
Abstract
Background
Single nucleotide polymorphism (SNP) arrays have facilitated discovery of genetic markers associated with complex traits in domestic cattle; thereby enabling modern breeding and selection programs. Genome-wide association analyses (GWAA) for growth traits were conducted on 10,837 geographically diverse U.S. Gelbvieh cattle using a union set of 856,527 imputed SNPs. Birth weight (BW), weaning weight (WW), and yearling weight (YW) were analyzed using GEMMA and EMMAX (via imputed genotypes). Genotype-by-environment (GxE) interactions were also investigated.
Results
GEMMA and EMMAX produced moderate marker-based heritability estimates that were similar for BW (0.36–0.37, SE = 0.02–0.06), WW (0.27–0.29, SE = 0.01), and YW (0.39–0.41, SE = 0.01–0.02). GWAA using 856K imputed SNPs (GEMMA; EMMAX) revealed common positional candidate genes underlying pleiotropic QTL for Gelbvieh growth traits on BTA6, BTA7, BTA14, and BTA20. The estimated proportion of phenotypic variance explained (PVE) by the lead SNP defining these QTL (EMMAX) was larger and most similar for BW and YW, and smaller for WW. Collectively, GWAAs (GEMMA; EMMAX) produced a highly concordant set of BW, WW, and YW QTL that met a nominal significance level (P ≤ 1e-05), with prioritization of common positional candidate genes; including genes previously associated with stature, feed efficiency, and growth traits (i.e., PLAG1, NCAPG, LCORL, ARRDC3, STC2). Genotype-by-environment QTL were not consistent among traits at the nominal significance threshold (P ≤ 1e-05); although some shared QTL were apparent at less stringent significance thresholds (i.e., P ≤ 2e-05).
Conclusions
Pleiotropic QTL for growth traits were detected on BTA6, BTA7, BTA14, and BTA20 for U.S. Gelbvieh beef cattle. Seven QTL detected for Gelbvieh growth traits were also recently detected for feed efficiency and growth traits in U.S. Angus, SimAngus, and Hereford cattle. Marker-based heritability estimates and the detection of pleiotropic QTL segregating in multiple breeds support the implementation of multiple-breed genomic selection.
Similar content being viewed by others
Background
Growth traits are commonly recorded and used as selection criteria within modern beef cattle breeding programs and production systems; primarily because of their correlation with increased overall meat production and other economically important traits [1,2,3,4]. Some of the most commonly investigated growth traits include birth weight (BW), weaning weight (WW) and yearling weight (YW); with BW considered as both a production indicator, and a primary selection criterion for improving calving ease by reducing dystocia events [1, 2, 5,6,7]. Moreover, while previous studies have demonstrated that low estimated breeding values (EBVs) for BW are associated with reductions in both calf viability [6] and growth rates [5, 7], increased dystocia rates may also occur if sires with high EBVs for BW are used in conjunction with dams that possess small pelvic size. Therefore, modern beef breeding programs and production systems generally strive to increase calving ease, and maximize other growth-related traits such as WW and YW, particularly considering the known correlations between growth traits and other economically important carcass and reproductive traits [3, 5, 7].
Given the increasing economic importance of growth traits in beef cattle, a number of studies have sought to identify quantitative trait loci (QTL) influencing bovine body weight, growth, and aspects of stature, including both linkage studies and modern genome-wide association analyses [2, 8,9,10,11,12,13]. Several recent studies have also established moderate heritability estimates for bovine growth traits in U.S. beef cattle including BW, WW, and YW [14,15,16,17], with a number of relevant QTL and positional candidate genes identified to date, including orthologous genes that affect both human and bovine height [2, 18,19,20,21,22]. Notably, with the advent of the bovine genome assembly [23], the development of the Illumina Bovine SNP50 and 778K HD assays [23, 24], and more recently, the demonstrated ability to impute high density genotypes with high accuracy [25], an industry-supported research framework [26] has emerged that allows for very large-sample studies to be conducted without the costs associated with directly ascertaining high density genotypes (≥ 778K) for all study animals.
Herein, we used 10,837 geographically diverse U.S. Gelbvieh beef cattle and a union set of 856,527 (856K) imputed array variants to conduct GWAA with marker-based heritability estimates for BW, WW, and YW. Additionally, we used thirty-year climate data and K-means clustering to assign all Gelbvieh beef cattle to discrete U.S. climate zones for the purpose of estimating genotype-by-environment (GxE) interactions for BW, WW, and YW. This study represents the largest, high-density, single breed report to date with both standard GWAA and GxE GWAA for BW, WW, and YW. Additionally, we also evaluate the general concordance of GWAAs conducted using two popular methods (GEMMA; EMMAX) [27,28,29]. The results of this study are expected to positively augment current beef cattle breeding programs and production systems, particularly for U.S. Gelbvieh cattle, but also serve to highlight the increasing potential for eliciting economic impacts from industry-supported research frameworks that were developed for enhancing U.S. food security.
Results and discussion
Heritability estimates for BW, WW, and YW in U.S. Gelbvieh beef cattle
Herein, we used two approaches to generate marker-based heritability estimates for all investigated traits. Specifically, standardized relatedness matrices produced with GEMMA (Gs) [27] and genomic relationship matrices (GRM) normalized via Gower’s centering approach and implemented in EMMAX [25, 28,29,30], were used to compare the chip or pseudo-heritability estimates for each investigated trait (Table 1). Notably, both approaches produced moderate heritability estimates with small standard errors for BW, WW, and YW; and heritability estimates for YW were highest among all investigated traits for U.S. Gelbvieh beef cattle. Moderate heritability estimates produced here using both approaches further support the expectation of positive economic gains resulting from the implementation of genomic selection [30].
GWAA for BW, WW, and YW in U.S. Gelbvieh beef cattle
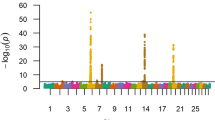
The results of our 856K single-marker analyses for BW (GEMMA; EMMAX) [27,28,29] are shown in Fig. 1 and in Figure S1 (Additional File 1), with detailed summary data for QTL detected by GEMMA and EMMAX described in Table 2 and Table S1, respectively. A comparison of GEMMA and EMMAX results revealed a concordant set of QTL defined by lead SNPs (i.e., the most strongly associated SNP within a QTL region) which met a nominal significance threshold (P ≤ 1e-05) [31] (Table 2, Table S1, Additional File 1, Additional File 2). Specifically, QTL signals for BW were detected on BTA6, BTA7, BTA14, and BTA20 across both analyses (Table 2, Table S1, Additional File 1), and included an array of positional candidate genes generally involved in diverse aspects of mammalian growth and development (i.e., CCSER1, ST18, RP1/XKR4, SLIT2, STC2, IBSP) as well as bovine growth (i.e., NCAPG, LCORL, KCNIP4, ARRDC3), stature (i.e., PLAG1), and production traits (i.e., IMPAD1/FAM110B, HERC6/PPM1K) [2, 13, 14, 18, 21, 22, 30, 32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60]. Interestingly, the lead SNP defining the BW QTL detected on BTA14 (14_25 Mb) was located in PLAG1, thereby further supporting the involvement of this gene in various aspects of bovine growth and stature across breeds [2, 14, 18, 21, 30, 32,33,34]. Additionally, all but one (i.e., NCAPG, exon 9) of the lead SNPs for the detected Gelbvieh BW QTL (GEMMA, EMMAX) were noncoding variants (Table 2, Table S1, Additional File 1). Genomic inflation factors and correlation coefficients for P-values obtained from all BW analyses are shown in Tables S2-S3 (Additional File 1).

Birth weight (BW) QTL. Manhattan plot with GEMMA -log10 P-values. Lead and supporting SNPs for QTL represented at or above the blue line (P ≤ 1e-05; −log10 P-values ≥ 5.00) for n = 10,837 U.S. Gelbvieh beef cattle. A summary of all markers passing the nominal significance threshold [31] is presented in Table 2
Single-marker analyses (856K) for WW in U.S. Gelbvieh beef cattle (GEMMA; EMMAX) revealed several of the same QTL detected for BW (Table 3, Fig. 2, Table S4, Figure S2, Additional File 1), thus providing statistical support for pleiotropic QTL located on BTA6 (i.e., NCAPG, CCSER1, KCNIP4, HERC6/PPM1K, LOC782905/SLIT2, LOC100336621/LOC104972717) as well as BTA14 (i.e., PLAG1, XKR4, IMPAD1/FAM110B). The lead SNPs for Gelbvieh BW and WW QTL detected on BTA20 (20_05 Mb) suggested proximal but independent causal mutations, thus implicating the potential involvement of at least three positional candidate genes (LOC104975192/STC2, ERGIC1). A detailed summary of lead and supporting SNPs for pleiotropic QTL is provided in Additional File 2. Beyond evidence for pleiotropy, four additional Gelbvieh WW QTL were also detected on BTA5 (5_60 Mb), BTA6 (6_31 Mb, 6_37 Mb) and BTA28 (28_37 Mb; Table 3, Fig. 2, Table S4, Figure S2, Additional File 1). Among the additional QTL detected, several positional candidate genes have been implicated in aspects of development (UNC5C, SNCA/GPRIN3) and immune function (SH2D4B) [61,62,63,64,65,66,67]. An investigation of all lead SNPs for the detected Gelbvieh WW QTL revealed 13 noncoding variants and one nonsynonymous variant (Table 3, Table S4, Additional File 1). Genomic inflation factors and correlation coefficients for P-values obtained from all WW analyses are presented in Tables S2 and S3 (Additional File 1).

Weaning weight (WW) QTL. Manhattan plot with GEMMA -log10 P-values. Lead and supporting SNPs for QTL represented at or above the blue line (P ≤ 1e-05; −log10 P-values ≥ 5.00) for n = 10,837 U.S. Gelbvieh beef cattle. A summary of all markers passing the nominal significance threshold [31] is presented in Table 3
Consistent with our analyses of BW and WW, our single-marker analyses (856K) for YW in U.S. Gelbvieh beef cattle again revealed evidence for pleiotropic QTL located on BTA6 and BTA14 (Table 4, Fig. 3, Table S5, Figure S3, Additional File 1). Specifically, the results obtained from our analyses of BW, WW, and YW revealed some common QTL signals for all investigated traits on BTA6 (6_36 Mb, 6_38 Mb, 6_39 Mb, 6_41 Mb, 6_42 Mb) and BTA14 (14_24 Mb, 14_25 Mb, 14_26 Mb). Likewise, the lead SNPs defining these QTL also resulted in the prioritization of the same positional candidate genes on BTA6 (i.e., LCORL, KCNIP4, HERC6/PPM1K, SLIT2, CCSER1) and BTA14 (i.e., PLAG1, IMPAD1/FAM110B, RP1/XKR4). Together with pleiotropic signals on BTA6 and BTA14, eight additional YW QTL were also detected; including one QTL (7_93 Mb) that was also found to influence Gelbvieh BW (Table 4, Table S5, Additional File 1). Positional candidate genes for these QTL have been implicated in diverse aspects of growth and development as well as bovine production traits (i.e., SNCA/GPRIN3, SLIT2, NSMAF, LOC101905238/ARRDC3), bovine milk traits (i.e., PPARGC1A), and chromatin modification (i.e., IWS1) [68,69,70,71]. Relevant to YW, it should also be noted that several of the pleiotropic QTL detected for U.S. Gelbvieh in this study have also been detected for mid-test metabolic weight in U.S. SimAngus beef cattle (6_39 Mb, 14_24 Mb, 14_25 Mb, 14_26 Mb) [30]. Moreover, Gelbvieh QTL (BW, YW) detected on BTA14 and BTA7 have also been detected for Angus residual feed intake (14_27 Mb), and Hereford average daily gain (7_93 Mb) [30]. An investigation of all lead SNPs for the detected Gelbvieh YW QTL revealed 16 noncoding variants (Table 4, Table S5, Additional File 1). Genomic inflation factors and correlation coefficients for P-values obtained from all YW analyses are shown in Tables S2-S3 (Additional File 1).

Yearling weight (YW) QTL. Manhattan plot with GEMMA -log10 P-values. Lead and supporting SNPs for QTL represented at or above the blue line (P ≤ 1e-05; −log10 P-values ≥ 5.00) for n = 10,837 U.S. Gelbvieh beef cattle. A summary of all markers passing the nominal significance threshold [31] is presented in Table 4
GxE GWAA for BW, WW, and YW in U.S. Gelbvieh beef cattle
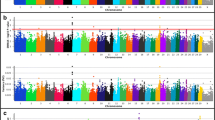
To investigate the potential for significant GxE interactions in relation to BW, WW, and YW in U.S. Gelbvieh beef cattle, we conducted six additional single-marker (856K) analyses using both GEMMA and EMMAX [27,28,29]. For all analyses, we included a variable for Gelbvieh geographic zone, which was generated via K-means clustering using thirty-year U.S. climate data, and treated as an interaction term (See Methods). Notably, a BW GxE QTL detected on BTA2 (2_32 Mb; lead SNP is intergenic) revealed multiple biologically relevant positional candidate genes, including GRB14, which has been shown to regulate insulin in mice [72], and FIGN, which has been associated with plasma folate levels in humans (Fig. 4, Table 5, Additional File 2) [73]. Importantly, maternal folate levels have been shown to influence human birthweight [74], and a role for insulin regulation in bovine feed efficiency and growth traits has also been described [30]. Beyond BTA2, BW GxE QTL were also detected on BTA17 (17_66 Mb) and BTA13 (13_67 Mb). Positional candidate genes for these QTL have been implicated in the removal of uracil residues from DNA and apoptosis (UNG) as well as human obesity (CTNNBL1) (Fig. 4, Table 5, Figure S4, Table S6, Additional File 1) [75, 76]. Examination of the lead SNPs for all GxE QTL detected for Gelbvieh BW (Table 5, Table S6, Additional File 1, Additional File 2) revealed three noncoding variants, which is suggestive of quantitative (i.e., regulatory) effects. Genomic inflation factors and correlation coefficients for P-values obtained from all GxE BW analyses are shown in Tables S2-S3 (Additional File 1).

Birth weight genotype-by-environment (BW GxE) QTL. Manhattan plot with GEMMA -log10 P-values. Lead and supporting SNPs for QTL represented at or above the blue line (P ≤ 1e-05; −log10 P-values ≥ 5.00) for n = 10,837 U.S. Gelbvieh beef cattle. A summary of all markers passing the nominal significance threshold [31] is presented in Table 5
Our analyses (GEMMA, EMMAX) to evaluate the potential for significant GxE interactions with respect to WW in U.S. Gelbvieh beef cattle produced evidence for one GxE QTL on BTA2 (2_18 Mb) which was only detected by GEMMA, and included relatively few supporting SNPs (P ≤ 1e-05, Table 6; Fig. 5, Figure S5, Additional File 1). The lead SNP defining this QTL was located in exon 304 of TTN, and encoded a nonsynonymous variant (Table 6, Fig. 5, Additional File 2). Interestingly, TTN is known to function as a myofilament system for skeletal and cardiac muscle, with mouse M-line deficient knockouts resulting in sarcomere disassembly as well as muscle atrophy and death [77,78,79].

Weaning weight genotype-by-environment (WW GxE) QTL. Manhattan plot with GEMMA -log10 P-values. Lead and supporting SNPs for QTL represented at or above the blue line (P ≤ 1e-05; −log10 P-values ≥ 5.00) for n = 10,837 U.S. Gelbvieh beef cattle. A summary of all markers passing the nominal significance threshold [31] is presented in Table 6
Analyses (GEMMA; EMMAX) to evaluate the potential for significant GxE interactions with respect to YW in U.S. Gelbvieh beef cattle revealed two GxE QTL with three positional candidate genes (LRAT/LOC101904475/FGG) on BTA17 (17_03 Mb), and one positional candidate gene on BTA5 (PHF21B at 116 Mb; P ≤ 1e-05, Table 7, Fig. 6, Table S7, Figure S6, Additional File 1, Additional File 2). The signal on BTA17 (i.e., GEMMA lead SNP in Intron 4 of LOC101904475 and supporting SNPs) was replicated by EMMAX (Figure S6, Additional File 1); but at a less stringent significance threshold (i.e. P < 6e-04). Notably, while the function of LOC101904475 remains unclear, LRAT is known to catalyze esterification of retinol (i.e., from Vitamin A) [80], and Vitamin A has been shown to promote growth in beef cattle as well as humans [81,82,83]. However, FGG is also an intriguing candidate, as fibrinogen has been shown to constrict blood vessels [84]. This vasoconstriction may alter the ability to cope with heat stress, but in the context of cattle production, the relationship between vasoconstriction and fescue toxicosis is perhaps more noteworthy. Fescue toxicosis is the result of ergot alkaloids produced by the endophytic fungus in fescue forage [85], especially the Kentucky 31 variety. One of the major symptoms of fescue toxicosis is vasoconstriction, thus variation in FGG expression levels may potentially alter cattle’s innate degree of vasoconstriction; perhaps further complicating both fescue toxicosis and heat stress. The other interesting positional candidate gene on BTA5 (PHF21B) is known to be involved in the modulation of stress responses, and the regulation of cellular division [86, 87].

Yearling weight genotype-by-environment (YW GxE) QTL. Manhattan plot with GEMMA -log10 P-values. Lead and supporting SNPs for QTL represented at or above the blue line (P ≤ 1e-05; −log10 P-values ≥ 5.00) for n = 10,837 U.S. Gelbvieh beef cattle. A summary of all markers passing the nominal significance threshold [31] is presented in Table 7
Conclusions
Herein, we present evidence for pleiotropic QTL influencing BW, WW, and YW in U.S. Gelbvieh beef cattle, and further confirm the involvement of PLAG1 in various aspects of bovine growth and stature across breeds [2, 14, 18, 21, 30, 32,33,34]. Additionally, we also present compelling evidence for QTL segregating in multiple breeds; with at least seven U.S. Gelbvieh growth QTL that were also detected for feed efficiency and growth traits in U.S. Angus, SimAngus, and Hereford beef cattle [30]. Despite the involvement of major genes such as NCAPG, PLAG1 and LCORL, more of the phenotypic variance in Gelbvieh BW, WW, and YW was explained by many other genome-wide loci (See Additional File 1, Additional File 2). Moreover, we demonstrate that most of the Gelbvieh QTL are detectable by two different large-sample analyses (GEMMA; EMMAX). However, some discordant QTL detected by the GxE GWAAs can also be attributed to differences in the model specifications for these analyses, as implemented by GEMMA and EMMAX (See Methods). While relatively few GxE QTL were detected, the identified GxE QTL harbor physiologically meaningful positional candidates. Moreover, the results of this study demonstrate that imputation to a union set of high-density SNPs (i.e., 856K) for use in large-sample analyses can be expected to facilitate future discoveries at a fraction of the cost associated with direct genotyping, which also underscores the present impact of genomic tools and resources developed by the domestic cattle research community.
Methods
Cattle phenotypes were received from the American Gelbvieh Association (pre-adjusted for age of animal [i.e. 205-day weight for WW] and age of dam as per breed association practice), and corresponding genotypes were transferred from their service provider Neogen GeneSeek. For GWAA analyses, the phenotypes were pre-adjusted for sex and contemporary group consisting of 5-digit breeder zip-code, birth year, and birth season (Spring, Summer, Fall, and Winter) using the mixed.solve() function from the rrBLUP package v4.4 [88] in R v3.3.3 [89].
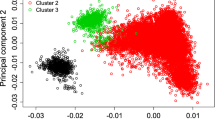
To group individuals into discrete climate zones, K-means clustering was performed on three continuous climate variables. Thirty-year normal values for temperature, precipitation, and elevation were drawn from the PRISM climate dataset [90]. Each one km square of the continental United States was assigned to one of nine climate zones using K-means clustering implemented in the RStoolbox R package [91, 92]. The optimal number of zones was identified using the pamk function from the R package fpc [93]. Individuals were assigned to zones based on the zip code of their breeder as recorded in the American Gelbvieh Association herdbook.
Quality control was performed on genotypes for 13,166 Gelbvieh individuals using PLINK 1.9 [94]. Individuals with call rates < 0.90 were removed on an assay-by-assay basis (For assay information see Additional File 3). Variants with call rates < 0.90 or Hardy-Weinberg Equilibrium (HWE) P-values <1e-20 were also removed. For this analysis, only autosomal chromosomes were analyzed. After filtering, genotypes for the 12,422 individuals that remained were merged using PLINK and then phased using EagleV2.4 [95]. Genotypes inferred by Eagle were removed with bcftools [96]. Imputation was performed with IMPUTE2 [97] using the “merge_ref_panels” flag. This allowed the phased haplotypes for 315 individuals genotyped on the Illumina HD (Illumina, San Diego, CA) and 559 individuals genotyped on the GGP-F250 (GeneSeek, Lincoln, NE) to be recursively imputed and treated as reference haplotypes. These reference haplotypes were used to impute the remaining 11,598 low-density genotypes from various assays (Additional File 3) to the shared number of markers between the two high-density research chips. The resulting dataset consisted of 12,422 individuals with 856,527 markers each (UMD3.1). To account for uncertainty in imputation, IMPUTE2 reports dosage genotypes. Hard-called genotypes were inferred from dosages using PLINK. When making hard-calls, PLINK treats genotypes with uncertainty > 0.1 as missing. This resulted in a hard-called dataset of 856,527 variants, which includes genotypes set as missing. Prior to the execution of all GWAAs (GEMMA; EMMAX), we filtered the Gelbvieh samples and all SNP loci as follows: Gelbvieh sample call rate filtering (< 90% call rate excluded); thereafter SNP filtering by call rate (> 15% missing excluded), MAF (< 0.01 excluded), polymorphism (monomorphic SNPs excluded), and HWE (excludes SNPs with HWE P < 1e-50), which resulted in 618,735 SNPs. Additionally, prior to all GWAAs (GEMMA; EMMAX) hard-called genotypes were numerically recoded as 0, 1, or 2, based on the incidence of the minor allele. Missing hard-called genotypes (i.e., that met our filtering criteria) were modeled as the SNP’s average value (0, 1, or 2) across all samples.
Using the numerically recoded hard-called genotypes and the adjusted Gelbvieh phenotypes, we employed GEMMA to conduct univariate linear mixed model GWAAs where the general mixed model can be specified as y = Wα + xβ + u + ϵ; where y represents a n-vector of quantitative traits for n-individuals, W is an n x c matrix of specified covariates (fixed effects) including a column of 1s, α is a c-vector of the corresponding coefficients including the intercept, x represents an n-vector of SNP genotypes, β represents the effect size of the SNP, u is an n-vector of random effects, and ϵ represents an n-vector of errors [27]. Moreover, it should also be noted that u ∼ MVNn(0, λτ−1Κ) and ϵ ∼ MVNn(0, λτ−1Ιn), where MVN denotes multivariate normal distribution, λτ−1 is the variance of the residual errors, λ is the ratio between the two variance components, Κ is a known n x n relatedness matrix, and Ιn represents an n x n identity matrix [27]. Using this general approach, GEMMA evaluated the alternative hypothesis for each SNP (H1 : β ≠ 0) as compared to the null (H0 : β = 0) by performing a likelihood ratio test with maximum likelihood estimates (−lmm 2) as follows:
\( {D}_{lrt}=2\mathit{\log}\frac{l_1\left(\hat{\lambda}1\right)}{l_0\left(\hat{\lambda}0\right)} \), with l1 and l0 being the likelihood functions for the null and alternative models, respectively, where \( \hat{\lambda} \) 0 and \( \hat{\lambda} \) 1 represent the maximum likelihood estimates for the null and the alternative models, respectively, and where P-values come from a \( {\mathcal{X}}^2 \) distribution, as previously described [27]. Herein, the only fixed-effect covariate specified for all GWAAs was an environmental variable (geographic zone for each individual). For all GxE GWAAs (−gxe command), the environmental variable (geographic zone for each individual) was treated as an interaction term, where the resulting P-values represent the significance of the genotype x environment interaction. Specifically, for the GxE GWAAs in GEMMA, the model is specified as y = Wα + xsnpβsnp + xenvβenv + xsnp × xenvβsnp × env + u + ϵ; where y represents a n-vector of quantitative traits for n-individuals, W is an n x c matrix of specified covariates (fixed effects) including a column of 1s, α is a c-vector of the corresponding coefficients including the intercept, xsnp represents an n-vector of SNP genotypes, βsnp represents the effect size of the SNP, xenv represents an n-vector of environmental covariates, βenv represents the fixed effect of the environment, βsnp × env is the interaction between SNP genotype and environment, u is an n-vector of random effects, and ϵ represents an n-vector of errors. GEMMA evaluated the alternative hypothesis for each interaction (H1 : βsnp × env ≠ 0) as compared to the null (H0 : βsnp × env = 0). Marker-based relatedness matrices (Gs) relating instances of the random effect specified to each of the growth phenotypes among all genotyped cattle were used to estimate the proportion of variance explained (PVE) by the hard-called genotypes in GEMMA, which is also commonly referred to as the “chip heritability” [27, 98]. For all investigated traits, single-marker P-values obtained from GEMMA (−lmm 2, −gxe) were used to generate Manhattan plots in R (manhattan command) and QTL were defined by ≥ 2 SNP loci with MAF ≥ 0.01 (i.e., a lead SNP plus at least one additional supporting SNP within 1 Mb) which also met a nominal significance threshold (P ≤ 1e-05) [30, 31].
Using hard-called genotypes and the adjusted Gelbvieh phenotypes, we performed a second set of GWAAs using a mixed linear model with variance component estimates, as implemented by EMMAX [28,29,30, 99,100,101]. Briefly, the general mixed model used in this approach can be specified as: y = Xβ + Zu + ϵ, where y represents a n × 1 vector of phenotypes, X is a n × q matrix of fixed effects, β is a q × 1 vector representing the coefficients of fixed effects, and Z is a n × t matrix relating the random effect to the phenotypes of interest [30, 99,100,101]. Herein, we must assume that \( Var(u)={\sigma}_g^2K \) and \( Var\left(\epsilon \right)={\sigma}_e^2I \), such that \( Var(y)={\sigma}_g^2 ZK{Z}^{\prime }+{\sigma}_e^2I \), however, in this study Z represents the identity matrix I, and K represents a kinship matrix of all Gelbvieh samples with hard-called genotypes. Moreover, to solve the mixed model equations using a generalized least squares approach, we must estimate the variance components (\( {\sigma}_g^2 \) and \( {\sigma}_e^2 \)) as previously described [28,29,30, 99, 100]. For this study, we estimated the variance components using the REML-based EMMA approach [29], with stratification accounted for and controlled using the genomic relationship matrix [25, 30], as computed from the Gelbvieh hard-called genotypes. Moreover, the only fixed-effect covariate specified for all GWAAs was an environmental variable (geographic zone for each individual). For all EMMAX GxE GWAAs utilizing hard-called genotypes, we used an implementation of EMMAX [29, 102] where interaction-term covariates may be specified; with the environmental variable (geographic zone for each individual) specified as the interaction term. The basis of this approach is rooted in full versus reduced model regression [99], where interaction-term covariates are included in the model as follows: each specified interaction-term covariate serves as one reduced-model covariate; each specified interaction-term covariate is also multiplied, element by element, with each SNP predictor (i.e., SNP × geographic zone) to create an interaction term to be included in the full model. Specifically, given n measurements of a Gelbvieh growth phenotype that is influenced by m fixed effects and n instances of one random effect, with one or more GxE effects (e) whereby the interaction is potentially with one predictor variable, we model this using a full and a reduced model. The full model can be specified as y = Xcβkc + Xiβki + Xkβkp + Xipβip + ufull + ϵfull, and the reduced model as y = Xcβkrc + Xiβkri + Xkβrkp + ureduced + ϵreduced, where y is an n-vector of observed phenotypes, Xc is an n × m matrix of m fixed-effect covariates, Xi is an n × e matrix of e fixed terms being tested for GxE interactions, Xk is an n-vector containing the covariate or predictor variable that may be interacting, and Xip is an n × e matrix containing the e interaction terms created by multiplying the columns of Xi element-by-element with Xk. All of the β terms correspond to the X terms as written above, and to the full or the reduced model, as specified, with u and ϵ representing the random effect and error terms, respectively. Like the EMMAX method without interactions [28, 29], we approximate this by finding the variance components once, using the parts of the above equations that are independent of Xk as follows: y = Xcβcvc + Xiβivc + uvc + ϵvc, where vc indicates the variance components. To estimate the variance components, we must again assume that \( Var\left({u}_{vc}\right)={\sigma}_g^2K \) and \( Var\left({\epsilon}_{vc}\right)={\sigma}_e^2I \), such that \( Var(y)={\sigma}_g^2K+{\sigma}_e^2I \). The EMMA technique can then be used to estimate the variance components \( {\sigma}_g^2 \) and \( {\sigma}_e^2 \) as well as a matrix B (and its inverse) such that \( B{B}^{\prime }=H=\frac{Var(y)}{\sigma_g^2}=K+\frac{\sigma_e^2}{\sigma_g^2}I \). Thereafter, for every marker (k) we can compute (as an EMMAX-type approximation) the full and reduced models as: B−1y = B−1Xcβkc + B−1Xiβki + B−1Xkβkp + B−1Xipβip + B−1(ufull + ϵfull) for the full model, where B−1(ufull + ϵfull) is assumed to be an error term proportional to the identity matrix, and as B−1Xcβkrc + B−1Xiβkri + B−1Xkβrkp + B−1(ureduced + ϵreduced) for the reduced model, where B−1(ureduced + ϵreduced) is assumed to be an error term proportional to the identity matrix. To estimate the significance of the full versus reduced model, an F-test was performed; with all analyses utilizing the EMMAX method [28, 29] (i.e., GWAAs, GxE GWAAs) produced and further evaluated by constructing Manhattan plots within SVS v8.8.2 (Golden Helix, Bozeman, MT). Moreover, while SVS explicitly computes the full model mentioned above and outputs all of its β values, it only performs an optimization of the reduced model computation, which is sufficient to determine the SSE of the reduced-model equation, and thereafter, estimate the full versus reduced model P-value via F-test. This optimization is used to solve: MB−1y = MB−1Xkβrkp + ϵMB, where M = (I − QQ′), and Q is derived from performing the QR algorithm, as QR = B−1 [Xc ∣ Xi]. All Gelbvieh QTL were defined by ≥ 2 SNP loci with MAF ≥ 0.01 (i.e., a lead SNP plus at least one additional supporting SNP within 1 Mb) which also met a nominal significance threshold (P ≤ 1e-05) [30, 31], and all EMMAX marker-based pseudo-heritability estimates were produced as previously described [28,29,30, 99, 100].
Genomic inflation factors (λ) for all analyses (GEMMA; EMMAX) were estimated from the observed and expected P-values using genABEL [103], and the relationships between the observed P-values were estimated (GEMMA versus EMMAX) via correlation coefficients (i.e., Pearson, Spearman) in R v3.3.3 [89].
Availability of data and materials
Data are available for non-commercial use via data use agreement (DUA) with the American Gelbvieh Association.
Abbreviations
- BW:
-
Birth Weight
- GWAA:
-
Genome-wide association analysis
- GxE:
-
Genotype-by-environment interaction
- QTL:
-
Quantitative Trait Locus
- WW:
-
Weaning Weight
- YW:
-
Yearling Weight
References
Barwick SA, Henzell AL. Development successes and issues for the future in deriving and applying selection indexes for beef breeding. Aust J Exp Agric. 2005;45:923. https://doi.org/10.1071/EA05068.
Utsunomiya YT, do Carmo AS, Carvalheiro R, Neves HH, Matos MC, Zavarez LB, et al. Genome-wide association study for birth weight in Nellore cattle points to previously described orthologous genes affecting human and bovine height. BMC Genet. 2013;14:52. https://doi.org/10.1186/1471-2156-14-52.
Brito Lopes F, da Silva MC, Magnabosco CU, Goncalves Narciso M, Sainz RD. Selection indices and multivariate analysis show similar results in the evaluation of growth and carcass traits in beef cattle. PLoS One. 2016;11:e0147180. https://doi.org/10.1371/journal.pone.0147180.
Meirelles SLC, Mokry FB, Espasandín AC, Dias MAD, Baena MM, de A. Regitano LC. Genetic parameters for carcass traits and body weight using a Bayesian approach in the Canchim cattle. Genet Mol Res. 2016;15. https://doi.org/10.4238/gmr.15027471.
Bourdon RM, Brinks JS. Genetic, environmental and phenotypic relationships among gestation length, birth weight, growth traits and age at first calving in beef cattle. J Anim Sci. 1982;55:543–53. https://doi.org/10.2527/jas1982.553543x.
Eriksson S, Näsholm A, Johansson K, Philipsson J. Genetic parameters for calving difficulty, stillbirth, and birth weight for Hereford and Charolais at first and later parities1. J Anim Sci. 2004;82:375–83. https://doi.org/10.2527/2004.822375x.
Cook BR, Tess MW, Kress DD. Effects of selection strategies using heifer pelvic area and sire birth weight expected progeny difference on dystocia in first-calf heifers. J Anim Sci. 1993;71:602–7. https://doi.org/10.2527/1993.713602x.
Spelman RJ, Huisman AE, Singireddy SR, Coppieters RJ, Arranz J, Georges M, et al. Quantitative trait loci analysis on 17 nonproduction traits in the New Zealand dairy population. J Dairy Sci. 2000;83:370. https://doi.org/10.3168/JDS.S0022-0302(00)74890-9.
Kneeland J, Li C, Basarab J, Snelling WM, Benkel B, Murdoch B, et al. Identification and fine mapping of quantitative trait loci for growth traits on bovine chromosomes 2, 6, 14, 19, 21, and 23 within one commercial line of Bos taurus1. J Anim Sci. 2004;82:3405–14. https://doi.org/10.2527/2004.82123405x.
Maltecca C, Weigel KA, Khatib H, Cowan M, Bagnato A. Whole-genome scan for quantitative trait loci associated with birth weight, gestation length and passive immune transfer in a Holstein · Jersey crossbred population. Anim Genet. 2008:27–34. https://doi.org/10.1111/j.1365-2052.2008.01793.x.
McClure MC, Morsci NS, Schnabel RD, Kim JW, Yao P, Rolf MM, et al. A genome scan for quantitative trait loci influencing carcass, post-natal growth and reproductive traits in commercial Angus cattle. Anim Genet. 2010;41:597–607. https://doi.org/10.1111/j.1365-2052.2010.02063.x.
Cole JB, Wiggans GR, Ma L, Sonstegard TS, Lawlor TJ, Crooker BA, et al. Genome-wide association analysis of thirty one production, health, reproduction and body conformation traits in contemporary U.S. Holstein cows. BMC Genomics. 2011;12:408. https://doi.org/10.1186/1471-2164-12-408.
Santiago GG, Siqueira F, Cardoso FF, Regitano LCA, Ventura R, Sollero BP, et al. Genomewide association study for production and meat quality traits in Canchim beef cattle. J Anim Sci. 2017;95:3381–90. https://doi.org/10.2527/jas.2017.1570.
Saatchi M, Schnabel RD, Taylor JF, Garrick DJ. Large-effect pleiotropic or closely linked QTL segregate within and across ten US cattle breeds. BMC Genomics. 2014;15.
Saatchi M, McClure MC, McKay SD, Rolf MM, Kim J, Decker JE, et al. Accuracies of genomic breeding values in American Angus beef cattle using K-means clustering for cross-validation. Genet Sel Evol. 2011;43.
Saatchi M, Schnabel RD, Rolf MM, Taylor JF, Garrick DJ. Accuracy of direct genomic breeding values for nationally evaluated traits in US Limousin and Simmental beef cattle. Genet Sel Evol. 2012;44.
Saatchi M, Garrick DJ, Tait RG, Mayes MS, Drewnoski M, Schoonmaker J, et al. Genome-wide association and prediction of direct genomic breeding values for composition of fatty acids in Angus beef cattle a. BMC Genomics. 2013;14.
Pryce JE, Hayes BJ, Bolormaa S, Goddard ME. Polymorphic regions affecting human height also control stature in cattle. Genetics. 2011;187:981–4.
Gudbjartsson DF, Walters GB, Thorleifsson G, Stefansson H, Halldorsson BV, Zusmanovich P, et al. Many sequence variants affecting diversity of adult human height. Nat Genet. 2008;40:609–15.
Weedon MN, Lango H, Lindgren CM, Wallace C, Evans DM, Mangino M, et al. Genome-wide association analysis identifies 20 loci that influence adult height. Nat Genet. 2008;40:575–83.
Pausch H, Flisikowski K, Jung S, Emmerling R, Edel C, Götz KU, et al. Genome-wide association study identifies two major loci affecting calving ease and growth-related traits in cattle. Genetics. 2011;187:289–97.
Abo-Ismail MK, Brito LF, Miller SP, Sargolzaei M, Grossi DA, Moore SS, et al. Genome-wide association studies and genomic prediction of breeding values for calving performance and body conformation traits in Holstein cattle. Genet Sel Evol. 2017;49.
Matukumalli LK, Lawley CT, Schnabel RD, Taylor JF, Allan MF, Heaton MP, et al. Development and characterization of a high density SNP genotyping assay for cattle. PLoS One. 2009;4:e5350. https://doi.org/10.1371/journal.pone.0005350.
Rincon G, Weber KL, Van Eenennaam AL, Golden BL, Medrano JF. Hot topic: performance of bovine high-density genotyping platforms in Holsteins and jerseys. J Dairy Sci. 2011;94:6116–21. https://doi.org/10.3168/JDS.2011-4764.
VanRaden PM. Efficient methods to compute genomic predictions. J Dairy Sci. 2008;91:4414–23. https://doi.org/10.3168/JDS.2007-0980.
Decker JE. Agricultural genomics: commercial applications bring increased basic research power. PLoS Genet. 2015;11:e1005621. https://doi.org/10.1371/journal.pgen.1005621.
Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet. 2012;44:821–4. https://doi.org/10.1038/ng.2310.
Segura V, Vilhjálmsson BJ, Platt A, Korte A, Seren Ü, Long Q, et al. An efficient multi-locus mixed-model approach for genome-wide association studies in structured populations. Nat Genet. 2012;44:825–30. https://doi.org/10.1038/ng.2314.
Kang HM, Sul JH, Service SK, Zaitlen NA, Kong S, Freimer NB, et al. Variance component model to account for sample structure in genome-wide association studies. Nat Genet. 2010;42:348–54. https://doi.org/10.1038/ng.548.
Seabury CM, Oldeschulte DL, Saatchi M, Beever JE, Decker JE, Halley YA, et al. Genome-wide association study for feed efficiency and growth traits in U.S. beef cattle. BMC Genomics. 2017;18:386. https://doi.org/10.1186/s12864-017-3754-y.
Wellcome Trust Case Control Consortium TWTCC. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–78. https://doi.org/10.1038/nature05911.
Lee SH, Choi BH, Lim D, Gondro C, Cho YM, Dang CG, et al. Genome-wide association study identifies major loci for carcass weight on BTA14 in Hanwoo (Korean cattle). PLoS One. 2013;8.
Utsunomiya YT, Milanesi M, Utsunomiya ATH, Torrecilha RBP, Kim ES, Costa MS, et al. A PLAG1 mutation contributed to stature recovery in modern cattle. Sci Rep. 2017;7.
Fink T, Tiplady K, Lopdell T, Johnson T, Snell RG, Spelman RJ, et al. Functional confirmation of PLAG1 as the candidate causative gene underlying major pleiotropic effects on body weight and milk characteristics. Sci Rep. 2017;7.
Eberlein A, Takasuga A, Setoguchi K, Pfuhl R, Flisikowski K, Fries R, et al. Dissection of genetic factors modulating fetal growth in cattle indicates a substantial role of the non-SMC condensin I complex, subunit G (NCAPG) gene. Genetics. 2009;183:951–64.
Setoguchi K, Furuta M, Hirano T, Nagao T, Watanabe T, Sugimoto Y, et al. Cross-breed comparisons identified a critical 591-kb region for bovine carcass weight QTL (CW-2) on chromosome 6 and the Ile-442-met substitution in NCAPG as a positional candidate. BMC Genet. 2009;10.
Lindholm-Perry AK, Kuehn LA, Oliver WT, Sexten AK, Miles JR, Rempel LA, et al. Adipose and muscle tissue gene expression of two genes (NCAPG and LCORL) located in a chromosomal region associated with cattle feed intake and gain. PLoS One. 2013;8:e80882. https://doi.org/10.1371/journal.pone.0080882.
Liu Y, Duan X, Chen S, He H, Liu X. NCAPG is differentially expressed during longissimus muscle development and is associated with growth traits in Chinese Qinchuan beef cattle. Genet Mol Biol. 2015;38:450–6.
Lyu S, Arends D, Nassar MK, Brockmann GA. Fine mapping of a distal chromosome 4 QTL affecting growth and muscle mass in a chicken advanced intercross line. Anim Genet. 2017;48:295–302.
Urbinati I, Stafuzza NB, Oliveira MT, Chud TCS, Higa RH, Regitano LC de A, et al. Selection signatures in Canchim beef cattle. J Anim Sci Biotechnol. 2016;7.
Buzanskas ME, Grossi DA, Ventura RV, Schenkel FS, Sargolzaei M, Meirelles SLC, et al. Genome-wide association for growth traits in canchim beef cattle. PLoS One. 2014;9:1–8.
Mosley JD, Shaffer CM, Van Driest SL, Weeke PE, Wells QS, Karnes JH, et al. A genome-wide association study identifies variants in KCNIP4 associated with ACE inhibitor-induced cough. Pharmacogenomics J. 2016;16:231–7.
Martínez R, Gómez Y, Martínez-Roch JF. Genome-wide association study on growth traits in Colombian creole breeds and crossbreeds with zebu cattle. Genet Mol Res. 2014;13:6420–32.
Lindholm-Perry AK, Kuehn LA, Smith TPL, Ferrell CL, Jenkins TG, Freetly HC, et al. A region on BTA14 that includes the positional candidate genes LYPLA1, XKR4 and TMEM68 is associated with feed intake and growth phenotypes in cattle. Anim Genet. 2012;43:216–9.
Jepsen MR, Kløverpris S, Mikkelsen JH, Pedersen JH, Füchtbauer EM, Laursen LS, et al. Stanniocalcin-2 inhibits mammalian growth by proteolytic inhibition of the insulin-like growth factor axis. J Biol Chem. 2015;290:3430–9.
Bolormaa S, Pryce JE, Reverter A, Zhang Y, Barendse W, Kemper K, et al. A multi-trait, meta-analysis for detecting pleiotropic polymorphisms for stature, fatness and reproduction in beef cattle. PLoS Genet. 2014;10.
Bouleftour W, Boudiffa M, Wade-Gueye NM, Bouët G, Cardelli M, Laroche N, et al. Skeletal development of mice lacking bone Sialoprotein (BSP) - impairment of long bone growth and progressive establishment of high trabecular bone mass. PLoS One. 2014;9.
Bellahcene A, Merville M-P, Castronovo V. Expression of Bone Sialoprotein, a Bone Matrix Protein, in Human Breast Cancer. Cancer Res. 1994;54:2823–6.
Cohen-Zinder M, Seroussi E, Larkin DM, Loor JJ. Everts-Van Der wind A, Lee JH, et al. identification of a missense mutation in the bovine ABCG2 gene with a major effect on the QTL on chromosome 6 affecting milk yield and composition in Holstein cattle. Genome Res. 2005;15:936–44.
An B, Xia J, Chang T, Wang X, Miao J, Xu L, et al. Genome-wide association study identifies loci and candidate genes for internal organ weights in simmental beef cattle. Physiol Genomics. 2018;50:523–31.
Saunders J, Wisidagama DR, Morford T, Malone CS. Maximal expression of the evolutionarily conserved Slit2 gene promoter requires Sp1. Cell Mol Neurobiol. 2016;36:955–64.
Holmes G, Niswander L. Expression of slit-2 and slit-3 during chick development. Dev Dyn. 2001;222:301–7. https://doi.org/10.1002/dvdy.1182.
Dallol A, Da Silva NF, Viacava P, Minna JD, Bieche I, Maher ER, et al. SLIT2, a human homologue of the Drosophila Slit2 gene, has tumor suppressor activity and is frequently inactivated in lung and breast cancers. Cancer Res. 2002;62:5874–80 http://www.ncbi.nlm.nih.gov/pubmed/12384551.
Yang J, Siqueira MF, Behl Y, Alikhani M, Graves DT. The transcription factor ST18 regulates proapoptotic and proinflammatory gene expression in fibroblasts. FASEB J. 2008;22:3956–67.
Han YJ, Chen Y, Liu Y, Liu XL. Sequence variants of the LCORL gene and its association with growth and carcass traits in Qinchuan cattle in China. J Genet. 2017;96:9–17. https://doi.org/10.1007/s12041-016-0732-0.
Al-Mamun HA, Kwan P, Clark SA, Ferdosi MH, Tellam R, Gondro C. Genome-wide association study of body weight in Australian merino sheep reveals an orthologous region on OAR6 to human and bovine genomic regions affecting height and weight. Genet Sel Evol. 2015;47.
Michot P, Chahory S, Marete A, Grohs C, Dagios D, Donzel E, et al. A reverse genetic approach identifies an ancestral frameshift mutation in RP1 causing recessive progressive retinal degeneration in European cattle breeds. Genet Sel Evol. 2016;48.
Do DN, Bissonnette N, Lacasse P, Miglior F, Sargolzaei M, Zhao X, et al. Genome-wide association analysis and pathways enrichment for lactation persistency in Canadian Holstein cattle. J Dairy Sci. 2017;100:1955–70.
Abo-Ismail MK, Vander Voort G, Squires JJ, Swanson KC, Mandell IB, Liao X, et al. Single nucleotide polymorphisms for feed efficiency and performance in crossbred beef cattle. BMC Genet. 2014;15:14. https://doi.org/10.1186/1471-2156-15-14.
Patel K, Scrimieri F, Ghosh S, Zhong J, Kim M-S, Ren YR, et al. FAM190A deficiency creates a cell division defect. Am J Pathol. 2013;183:296–303. https://doi.org/10.1016/j.ajpath.2013.03.020.
Siddiqui IJ, Pervaiz N, Abbasi AA. The Parkinson disease gene SNCA: evolutionary and structural insights with pathological implication. Sci Rep. 2016;6.
Doxakis E. Post-transcriptional regulation of α-synuclein expression by mir-7 and mir-153. J Biol Chem. 2010;285:12726–34.
Brito LF, Kijas JW, Ventura RV, Sargolzaei M, Porto-Neto LR, Cánovas A, et al. Genetic diversity and signatures of selection in various goat breeds revealed by genome-wide SNP markers. BMC Genomics. 2017;18:1–20.
Kuemmerle JM, Theiss F, Okoniewski MJ, Weber FA, Hemmi S, Mirsaidi A, et al. Identification of novel equine (Equus caballus) tendon markers using RNA sequencing. Genes (Basel). 2016;7:1–14.
Round J, Stein E. Netrin signaling leading to directed growth cone steering. Curr Opin Neurobiol. 2007;17:15–21. https://doi.org/10.1016/j.conb.2007.01.003.
Sugimoto M, Gotoh Y, Kawahara T, Sugimoto Y. Molecular effects of polymorphism in the 3’UTR of unc-5 homolog C associated with conception rate in Holsteins. PLoS One. 2015;10.
Lapinski PE, Oliver JA, Bodie JN, Marti F, King PD. The T-cell-specific adapter protein family: TSAd, ALX, and SH2D4A/SH2D4B. Immunol Rev. 2009;232:240–54.
Qrafli M, Asekkaj I, Bourkadi JE, El Aouad R, Sadki K. New variant identified in major susceptibility locus to tuberculosis on chromosomal region 8q12-q13 in Moroccan population: A case control study. BMC Infect Dis. 2017;17.
Gérard A, Ségéral E, Naughtin M, Abdouni A, Charmeteau B, Cheynier R, et al. The Integrase cofactor LEDGF/p75 associates with Iws1 and Spt6 for Postintegration silencing of HIV-1 gene expression in latently infected cells. Cell Host Microbe. 2015;17:107–17.
Khatib H, Zaitoun I, Wiebelhaus-Finger J, Chang YM, Rosa GJM. The association of bovine PPARGC1A and OPN genes with milk composition in two independent Holstein cattle populations. J Dairy Sci. 2007;90:2966–70.
Weikard R, Kuhn C, Goldammer T, Freyer G, Schwerin M. The bovine PPARGC1A gene: molecular characterization and association of an SNP with variation of milk fat synthesis. Physiol Genomics. 2005:1–13.
Holt LJ, Siddle K. Grb10 and Grb14: enigmatic regulators of insulin action - and more? Biochem J. 2005;388:393–406.
Hazra A, Kraft P, Lazarus R, Chen C, Chanock SJ, Jacques P, et al. Genome-wide significant predictors of metabolites in the one-carbon metabolism pathway. Hum Mol Genet. 2009;18:4677–87.
Iyengar L, Rajalakshmi K. Effect of folic acid supplement on birth weights of infants. Am J Obstet Gynecol. 1975;122:332–6.
Pulukuri SMK, Knost JA, Estes N, Rao JS. Small interfering RNA-directed knockdown of uracil DNA glycosylase induces apoptosis and sensitizes human prostate cancer cells to genotoxic stress. Mol Cancer Res. 2009;7:1285–93.
Liu Y-J, Liu X-G, Wang L, Dina C, Yan H, Liu J-F, et al. Genome-wide association scans identified CTNNBL1 as a novel gene for obesity. Hum Mol Genet. 2008;17:1803–13. https://doi.org/10.1093/hmg/ddn072.
Labeit S, Fautel M, Lakey A, Trinick J. Towards a molecular understanding of titin. EMBO J. 1992;11:1711–6.
Linke WA, Stockmeier MR, Ivemeyer M, Hosser H, Mundel P. Characterizing titin’s I-band Ig domain region as an entropic spring. J Cell Sci. 1998:1567–74.
Neagoe C, Kulke M, Del Monte F, Gwathmey JK, De Tombe PP, Hajjar RJ, et al. Titin isoform switch in ischemic human heart disease. Circulation. 2002;106:1333–41.
Liu L, Gudas LJ. Disruption of the lecithin:retinol acyltransferase gene makes mice more susceptible to vitamin A deficiency. J Biol Chem. 2005;280:40226–34.
Wang B, Nie W, Fu X, de Avila JM, Ma Y, Zhu MJ, et al. Neonatal vitamin A injection promotes cattle muscle growth and increases oxidative muscle fibers. J Anim Sci Biotechnol. 2018;9.
Harris CL, Wang B, Deavila JM, Busboom JR, Maquivar M, Parish SM, et al. Vitamin A administration at birth promotes calf growth and intramuscular fat development in Angus beef cattle. J Anim Sci Biotechnol. 2018;9.
West KP, LeClerq SC, Shrestha SR, Wu LS-F, Pradhan EK, Khatry SK, et al. Effects of vitamin A on growth of vitamin A-deficient children: field studies in Nepal. J Nutr. 1997;127:1957–65.
Lominadze D, Tsakadze N, Sen U, Falcone JC, D’Souza SE. Fibrinogen and fragment D-induced vascular constriction. Am J Physiol - Hear Circ Physiol. 2005;288(3):57–3.
Paterson J, Forcherio C, Larson B, Samford M, Kerley M. The effects of fescue toxicosis on beef cattle productivity. J Anim Sci. 1995;73:889–98.
Wong M-L, Arcos-Burgos M, Liu S, Vélez JI, Yu C, Baune BT, et al. The PHF21B gene is associated with major depression and modulates the stress response. Mol Psychiatry. 2017;22:1015–25. https://doi.org/10.1038/mp.2016.174.
Bertonha FB, Barros Filho Mde C, Kuasne H, dos Reis PP, da Costa Prando E, JJAM M, et al. PHF21B as a candidate tumor suppressor gene in head and neck squamous cell carcinomas. Mol Oncol. 2015;9:450–62. https://doi.org/10.1016/j.molonc.2014.09.009.
Endelman JB. Ridge regression and other kernels for genomic selection with R package rrBLUP. Plant Genome. 2011;4:250–5.
R Core Team. R: The R Project for Statistical Computing. R Foundation for Statistical 844 Computing. 2017. https://www.r-project.org/.
PRISM. Climate Group. PRISM. 2011; http://prism.oregonstate.edu/. .
Leutner B, Horning N. Tools for Remote Sensing Data Analysis [R package RStoolbox version 0.1.10]. 2017. https://cran.r-project.org/web/packages/RStoolbox/index.html. Accessed 27 Aug 2019.
R Core Team. R: The R Project for Statistical Computing. 2013. https://www.r-project.org/. Accessed 27 Aug 2019.
Hennig C. CRAN - package fpc. 2013. https://cran.r-project.org/web/packages/fpc/index.html.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. https://doi.org/10.1086/519795.
Loh P-R, Danecek P, Palamara PF, Fuchsberger C, Reshef YA, Finucane HK, et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat Genet. 2016;48:1443–8. https://doi.org/10.1038/ng.3679.
Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–93. https://doi.org/10.1093/bioinformatics/btr509.
Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet. 2007;39:906–13. https://doi.org/10.1038/ng2088.
Zhou X, Stephens M. Efficient multivariate linear mixed model algorithms for genome-wide association studies. Nat Methods. 2014;11:407–9. https://doi.org/10.1038/nmeth.2848.
Neibergs HL, Seabury CM, Wojtowicz AJ, Wang Z, Scraggs E, Kiser JN, et al. Susceptibility loci revealed for bovine respiratory disease complex in pre-weaned Holstein calves. BMC Genomics. 2014;15:1164. https://doi.org/10.1186/1471-2164-15-1164.
Davis BW, Seabury CM, Brashear WA, Li G, Roelke-Parker M, Murphy WJ. Mechanisms underlying mammalian hybrid sterility in two feline interspecies models. Mol Biol Evol. 2015;32:2534–46. https://doi.org/10.1093/molbev/msv124.
Hyun MK, Zaitlen NA, Wade CM, Kirby A, Heckerman D, Daly MJ, et al. Efficient control of population structure in model organism association mapping. Genetics. 2008;178:1709–23.
Vihjal B. "mixmogam" 2012. https://github.com/bvilhjal/mixmogam/tree/a40f3e2c95aa17d22243ae2865aee6176ae821a6.
Aulchenko YS, Ripke S, Isaacs A, van Duijn CM. GenABEL: an R library for genome-wide association analysis. Bioinformatics. 2007;23:1294–6. https://doi.org/10.1093/bioinformatics/btm108.
Acknowledgements
We sincerely appreciate the American Gelbvieh Association and Gelbvieh breeders for sharing the genotypes and phenotypes used in this research.
Funding
This project was supported by Agriculture and Food Research Initiative Competitive Grant no. 2016–68004-24827 from the USDA National Institute of Food and Agriculture.
Author information
Authors and Affiliations
Contributions
JED and CMS conceptualized and designed the research. RDS managed data acquisition, storage, and retrieval. TNR performed initial quality control, phasing and imputation. SMN pre-adjusted phenotypes. DLO provided programming support for data formatting and management. JLS, MLW, and CMS performed GWAAs. JLS, JED, and CMS interpreted results. JLS, JED, and CMS wrote initial version of the manuscript, which was edited by all authors.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Prior to the planning and onset of the current study, data were collected under typical production practices and reported to the American Gelbvieh Association, for inclusion in an existing data repository; thus an animal care and use protocol is not necessary. Staff at the American Gelbvieh Association have read and approved the manuscript.
Consent for publication
Staff at the American Gelbvieh Association have provided the authors with consent to publish.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
EMMAX birth weight (BW) analysis. Figure S2. EMMAX weaning weight (WW) analysis. Figure S3. EMMAX yearling weight (YW) analysis. Figure S4. EMMAX birth weight (BW) genotype-by-environment (GxE) analysis. Figure S5. EMMAX weaning weight (WW) genotype-by-environment (GxE). Figure S6. EMMAX yearling weight (YW) genotype-by-environment (GxE) analysis. Table S1. Summary of QTL detected by EMMAX for BW in U.S. Gelbvieh cattle. Table S2. Genomic inflation factors (λ) calculated using observed P-values and expected P-values. Table S3. Correlation coefficients for GEMMA versus EMMAX P-values. Table S4. Summary of QTL detected by EMMAX for WW in U.S. Gelbvieh cattle. Table S5. Summary of QTL detected by EMMAX for YW in U.S. Gelbvieh cattle. Table S6. Summary of GxE QTL detected by EMMAX for BW in U.S. Gelbvieh cattle.Table S7. Summary of GxE QTL detected by EMMAX for YW in U.S. Gelbvieh cattle.
Additional file 2.
Summary of lead and supporting SNPs from analyses for BW, WW, YW, BW GxE, WW GxE, and YW GxE using 778K imputed genotypes, including QTL chromosome, base pair, and rounded Mb.
Additional file 3.
Summary of SNP panels used in analyses, including number of SNPs and individuals available before and after filtering.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Smith, J.L., Wilson, M.L., Nilson, S.M. et al. Genome-wide association and genotype by environment interactions for growth traits in U.S. Gelbvieh cattle. BMC Genomics 20, 926 (2019). https://doi.org/10.1186/s12864-019-6231-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-019-6231-y