (Hamblin et al. 2007). However, in contrast to results of Hamblin et al. (2007) and Jones et al. (2007), who observed across all genotypes no significant correlation and only moderate correlations for sets of inbreds related by pedigree, we observed a very high correlation. This difference might be explained by the fact that the former studies were based on a relatively small number of inbreds as well as fewer markers compared to our study.
In addition to the absolute genetic distance estimates, the variance associated with these estimates is an important criterion for marker applications. Therefore, we compared the CV of MRD estimates calculated from SSRs and SNPs using a bootstrap procedure. For all fractions of the total numbers of markers examined, we observed a higher CV for the SSRs than for the SNP markers (Fig. 4). Considering the comparable genotyping costs for both two data sets, our result suggested that based on the same budget for genotyping, MRD can be more precisely estimated with SNPs than with SSRs.
Furthermore, we observed for both marker types a plateau indicating that above a certain number of markers the precision gained from the additional markers was decreasing. As the number of markers moved below this threshold, the CV began to increase (and precision decreased) at a greater rate. This result is in accordance with findings of Pejic et al. (1998) and Garcia et al. (2004) and indicates that genotyping with more than such a number of markers does only marginally improve the precision of MRD estimates.
Under the assumption that a CV of 1% is sufficient for the estimation of genetic distances, our results suggested that about 270 SSRs or 3,150 SNPs (ratio SSR:SNP 1:11) are required to reach this precision. This is in good accordance with (a) the theoretical consideration of Laval et al. (2002) and Vignal et al. (2002), according to whom (k − 1) times more biallelic markers are needed to achieve the same genetic distance precision as a set of microsatellites with k alleles per locus as well as (b) the empirical simulations of Yu et al. (2009) who suggested that a SSR:SNP ratio of 1:10 is required for robust kinship estimates. Based on the genotyping costs underlying our study, the financial resources required for a SSR data set with 270 markers are about twice as high as those for a SNP data set with 3,150 markers. In addition to the number of markers used for estimation of MRD, also their positions in the genome influence the CV.
We observed for both marker types a slightly lower CV for the stratified sampling than for the random sampling strategy. This observation suggested that by choosing markers equally distributed across the genome, it is possible to reduce their number compared to randomly distributed markers and achieve the same level of precision in MRD. Alternatively, a higher precision can be obtained with the same number of markers if they are chosen as not randomly distributed.
Comparison of marker-based distances with distances calculated from pedigree records
A significant correlation between f values and MRD estimates, for both SSR and SNP markers was observed (Supplementary material S2). The correlation coefficient found (Table 3) was slightly lower than that observed by Liu et al. (2003). However, Smith et al. (1997) and Bernardo et al. (2000) found with 0.81 and 0.92 considerably higher correlation coefficients between SSR distances and f. The low correlation in our study is probably due to the fact that our f values were very unevenly distributed, with almost 80% of the pair-wise 1 − f estimates between 0.96 and 1.00 (Fig. 1). Therefore, our results suggest that marker-based distances are more appropriate for assessment of genetic relationship between maize inbreds than distances calculated from pedigree records.
Conclusions
The results of our study indicated that for the assignment of inbreds to sub-groups using STRUCTURE, the highest membership probability criterion has to be applied for SNP data in order to get sub-groups which are identical to those estimated from SSR data. However, the same conclusions regarding the structure and the diversity of heterotic pools can be drawn from both markers types. Nevertheless, computer simulations have to be performed in order to draw conclusions about the most favorable marker system for assessing population structure in an association-mapping context. Finally, our findings indicated that under the assumption of a fixed budget, MRD and D could be more precisely estimated with SNPs than with SSRs.
References
Bernardo R (2002) Breeding for quantitative traits in plants. Stemma Press, Woodbury, p 41, p 249
Bernardo R, Romero-Severson J, Ziegle J, Hauser J, Joe L, Hookstra G, Doerge RW (2000) Parental contribution and coefficient of coancestry among maize inbreds: pedigree, RFLP, and SSR data. Theor Appl Genet 100:552–556
Botstein D, White RL, Skalnick MH, Davies RW (1980) Construction of a genetic linkage map in man using restriction fragment length polymorphism. Am J Hum Genet 32:314–331
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Garcia AAF, Benchimol LL, Barbosa AMM, Geraldi IO, Souza CL Jr, de Souza AP (2004) Comparison of RAPD, RFLP, AFLP and SSR markers for diversity studies in tropical maize inbred lines. Genet Mol Biol 27:579–588
Gower JC (1966) Some distance properties of latent root and vector methods used in multivariate analysis. Biometrika 53:325–338
Hallauer AR, Miranda JBF (1988) Quantitative genetics in maize breeding, 2nd edn. Iowa State University Press, Ames
Hamblin MT, Warburton ML, Buckler ES (2007) Empirical comparison of simple sequence repeats and single nucleotide polymorphisms in assessment of maize diversity and relatedness. PLoS ONE 2(12):e1367
International Seed Federation (2008) Guidelines for the handling of a dispute on essential derivation of maize lines
Jones ES, Sullivan H, Bhattramakki D, Smith JSC (2007) A comparison of simple sequence repeat and single nucleotide polymorphism marker technologies for the genotypic analysis of maize (Zea mays L.). Theor Appl Genet 115:361–371
Laval G, San Cristobal M, Chevalet C (2002) Measuring genetic distances between breeds: use of some distances in various short term evolution models. Genet Sel Evol 34:481–507
Lee M, Sharopova N, Beavis WD, Grant D, Katt M, Blair D, Hallauer A (2002) Expanding the genetic map of maize with the intermated B73 × Mo17 (IBM) population. Plant Mol Biol 48:453–461
Liu K, Goodman M, Muse S, Smith JS, Buckler E, Doebley J (2003) Genetic structure and diversity among maize inbred lines as inferred from DNA microsatellites. Genetics 165:2117–2128
Lübberstedt T, Melchinger AE, Dußle C, Vuylsteke M, Kuiper M (2000) Relationships among early European maize inbreds: IV Genetic diversity revealed with AFLP markers and comparison with RFLP, RAPD, and pedigree Data. Crop Sci 40:783–791
Malécot G (1948) Les mathématiques de l’hérédité. Masson & Cie, Paris
Maurer HP, Knaak C, Melchinger AE, Ouzunova M, Frisch M (2006) Linkage disequilibrium between SSR markers in six pools of elite lines of an European breeding program for hybrid maize. Maydica 51:269–279
Melchinger AE (1999) Genetic diversity and heterosis. In: Coors JG, Pandey S (eds) The genetics and exploitation of heterosis in crops. ASA. CSSA, and SSSA, Madison, pp 99–118
Melchinger AE, Gumber RK (1998) Overview of heterosis and heterotic groups in agronomic crops. In: Lamkey KR, Staub JE (eds) Concepts and breeding of heterosis in crop plants. CSSA Spec. Publ. 25. CSSA, Madison, pp 29–44
Melchinger AE, Messmer MM, Lee M, Woodmana WL, Lamkey KR (1991) Diversity and relationships among U.S. maize inbreds revealed by restriction fragment length polymorphisms. Crop Sci 31:669–678
Messmer MM, Melchinger AE, Boppenmaier J, Brunklaus-Jung E, Herrmann RG (1992) Relationships among early European maize (Zea mays L.) inbreds: I Genetic diversity among flint and dent lines revealed by RFLPs. Crop Sci 32:1301–1309
Messmer MM, Melchinger AE, Hermann RG, Boppenmeier J (1993) Relationship among early European maize inbreds. II Comparison of pedigree and RFLP data. Crop Sci 33:944–950
Nei M (1987) Molecular evolutionary genetics. Colombia University Press, New York
Pejic I, Ajmone-Marsan P, Morgante M, Kozumplick V, Castiglioni P, Taramino G, Motto M (1998) Comparative analysis of genetic similarity among maize inbred lines detected by RFLPs, RAPDs, SSRs, and AFLPs. Theor Appl Genet 97:1248–1255
Pritchard JK, Stephens P, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
R Development Core Team (2006) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Rebourg C, Gouesnard B, Charcosset A (2001) Large scale molecular analysis of traditional European maize populations. Relationships with morphological variation. Heredity 86:574–587
SAS Institute Inc. (2000–2004) SAS 9.1.3 Help and Documentation, Cary, NC
Schnell FW (1992) Maiszüchtung und die Züchtungforschung in der Bundesrepublik Deutschland. Vortr Pflanzenzücht 22:27–44
Smith JSC, Ertl DS, Orman BA (1995) Identification of maize varieties. In: Wrigley CW (ed) Identification of food grain varieties. Am. Assoc. Cereal Chemists, St Paul, pp 253–264
Smith JSC, Chin ECL, Shu H, Smith OS, Wall SJ, Senior ML, Mitchell SE, Kresovich S, Ziegle J (1997) An evaluation of the utility of SSR loci as molecular markers in maize (Zea mays L.): comparison with data from RFLPs and pedigree. Theor Appl Genet 95:163–173
Stich B, Melchinger AE, Frisch M, Maurer HP, Heckenberger M, Reif JC (2005) Linkage disequilibrium in European elite maize germplasm investigated with SSRs. Theor Appl Genet 111:723–730
Troyer AF (1999) Background of US: Hybrid Corn. Crop Sci 39:601–626
Vignal A, Milan D, SanCristobal M, Eggen A (2002) A review on SNP and other types of molecular markers and their use in animal genetics. Genet Sel Evol 34:275–305
Wright S (1965) The interpretation of population structure by F-statistics with special regard to systems of mating. Evolution 19:395–420
Wright S (1978) Evolution and genetics of populations, vol IV. The University of Chicago Press, Chicago, p 91
Yu J, Pressoir G, Briggs WH, Bi IV, Yamsaki M, Doebley JF, McMullen MD, Gaut BS, Nielsen DM, Holland JB, Kresovich S, Buckler ES (2006) A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38:203–208
Yu J, Zhang Z, Zhu C, Tabanao DA, Pressoir G, Tuinstra MR, Kresovich S, Todhunter RJ, Buckler ES (2009) Simulation appraisal of the adequacy of number of background markers for relationship estimation in association mapping. Plant Genome 2:63–77
Acknowledgments
The authors thank the associate editor J. Yu and two anonymous reviewers for their valuable suggestions. The authors thank Limagrain Verneuil Holding for providing the genotyping data, J.P. Martinant for the information concerning SNPs genotyping, as well as P. Bertaux, T. Ronsin, and P. Flament for their comments on an earlier version of this manuscript. Funding for B. Stich was provided by the Max Planck Society.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by J. Yu.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Van Inghelandt, D., Melchinger, A.E., Lebreton, C. et al. Population structure and genetic diversity in a commercial maize breeding program assessed with SSR and SNP markers. Theor Appl Genet 120, 1289–1299 (2010). https://doi.org/10.1007/s00122-009-1256-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-009-1256-2