
Abstract
Background
Genome-wide association analysis is a powerful tool for annotating phenotypic effects on the genome and knowledge of genes and chromosomal regions associated with dairy phenotypes is useful for genome and gene-based selection. Here, we report results of a genome-wide analysis of predicted transmitting ability (PTA) of 31 production, health, reproduction and body conformation traits in contemporary Holstein cows.
Results
Genome-wide association analysis identified a number of candidate genes and chromosome regions associated with 31 dairy traits in contemporary U.S. Holstein cows. Highly significant genes and chromosome regions include: BTA13's GNAS region for milk, fat and protein yields; BTA7's INSR region and BTAX's LOC520057 and GRIA3 for daughter pregnancy rate, somatic cell score and productive life; BTA2's LRP1B for somatic cell score; BTA14's DGAT1-NIBP region for fat percentage; BTA1's FKBP2 for protein yields and percentage, BTA26's MGMT and BTA6's PDGFRA for protein percentage; BTA18's 53.9-58.7 Mb region for service-sire and daughter calving ease and service-sire stillbirth; BTA18's PGLYRP1-IGFL1 region for a large number of traits; BTA18's LOC787057 for service-sire stillbirth and daughter calving ease; BTA15's CD82, BTA23's DST and the MOCS1-LRFN2 region for daughter stillbirth; and BTAX's LOC520057 and GRIA3 for daughter pregnancy rate. For body conformation traits, BTA11, BTAX, BTA10, BTA5, and BTA26 had the largest concentrations of SNP effects, and PHKA2 of BTAX and REN of BTA16 had the most significant effects for body size traits. For body shape traits, BTAX, BTA19 and BTA3 were most significant. Udder traits were affected by BTA16, BTA22, BTAX, BTA2, BTA10, BTA11, BTA20, BTA22 and BTA25, teat traits were affected by BTA6, BTA7, BTA9, BTA16, BTA11, BTA26 and BTA17, and feet/legs traits were affected by BTA11, BTA13, BTA18, BTA20, and BTA26.
Conclusions
Genome-wide association analysis identified a number of genes and chromosome regions associated with 31 production, health, reproduction and body conformation traits in contemporary Holstein cows. The results provide useful information for annotating phenotypic effects on the dairy genome and for building consensus of dairy QTL effects.
Similar content being viewed by others

Background
Genome-wide association studies (GWAS) using single nucleotide polymorphism (SNP) markers provide a powerful approach for annotating phenotypic effects or mapping QTL of important dairy traits on the genome. Dense genome coverage allows detection of QTLs with greater accuracy than was previously possible [1–3]. Combined with bovine whole-genome sequence information [4, 5], many SNP effects can be readily localized to specific genes or gene regions. Such QTL detection provides valuable information for understanding genetic mechanisms underlying dairy phenotypes and for identifying causal polymorphisms that lead to more rapid genetic improvement using genome selection [6, 7] or gene-based selection [8]. Several dairy GWAS using the bovine 50 k SNP chip [1–3] have been reported, including a study of U.S. Holstein bulls for 27 dairy traits that focused on the size and distribution of QTL effects [9], a study of milk traits in Danish Jersey bulls [10], a study of fertility traits in Danish and Swedish Holstein bulls [11], and a genome-wide candidate gene study using 1,536 SNP markers of candidate genes of Canadian Holstein bulls for association analysis with 17 type and functional traits [12]. These genome-wide studies contributed considerable new information over the many QTL studies based on microsatellite markers as compiled at the Cattle QTL Database [13] and contribute towards building a consensus on dairy QTL effects.
In this study, we conducted genome-wide association analysis of 31 production, health, reproduction and body conformation traits of contemporary U.S. Holstein cows. We used the bovine 50 k SNP panel to identify SNP markers, genes and chromosome regions on the 29 bovine autosomes and the X chromosome associated with these 31 economically important dairy traits. Production, health and reproduction are fundamental dairy functions while body conformation (type) traits are related to functionality of the cow's body and are related to value of the cow as a show animal.
Results
Overview of SNP effects
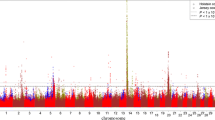
A global view of all additive SNP effects for each trait is presented in Manhattan plots [14, 15] in Figure S1 (Additional file 1), which shows that a large number of additive SNP effects reached 5% genome-wide significance with the Bonferroni correction (P value < 10-6.4) for each trait. Therefore, only the top 100 effects for each trait are reported for a total of 3,100 effects of 1,586 SNPs with 573 (36%) in 486 genes based on the Btau_4.0 and UMD 3.0 genome assemblies. Of the 45,878 SNPs that were genotyped, 16,516 (36%) were in 7,434 genes. The majority of the 1,586 SNPs each affected one trait, whereas 27 SNPs each affected 10 or more traits (Figure 1). All SNP effects in this report were additive as was expected because PTAs predict only additive genetic merit. Genotypic effects for SNPs had nearly identical P values to those of additive SNP effects. Therefore, P values of additive effects were used to rank SNP markers for each trait. Permutation tests of all 45,878 SNPs using 1,000 permutes and the cutoff P value for the top 100 most significant effects for each trait produced no observed false positive effects.

Distribution of 1,586 SNPs by number of traits affected for each of 31 production, health, reproduction and body conformation traits of contemporary U.S. Holsteins.
Detailed test results of the 3,100 effects are given in Table S1 (Additional File 2); complete QTL maps are shown in Figure S2 (Additional file 3) for the 13 production, health and reproduction traits, and are shown in Figure S3 (Additional File 4) for the 18 body conformation traits. Detailed characterization of the top 20 effects of each trait are given in Table S2 (Additional File 5), including actual SNP alleles, UMD 3.0 and Btau_4.0 SNP positions, the favorable allele, the favorable allele frequency, gene or gene region of the SNP, observed P-value, and the effect size with standard deviation.
The 3,100 effects of the 1,586 SNPs were distributed over all 29 Bos taurus (BTA) autosomes and the X chromosome but the distribution was uneven, with certain chromosomes having large numbers of SNP effects, and different chromosomes generally were associated with different traits (Table 1, Table 2). In Table 2, the 18 body conformation traits were divided into six trait groups: body size, body shape, udder, teats, feet and legs, and final score. "Body size" includes four traits; stature, strength, body depth, and rump width. "Body shape" includes two traits; dairy form and rump angle. "Udder" includes four traits; fore udder attachment, rear udder height, udder depth, and udder cleft. "Teats' includes three traits; front teat placement, rear teat placement and teat length. "Feet and legs" includes four traits; foot angle, rear legs (side view), rear legs (rear view) and feet/legs score. Definition and graphical illustration of each body conformation trait are available online [16, 17]. Chromosomes with a large number of effects for a trait did not necessarily have the most significant effect associated with that trait. For example, BTA18 did not have the largest number of SNP effects for any trait but did have a SNP (BFGL-NGS-117985) that had the most significant effect for five traits (Additional File 2: Table S1).
The top 100 SNPs for each trait accounted for 38% to 56% of the PTA variation (Table 1, Table 2). Significance levels varied by trait. Among production, health and reproduction traits, sire calving ease had the highest significance (smallest cutoff for P value; P < 10-38) and milk yield had the lowest significance (P < 10-12) (Table 1). For body conformation traits, stature had the highest significance (P < 10-21) and rump angle had the lowest significance (P < 10-11) (Table 2).
SNP effects in gene clusters, localized effect concentrations, highly significant genes
SNP effects for daughter pregnancy rate, somatic cell score and productive life overlapped with a large gene cluster of approximately 1,166 genes in a 15.4 Mb region of BTA7 (Figure 2A; Additional File 6: Figure S4A). The insulin receptor (INSR) gene in this cluster was 1.5 kb from the SNP with the most significant effect on somatic cell score and daughter pregnancy rate, and was third most significant for productive life. A narrow 2.81 Mb region of BTA14 with approximately 125 genes (Figure 2B; Additional File 6: Figure S4B) had 19 SNP effects for fat percentage, one SNP effect for milk yield associated with the vacuolar protein sorting 2 homolog (VPS28) gene, and one SNP effect each for fat yield and protein percentage in the NIK and IKKβ binding protein (NIBP) gene. A SNP in the diacylglycerol O-acyltransferase homolog 1 (DGAT1) gene (Figure 3A) had the most significant effect for fat percentage followed by a SNP in NIBP, which was the largest (387.23 kb) gene in this cluster (Figure 3B). The 15.82 Mb region of BTA18 with approximately 1,322 genes (Figure 2C; Additional File 6: Figure S4C) had SNP effects for many traits but was most pronounced for service-sire and daughter calving ease and service-sire stillbirth. The peptidoglycan recognition protein 1 (NCBI's PGLYRP1 or ENSEMBL's PGRP) gene and IGF-like family member 1 (IGFL1) gene in this cluster flanked a SNP that was highly significant for many traits. This SNP had the top effect for fat and protein yields, service-sire and daughter calving ease, and net merit; the eighth most significant effect for milk yield and service-sire stillbirth; the 16th most significant effect for productive life, and the 25th most significant effect for fat and protein percentages. The zinc finger protein 415-like gene (LOC787057) was most significant for service-sire stillbirth and second most significant for daughter calving ease. The 211.67 kb MOCS1-LRFN2 region of BTA23 (Figure 2D; Additional File 7: Figure S5A) included several SNPs with significant effects on daughter stillbirth, while a BTA15 marker between two CD82 genes had the most significant effect for daughter stillbirth (Additional File 7: Figure S5B). The MOCS1 gene is related to early infant death in humans [18, 19]. The most significant effect for milk yield was near the guanine nucleotide binding protein, alpha stimulating (GNAS) locus of BTA13 (Additional File 7: Figure S5C).

Map of SNP position (Mb) on Bos taurus chromosomes 7 (A), 14 (B), 18 (C), and 23 (D) by P -value for 725 SNPs that comprise the top 100 effects for each of 13 production, health, and reproduction traits of contemporary U.S. Holsteins.

A 2.81 Mb gene cluster on Bos taurus chromosome 14 that was associated with significant effects of single nucleotide polymorphisms for fat percentage. Red arrow, fat percentage effect; gold arrow, milk yield effect; green arrow, fat yield effect. A) 1 Mb region with A5D786, CYHR1, VPS28, and DGAT1 genes; A5D786 was second largest gene in the cluster and contained CYHR1 and VPS28. B) 1 Mb region with NIBP, the largest gene in the cluster.
Additional gene clusters with SNP effects were also observed on BTA3, BTA5, BTA7, BTA10, BTA17, BTA21, BTA23, BTA26, BTA29, and BTAX (Additional File 6: Figure S4D-N). Additional localized concentrations of SNP effects were also observed at 45 Mb on BTA3 and from 1 to 15 Mb on BTA13 for milk yield, at 135 Mb on BTA1 and at 91 Mb on BTA3 for daughter pregnancy rate, at 14 and 34 Mb on BTA17 for daughter calving ease, and at 49.5 Mb on BTA26 for fat yield, fat and protein percentages, and daughter calving ease.
For body conformation traits, BTAX's phosphorylase kinase, alpha 2 (liver) gene (PHKA2) was highly significant for body size traits. The top SNP effects in the 80 to 90 Mb region of BTA11 predominantly affected stature, strength, body depth, rear udder height, teat length, rear legs (rear view), feet/legs score, and final score (Figure 4A). The renin gene (REN) at the top telomere region of BTA16 was highly significant for 12 traits (the largest number of conformation traits affected by one gene). The 65 to 75 Mb region of BTA16 (Figure 4B) had a concentration of SNP effects for rear legs (side view). The 39 Mb region and the O-6-methylguanine-DNA methyltransferase (MGMT) gene region on BTA26 (Figure 4C) had an effect concentration for feet/legs traits. The 125 to 145 Mb region of BTAX (Figure 4D) had significant effects for body size traits, foot angle, rear legs (rear view), and final score. Other chromosomes with local effect concentrations included the 30 to 35 Mb region of BTA2 for dairy form, rear udder height, and feet/legs score; the 19 to 21 Mb region of BTA13 for foot angle and feet/legs score; the 56.2 to 57.6 Mb region of BTA19 for rump width; the 10 to 20 Mb region of BTA20 for udder depth and foot angle; and the 20 to 22 Mb region of BTA21 for teat length.

Map of SNP position (Mb) on Bos taurus chromosomes 11 (A), 16 (B), 26 (C) and X (D) by P -value for 1,005 SNPs that comprise the top 100 effects for each of 18 conformation traits of contemporary U.S. Holsteins.
For the 31 traits, the X chromosome had the largest number of SNP effects (Table 1, Table 2) and most of these effects were in four regions; 5 to 25 Mb, 50 to 60 Mb, 85 to 105 Mb, and 125 to 140 Mb (Additional File 3: Figure S2; Additional File 4: Figure S3). The most significant X chromosome SNP effects were associated with daughter pregnancy rate, body size, dairy form, rear legs (rear view), productive life, udder attachment, rear teat placement, fat yield, protein yield, net merit, and final score of body conformation traits (Additional file 2: Table S1). Eighteen significant SNP markers were in the pseudo-autosomal region defined by UMD 3.0. However, based on male heterozygosity, only eight of the 18 markers could be truly in the pseudo-autosome region. Male heterozygosity for ten of these 18 SNPs (marked in yellow in Additional file 2: Table S1) were inconsistent with the pseudo-autosomal assignments because they had zero or a small number of heterozygous genotypes among 483 males (not included in this report). The region from 140,525,988 bp to 143,832,372 bp had 28 markers. Of these, one marker had three male heterozygous genotypes, two markers each had one male heterozygous genotype, and the other 25 markers had no male heterozygous genotypes. Similarly, of the 8 markers in the region from 139,306,649 bp to 139,975,594 bp, only one marker had 15 male heterozygous genotypes while the other seven markers had no male heterozygous genotype.
The second largest number of SNP effects for production, health and reproduction traits (Table 1) were in the 10 to 35 Mb region of BTA17 (Figure S1). These included SNP effects for daughter calving ease, sire calving ease and protein percentage, but most of the effects fell in gene-sparse areas.
Summary of SNP effects by trait
The results in this study generally point to polygenic genetic mechanisms for all 31 dairy traits. However, some chromosome regions and genes had more striking association(s) with the traits in terms of statistical significance and known relevant biology, making these regions and genes more likely candidates for causal effects. The following is a brief summary of such regions and genes.
Milk, fat and protein yields
Milk, fat and protein yields had a tendency of sharing common SNP effects. The 58 Mb region of BTA13 with four genes near GNAS was highly significant for the three yield traits (Additional File 5: Table S2.1 to S2.3). A BTA1 region 220 kb upstream of FKBP2, which plays a role in immunoregulation and basic cellular processes involving protein folding and trafficking, was highly significant for fat yield, protein yield and protein percentage. The 47 Mb region of BTA3 had a strong effect for milk yield, while the 54 Mb region of BTA18 near PGLYRP1-IGFL1 and the 146 Mb X chromosome region in the DOCK11-IL13RA1-AF074402-LOC616260 gene cluster could be targets for fat and protein yields.
Fat and protein percentages
This study specifically identified a 2.81 Mb BTA14 gene cluster spanning DGAT1-NIBP (Figure 2B, Figure 3) as a region with a heavy concentration of SNP effects for fat percentage, accounting for 8% of the PTA variation. Within this cluster, DGAT1 had the most significant and NIBP had the second most significant SNP effects for fat percentage (Additional File 5: Table S2.4). The VPS28 gene, which is about 100 kb upstream of DGAT1 and is near the left end of the 2.81 Mb DGAT1-NIBP region, had a highly significant effect for milk yield, while NIBP, which is at the right end of this 2.81 Mb region, had a SNP at position 4.468 Mb (2.71 Mb according to Btau_4.0) with a highly significant effect for fat yield, fat percentage and protein percentage (Figure 3). The 49 Mb BTA26 region near MGMT had highly significant effects on protein percentage as well as protein yield, and the 71 Mb BTA6 region near PDGFRA included highly significant SNPs affecting protein percentage (Additional File 5: Table S2.5).
Productive life, somatic cell score, daughter pregnancy rate
These three traits had two common regions and some trait-specific regions. The first common region for these three traits was the 15.4 Mb BTA7 gene cluster of approximately 1,166 genes (Figure 2A). Although the exact location of the causal effect may be difficult to dissect for such a tightly linked gene cluster (7.5 genes per 100 kb), our data placed the most significant effect in this region near INSR (Additional File 5: Table S2.6 to S2.8; Figure 2A; Additional File 6: Figure S4A). The second common region was the 106 Mb BTAX region near LOC520057 (similar to type 1 protein phosphatase inhibitor). Trait-specific regions include the MIR2353-STK39 and LRP1B (low density lipoprotein-related protein 1B) regions of BTA2 for somatic cell score, the 129 to 141 Mb region of BTA1, the 90 Mb region of BTA3 for daughter pregnancy rate, and the ATP1B4 (ATPase, Na+/K+ transporting, beta 4 polypeptide) and GRIA3 (glutamate receptor 3) genes on BTAX for daughter pregnancy rate.
Productive life measures the cow's longevity in the herd and is affected by production, health and reproduction. Somatic cell score is a measure of udder health and daughter pregnancy rate is a measure of cow fertility. The fact that productive life shared many common SNP effects with somatic cell scores and daughter pregnancy rate and did not share many SNP effects with production and calving traits indicates productive life was more genetically related with health and fertility traits than with production and calving traits.
Service-sire calving ease, daughter calving ease, service-sire stillbirth
These three traits shared a common region on BTA18 in the 15.82 Mb gene cluster with approximately 1,322 genes (Figure 2C; Additional File 6: Figure S4C). The PGLYRP1-IGFL1 region and LOC787057 had the most significant effects for these three traits (Additional File 5: Table S2.9-S2.11).
Daughter stillbirth
Nine of ten SNPs in the 211.67 kb MOCS1-LRFN2 region of BTA23 were among the top 100 SNPs that were significant for daughter stillbirth (Figure 2D), which is a measure of the cow's effect on calf stillbirth. The SNP marker between two CD82 genes on BTA15 (Additional File 2: Figure S1; Additional File 5: Table S2.12) had the most significant effect. The second most significant SNP effect was in the dystonin gene (DST) on BTA23.
Lifetime net merit index
Highly significant chromosome regions and genes for this composite trait involved the significant regions and genes for milk, fat and protein yields, protein percentage, service sire calving ease, daughter calving ease, and service sire stillbirth (Additional File 5: Table S2.13).
Body size traits - stature, strength, body depth, rump width
Stature and body depth shared many common SNP markers on BTAX, BTA11 and BTA5, while strength and rump width shared common SNP effects on BTAX and BTA16 (Additional File 5: Table S2.14 to S2.17). The PHKA2 gene on BTAX (Additional File 8: Figure S6A) was highly significant for all four body size traits, and the BTAX region from the gem (nuclear organelle) associated protein 8 gene (GEMIN8) to the glycoprotein M6B gene (GPM6B) (Figure 4D) included SNPs among the top 20 effects for all four body size traits.
The largest numbers of significant SNP effects for stature were found on BTA11 and BTAX, with 30 and 14 effects, respectively (Table 1). Genes on BTA11 that included or were near SNPs with the top 20 effects were lipin 1 (LPIN1; second most significant SNP effect), tribbles homolog 2 (TRIB2; third and fourth SNP effects), odd-skipped related 1 (OSR1), aromatic-preferring amino acid transporter-like (LOC529399), and neuroblastoma amplified sequence (LOC521982) (Additional File 5: Table S2.14; Additional File 8: Figure S6B).
The REN gene on BTA16 was highly significant for body strength (ranked #2), depth (ranked #4) and rump width (ranked #1). This gene was located in the middle of a 70-kb gene cluster with five tightly linked genes: SRY (sex determining region Y)-box 13 (SOX13), ethanolamine kinase 2 (ETNK2), REN, KiSS-1 metastasis-suppressor (KISS1), and golgi transport 1A (GOLT1A) (Additional File 8: Figure S6C). The pleckstrin homology domain containing, family A member 6 (PLEKHA6) gene was 70 kb downstream of these five tightly linked genes and had the ninth most significant SNP effect for strength.
The most significant BTA13 SNP effect for strength was ranked seventh for the trait and was in the pitrilysin metallopeptidase 1 gene (PITRM1), which also was linked to two of the top 20 effects for stature and three of the top 20 effects for body depth. A SNP on BTA23 at position 18,197,600 bp had the second most significant effect for body depth and was the third most significant for strength.
Strength and rump width are related to body width but likely involved different genes because <25% of the top 100 SNPs (Additional File 2: Table S1) and only four of the top 20 SNPs were in common for the two traits. In addition, none of the significant SNP effects on BTA19 for strength were among the top 20 for rump width. Of the seven BTA19 SNPs among the top 20 for rump width, five were within genes; GPRC5C (ranked 3rd), RNF157 (ranked 5th), SRP68 (ranked 8th), LOC789539 (ranked 11th), and OTOP3 (ranked 15th). The six most significant BTA19 SNPs were in a 1.8 Mb region of 57.2 to 59.0 Mb (Additional File 8: Figure S6D).
Body shape traits - dairy form and rump angle
Although both dairy form and rump angle are measures of the cow's body shape, these two traits likely involved different genes. The top 20 SNP effects for dairy form involved eight chromosomes (Additional File 5: Table S2.18). The most significant SNP for dairy form was 30.9 kb upstream of LOC520059, the same SNP that was highly significant for daughter pregnancy rate and productive life. The top 20 effects for dairy form only had four SNPs in genes; immunoglobulin superfamily, member 5 (LOC511594) of BTA1, myocyte enhancer factor 2C (MEF2C) of BTA7, and phospholipase A2, group IVF (PLA2G4F) and calpain 3 (p94) (CAPN3) of BTA10. For rump angle, BTA5 and BTA9 had the most significant effects (Additional File 5: Table S2.19). Two of the top 20 effects for rump angle were SNPs located in the NADPH oxidase 4 (NOX) and SH3 and multiple ankyrin repeat domains 2 genes (LOC618649) on BTA29.
Udder traits - fore udder attachment, rear udder height, udder depth, udder cleft
Fore udder attachment shared some common SNP effects with body size traits and udder height, but essentially had no common SNP effects with udder depth and udder cleft (Additional File 5: Table S2.20-S2.23). Fore udder attachment also shared some effects with teat traits, daughter pregnancy rate, somatic cell score and productive life. A SNP in REN of BTA16 and a SNP in PHKA2 of BTAX were most significant for udder attachment. Four SNPs in the BTA19 region associated with rump width were among the top 20 effects for fore udder attachment. Two BTA2 SNPs that were 19.2 to 58.3 kb upstream of the T-box, brain, 1 (TBR1) gene had the first and fourth most significant effects, and two BTA10 SNPs in the adenomatous polyposis coli (APC) and CAPN3 genes had the second and third most significant effects, respectively. The most significant effects for udder depth were located on BTA25, BTA22, BTA7, BTA26, BTA20 and BTA23. The SNP 1.5 kb downstream of INSR on BTA7 was the tenth most significant SNP for udder depth (Additional File 5: Table S2.23). The most significant effects for udder cleft were located on BTA22, BTA7, and BTA25. Six of the top 20 SNP effects for udder cleft (Additional File 5: Table S2.24) were found on BTA7. Two of the top 20 effects for udder cleft were BTA6 SNPs in the leucine zipper-EF-hand containing transmembrane protein 1 (LETM1) and Wolf-Hirschhorn syndrome candidate 2 (WHSC2) genes. The same BTA6 and BTA7 SNP markers were also highly significant for teat placement traits, which indicated that udder cleft and teat placement involved some common genes. The tenth most significant SNP for udder cleft was on BTA19 SNP and was just downstream from a gene cluster that affected rump width and fore udder attachment (Additional File 8: Figure S6D).
Teat traits - front teat placement, rear teat placement, teat length
Front and rear teat placements involved different and common SNP effects. Teat length and teat placement traits appeared to have been associated with different genes. Two BTA6 SNPs in the LETM1 and WD repeat and FYVE domain containing 3 (WDFY3) genes were the top two most significant SNPs for front teat placement and were among the top 20 effects for rear teat placement (Additional File 5: Table S2.25 to S2.27). The LETM1 SNP was also ranked sixth in significance for udder cleft. A relatively gene-sparse region of BTA7, 347.5-412.1 kb upstream from the centrin EF-hand protein 3 gene (CETN3), was highly significant for both rear teat placement and udder cleft. The TAF1 RNA polymerase II, TATA box binding protein-associated factor, 250 kDa gene (TAF1) on BTAX had the second most significant SNP effect for rear teat placement and the 16th for udder cleft. The GPRC5C gene on BTA19 (Additional File 8: Figure S6D) had the tenth most significant SNP for rear teat placement and the second for udder cleft. These results indicate that the same chromosome regions were involved in rear teat placement and udder cleft and that the LETM1 and WHSC2 genes on BTA6 had a major role in udder cleft and teat placement traits. The most significant SNP effect for teat length was on BTA11, 98.5 kb downstream from LOC615674, a ribosomal protein L36-like gene, followed by a BTA26 SNP 80.8 kb upstream from MGMT. The three BTA21 SNPs among the top 20 effects for teat length were in a gene cluster (Additional File 8: Figure S6E), with one SNP in the hypothetical protein LOC613997 (MGC129355; ENSEMBL CO038) and one SNP in the abhydrolase domain containing 2 gene (ABHD2).
Feet/legs traits - foot angle, rear legs (side view), rear legs (rear view), feet/legs score
Three BTA26 SNPs that spanned a 1.09 Mb region in or upstream from MGMT had the top three effects for foot angle, and another four BTA26 SNPs were also among the top 20 effects for foot angle (Additional File 5: Table S2.28). BTA1 had the most significant SNP for rear legs (side view), whereas BTA18 had the largest number of significant SNPs (five effects), followed by BTA1, BTA16, and BTAX with three effects each (Additional File 5: Table S2.29). The top 20 effects for rear legs (rear view) involved only four chromosomes: BTA11, BTAX, BTA20, and BTA26. The most significant SNP was on BTAX, followed by three BTA11 SNPs. The most significant SNP for foot angle and for feet/legs score was in MGMT on BTA26 (Additional File 5: Table S2.27, Table S2.30). This SNP was the tenth most significant SNP for rear legs (rear view). The side and rear views of the legs apparently were associated with different sets of chromosome and gene regions. Of the top 20 effects, BTA26 and BTA12 had the most SNPs (five each), followed by BTA5 and BTAX (four each). The top 20 SNP effects for feet/legs score were predominantly the same as those for foot angle and rear legs (rear view).
Final score (also known as PTA type)
The most significant SNP for final score (Additional File 5: Table S2.31) was a BTAX SNP in PHKA2, which was also the most significant SNP for stature, strength, and body depth, the second most significant for rump width and fore udder attachment, and the 11th most significant for rear udder height. The second most significant SNP for final score was in BTA16's REN, which was among the top 20 effects for five other conformation traits. The third most significant SNP for final score was in BTA10's APC, which was second most significant for rear udder height, eighth for fore udder attachment, and ninth for rump width.
Discussion
Comparison with reported results
Most previously reported QTL locations were based on genetic distances in units of Morgan (or centiMorgan) rather than the physical distances (Mb or kb) for QTL locations reported in this study. Because exact and complete translation of bovine genetic distances into physical distances is not available, the results in this study could be compared only with studies that reported genes or markers with known physical locations.
This study confirmed widely reported findings that the BTA14 region containing DGAT1 was important for fat percentage [20] and provided some evidence of QTL effects in this region on milk and fat yields and protein percentage. This study specifically identified a 2.81 Mb BTA14 gene cluster with 125 genes with a concentration of SNP effects for fat percentage (Figure 2B), with A5D786 near the left end (Figure 3A) and NIBP at the right end (Figure 3B). The largest and second largest genes in this cluster were NIBP and A5D786, respectively.
Two significant SNP effects for calving traits were found in a region on BTA18 that Cole et al. [9] had reported to be associated with calving traits for U.S. Holsteins. That region included BTA-29287-no-rs that ranked 4th for service-sire calving ease, 2nd for daughter calving ease, and 1st for service-sire stillbirth and ARS-BFGL-BAC-36087 that ranked 92nd for daughter stillbirth (Additional file 2: Table S1). The most significant genes for calving traits identified in our study were PGLYRP1-IGFL1 and LOC787057. The PGLYRP1-IGFL1 region was 360 kb upstream and LOC787057 was 124 kb downstream from SIGLEC12 reported in Cole et al. [9]. Results from this study and from Cole et al. [9] indicate the 0.36 Mb region of PGLYRP1-IGFL1-SIGLEC12-LOC787057 should be an interesting target for factors that affect calving traits. Other dairy GWAS [10–12] had only a small number of traits that overlapped with this study. The PGLYRP1-IGFL1 region we identified was 150 kb upstream from LOC538513 that was reported to be associated with a direct calving ease effect. We also identified a SNP at BTAU_4.0 136,742,669 bp of BTA1 that ranked 69th for daughter pregnancy effect and this was close to a previously reported fertility effect at position 136,499,200 bp [12].
Gene association with conformation traits
The SNP significance tests showed that different traits generally were associated with different genes or gene regions, but many of the traits also shared common genes or gene regions (Additional File 2: Table S1). Traits within a phenotype group (e.g., body size or udder conformation) had a greater tendency to have genes or gene regions with significant SNPs in common than did traits in different phenotype groups.
All four traits related to body size (stature, strength, body depth, and rump width) had top-20 SNPs in BTA16's REN, BTAX's PHKA2 and BTAX's GPM6B. The REN gene is part of the renin-angiotensin system that regulates cellular growth in response to developmental, physiological, and pathological processes [21]. In humans, PHKA2 is responsible for glycogen storage diseases [22]. These known biological functions of REN and PHKA2 are consistent with the highly significant effects of REN and PHKA2 on body size traits. The two body height traits (stature and body depth) had many significant SNPs with overlapping gene regions of BTA11. The two body width traits (strength and rump width) also had top-20 SNPs on chromosomes other than BTA16 and BTAX (e.g., BTA13 and BTA26 for strength and BTA19 for rump width). The two body shape traits (dairy form and rump angle) also had top-20 SNPs on BTA3, BTA7, BTA10 (for dairy form) and BTA5 and BTA9 (for rump angle).
Four udder traits (fore udder attachment, rear udder height, udder depth, and udder cleft) had top-20 SNPs with gene regions in common. Fore udder attachment and udder depth had BTA22 SNPs near the succinate-CoA ligase, GDP-forming, beta subunit (SUCLG2) gene, a BTA7 SNP in the CREB regulated transcription coactivator 1 (CRTC1) gene, and a BTA23 SNP in the collagen, type XXI, alpha 1 (COL21A1) gene. Rear udder height shared only one top 20 SNP on BTA7 with udder cleft but shared two BTA11 SNPs with body depth. Many top-20 SNPs in gene regions overlapped for udder cleft and teat placement traits; four BTA7 SNPs (with three near CETN3), two BTA6 SNPs in LETM1 and WHSC2, one BTA19 SNP in GPRC5C, and one BTAX SNP in TAF1. Udder cleft appears to be more related genetically to teat placement traits than to other udder traits. It is interesting to note that the most significant genes for body size, REN and PHKA2, also were most significant for fore udder attachment and were highly significant for rear udder height. Significant SNPs for front and rear teat placement had considerable effect overlap with udder cleft, indicating that teat positions and udder shape may share common genes. In contrast, teat length had little gene region overlap with SNPs for teat position or udder shape, indicating the likely involvement of different genes for these traits.
The three individual feet/legs traits (excluding feet/legs score) mostly involved different genes. The MGMT gene of BTA26 was most significant for foot angle and feet/legs score and was among the top 20 effects for rear legs (rear view). Most of the highly significant effects for rear legs (rear view) were on BTA11 and BTA18 while BTA1, BTA18 and BTA16 had most of the top 20 effects for rear legs (side view). Feet/legs score had more effects that overlapped with foot angle than with the two rear leg traits.
Effect of X chromosome
Only limited research on the X chromosome is available, compared to the extensive literature on autosomes. In this study, the X chromosome had the largest number of significant SNP effects (Table 1) and the highest chromosomal frequency for the top 20 SNP effects for fat yield (along with BTA13), protein yield, protein percentage, productive life, somatic cell score (along with BTA2, BTA6, and BTA7), daughter pregnancy rate, service-sire stillbirth, and net merit. This was observed even though the X chromosome had the smallest number of SNPs among all chromosomes (1.67% for X chromosome compared with 3.33% per chromosome if the 45,878 SNPs had been distributed equally over all 30 chromosomes).
The most significant X chromosome effects based on effect ranking relative to autosome effect were on daughter pregnancy rate, productive life, protein percentage, somatic cell score and fat yield. The 106 Mb BTAX region near LOC520057 contained significant SNPs for daughter pregnancy rate, productive life and somatic cell score. Type 1 protein phosphatase has been implicated in the control of a range of cellular processes, including the cell cycle, gene expression, cell adhesion, and glycogen metabolism [23, 24]. Two interesting gene clusters bracketed the LOC520057 gene. The NDP-MAOA-MAOB gene cluster was about 1.2 Mb upstream and MAOB plays an important role in the metabolism of neuroactive and vasoactive amines in the central nervous system and peripheral tissues [25]. The LOC100138543-MID1IP1-MIR2488 cluster was about 1.0 Mb downstream and LOC100139224 is the largest and most complicated enzyme of the electron transport chain [26]. The MID1IP1 portion of this cluster is MID1 interacting protein 1 gastrulation specific G12 homolog (zebrafish). Gastrulation is an early phase of embryonic development so MID1IP1's biology could be relevant to daughter pregnancy rate and productive life. The MIR2488 portion is a microRNA involved in post-transcriptional regulation of gene expression in multicellular organisms and affects both the stability and translation of mRNAs [26]. In addition, daughter pregnancy rate was associated with SNPs in the ATP1B4 and GRIA3 genes on BTAX. The protein encoded by ATP1B4 interacts with the nuclear transcriptional coregulator SKIP and may be involved in the regulation of TGF-beta signaling [25]. Glutamate receptors are the predominant excitatory neurotransmitter receptors [25].
Significance and frequency
Most of the top 20 SNP effects had the minor allele (frequency of <0.5) as the favorable allele (Additional File 5: Table S2), including the most significant SNP for fat percentage (ARS-BFGL-NGS-4939), which was located in DGAT1. Frequencies of favorable alleles were particularly low for protein yield and percentage, service-sire and daughter calving ease, and net merit. Daughter stillbirth was the only exception with major alleles (frequency of ≥0.5) as the favorable alleles for over half of the top 20 effects. Minor alleles were the favorable allele primarily because they were associated with high PTAs. For example, cows that were homozygous for the minor allele of BTA18's BFGL-NGS-117985, which was among the top 100 significant SNP effects for 10 of the thirteen production, health, and reproduction traits, had a PTA for fat yield of ≥15 kg, whereas cows homozygous for the major allele had PTAs between -21 and 41 kg, with a mean of 9 kg (Figure 5). The frequency of the favorable allele of this marker was 9.1% in the 1,654 contemporary U.S. Holstein cows. Because of low allele frequencies, some of the highly favorable associations with minor alleles could be due to sampling rather than biological effects.

Distribution of cows for predicted transmitting ability (PTA) for fat yield by allele combination for SNP BFGL-NGS-117985 in the PGLYRP - IGFL1 region of Bos taurus chromosome 18. This SNP explained 13.68% of PTA variation (R2 = 0.1368) of fat yield, was among the top 20 significant SNP effects for fat and protein yields, service-sire and daughter calving ease, service-sire stillbirth, net merit (ranked 1st), milk yield (ranked 9th), and productive life (ranked 16th), and was among the top 100 effects for fat and protein percentages. A) All cows. B) Cows homozygous for major allele of SNP. C) Cows heterozygous for SNP. D) Cows homozygous for minor allele of SNP.
The majority of the 1,005 SNPs for body conformation traits had intermediate allele frequencies. Only 94 of the 1,005 SNPs had a minor allele frequency of <0.10 (Additional File 2: Table S1). Because an intermediate value may be optimal for many conformation traits, few SNPs were driven towards fixation. The intermediate frequencies should allow considerable flexibility in genetic selection for improving conformation traits and associated functionality.
Sensitivity of association results to PTA variations
PTA values from different individuals had different accuracies measured by reliability [27]. Results reported in this study were based on PTA values without being adjusted by the reliability of each PTA value. This approach allowed the use of all PTA values including PTA values with zero estimates of reliability. To study the effects of different accuracies on the SNP results, we also analyzed the data using a weighted least squares analysis, with the reliability as the weight of each PTA value. The results from this weighted least squares analysis were similar to the original analysis without considering PTA accuracies (Additional File 9: Table S3). The four calving traits had 360 individuals with zero reliability values, but the weighted least squares results were similar to the original results in terms of effect ranking and statistical significance, particularly for the top 50 effects. For daughter calving ease, the original least squares effects ranked below 50th had relatively poor overlap with the weighted least squares effects.
Conclusions
Genome-wide association analysis of U.S. contemporary Holstein cows produced comprehensive descriptions of genes and chromosome regions associated with 31 production, health, reproduction and body conformation phenotypes and provided a large quantity of genome annotation details for phenotypic effects based on the latest bovine genome sequencing results and SNP chip development. The results of this study should significantly contribute to the process of building consensus of dairy QTL effects. The results support the polygenic hypothesis for all 31 traits in this study. Production, health and reproduction traits involved more gene clusters of tightly linked genes than body conformation traits, indicating that genetic mechanisms of production, health and reproduction were more complex than those of body conformation traits.
Methods
Phenotypic data, study population and SNP genotyping
Thirty one dairy traits, including 13 production, health and reproduction traits and 18 body conformation traits were studied. Traditional predicted transmitting abilities (PTAs) for each trait calculated by the U.S. Department of Agriculture (Beltsville, MD) were phenotypic data for association with SNPs. The 13 production, health and reproduction traits were milk, fat and protein yields, fat and protein percentages, productive life, somatic cell score, daughter pregnancy rate, service-sire and daughter calving ease, service-sire and daughter stillbirth, and a genetic-economic index for lifetime net merit. The 18 conformation traits as defined by Holstein Association USA and the World Holstein Friesian Federation [16, 17] were stature (distance from the top of the spine in between the hips to the ground), strength (inside surface between the top of the front legs; also known as chest width), body depth (distance at the last rib from the top of the spine to the bottom of the barrel), dairy form (angle and openness of the ribs combined with the flatness of bone to avoid coarseness; also known as angularity), rump angle (angle of the rump structure from the hips to the pins), rump width (distance between the most posterior point of the pin bones; also known as thurl width), fore udder attachment (strength of the attachment of the fore udder to the abdominal wall), rear udder height (distance from the bottom of the vulva to milk-secreting tissue in relation to the height of the animal), udder depth (distance from the lowest part of the udder floor to the hock), udder cleft (depth of the cleft at the base of the rear udder; also known as central ligament), front teat placement (position of the front teat relative to the center of the udder quarter), rear teat placement (position of the rear teat relative to the center of the udder quarter), teat length (length of the front teat), foot angle (angle at the front of the rear right hoof measured from the floor to the hairline), rear legs (side view) (angle measured at the front of the hock; also known as set of rear legs), rear legs (rear view) (direction of feet when viewed from the rear of the animal), feet/legs score (an overall score assigned by a classifier based on side and rear views of rear legs, locomotion, feet, thurl position, hocks, bone, and pasterns), and final score (an overall conformation score assigned by a classifier based on front end and body capacity, dairy strength, rump, feet and legs and udder; also known as PTA type).
The design of the study population aimed at having a broad representation of contemporary U.S. Holstein cows. The 1,654 cows in the study population included elite and average Holstein cows for which DNA was supplied by Genetic Visions (Middleton, WI), Genex Cooperative (Shawano, WI), Holstein Association USA (Brattleboro, VT), Iowa State University (Ames, IA), Pennsylvania State University (University Park, PA), the University of Florida (Gainesville, FL), the University of Minnesota (St. Paul, MN), and Virginia Polytechnic Institute and State University (Blacksburg, VA).
A total of 45,878 SNP markers from the BovineSNP50 BeadChip (Illumina, San Diego, CA) were selected for a dual purpose research of association analysis in this study and a selection signature analysis [28]. This SNP set required an allele frequency difference of ≥2% between the study population and a group of 301 Holstein cattle that have remained unselected since 1964 to allow identification of near-fixed alleles in the contemporary population due to selection. Of the 45,878 SNP markers, 45,461 had known chromosome positions with mean marker spacing of 58.45 kb. Extraction of DNA and SNP genotyping were performed at the Bovine Functional Genomics Laboratory (Agricultural Research Service, U.S. Department of Agriculture, Beltsville, MD). Marker genotypes were scored using GenomeStudio software (version 1.1.9; Illumina, San Diego, CA).
Data analyses
Statistical tests of SNP effects were conducted using the epiSNP computer package [29, 30]. The epiSNP package implements the extended Kempthorne model that allows linkage disequilibrium between SNPs and Hardy-Weinberg disequilibrium for each SNP [31]. Normality of phenotypic residuals of each trait was evaluated using the R package [32] and residual values for all traits were found to satisfy the bell shaped normal distribution. Since PTA values are predicted additive genetic effects after removing fixed non-genetic effects such as herd-year-season, the statistical model did not need to consider fixed non-genetic effects. The statistical model for testing SNP-phenotype association used a single-locus model: PTA = μ + g + e, where μ = common mean, g = SNP genotypic effect, and e = random residual. Based on estimates of SNP genotypic values from least squares (LS) regression, the epiSNP package tests three effects for each locus by default; the marker genotypic effect, additive and dominance effects. The marker genotypic effect was tested using F-test, while additive and dominance effects were tested using t-test by the following t-statistic [31]:  , where si is a function of marginal and conditional probabilities calculated from SNP genotypic frequencies and is a row vector of contrast coefficients of the SNP genotypic effects for defining additive or dominance effect, and
, where si is a function of marginal and conditional probabilities calculated from SNP genotypic frequencies and is a row vector of contrast coefficients of the SNP genotypic effects for defining additive or dominance effect, and  is a column vector of LS estimates of three SNP genotypic effects. Although we did not expect to detect dominance effects because PTA values are estimated additive genetic effects, the test of dominance effects provided a check on whether the statistical tests produced unexpected genetic effects. The results were as expected. Only spurious dominance effects were observed and no dominance effect was among the top 100 effect for any trait.
is a column vector of LS estimates of three SNP genotypic effects. Although we did not expect to detect dominance effects because PTA values are estimated additive genetic effects, the test of dominance effects provided a check on whether the statistical tests produced unexpected genetic effects. The results were as expected. Only spurious dominance effects were observed and no dominance effect was among the top 100 effect for any trait.
The PTA values from different individuals had different accuracies measured by reliability [27]. The statistical analysis described above did not consider different PTA accuracies of different individuals but allowed the use of all PTA values including PTA values with zero estimates of reliability. To study the effects of different accuracies on SNP test results, we analyzed the data using a weighted least squares analysis (WLS), with the reliability as the weight of each PTA value. This approach gave more weight to PTA values with higher reliability values and ignored PTA values with zero reliability estimates. The was estimated by (X'WX)-1(X'Wz)-1, where  is a column vector of WLS estimates of three SNP genotypic effects, W is diagonal matrix with reliability estimates as the diagonal elements, X is the model matrix for PTA values as deviation from the common mean, and z is a column vector of PTA values as deviation from the common mean. The t-test under WLS replaced (X'X)-1 in the standard deviation of with (X'WX)-1.
is a column vector of WLS estimates of three SNP genotypic effects, W is diagonal matrix with reliability estimates as the diagonal elements, X is the model matrix for PTA values as deviation from the common mean, and z is a column vector of PTA values as deviation from the common mean. The t-test under WLS replaced (X'X)-1 in the standard deviation of with (X'WX)-1.
A genome-wide 5% type-I error with the Bonferroni correction was considered as the threshold P value (10-6.4) for genome-wide significance. The contribution of the top 100 SNP effects for each trait was measured by the coefficient of determination (R2) and calculated using the linear regression procedure (PROC REG) of SAS [33]. Gene and SNP locations were identified based on the University of Maryland bovine genome assembly (UMD 3.0) [5, 34]. Location of SNPs based on the Baylor College of Medicine bovine genome assembly Build 4.0 (Btau_4.0) from NCBI [24] and ENSEMBL [35] are noted in the results. Figures of gene clusters were from ENSEMBL based on Btau_4.0 because such figures based on the UMD assembly were not available.
Abbreviations
- GWAS :
-
genome-wide association study
- SNP :
-
single nucleotide polymorphism
- bp :
-
base pair
- kb :
-
kilo base pairs = 1000 base pairs
- Mb :
-
mega bases pairs = 1000 kb = 1 million base pairs
- MAF :
-
minor allele frequency
- PTA :
-
predicted transmitting ability
- QTL :
-
quantitative trait locus
- LS :
-
least squares
- WLS :
-
weighted least squares
- R 2 :
-
coefficient of determination
- Chr :
-
chromosome
- MY :
-
milk yield
- FY :
-
fat yield
- PY :
-
protein yield
- FPC :
-
fat percentage
- PPC :
-
protein percentage
- PL :
-
productive life
- SCS :
-
somatic cell score
- DPR :
-
daughter pregnancy rate
- SCE :
-
service-sire calving ease
- DCE :
-
daughter calving ease
- SSB :
-
service-sire stillbirth
- DSB :
-
daughter stillbirth
- NM :
-
net merit
- STA :
-
stature
- STR :
-
strength
- BD :
-
body depth, RW: rump width
- DF :
-
dairy form
- RA :
-
rump angle
- FUA :
-
fore udder attachment
- RUH :
-
rear udder height
- UD :
-
udder depth
- UC :
-
udder cleft
- FTP :
-
front teat placement
- RTP :
-
rear teat placement
- TL :
-
teat length
- FA :
-
foot angle
- RLS :
-
rear legs (side view)
- RLR :
-
rear legs (rear view)
- FL :
-
feet/legs score
- FS :
-
final score.
References
Van Tassell CP, Smith TPL, Matukumalli LK, Taylor JF, Schnabel RD, Lawley CT, Haudenschild CD, Moore SS, Warren WC, Sonstegard TS: SNP discovery and allele frequency estimation by deep sequencing of reduced representation libraries. Nat Methods. 2008, 5: 247-252. 10.1038/nmeth.1185.
Matukumalli LK, Lawley CT, Schnabel RD, Taylor JF, Allan MF, Heaton MP, O'Connell J, Moore SS, Smith TPL, Sonstegard TS, Van Tassell CP: Development and characterization of a high density SNP genotyping assay for cattle. PLoS ONE. 2009, 4: e5350-10.1371/journal.pone.0005350.
Wiggans GR, Sonstegard TS, VanRaden PM, Matukumalli MK, Schnabel RD, Taylor JF, Schenkel FS, Van Tassell CP: Selection of single-nucleotide polymorphisms and quality of genotypes used in genomic evaluation of dairy cattle in the United States and Canada. J Dairy Sci. 2009, 92: 3431-3436. 10.3168/jds.2008-1758.
Bovine HapMap Consortium: Genome-wide survey of SNP variation uncovers the genetic structure of cattle breeds. Science. 2009, 324: 528-532.
Zimin AV, Delcher AL, Florea L, Kelley DR, Schatz MC, Puiu D, Hanrahan F, Pertea G, Van Tassell CP, Sonstegard TS, Marçais G, Roberts M, Subramanian P, Yorke JA, Salzberg SL: A whole-genome assembly of the domestic cow, Bos taurus. Genome Biol. 2009, 10 (4): R42-10.1186/gb-2009-10-4-r42.
Meuwissen THE, Hayes BJ, Goddard ME: Prediction of total genetic value using genome-wide dense marker maps. Genetics. 2001, 157: 1819-1829.
VanRaden PM: Efficient methods to compute genomic predictions. J Dairy Sci. 2008, 91: 4414-4423. 10.3168/jds.2007-0980.
Goddard ME, Hayes BJ, Meuwissen THE: Genomic Selection in Farm Animal Species - Lessons Learnt and Future Perspectives. Proc 9th World Cong Genet Appl Livest Prod Leipzig, Germany. 2010, [http://www.kongressband.de/wcgalp2010/assets/pdf/0701.pdf]
Cole JB, VanRaden PM, O'Connell JR, Van Tassell CP, Sonstegard TS, Schnabel RD, Taylor JF, Wiggans GR: Distribution and location of genetic effects for dairy traits. J Dairy Sci. 2009, 92: 2931-2946. 10.3168/jds.2008-1762.
Mai MD, Sahana G, Christiansen FB, Guldbrandtsen B: A genome wide association study for milk production traits in Danish Jersey cattle. J Anim Sci. 2010, 88 (11): 3522-3528. 10.2527/jas.2009-2713. , [http://www.animalgenome.org/cgi-bin/QTLdb/BT/index]
Sahana G, Guldbrandtsen B, Bendixen C, Lund MS: Genome-wide association mapping for female fertility traits in Danish and Swedish Holstein cattle. Anim Genet. 2010, 41: 579-588. 10.1111/j.1365-2052.2010.02064.x.
Kolbehdari D, Wang Z, Grant JR, Murdoch B, Prasad A, Xiu Z, Marques E, Stothard P, Moore SS: A whole-genome scan to map quantitative trait loci for conformation and functional traits in Canadian Holstein bulls. J Dairy Sci. 2008, 91: 2844-2856. 10.3168/jds.2007-0585.
Cattle Quantitative Trait Locus (QTL) database. Release 14, April 20, 2011, [http://www.animalgenome.org/cgi-bin/QTLdb/BT/index]
Zhao JH: gap: Genetic analysis package. J Stat Softw . 2007, 23 (i08): -[http://www.jstatsoft.org/v23/i08/paper]
Wang S, Dvorkin D, Da Y: SNPEVG: A graphical tool for SNP effect viewing and graphing. Version 2.1, June 14, 2011, [http://animalgene.umn.edu/snpevg/index.html]
Holstein Association USA: Linear Descriptive Traits. 2009, [http://www.holsteinusa.com/pdf/print_material/linear_traits.pdf]
World Holstein Friesian Federation: International type evaluation of dairy cattle. VP/05.0393/AH/GvO 14, June 2005, [http://www.whff.info/info/typetraits/type_en_2005-2.pdf]
Arenas M, Fairbanks LD, Vijayakumar K, Carr L, Escuredo E, Marinaki AM: An unusual genetic variant in the MOCS1 gene leads to complete missplicing of an alternatively spliced exon in a patient with molybdenum cofactor deficiency. J Inherit Metab Dis. 2009, 4: 560-569.
Leimkühler S, Charcosset M, Latour P, Dorche C, Kleppe S, Scaglia F, Szymczak I, Schupp P, Hahnewald R, Reiss J: Ten novel mutations in the molybdenum cofactor genes MOCS1 and MOCS2 and in vitro characterization of a MOCS2 mutation that abolishes the binding ability of molybdopterin synthase. Hum Genet. 2005, 117 (6): 565-570. 10.1007/s00439-005-1341-9.
Grisart B, Farnir F, Karim L, Cambisano N, Kim J-J, Kvasz A, Mni M, Simon P, Frère J-M, Coppieters W, Georges M: Genetic and functional confirmation of the causality of the DGAT1 K232A quantitative trait nucleotide in affecting milk yield and composition. Proc Natl Acad Sci USA. 2004, 101: 2398-2403. 10.1073/pnas.0308518100.
Dostal DE, Baker KM: The cardiac renin-angiotensin system: conceptual, or a regulator of cardiac function?. Circ Res. 1999, 85: 643-50.
Hendrickx J, Lee P, Keating JP, Carton D, Sardharwalla IB, Tuchman M, Baussan C, Willems PJ: Complete genomic structure and mutational spectrum of PHKA2 in patients with X-Linked liver glycogenosis Type I and II. Am J Hum Genet. 1999, 64: 1541-1549. 10.1086/302399.
Cohen P: The structure and regulation of protein phosphatases. Annu Rev Biochem . 1989, 58: 453-508. 10.1146/annurev.bi.58.070189.002321.
Shenolikar S: Protein serine/threonine phosphatases -- new avenues for cell regulation. Annu Rev Cell Biol. 1994, 10: 55-86. 10.1146/annurev.cb.10.110194.000415.
National Center for Biotechnology Information (NCBI). [http://www.ncbi.nlm.nih.gov]
Brandt U: Energy converting NADH:quinone oxidoreductase (complex I). Annual Review of Biochemistry. 2006, 75: 69-92. 10.1146/annurev.biochem.75.103004.142539.
VanRaden PM, Wiggans GR: Derivation, calculation, and use of national animal model information. J Dairy Sci. 1991, 74: 2737-2746. 10.3168/jds.S0022-0302(91)78453-1.
Sonstegard TS, Ma L, Van Tassell CP, Kim E-S, Cole JB, Wiggans GR, Crooker BA, Mariani BD, Matukumalli LK, Garbe JR, Fahrenkrug SC, Liu G, Da Y: Forty years of artificial selection in U.S. Holstein cattle had genome-wide signatures. Poster presentation at 9th World Congr Genet Appl Livest Prod, Leipzig, Germany. 2010, [http://www.aipl.arsusda.gov/publish/presentations/WC9_10/WC9_10_yang_da.pdf]
Ma L, Runesha HB, Dvorkin D, Garbe JR, Da Y: Parallel and serial computing tools for testing single-locus and epistatic SNP effects of quantitative traits in genome-wide association studies. BMC Bioinformatics. 2008, 9: 315-10.1186/1471-2105-9-315.
Ma L, Yang J, Runesha HB, Tanaka T, Ferrucci L, Bandinelli S, Da Y: Genome-wide association analysis of total cholesterol and high-density lipoprotein cholesterol levels using the Framingham Heart Study data. BMC Med Genet. 2010, 11: 55-
Mao Y, London NR, Ma L, Dvorkin D, Da Y: Detection of SNP epistasis effects of quantitative traits using an extended Kempthorne model. Physiol Genomics. 2006, 28: 46-52. 10.1152/physiolgenomics.00096.2006.
R Development Core Team: R: A language and environment for statistical computing. R Foundation for Statistical Computing. Vienna, Austria. 2008, ISBN 3-900051-07-0, [http://www.R-project.org]
SAS 9.1.3. 2006, SAS Institute, Cary, NC
Zimin AV, Puiu D, Marcais G, Delcher A, Yorke JA, Salzberg SL: The latest high-quality bovine genome assembly, UMD Bos taurus 3.0 [abstract]. Plant Anim Genome XVIII Conf Abstr [Online]. 2010, International Plant & Animal Genome Conference. http://www.intl-pag.org/18/abstracts/W17_PAGXVIII_135.html (Last accessed 9/24/2010)
ENSEMBL Genome Browser. Release 63, June 2011, [http://www.ensembl.org/index.html]
Acknowledgements
This project was supported by National Research Initiative Competitive Grant no. 2008-35205-18846 from the USDA National Institute of Food and Agriculture and by a financial contribution from Holstein Association USA. Data analysis was supported in part by National Research Initiative Competitive Grant no. 2008-35205-18846, Agriculture and Food Research Initiative Competitive Grant no. 2011-67015-30333 from the USDA National Institute of Food and Agriculture, project MN-16-043 of the Agricultural Experiment Station at the University of Minnesota, and the Minnesota Supercomputer Institute. The assistance of M. Cowan (Genetic Visions), R. Wilson (Genex Cooperative), C. Dechow (Pennsylvania State University), D. Spurlock (Iowa State University), A. de Vries (University of Florida), and B. Cassell (Virginia Polytechnic Institute and State University) in obtaining DNA samples is appreciated as is manuscript review by S. Hubbard (Agricultural Research Service, USDA). The authors wish to thank two anonymous reviewers and the Associate Editor for constructive comments and suggestions.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
YD, TSS, GRW, JBC, CPVT and BAC organized and implemented this study. JBC, GRW and YD led the manuscript preparation. TSS directed the genotyping work. YD, LM, JY and SW led the data analysis. TJL provided the Holstein body conformation data and input in interpreting the results. LKM and TSS provided support for UMD 3.0 bovine genome assembly. All authors read and approved this manuscript.
Electronic supplementary material
12864_2010_3570_MOESM1_ESM.PDF
Additional file 1:Figure S1. Global view of P-values of 45,878 SNP effects per trait for 31 production, health, reproduction and body conformation traits by Mahattan plot. MY, milk yield; FY, fat yield; PY, protein yield; FPC, fat percentage; PPC, protein percentage; SCS, somatic cell score; DPR, daughter pregnancy rate; PL, productive life; SCE, sire calving ease; DCE, daughter calving ease; SSB, sire stillbirth; DSB, daughter stillbirth; NM, net merit; STA, stature; STR, strength; BD, body depth; RW, rump width; DF, dairy form; RA, rump angle; FUA, fore udder attachment; RUH, rear udder height; UD, udder depth; UC, udder cleft; FTP, front teat placement; RTP, rear teat placement; TL, teat length; FA, foot angle; RLS, rear legs (side view); RLR, rear legs (rear view); FL, feet and legs; FS, final score. (PDF 480 KB)
12864_2010_3570_MOESM2_ESM.XLSX
Additional file 2:Table S1. Output file of all 1,586 SNP markers with 1,300 effects on 31 dairy traits by chromosome (Chr). Chr30 is the X chromosome, and Chr32 indicates markers with unknown chromosome locations. MY, milk yield; FY, fat yield; PY, protein yield; FPC, fat percentage; PPC, protein percentage; SCS, somatic cell score; DPR, daughter pregnancy rate; PL, productive life; SCE, sire calving ease; DCE, daughter calving ease; SSB, sire stillbirth; DSB, daughter stillbirth; NM, net merit; STA, stature; STR, strength; BD, body depth; RW, rump width; DF, dairy form; RA, rump angle; FUA, fore udder attachment; RUH, rear udder height; UD, udder depth; UC, udder cleft; FTP, front teat placement; RTP, rear teat placement; TL, teat length; FA, foot angle; RLS, rear legs (side view); RLR, rear legs (rear view); FL, feet and legs; FS, final score. Columns B and C show UMD 3.0 chromosome and positions, columns D and E show Btau_4.0 chromosome and position, respectively; column G is A1F, A1 allele frequency; column H is MAF, minor allele frequency; columns J to AN are P values (top 100 effects in red); columns AO to CY are t values (those corresponding to top 100 effects for P values in blue); column CZ is N, number of traits for which SNP was among top 100. (XLSX 1 MB)
12864_2010_3570_MOESM3_ESM.PDF
Additional file 3:Figure S2. Map of SNP position (Mb) by P-value and Bos taurus chromosome for 725 SNPs that comprise the top 100 effects for each of 13 production, health, and reproduction traits of contemporary U.S. Holsteins. (PDF 193 KB)
12864_2010_3570_MOESM4_ESM.PDF
Additional file 4:Figure S3. Map of SNP position (Mb) by P-value and Bos taurus chromosome for 1,005 SNPs that comprise the top 100 effects for each of 18 conformation traits of contemporary U.S. Holsteins. (PDF 196 KB)
12864_2010_3570_MOESM6_ESM.PDF
Additional file 6:Figure S4. Gene clusters that overlapped localized concentrations of SNP effects or contained significant SNP effects on Bos taurus (BTA) chromosomes. A) BTA7 15.4 Mb gene cluster (Btau_4.0:4807980-20004663) of ~1,166 genes (not counting pseudo and RNA genes) with a mean of 75 genes/Mb. Left end was LSM4-JUND region; right end was LSM7-SPPL2B-OAZ1 region. This cluster included ARS-BFGL-NGS-4774 [1.5 kb downstream from INSR (Q95M43; highlighted in green)], which was significant for somatic cell score (1st) and daughter pregnancy rate (1st), productive life (3rd), and net merit (78th)] as well as all 10 significant SNP effects on this chromosome for productive life, 15 of the 16 significant SNP effects for daughter pregnancy rate, and 6 of the 12 significant SNP effects for somatic cell score. B) BTA14 2.81 Mb gene cluster (Btau_4.0:50872-2,859,132) of ~125 genes with a mean of 44 genes/Mb; NIBP (highlighted in green) was the largest gene (387.23 kb) in the cluster. This cluster included 19 significant SNP effects for fat percentage [SNP in DGAT1 was 1st and SNP in NIBP was 2nd], one SNP in VPS28 with effect for milk yield, and one SNP in NIBP with effects for fat yield and protein percentage. C) BTA18 15.82 Mb gene cluster (Btau_4.0:48755332-64574451) of ~1,322 genes with a mean of 83 genes/Mb. The PGLYRP1(PGRP)-IGFL1 (highlighted in green) region had the most significant SNP effects in the cluster [fat and protein yields, service-sire and daughter calving ease, and net merit (1st); service-sire stillbirth (8th); milk yield (9th); productive life (16th); and fat and protein percentages (25th)]. D) BTA3 gene cluster with significant SNPs for fat yield [BFGL-NGS-113990 (49th) and INRA-304 (53rd)]. E) BTA5 gene cluster with ARS-BFGLNGS-36745 (associated with SREBF2) and ARS-BFGL-NGS-71946 in LOC535121, which were among top 100 SNP effects for fat and protein percentages and service-sire calving ease and stillbirth, as well as other significant SNPS for service-sire stillbirth [ARS-BFGL-NGS-2337 in TCF20 (39th) and ARS-BFGL-NGS-7380 in SEPT3 (76th). F) BTA7 gene cluster with ARS-BFGL-NGS-76638 in RGS14, which was significant for somatic cell score (77th). G) BTA10 gene cluster with Hapmap41316-BTA-62253, which was significant for milk yield (60th). H) BTA13 gene cluster with significant SNPs for milk yield [ARS-BFGL-BAC-16372 (65th) and Hapmap41228-BTA-32897 (87th)]. I) BTA17 gene cluster with significant SNPs for milk yield [(ARS-BFGL-NGS-17192 (39th) and BTB-01992588 (63rd)] and fat percentage [ARS-BFGL-NGS-34106 (19th) and Hapmap40427-BTA-41914 (78th)]. J) BTA21 gene cluster with significant SNP effects for service-sire calving ease [BFGL-NGS-113671 (87th) and service-sire stillbirth [BFGL-NGS-116152 (46th), Hapmap39755-BTA-52639 (73rd), and BFGL-NGS-113671 (83rd). K) BTA23 gene cluster with ARS-BFGL-NGS-72191 in ZNF192 and BTA-68781-no-rs, which were among top 100 SNP effects for fat yield, protein yield and percentage, service-sire calving ease, and net merit. L) BTA26 gene cluster with BFGL-NGS-111739, the 14th most significant SNP for service-sire stillbirth. M) BTA29 gene cluster with ARS-BFGL-NGS-24998 in LOC787296, 49th most significant SNP for daughter stillbirth. N) BTAX gene cluster with ARS-BFGL-NGS-42972 in TIMP1, 45th most significant SNP for service-sire stillbirth. (PDF 1 MB)
12864_2010_3570_MOESM7_ESM.PDF
Additional file 7:Figure S5. Examples of gene regions associated with significant effects of SNPs for daughter stillbirth and milk yield. A) The 211.67 kb MOCS1-LRFN2 region of Bos taurus (BTA) chromosome 23 with nine highly significant effects (red arrows) for predicted transmitting ability (PTA) for daughter stillbirth. B) The most significant SNP effect (red arrow) for daughter stillbirth was 23.9 kb upstream from the second CD82 gene on BTA15. C) The BTA13 region with the most significant SNP effect (19.7 kb downstream from GNAS2 or GNAS) for PTA for milk yield. (PDF 698 KB)
12864_2010_3570_MOESM8_ESM.PDF
Additional file 8:Figure S6. Examples of gene regions associated with significant SNP effects for body conformation traits; a boxed gene contained at least one top-100 effect, and numbers above red arrows (significant SNP effects) indicate rank of SNP effect. A) The 1 Mb region of Bos taurus (BTA) chromosome X with the most significant SNP effect (LOC515732 is PHKA2) for predicted transmitting ability (PTA) for stature. B) The 10.2 Mb region of BTA 11 with eight of the top 20 SNP effects for stature. C) The 1 Mb region of BTA16 with the most significant SNP effect (REN) for PTA for strength. D) The 1.75 Mb region of BTA19 with five genes with highly significant SNP effects for rump width and udder cleft. E) A BTA21 gene cluster with three genes associated with teat length. (PDF 1 MB)
12864_2010_3570_MOESM9_ESM.XLSX
Additional file 9:Table S3. Output file of 725 SNP markers with top 100 effects per trait from least squares analysis and weighted least squares analysis. MY, milk yield; FY, fat yield; PY, protein yield; FPC, fat percentage; PPC, protein percentage; SCS, somatic cell score; DPR, daughter pregnancy rate; PL, productive life; SCE, sire calving ease; DCE, daughter calving ease; SSB, sire stillbirth; DSB, daughter stillbirth; NM, net merit. Subscript 'w' indicates P-values from weighted least squares analysis. Trait name with subscript 'w' indicates tests using weighted least squares, and trait name without subscript indicates tests using least squares. (XLSX 218 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Cole, J.B., Wiggans, G.R., Ma, L. et al. Genome-wide association analysis of thirty one production, health, reproduction and body conformation traits in contemporary U.S. Holstein cows. BMC Genomics 12, 408 (2011). https://doi.org/10.1186/1471-2164-12-408
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-12-408