
Abstract
Backgrounding
Stayability, which may be defined as the probability of a cow remaining in the herd until a reference age or at a specific number of calvings, is usually measured late in the animal’s life. Thus, if used as selection criteria, it will increase the generation interval and consequently might decrease the annual genetic gain. Measuring stayability at an earlier age could be a reasonable strategy to avoid this problem. In this sense, a better understanding of the genetic architecture of this trait at different ages and/or at different calvings is important. This study was conducted to identify possible regions with major effects on stayability measured considering different numbers of calvings in Nellore cattle as well as pathways that can be involved in its expression throughout the female’s productive life.
Results
The top 10 most important SNP windows explained, on average, 17.60% of the genetic additive variance for stayability, varying between 13.70% (at the eighth calving) and 21% (at the fifth calving). These SNP windows were located on 17 chromosomes (1, 2, 4, 6, 7, 8, 9, 10, 11, 12, 13, 14, 18, 19, 20, 27, and 28), and they harbored a total of 176 annotated genes. The functional analyses of these genes, in general, indicate that the expression of stayability from the second to the sixth calving is mainly affected by genetic factors related to reproductive performance, and nervous and immune systems. At the seventh and eighth calvings, genes and pathways related to animal health, such as density bone and cancer, might be more relevant.
Conclusion
Our results indicate that part of the target genomic regions in selecting for stayability at earlier ages (from the 2th to the 6th calving) would be different than selecting for this trait at later ages (7th and 8th calvings). While the expression of stayability at earlier ages appeared to be more influenced by genetic factors linked to reproductive performance together with an overall health/immunity, at later ages genetic factors related to an overall animal health gain relevance. These results support that selecting for stayability at earlier ages (perhaps at the second calving) could be applied, having practical implications in breeding programs since it could drastically reduce the generation interval, accelerating the genetic progress.
Similar content being viewed by others


Background
Reproductive efficiency in beef cattle is one of the main factors defining the number of animals available for selection and slaughter. It helps to explain why reproductive traits have, generally, the greatest importance in economic selection indexes. Brumatti et al. [1], for example, verified that reproductive traits have between four and thirteen times more economic impact than growth traits and, stayability being one of the most important traits on beef herd profitability. Stayability (STAY) may be defined as the probability of a cow remaining in the herd until a reference age or a specific number of calving, since it had the opportunity to reach that age or calving number [2]. Indeed, the maintenance of cows and heifers for replacement is responsible for a large part of the cost in beef cattle production systems [3] and this cost increases with low reproductive rates [4]. Increasing female’s productive longevity can reduce heifer replacement costs [5]. The idea is that the cow must remain in the herd at least to pay its raising and maintenance cost, and STAY is a common trait used to measure the period that the female remains productive in the herd.
Selection for STAY is difficult because it usually presents low heritability, ranging between 0.08 and 0.14 [6, 7], in addition to be a sex-limited trait measured late in female’s life, normally at 76 months of age. All these factors decrease the expected genetic gain when this trait is included as selection criterion. Therefore, selecting for STAY at earlier ages would be an alternative to increase the rate of the genetic gain for this trait, with the hypothesis that most of the genetic factors with major effects on this trait act throughout female’s productive life.
It is worth mentioning that some genome-wide association studies have already been applied to identify genetic variants associated to STAY in cattle [6, 8, 9], but, to our knowledge, none considered STAY at different calvings. Therefore, this study was conducted to identify possible genomic regions and pathways related to genes with effects on the expression of STAY throughout female’s productive life, in a commercial Nellore population. The results of this study may provide a better understanding of the expression of this trait at different stages of animal life.
Materials and methods
Phenotypic dataset
Information of 2,084,880 Nellore animals, of which 672,000 were dams, born between 1984 and 2017 and belonging to 583 farms and three breeding programs (DeltaGen, Paint and CIA de Melhoramento) were utilized in the present study. In the reproductive management, females between fourteen and eighteen months of age participate in their first breeding season for 60 days in order to identify sexually precocious females. The heifers that did not conceive, non-precocious, have another chance to breeding when they are 22 months of age, on average. But if failing to calve after this second breeding season, these non-precocious heifers are culled.
STAY was evaluated from the second to the eighth calving. Only females with first calving until 40 months of age were phenotyped for STAY. For each calving, STAY was a binary trait in which the value 2 was attributed for females that had the respective calving and 1 for those that had the opportunity of mating but did not calve. If the interval between the date of the last calving of a female and the date of the last calving recorded in the dataset was smaller than 500 days, the subsequent records for that female were considered as missing. This is important to take into account that young females did not have the opportunity to express their phenotypes for STAY at older ages. More details with examples about phenotypic definition are available at Morales et al. [10].
Contemporary groups (CG) were defined as: herd, year and season of birth, management group at weaning and at yearling and precocity score. The birth seasons were defined as dry (March to August) and rainy (September to February) and the precocity scores were: 2 (precocious) for cows with age at first calving until 31 months and 1 (non-precocious) for females with age at first calving higher than 31 months. CGs without variability as well as with less than three animals were excluded. The percentage of precocious females in each measure of STAY was 34.4%, 23.3%, 18.4%, 17.0%, 15.6%, 14.5% and 15.5% for the second, third, fourth, fifth, sixth, seventh and eighth calving, respectively.
The final database used in the analyses had 195,452; 161,261; 130,236; 103,043; 79,844; 62,663 and 47,045 females with phenotypic information for the second; third; fourth; fifth; sixth; seventh and eighth calving, respectively. The percentage of females with precocity score of 2 (success) for STAY was 50.7%, 33.0%, 23.2%, 17.2%, 13.4%, 10.5% and 8.6%, for second to eighth calving, respectively.
Genotypic dataset
In total, 2,021 females and 949 sires had genotyped information from a panel with 777,962 SNP markers (Illumina BovineHD BeadChip). All SNPs were mapped according to the ARS-UCD1.2 reference map [11]. Females with genotypic information were born between 2007 and 2013 and the sires between 1993 and 2011. The genotype quality control was performed considering only autosomal markers and following SNP marker exclusion criteria: MAF lower than 2%; call rate lower than 0.90; highly correlated SNPs (r 2 > 0.995) and markers that show deviation of heterozygosity higher than 0.15 in comparison to the expected heterozygosity according to the Hardy-Weinberg equilibrium. In addition, animals with call rate lower than 0.90 were excluded. After quality control, 3.849 genotyped animals and 474.640 markers remained. All data editing was performed using R software version 4.3 [12].
Genome-wide association studies
Stay at different calving’s were evaluated through a single-trait generalized linear mixed animal model considering a probit function using the program THRGIBBS1F90 from blupf90 suite [13]. The general model can be described as follows:
where ɳ represents the linear predictor; β is a vector of fixed effect of CG; a is a vector of animal additive effect, assuming a ~ N(0, H \({\sigma }_{a}^{2}\)), where H is a relationship matrix that combines pedigree (A) and genomic information (G) and \({\sigma }_{a}^{2}\) corresponds to the additive genetic variance; e is a vector of residuals, assuming e ~ N (0, I \({\sigma }_{e}^{2}\)), where I is an identity matrix and \({\sigma }_{e}^{2}\) is the residual variance. The \(\mathbf{X}\), and W are incidence matrices relating \(\mathbf{y}\) to the fixed effects (β), and to the additive genetic effects (\(\mathbf{a}\)), respectively.
The inverse of the H matrix used to solve the mixed model equations was obtained according to [14]:
where \({\mathbf{G}}^{-1}\) corresponds to the inverse of the genomic relationship matrix and \({\mathbf{A}}_{22}^{-1}\) is the inverse of the pedigree relationship matrix for the genotyped animals. The G matrix was obtained according to [15]:
where Z is a matrix of genotypes adjusted for allele frequencies; D is a diagonal matrix with weights for SNP effects assuming; and q is a normalization factor.
Variance components were estimated by Bayesian inference via Gibbs sampling. A total of 300,000 Gibbs samples were generated, considering a burn-in of 30,000 iterations and samples being stored every ten iterations. The convergence analysis was verified through graphic inspection as well as based on the [16] and [17] tests, using the boa package version 1.1.8 [18].
The SNP effects were obtained using the postGSf90 software [19], following the method described in [20]:
where, \(\hat{\mathbf{u}}_{\mathbf{s}}\) is a vector with SNP effects, D is a diagonal matrix containing the weights for each SNP marker; Z is a matrix with genotypes; \(\hat{\text{a}}\) is the vector of genomic estimated breeding values (GEBV) for genotyped animals. The SNP effects estimation was performed considering two iterations. In the first iteration, D was an identity matrix. In the second iteration, the D matrix was updated with weights (\({d}_{i}\)) calculated in the first iteration as in [21]: \({d}_{i} = \hat{\text{u}}_{i}^{2}*2{p}_{i} \left(1- {p}_{i}\right)\), where \(\hat{\text{u}}_{i}^{2}\) is the square of the effect for the ith SNP and \({p}_{i}\) is the frequency of the second allele of the ith SNP. The GEBV (â) were also updated for the second iteration [20].
Genomic regions with major effects on STAY at different calving were identified based on the proportion of variance explained by windows with 100 consecutive SNPs. The proportion of genetic variance explained by each SNP-window was calculated as follow:
where \({a}_{i}\) is the genetic value of the ith SNP-window; \({{\sigma }_{a}}^{2}\) is the total genetic variance; \({Z}_{j}\) is the vector with genotype for the jth SNP for all animals; and \(\hat{\text{u}}_{j}\) is the estimated effect for each marker within the SNP-window.
The identification of genes in the top ten SNP-windows with the highest percentage of the additive genetic variance was performed using the program ENSEMBL [22], considering the Bos Taurus ARS-UCD1.2 assembly [11] as reference. The list of genes was then submitted to the software DAVID v6.7 [23] to identify over-represented gene ontology terms and pathways associated with STAY. This enrichment analysis was done considering a p-value < 0.05 as significant.
Results and discussion
The heritability estimates were 0.17, 0.15, 0.17, 0.17, 0.16, 0.17 and 0.17 for second, third, fourth, fifth, sixth, seventh and, eighth calving, respectively. These values are similar or lower to those reported in the literature for stayability measured at 76 months in Nellore (0.14) and Nellore-Angus crossbred (0.12) [24, 25]. Our estimates are higher than those obtained for stayability in consecutive calvings in Hereford cattle (from 0.05 to 0.08), without genomic information [26].
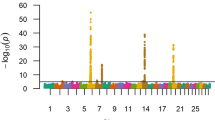
The top ten SNP-windows explained, on average, 17.60% of the genetic additive variance for STAY, varying between 13.70% and 21% according to the calving considered (Figs. 1, 2, 3, 4, 5, 6 and 7). The major SNP-windows are located on chromosomes 1, 2, 4, 6, 7, 8, 9, 10, 11, 12, 13, 14, 18, 19, 20, 27, and 28. Some of these chromosomes have been already reported by Teixeira et al. [24] in Nellore and Speidel et al. [25] in Angus, for STAY at a specific age. Teixeira et al. [24] reported genomic regions associated with STAY at 65 months of age on chromosomes 1, 2, 5, 6, 9, 20, and X. Speidel et al. [25], defining STAY of the female to produce 5 consecutive calves by 6 years of age, reported SNPs on chromosomes 6, 8, 9, 12, 15, 18, 22, and 23.

Manhattan plot for stayability measured at second calving in Nelore cattle

Manhattan plot for stayability measured at third calving in Nelore cattle

Manhattan plot for stayability measured at fourth calving in Nelore cattle

Manhattan plot for stayability measured at fifth calving in Nelore cattle

Manhattan plot for stayability measured at sixth calving in Nelore cattle

Manhattan plot for stayability measured at seventh calving in Nelore cattle

Manhattan plot for stayability measured at eighth calving in Nelore cattle
A total of 176 genes, associated with STAY, were identified (Table 1) and some regions were reported in other studies for STAY at a specific age. For instance, the BTA9:40.7–41.7 Mb region, identified here for the seventh calving, overlaps with Teixeira’s [23]. Also, the BTA6:13.0-13.4 Mb, BTA8:45.9–46.2, and BTA18:59.3–60.4 Mb regions identified for the eighth, fourth and eighth calvings, respectively, are very close (distance < 1 Mb) to SNP regions reported by Speidel et al. [25]. It is worth mentioning that caution should be taken in comparisons between our and the aforementioned studies due to differences on STAY definitions, methodology of analysis and also genome reference. In both studies, [24] and [25], the reference genome was the UMD 3.1, an earlier and less accurate assembly than the ARS-UCD1.2 used in the present study [27].
Many regions were associated with STAY in more than one calving (Table 1), indicating that part of the genetic factors may influence the expression of STAY throughout female’s productive life. This corroborates the results observed in Nellore [28] and in Canadian Simmentals [29], where moderate to high genetic correlation estimates between STAY at different calvings were found.
The BTA6:48.27–48.79 Mb region was associated with STAY from second to seventh calving (Table 1). A search in the QTL database [30] showed that this region harbors QTLs affecting average daily gain, mature weight, body temperature, fertility, and resistance to ectoparasites. Additionally, this genomic region was associated with reproduction and milk yield traits in N’Dama cattle [31]. The gene KCNIP4 (Potassium Voltage-Gated Channel Interacting Protein 4), located in the BTA6:40–42 Mb SNP-window, was associated with calving ease in Holstein cows [32]. So, our results show that the BTA6:42-49 Mb might affect STAY throughout female’s productive life indicating that this genomic region has pleiotropic effects on adaptive and reproductive traits.
The BTA11:14-19 Mb was associated with STAY in the fourth, fifth, sixth and seventh calving (Table 1). This region shows QTLs associated with calving ease and daily weight gain. This genomic region contains the MEMO1 (mediator of cell motility 1) gene which has been associated with somatic cell count in Holstein cattle [33]. The MEMO1 gene, mediated by the c-Src (Src) kinase, controls the Estrogen Receptor alpha, a hormone widely expressed in ovarian tissues and fallopian tubes, being responsible for stimulating uterine myometrium growth, preparing it for calving [34].
Identified for the second and the third calving, the windows region on BTA4 (34.9–35.6 Mb) (Table 1) encloses the gene SEMA3D (Semaphorin 3 D), which was differentially expressed in nervous system cells during the embryonic development of broilers [35]. The SEMA3D gene, in humans, is associated with Kallmann Syndrome, a condition characterized by delayed or absent puberty and an impaired sense of smell [36]. Also, this gene was associated with embryonic development in chickens [35] and with maternal calving difficulty in bovines [37].
The region BTA7: 106.8–107.0 Mb was associated to third, fourth, fifth and sixth calving. In this region is the FBXL17 (F-Box and Leucine Rich Repeat protein 17) gene that is related to recycling processes in part of proteosoma 26 S. This gene was associated to conformation (morphological trait) in Nellore cattle [38] and transcript on skeletal muscle at different ages [39]. The BTA8:78.3–79.4 Mb region, associated to STAY in the fourth and fifth calving (Table 1), harbors the gene TUT7 (terminal uridylyl transferase 7) that is involved with pos-transcriptional regulation of immune response cells (macrophages). After parturition, the cow is more susceptible to infections and diseases on genital tract such as ovarian and uterus [40] and the loss of immune response increases the animal susceptibility to diseases [41]. The TUT7 gene also plays a role in oocyte maturation and fertility and is involved in microRNA induced gene silencing through uridylation of deadenylated miRNA targets [42].
Identified for the fifth and sixth calving, the BTA27: 2.12–21.8 Mb region overlapped QTLs associated to dystocia and calving ease [30]. This region encloses the TUSC3 (tumor suppressor candidate 3) gene that was upregulated in placenta of women with HELLP syndrome which is associated with abortion and fetal morbidity [43]. The BTA19:43,9–44,3 Mb region was associated to STAY at the seventh and eighth calving (Table 1). The genes in this region are related to reproduction, energy metabolism and bone formation factors, which could be directly associated to cow’s longevity. The genes ETV4 (ETS variant 4) and ATXN7L3 (Ataxin 7 like 3), were differentially expressed in cattle oocytes during the folliculogenesis and ovulation, suggesting that these genes have a direct influence in females’ reproductive ability, especially during the formation and production of oocytes [44, 45]. The gene ETV4 is crucial for embryonic stem cells properties such as proliferation and expression of stem cell-related genes [46]. The SOST (sclerostin) gene was widely associated with bone formation and density bone in humans [47,48,49], through expression of sclerostin and have a negative regulator of bone formation [49]. The UBTF (upstream binding transcription factor), is strongly associated with neurogenerative disease in childhood [50,51,52].
The region BTA11:4.1– 4.6 Mb, identified for the sixth and seventh calving, harbors the TSGA10 (testis specific 10) gene that was associated with migration, differentiation, and cellular division in the initial phase of embryos development [53]. Associated to sixth and seventh calving, the BTA9:43.3–43.6 Mb region includes the ATG5 (Autophagy related 5) gene that has an autophagic function, mainly in pre-implantation ovaries. This autophagic function activity was founded in abundance in ovaries with heat stress conditions in pigs [54]. This gene was associated with the development of embryos after four- and eight-cell stages in mouse [55]. The RTN4IP1 (Reticulon 4 Interacting Protein 1) gene, also located in this genomic region, was related to breast cancer being upregulated in affected individuals compared to healthy [56, 57]. The BTA1:66.3–66.6 Mb was associated to STAY in the fifth, sixth and eighth calvings (Table 1). This region surrounds a QTL associated with puberty age and daily weight gain in bovine [30].
Seven of the identified genomic regions were specifically associated with STAY at the second calving (Table 1). Among these regions, the BTA2:135,4–136,1 Mb harbors the genes MFAP2 (Microfibril associated protein 2) and ATP13A2 (ATPase cation transporting 13A2). The MFAP2 was previously related to infertility or failures in embryo implantation in humans [58]. The ATP13A2 acts in the transport of zinc in nervous cells [59]. To remain in the herd, a cow must maintain a regular reproductive performance which can be directly influenced by the hormonal regulation acting on the pituitary-hypothalamus axis [60]. The BTA10:58–59 Mb region was associated with tick resistance in F1 Gyr X Holstein animals [61]. This region contains the genes LYSMD2 (LysM domain containing 2) and MYO5C (Myosin VC). The LYSMD2 was related to growth, cell adhesion and nervous signaling in live organisms [62]. A copy number variation in this gene sequence was associated, in humans, with aromatase excess syndrome, a disorder that causes prepubertal onset gynecomastia, hypogonadotropic hypogonadism, and short height in men. In women are usually asymptomatic, although macromastia, irregular menses, have been reported in a few individuals [63]. The MYO5C gene is related to the transport of melanin into melanocytes [64], and the degree of pigmentation is an important indicator of animal adaptation in tropical environments [65]. It is expected that females more adapted to tropical environments have longer reproductive life in the herd.
The BTA2:119.0–119.4 Mb (Table 1) was identified for the third calving, surround the genes B3GNT7 (UDP-GlcNAc:BetaGal Beta-1,3-N-Acetylglucosaminyltransferase 7) and NMUR1 (neuromedin U receptor 1). The B3GNT7 was found co-expressed with genes related to a healthy human placenta with no possibility of premature calving [66]. The NMUR1 was associated with environmental acclimation in sheep [67]. Also identified for the third calving, the BTA6:83.6–84.2 Mb region encloses the TMPRSS11F (transmembrane serine protease 11 F) gene, that was associated to immune response to mastitis [68] and somatic cell score [69] in dairy cattle and the BTA20:63.9–64.2 Mb region harbors the SEMA5A (semaphoring 5 A) gene, which was associated to antibody response in chickens [70]. All these results indicate the importance of adaptive traits for STAY mainly at the beginning of female’s reproductive life.
The region BTA8:45.9–46.3 Mb identified for the fourth calving, surround the MAMDC2 (MAM Domain Containing 2) gene and was up regulated in endometrium from fertile women when were stimulated by progesterone [71]. This situation is common in beef cattle by time-fixed artificial insemination, widely used in the herds in our datasets. The EYA2 (EYA Transcriptional Coactivador and Phosphatase 2) gene, located on BTA13:75.2–75,7 Mb, was reported as transcript on endometrium [72] and epithelial ovarian [73]. These findings suggest that reproduction factors have a major importance to keep a cow in the herd until the fourth calving.
A total of 35 regions were found to be associated to STAY in only one of the intermediate calvings (fifth or sixth). Identified for the fifth calving, the BTA9: 58.4–58.9 Mb harbors the 7SK gene that was associated to reproductive traits in horses [74], and beef cattle [75]. Moreover, the BTA12:60,7–61,4 Mb encloonses the HTATSF1 (HIV-1 Tat Specific Factor 1) gene that was related to estrogen receptor of precocious puberty women [76] and transcript in preimplantation of mouse embryos in 2-cell stage [77]. The 7SK gene plays important role in growth control of primordial germ cells [78]. For the sixth calving, it was identified the BTA20:25.3–25.7 Mb region with the genes FST (Follistatin) and NDUFS4 (NADH:Ubiquinone Oxidoreductase Subunit S4). The FST plays a role on formation of granulosa cells [79], having reduced expression in ovarian of pigs selected to increase ovulation rate. This gene was associated with heifer and cow conception rate [80]. The NDUFS4 gene has been associated with progressive neurodegenerative disorder (Leigh syndrome) [81,82,83,84,85], which could lead to carriers’ death in initial period of life in humans and mouse. For the fifth and sixth calvings, our results showed genes with reproductive and nervous systems functions.
For STAY in the last calvings (seventh and eighth), 84 genes were identified. The BTA9: 41.3–41.8 Mb, associated with the seventh calving, harbors the FOXO (Forkhead Box) gene which was associated with longevity in humans [86] and in sheep [87]. This gene is related to apoptosis on granulosa cells and it was downregulated in primary ovarian when different levels of LH (Luteinizing hormone) and FSH (Follicle-stimulating hormone) concentration were used in chickens [88]. Associated to STAY in the eighth calving, the region BTA6:13.0–13.4 Mb, harbors genes such as: TIFA (TRAF interacting protein with forkhead associated domain), ALPK1 (Alpha Kinase 1) and FAM241A (family with sequence similarity 241 member A). The TIFA gene was related with oxidative stress affecting the innate immune response [89]. Inflammatory mediators play a vital role in maintaining tissue homeostasis in the female reproductive tract, mainly during the ovulation process, which involves the rupture of the dominant follicle, and innate immunity and inflammation contribute to this process in the ovary [90]. The ALPK1 gene was associated to adnexal carcinomas in reproductive system (ovaries and fallopian tubes) and tumor of cutaneous origin [91]. The FAM241A gene transcript in endometrium was related to reproductive disease and endometriosis in humans [92]. These last two genes show a function in reproductive performance. The BTA6: 15,0–15,5 Mb was also associated to STAY in the eighth calving. Located in this region, the ELOVL6 (ELOVL Fatty Acid Elongase 6) gene was associated to lipid’s metabolism in cattle [93], pigs [94, 95] and chickens [96]. Indeed, there is a close connection among fat metabolism, reproduction and lifespan, where alterations of fat content and/or composition are interconnected with reproductive system regulation contributing to extend longevity by influencing the overall metabolism of animals [97].
The list of genes identified in the present study was enriched for pathways related to feeding behavior (GO:0007631), digestion (GO:0007586), neuropeptide hormone activity (GO:0005184), G-protein coupled receptor binding (GO:0001664), regulation of appetite (GO:0032098) and Pancreatic hormone-like (IPR001955) (Table 2). Some of the genes involved in these pathways (PPY, PYY and PYY2) are related to energy metabolism, feed intake and satiety process through the metabolism of ghrelin hormone [98, 99]. The ghrelin hormone participates in the reproductive tissue hormonal regulation with positive feedback on pituitary and hypothalamus [100]. A modulation of PYY on the secretion of LH hormone was identified in mice [101]. These pathways affect animal’s reproductive performance by inducing the pituitary-hypothalamus axis to process reproductive hormone production. In humans, genes involved in these pathways also were related to increased food intake and elevated in nervous anorexy [102]. The related GO terms and pathways also refer to the importance of nutrition and digestion to STAY. Research has shown that inadequate nutrition can negatively impact the reproductive performance of dairy cows, including delayed onset of puberty, reduced conception rates, decreased pregnancy rates, and increased embryonic loss [103, 104]. As a quantitative and reproductive trait, STAY has strong influence from environment, with low heritability estimates [28]. Thus, our findings support the importance of nutrition and feed behavior on STAY at different calvings.
Some of the enriched pathways (IPR003877: SPla/RYanodine receptor SPRY, IPR013320:Concanavalin A-like lectin/glucanase, SM00449:SPRY, IPR001870:B30.2/SPRY domain) shared TRIM family genes (Table 2) that have important function on elimination of shorted-lived regulatory proteins such as cell cycle regulation, cellular signalling and DNA repair and are related to development and progression of tumor [105]. These pathways also include the HERC family, which has been related to human diseases like cancer and neurological disorders [106]. Probably, these genes related to nervous system or neurological system could be associated to regulation of hormonal metabolites.
The concanavalin A-like domain, in bovine, plays an important role in sugar recognition and binding, and can have implications for milk production and immune defense [107]. This domain is a carbohydrate-binding found in a variety of proteins, including lectins, which are proteins that can bind to specific sugars. In bovines, the concanavalin A-like domain is found in several proteins, including the mannose-binding lectin (MBL), a serum protein that is part of the innate immune system and can recognize and bind to sugar molecules on the surface of pathogens, leading to their opsonization and clearance [107, 108].
The SPRY domain is a protein interaction module that is found in a wide range of eukaryotic proteins, with diverse functions [109]. It plays a critical role in several important signaling pathways, including RNA processing, regulation of histone H3 methylation, innate immunity, and embryonic development [110, 111].
Significantly enriched terms, families and pathways are GO:0051537 ~ 2 iron; 2 sulfur cluster binding; transit peptide:Mitochondrion; IPR015943:WD40/YVTN repeat-like-containing domain; IPR003954:RNA recognition motif domain; eukaryote; and SM00361:RRM_1. Iron is an essential molecule that plays a crucial role in cellular processes, including energy metabolism, DNA synthesis, and oxygen transport. The mitochondria play an essential role in iron metabolism, as they are involved in heme and iron-sulfur cluster biogenesis. In reproductive tissues, iron is necessary for gamete maturation and embryo development. During pregnancy, iron requirements increase due to the growth and development of the fetus, placenta, and expansion of the maternal blood volume [112, 113]. The RRM_1 domain is a highly conserved RNA-binding motif found in a variety of eukaryotic proteins involved in RNA metabolism [114]. Some studies suggest that RRM_1 may play a role in iron homeostasis through its regulation of iron-binding proteins. The transit peptide and RRM_1 domain contributes to the overall health and reproductive success of bovine, by facilitating proper mitochondrial, RNA function, and iron metabolism [115]. Any disruption in these processes can lead to negative effects on reproductive functions.
Conclusions
Many of the genomic regions identified in this study were associated to STAY in more than one calving, indicating that part of the genetic control of this trait acts throughout the female’s productive life. The in silico functional analyses of the genes found in this study, indicate that while the expression of stayability at earlier ages (from the 2th to the 6th calving) appeared to be more influenced by genetic factors linked to reproductive performance together with an overall health/immunity, at later ages (7th and 8th calvings) genetic factors related to an overall animal health gain more relevance. These results could support that selecting for stayability at earlier ages (perhaps at the second calving) should be applied instead of selecting for this trait at later ages, having practical implications in breeding programs since it could drastically reduce the generation interval, accelerating the genetic progress.
Availability of data and materials
The data that support the findings of this study are available from Alliance Nellore breeding program (https://gensys.com.br) but restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available. Data are however available from the authors (L.G. Albuquerque, galvao.albuquerque@unesp.br) upon reasonable request and with permission of Alliance Nellore breeding program (https://gensys.com.br).
Change history
26 February 2024
A Correction to this paper has been published: https://doi.org/10.1186/s12864-024-10098-4
References
Brumatti RC, Ferraz JBS, Eler JP, Formigonni EIB. Desenvolvimento De índice de seleção em gado corte sob o enfoque de um modelo bioeconômico. Arch De Zootecnia. 2011;60:205–13.
Hudson GFS, Van Vleck LD. Relationship between production and stayability in Holstein cattle. J Dairy Sci. 1981;64:2246–50.
Newman S, Morris CA, Baker RL, Nicoll GB. Genetic improvement of beef cattle in New Zealand: breeding objectives. Livest Prod Sci. 1992;32:111–30.
Formigoni IB, Ferraz JBS, Silva JAIIV, Eler JP, Brumatti RC. Valores econômicos para habilidade de permanência e probabilidade de prenhez aos 14 meses em bovinos de corte. Arq Bras Med Vet Zootec. 2005;57(suppl 2):220–6.
Rizzo ECA, Neto FRA, Diaz IDPS, Dias MM, Costa RB, Ventura HT, et al. Genetic association of productive and reproductive traits with stayability in Nellore cattle: analysis using bayesian models. Genet Mol Res. 2015;14:14956–66.
Engle BN, Herring AD, Sawyer JE, Riley DG, Sanders JO, Gill CA. Genome-wide association study for stayability measures in nellore–angus crossbred cows. J Anim Sci. 2018;96:1205–14.
Van Melis MH, Eler JP, Oliveira HN, Rosa GJM, Silva JAIIV, Ferraz JBS, et al. Study of stayability in Nellore cows using a threshold model. J Anim Sci. 2007;85:1780–6.
Teixeira DBA, Fernandes GA, Dos Santos Silva DB, Costa RB, Takada L, Gordo DGM, et al. Genomic analysis of stayability in Nellore cattle. PLoS One. 2017;12:1–10.
Speidel SE, Buckley BA, Boldt RJ, Enns RM, Lee J, Spangler ML, et al. Genome-wide association study of stayability and heifer pregnancy in Red Angus cattle. J Anim Sci. 2018;96:846–53.
da Silva Morales D, Silva DO, Ayres DR, Junior MLS, Bignardi AB, Ventura RV, et al. Genetic associations between stayability to consecutive calvings and traits of economic interest in taurine and zebu breeds. J Anim Breed Genet. 2023. https://doi.org/10.1111/jbg.12827.
Rosen BD, Bickhart DM, Schnabel RD, Koren S, Elsik CG, Tseng E, et al. De novo assembly of the cattle reference genome with single-molecule sequencing. Gigascience. 2020;9:1–9.
R Core Team. R: A language and environment for statistical computing. 2021.
Misztal I, Tsuruta S, Strabel T, Auvray B, Druet T, Lee DH. BLUPF90 and related programs (BGF90). In: Proceedings of 7th World Congress on Genetics Applied to Livestock Production, Montpellier, France; 2002.
Legarra A, Aguilar I, Misztal I. A relationship matrix including full pedigree and genomic information. J Dairy Sci. 2009;92:4656–63.
VanRaden PM. Efficient methods to compute genomic predictions. J Dairy Sci. 2008;91:4414–23.
Geweke J. Evaluating the accuracy of sampling-based approaches to the calculation of posterior moments. In: Bernardo JM, Berger JO, Dawid AP, Smith AFM, editors. Bayesian Statistics. 4th ed. Oxford: Clarendon Press; 1992.
Raftery AL, Lewis S. How many iterations in the Gibbs sampler. Bayesian stat. 1992;4:2:763–73.
Smith BJ. boa: an R Package for MCMC output convergence assessment and posterior inference. J Stat Softw. 2007;21:1–27.
Aguilar I, Misztal I, Tsuruta S, Legarra A, Wang H. PREGSF90 – POSTGSF90: computational tools for the implementation of single-step genomic selection and genome-wide association with ungenotyped individuals in BLUPF90 Programs. Vancouver: 10th World Congress of Genetics Applied to Livestock Production; 2014.
Wang H, Misztal I, Aguilar I, Legarra A, Muir WM. Genome-wide association mapping including phenotypes from relatives without genotypes. Genet Res (Camb). 2012;94:73–83.
Zhang Z, Liu J, Ding X, Bijma P, de Koning DJ, Zhang Q. Best linear unbiased prediction of genomic breeding values using a trait-specific marker-derived relationship matrix. PLoS ONE. 2010;5:1–8.
Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, et al. Ensembl 2018. Nucleic Acids Res. 2018;46:D754-761.
Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57.
Barreto Amaral Teixeira D, Alves Fernandes Júnior G, Beraldo dos Santos Silva D, Bermal Costa R, Takada L, Gustavo Mansan Gordo D, et al. Genomic analysis of stayability in Nellore cattle. PLoS ONE. 2017;12:e0179076.
Speidel SE, Buckley BA, Boldt RJ, Enns RM, Lee J, Spangler ML, et al. Genome-wide association study of stayability and heifer pregnancy in Red Angus cattle. Journal of Animal Science. 2018;96:846–53.
Silva DO, Morales DdaS, Santana ML Jr, Ayres DR, Bignardi AB, Carvalheiro R, et al. Genetic analyses of stayability to consecutive calvings in taurine and crossbred (Bos indicus x Bos taurus) cattle. Livest Sci. 2021;244:104331.
Rosen BD, Bickhart DM, Schnabel RD, Koren S, Elsik CG, Tseng E, et al. De novo assembly of the cattle reference genome with single-molecule sequencing. Gigascience. 2020;9:9.
Silva DO, Santana ML, Ayres DR, Menezes GRO, Silva LOC, Nobre PRC, et al. Genetic parameters for stayability to consecutive calvings in Zebu cattle. Animal. 2018;12:1807–14.
Jamrozik J, McGrath S, Kemp RA, Miller SP. Estimates of genetic parameters for stayability to consecutive calvings of Canadian simmentals by random regression models. J Anim Sci. 2013;91:3634–43.
Hu Z-L, Park CA, Wu X-L, Reecy JM. Animal QTLdb: an improved database tool for livestock animal QTL/association data dissemination in the post-genome era. Nucleic Acids Res. 2013;41:D871-879.
Taye M, Lee W, Jeon S, Yoon J, Dessie T, Hanotte O, et al. Exploring evidence of positive selection signatures in cattle breeds selected for different traits. Mamm Genome. 2017;28:528–41.
Cole JB, Wiggans GR, Ma L, Sonstegard TS, Lawlor TJ, Crooker BA, et al. Genome-wide association analysis of thirty one production, health, reproduction and body conformation traits in contemporary U.S. Holstein cows. BMC Genomics. 2011;12:408.
Wang X, Ma P, Liu J, Zhang Q, Zhang Y, Ding X, et al. Genome-wide association study in Chinese holstein cows reveal two candidate genes for somatic cell score as an indicator for mastitis susceptibility. BMC Genet. 2015;16:111.
Frei A, MacDonald G, Lund I, Gustafsson J-Å, Hynes NE, Nalvarte I. Memo interacts with c-Src to control estrogen receptor alpha sub-cellular localization. Oncotarget. 2016;7:56170–82.
Bao ZZ, Jin Z. Sema3D and Sema7A have distinct expression patterns in chick embryonic development. Dev Dyn. 2006;235:2282–9.
Garaffo G, Provero P, Molineris I, Pinciroli P, Peano C, Battaglia C, et al. Profiling, bioinformatic, and functional data on the developing olfactory/GnRH system reveal cellular and molecular pathways essential for this process and potentially relevant for the Kallmann syndrome. Front Endocrinol (Lausanne). 2013;4:203.
Purfield DC, Bradley DG, Evans RD, Kearney FJ, Berry DP. Genome-wide association study for calving performance using high-density genotypes in dairy and beef cattle. Genet Selection Evol. 2015;47:47.
Carreño LOD, da Conceição Pessoa M, Espigolan R, Takada L, Bresolin T, Cavani L, et al. Genome association study for visual scores in Nellore cattle measured at weaning. BMC Genomics. 2019;20:150.
Sadkowski T, Jank M, Oprzadek J, Motyl T. Age-dependent changes in bovine skeletal muscle transcriptomic profile. J Physiol Pharmacol. 2006;57(Suppl 7):95–110.
Sheldon IM, Lewis GS, LeBlanc S, Gilbert RO. Defining postpartum uterine disease in cattle. Theriogenology. 2006;65:1516–30.
Kozlowski E, Wasserman GA, Morgan M, O’Carroll D, Ramirez N-GP, Gummuluru S, et al. The RNA uridyltransferase Zcchc6 is expressed in macrophages and impacts innate immune responses. PLoS ONE. 2017;12:e0179797.
Lim J, Ha M, Chang H, Kwon SC, Simanshu DK, Patel DJ, et al. Uridylation by TUT4 and TUT7 marks mRNA for degradation. Cell. 2014;159:1365–76.
Kang B-Y, Tsoi S, Zhu S, Su S, Kay HH. Differential gene expression profiling in HELLP syndrome placentas. Reproductive Sci. 2008;15:285–94.
Eo J, Han K, Murphy M, Song K, Lim H. Etv5, an ETS transcription factor, is expressed in granulosa and cumulus cells and serves as a transcriptional regulator of the cyclooxygenase-2. J Endocrinol. 2008;198:281–90.
Nivet A-L, Dufort I, Gilbert I, Sirard M-A. Short-term effect of FSH on gene expression in bovine granulosa cells in vitro. Reprod Fertil Dev. 2018;30:1154.
Akagi T, Kuure S, Uranishi K, Koide H, Costantini F, Yokota T. ETS-related transcription factors ETV4 and ETV5 are involved in proliferation and induction of differentiation-associated genes in embryonic stem (ES) cells. J Biol Chem. 2015;290:22460–73.
Delgado-Calle J, Sañudo C, Bolado A, Fernández AF, Arozamena J, Pascual-Carra MA, et al. DNA methylation contributes to the regulation of sclerostin expression in human osteocytes. J Bone Miner Res. 2012;27:926–37.
Velázquez-Cruz R, Jiménez-Ortega RF, Parra-Torres AY, Castillejos-López M, Patiño N, Quiterio M, et al. Analysis of association of MEF2C, SOST and JAG1 genes with bone mineral density in mexican-mestizo postmenopausal women. BMC Musculoskelet Disord. 2014;15:400.
van Bezooijen RL, ten Dijke P, Papapoulos SE, Löwik CW. SOST/sclerostin, an osteocyte-derived negative regulator of bone formation. Cytokine Growth Factor Rev. 2005;16:319–27.
Edvardson S, Nicolae CM, Agrawal PB, Mignot C, Payne K, Prasad AN, et al. Heterozygous de novo UBTF gain-of-function variant is associated with neurodegeneration in childhood. Am J Hum Genet. 2017;101:267–73.
Toro C, Hori RT, Malicdan MCV, Tifft CJ, Goldstein A, Gahl WA, et al. A recurrent de novo missense mutation in UBTF causes developmental neuroregression. Hum Mol Genet. 2018;27:691–705.
Sedláčková L, Laššuthová P, Štěrbová K, Haberlová J, Vyhnálková E, Neupauerová J, et al. UBTF mutation causes complex phenotype of neurodegeneration and severe epilepsy in childhood. Neuropediatrics. 2019;50:057–60.
Behnam B, Modarressi MH, Conti V, Taylor KE, Puliti A, Wolfe J. Expression of Tsga10 sperm tail protein in embryogenesis and neural development: from cilium to cell division. Biochem Biophys Res Commun. 2006;344:1102–10.
Hale BJ, Hager CL, Seibert JT, Selsby JT, Baumgard LH, Keating AF, et al. Heat stress induces autophagy in pig ovaries during follicular development. Biol Reprod. 2017;97:426–37.
Tsukamoto S, Kuma A, Murakami M, Kishi C, Yamamoto A, Mizushima N. Autophagy is essential for preimplantation development of mouse embryos. Sci (1979). 2008;321:117–20.
Sareyeldin RM, Gupta I, Al-Hashimi I, Al-Thawadi HA, Al Farsi HF, Vranic S, et al. Gene expression and miRNAs profiling: function and regulation in human epidermal growth factor receptor 2 (HER2)-positive breast cancer. Cancers (Basel). 2019;11:646.
Savci-Heijink CD, Halfwerk H, Koster J, Horlings HM, van de Vijver MJ. A specific gene expression signature for visceral organ metastasis in breast cancer. BMC Cancer. 2019;19:333.
Turkyilmaz E, Guner H, Erdem M, Erdem A, Biri AA, Konac E, et al. NLF2 gene expression in the endometrium of patients with implantation failure after IVF treatment. Gene. 2012;508:140–3.
Murphy KE, Cottle L, Gysbers AM, Cooper AA, Halliday GM. ATP13A2 (PARK9) protein levels are reduced in brain tissue of cases with Lewy bodies. Acta Neuropathol Commun. 2013;1:11.
Luo W, Gumen A, Haughian JM, Wiltbank MC. The role of luteinizing hormone in regulating gene expression during selection of a dominant follicle in cattle. Biol Reprod. 2011;84:369–78.
Otto PI, Guimarães SEF, Verardo LL, Azevedo ALS, Vandenplas J, Soares ACC, et al. Genome-wide association studies for tick resistance in Bos taurus × Bos indicus crossbred cattle: a deeper look into this intricate mechanism. J Dairy Sci. 2018;101:11020–32.
Goto S, Cao F, Kono T, Ogawa H. Microarray analysis of differentially expressed genes in inner cell mass and trophectoderm of parthenogenetic embryos. J Mamm Ova Res. 2016;33:45–54.
Fukami M, Tsuchiya T, Vollbach H, Brown KA, Abe S, Ohtsu S, et al. Genomic basis of aromatase excess syndrome: recombination- and replication-rediated rearrangements leading to CYP19A1 overexpression. J Clin Endocrinol Metab. 2013;98:E2013-2021.
Rodriguez OC, Cheney RE. Human myosin-Vc is a novel class V myosin expressed in epithelial cells. J Cell Sci. 2002;115:991–1004.
Bertipaglia ECA, da Silva RG, Cardoso V, Fries LA. Hair coat characteristics and sweating rate of Braford cows in Brazil. Livest Sci. 2007;112:99–108.
Buckberry S, Bianco-Miotto T, Bent SJ, Clifton V, Shoubridge C, Shankar K, et al. Placental transcriptome co-expression analysis reveals conserved regulatory programs across gestation. BMC Genomics. 2017;18:10.
Yurchenko AA, Deniskova TE, Yudin NS, Dotsev AV, Khamiruev TN, Selionova MI, et al. High-density genotyping reveals signatures of selection related to acclimation and economically important traits in 15 local sheep breeds from Russia. BMC Genomics. 2019;20:294.
Chen X, Cheng Z, Zhang S, Werling D, Wathes DC. Combining genome wide association studies and differential gene expression data analyses identifies candidate genes affecting mastitis caused by two different pathogens in the dairy cow. Open J Anim Sci. 2015;05:358–93.
Abdel-Shafy H, Bortfeldt RH, Tetens J, Brockmann GA. Single nucleotide polymorphism and haplotype effects associated with somatic cell score in German holstein cattle. Genet Selection Evol. 2014;46:35.
Lillie M, Sheng Z, Honaker CF, Dorshorst BJ, Ashwell CM, Siegel PB, et al. Genome-wide standing variation facilitates long-term response to bidirectional selection for antibody response in chickens. BMC Genomics. 2017;18:99.
Young SL, Savaris RF, Lessey BA, Sharkey AM, Balthazar U, Zaino RJ, et al. Effect of randomized serum progesterone concentration on secretory endometrial histologic development and gene expression. Hum Reprod. 2017;32:1903–14.
Punyadeera C, Dassen H, Klomp J, Dunselman G, Kamps R, Dijcks F, et al. Oestrogen-modulated gene expression in the human endometrium. Cell Mol Life Sci. 2005;62:239–50.
Zhang L, Yang N, Huang J, Buckanovich RJ, Liang S, Barchetti A, et al. Transcriptional coactivator Drosophila eyes absent homologue 2 is up-regulated in epithelial ovarian cancer and promotes tumor growth. Cancer Res. 2005;65:925–32.
Asadollahpour Nanaei H, Ayatollahi Mehrgardi A, Esmailizadeh A. Whole-genome sequence analysis reveals candidate genomic footprints and genes associated with reproductive traits in Thoroughbred horse. Reprod Domest Anim. 2020;55:200–8.
Taye M, Lee W, Caetano-Anolles K, Dessie T, Hanotte O, Mwai OA, et al. Whole genome detection of signature of positive selection in African cattle reveals selection for thermotolerance. Anim Sci J. 2017;88:1889–901.
Bessa DS, Maschietto M, Aylwin CF, Canton APM, Brito VN, Macedo DB, et al. Methylome profiling of healthy and central precocious puberty girls. Clin Epigenetics. 2018;10:146.
Zeng F, Schultz RM. RNA transcript profiling during zygotic gene activation in the preimplantation mouse embryo. Dev Biol. 2005;283:40–57.
Okamura D, Maeda I, Taniguchi H, Tokitake Y, Ikeda M, Ozato K, et al. Cell cycle gene-specific control of transcription has a critical role in proliferation of primordial germ cells. Genes Dev. 2012;26:2477–82.
Gladney CD, Bertani GR, Johnson RK, Pomp D. Evaluation of gene expression in pigs selected for enhanced reproduction using differential display PCR and human microarrays: I. Ovarian follicles1,2. J Anim Sci. 2004;82:17–31.
Ortega MS, Denicol AC, Cole JB, Null DJ, Hansen PJ. Use of single nucleotide polymorphisms in candidate genes associated with daughter pregnancy rate for prediction of genetic merit for reproduction in Holstein cows. Anim Genet. 2016;47:288–97.
LEIGH D. Subacute necrotizing encephalomyelopathy in an infant. J Neurol Neurosurg Psychiatry. 1951;14:216–21.
Quintana A, Zanella S, Koch H, Kruse SE, Lee D, Ramirez JM, et al. Fatal breathing dysfunction in a mouse model of Leigh syndrome. J Clin Invest. 2012;122:2359–68.
Adjobo-Hermans MJW, de Haas R, Willems PHGM, Wojtala A, van Emst-de Vries SE, Wagenaars JA, et al. NDUFS4 deletion triggers loss of NDUFA12 in Ndufs4 mice and Leigh syndrome patients: a stabilizing role for NDUFAF2. Biochim Biophys Acta Bioenerg. 2020;1861:148213.
Anderson SL, Chung WK, Frezzo J, Papp JC, Ekstein J, DiMauro S, et al. A novel mutation in NDUFS4 causes Leigh syndrome in an Ashkenazi jewish family. J Inherit Metab Dis. 2008;31:461–7.
Petruzzella V. A nonsense mutation in the NDUFS4 gene encoding the 18 kDa (AQDQ) subunit of complex I abolishes assembly and activity of the complex in a patient with leigh-like syndrome. Hum Mol Genet. 2001;10:529–35.
Flachsbart F, Dose J, Gentschew L, Geismann C, Caliebe A, Knecht C, et al. Identification and characterization of two functional variants in the human longevity gene FOXO3. Nat Commun. 2017;8:2063.
Byun SO, Forrest RH, Zhou H, Frampton CM, Hickford JGH. Ovine forkhead box class O 3 (FOXO3) gene variation and its association with lifespan. Mol Biol Rep. 2013;40:3829–34.
Cui C, Han S, Yin H, Luo B, Shen X, Yang F, et al. FOXO3 is expressed in ovarian tissues and acts as an apoptosis initiator in granulosa cells of chickens. Biomed Res Int. 2019;2019:1–9.
Lin T-Y, Wei T-YW, Li S, Wang S-C, He M, Martin M, et al. TIFA as a crucial mediator for NLRP3 inflammasome. Proc Natl Acad Sci. 2016;113:15078–83.
Sheldon IM, Cronin JG, Healey GD, Gabler C, Heuwieser W, Streyl D, et al. Innate immunity and inflammation of the bovine female reproductive tract in health and disease. Reproduction. 2014;148:R41-51.
Rashid M, van der Horst M, Mentzel T, Butera F, Ferreira I, Pance A, et al. ALPK1 hotspot mutation as a driver of human spiradenoma and spiradenocarcinoma. Nat Commun. 2019;10:2213.
Mortlock S, Kendarsari RI, Fung JN, Gibson G, Yang F, Restuadi R, et al. Tissue specific regulation of transcription in endometrium and association with disease. Hum Reprod. 2020;35:377–93.
Junjvlieke Z, Khan R, Mei C, Cheng G, Wang S, Raza SHA, et al. Effect of ELOVL6 on the lipid metabolism of bovine adipocytes. Genomics. 2020;112:2282–90.
Corominas J, Ramayo-Caldas Y, Puig-Oliveras A, Pérez-Montarelo D, Noguera JL, Folch JM, et al. Polymorphism in the ELOVL6 gene is associated with a major QTL effect on fatty acid composition in pigs. PLoS ONE. 2013;8:e53687.
Zhang Y, Zhang J, Gong H, Cui L, Zhang W, Ma J, et al. Genetic correlation of fatty acid composition with growth, carcass, fat deposition and meat quality traits based on GWAS data in six pig populations. Meat Sci. 2019;150:47–55.
Claire D’Andre H, Paul W, Shen X, Jia X, Zhang R, Sun L, et al. Identification and characterization of genes that control fat deposition in chickens. J Anim Sci Biotechnol. 2013;4:43.
Hansen M, Flatt T, Aguilaniu H. Reproduction, fat metabolism, and life span: what is the connection? Cell Metab. 2013;17:10–9.
Leiter AB, Toder A, Wolfe HJ, Taylor IL, Cooperman S, Mandel G, et al. Peptide YY. Structure of the precursor and expression in exocrine pancreas. J Biol Chem. 1987;262:12984–8.
Batterham RL, Heffron H, Kapoor S, Chivers JE, Chandarana K, Herzog H, et al. Critical role for peptide YY in protein-mediated satiation and body-weight regulation. Cell Metab. 2006;4:223–33.
Fernandez-Fernandez R, Martini AC, Navarro VM, Castellano JM, Dieguez C, Aguilar E, et al. Novel signals for the integration of energy balance and reproduction. Mol Cell Endocrinol. 2006;254–255:127–32.
Raposinho PD, Broqua P, Pierroz DD, Hayward A, Dumont Y, Quirion R, et al. Evidence that the inhibition of luteinizing hormone secretion exerted by central administration of neuropeptide Y (NPY) in the rat is predominantly mediated by the NPY-Y5 receptor subtype1. Endocrinology. 1999;140:4046–55.
Fagerberg L, Hallström BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteom. 2014;13:397–406.
Butler WR. Nutritional interactions with reproductive performance in dairy cattle. Anim Reprod Sci. 2000;60–61:449–57.
Lucy MC. Reproductive loss in high-producing dairy cattle: where will it end? J Dairy Sci. 2001;84:1277–93.
Hatakeyama S. TRIM proteins and cancer. Nat Rev Cancer. 2011;11:792–804.
Sánchez-Tena S, Cubillos-Rojas M, Schneider T, Rosa JL. Functional and pathological relevance of HERC family proteins: a decade later. Cell Mol Life Sci. 2016;73:1955–68.
Lu J. Collectins and ficolins: sugar pattern recognition molecules of the mammalian innate immune system. Biochimica et Biophysica Acta (BBA). Gen Subj. 2002;1572:387–400.
Sá SR, Silva Junior AG, Lima-Neto RG, Andrade CAS, Oliveira MDL. Lectin-based impedimetric biosensor for differentiation of pathogenic candida species. Talanta. 2020;220:121375.
Woo J-S, Suh H-Y, Park S-Y, Oh B-H. Structural basis for protein recognition by B30.2/SPRY domains. Mol Cell. 2006;24:967–76.
D’Cruz AA, Babon JJ, Norton RS, Nicola NA, Nicholson SE. Structure and function of the SPRY/B30.2 domain proteins involved in innate immunity. Protein Sci. 2013;22:1–10.
Diaz-Granados A, Petrescu A-J, Goverse A, Smant G. SPRYSEC effectors: a versatile protein-binding platform to disrupt plant innate immunity. Front. Plant Sci. 2016;7:1575.
Ganz T, Nemeth E. Iron homeostasis in host defence and inflammation. Nat Rev Immunol. 2015;15:500–10.
Lill R, Mühlenhoff U. Maturation of Iron-Sulfur proteins in eukaryotes: mechanisms, connected processes, and diseases. Annu Rev Biochem. 2008;77:669–700.
Jordheim LP, Sève P, Trédan O, Dumontet C. The ribonucleotide reductase large subunit (RRM1) as a predictive factor in patients with cancer. Lancet Oncol. 2011;12:693–702.
Maris C, Dominguez C, Allain FH-T. The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J. 2005;272:2118–31.
Acknowledgements
The authors are thankful to FAPESP, CNPq and CAPES for financial support. We also thank the commercial breeding programs contributing to the Alliance Nellore dataset (https://gensys.com.br/sumario/) for providing the data dataset used in this work.
Funding
This research was supported by the São Paulo Research Foundation (FAPESP – Grants #2009/16118-5; #2017/24272-0; #2017/10630-2; #2018/20026-8); the National Council for Science and Technological Development (CNPq - Grant #559631/2009-0) and the Coordination for the Improvement of Higher Education Personnel (CAPES; financial code 001).
Author information
Authors and Affiliations
Contributions
Conceptualization, L.G.A.; methodology, D.O.S., G.A.F.J., L.G.A; investigation, D.O.S., G.A.F.J., L.F.S.F., L.F.M.M., T.B., R.C., L.G.A; writing-original draft preparation, D.O.S., G.A.F.J., L.F.S.F., L.F.M.M., T.B., R.C., L.G.A; writing-review and editing, D.O.S., G.A.F.J., L.F.S.F., L.F.M.M., T.B., R.C., L.G.A; visualization, D.O.S.; supervision, L.G.A. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of this article was revised: the names of Gerardo Alves Fernandes Júnior and Lucia Galvão de Albuquerque were incorrectly published as ‘Gerardo Alves Fernandes’ and ‘Lucia Galvão Albuquerque’ respectively.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Silva, D.O., Fernandes Júnior, G.A., Fonseca, L.F.S. et al. Genome-wide association study for stayability at different calvings in Nellore beef cattle. BMC Genomics 25, 93 (2024). https://doi.org/10.1186/s12864-024-10020-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-024-10020-y