
Abstract
Background
Increasing the yield of barley (Hordeum vulgare L.) is a main breeding goal in developing barley cultivars. A high density genetic linkage map containing 1894 SNP and 68 SSR markers covering 1375.8 cM was constructed and used for mapping quantitative traits. A late-generation double haploid population (DH) derived from the Huaai 11 × Huadamai 6 cross was used to identify QTLs and QTL × environment interactions for ten traits affecting grain yield including length of main spike (MSL), spikelet number on main spike (SMS), spikelet number per plant (SLP), grain number per plant (GP), grain weight per plant (GWP), grain number per spike (GS), thousand grain weight (TGW), grain weight per spike (GWS), spike density (SPD) and spike number per plant (SP).
Results
In single environment analysis using composite interval mapping (CIM), a total of 221 QTLs underlying the ten traits were detected in five consecutive years (2009–2013). The QTLs detected in each year were 50, 48, 41, 41 and 41 for the year 2009 to 2013. The QTLs associated with these traits were generally clustered on chromosome 2H, 4H and 7H.
In multi-environment analysis, a total of 111 significant QTLs including 18 for MSL, 16 for SMS, 15 for SPD, 5 for SP, 4 for SLP, 14 for TGW, 5 for GP, 11 for GS, 8 for GWP, and 15 for GWS were detected in the five years. Most QTLs showed significant QTL × environment interactions (QEI), nine QTLs (qIMSL3-1, qIMSL4-1, qIMSL4-2, qIMSL6-1, qISMS7-1, qISPD2-7, qISPD7-1, qITGW3-1 and qIGWS4-3) were detected with minimal QEI effects and stable in different years. Among 111 QTLs,71 (63.40 %) QTLs were detected in both single and multiple environments.
Conclusions
Three main QTL cluster regions associated with the 10 agronomic traits on chromosome 2H, 4H and 7H were detected. The QTLs for SMS, SLP, GP and GWP were located in the region near Vrs1 on chromosome 2H. The QTLs underlying SMS, SPD and SLP were clustered on chromosome 4H. On the terminal of chromosome 7H, there was a QTL cluster associated with TGW, SPD, GWP and GWS. The information will be useful for marker-assisted selection (MAS) in barley breeding.
Similar content being viewed by others

Background
Barley (Hordeum vulgare L.) is one of the world’s earliest domesticated crops and ranks the fourth cereal crop after maize, rice and wheat. It is widely used in many ways, including animal feed, malting and brewing. High-grain yield is the main breeding goal in developing cultivars. Due to most agronomic traits related to grain yield are controlled by quantitative trait loci (QTL), it is difficult to dissect the genetic and molecular basis of complex grain yield traits. Genome-analysis tools are useful for dissecting complex traits and manipulating determinants of multiple traits in breeding procedures [1–3].
QTL analysis has been widely applied to all crops including barley [4–7]. Since the first barley genetic map was constructed from RFLP marker, barley breeders have constructed many genetic maps using various genetic markers, including RFLP, AFLP, SSR and DArT [8–15]. These maps have been employed to identify, locate and estimate the phenotypic effects of QTLs underlying economically important traits [11, 12, 16–23]. Multiple environment trials, especially for grain yield traits, are commonly used to assess the performance of genotypes across a range of locations and years. Some QTLs are sensitive to environment and may have different effects in different years, with strong QTL × environment interaction (QEI). Therefore,identification QTL for grain yield trait in multiple environment and QTL × environment interaction to find crucial stable QTL is of vital importance for applying them in marker-assisted selection (MAS). With availability of genome sequence data and development of next generation sequencing (NGS) for rapid identification and scoring of genetic markers, single nucleotide polymorphism (SNP) markers have been widely used for constructing haplotype maps and genome-wide association studies [24–28]. Restriction site associated DNA (RAD) was first described as a tool for genetic mapping in fish and fungi by Baird et al. [29]. This newly developed sequencing technology can increase the data generated via short-read sequencing using restricted enzyme digested DNA to reduce complexity of genome compared to genome de novo sequencing and re-sequencing [29–32]. It has rapidly become a popular method for quick SNP discovery, linkage map construction [33, 34], and QTL mapping [35, 36].
Grain yield is directly determined by number of tillers per plant, number of grains per spike and thousand grain weight. Agronomic traits, such as spike density, main spike length, spike number per plant, grain weight per spike could indirectly affect the yield [5, 37–40]. QTLs underlying these important yield component traits are rarely analyzed in a cross between two-rowed and six-rowed barley. Huaai 11 is a new source of dwarfing discovered in our research group, and dwarf gene was mapped on the long arm of chromosome 7H [41]. With application of this population, our group constructed genetic map with 153 SSR markers to dissect phenotypic effect of QTL for agronomic traits, morphological and physiological traits of flag leaf [42, 43]. However, previous linkage map constructed based on 153 SSR markers is not able to provide precise and complete information about the numbers and location of QTL. The high density map is essential for precisely QTL identification and fine mapping of agronomic traits associated with grain yield [44]. The objective of this study was to use an ultra-high density SNP map containing 1894 SNP and 68 SSR markers to identify QTL underlying 10 agronomic traits related to grain yield.
Methods
Plant material and experimental design
The Huaai 11 was discovered from the barley landrace Dofu Bai Qing Ke and collected by Professor Sun Dongfa in 1993. Huadamai 6 is an elite brewing barley cultivar developed by Huazhong Agricultural University and available in China. The mapping population consists of 122 doubled haploid (DH) lines derived from a cross between the six-rowed dwarfing barley cultivar Huaai 11 (height is about 40 cm) and the two-rowed barley cultivar Huadamai 6 (height is about 85 cm) using anther culture [41]. The field trials were performed on the experimental farm of Huazhong Agricultural University, Wuhan (30°33’N) in five consecutive years (2009 to 2013). Each of the DH and parental lines were grown in three rows with a plot of lines of 1.5 m in length. The six seeds from each line were grown in each row, and space of the plant was 0.1 m.
DNA isolation and genotype
Genomic DNA was extracted from seeding leaf using CTAB method, and treated with RNase to remove residual RNA. All lines from mapping population and two parents were genotyped at the Personal Biotechnology Co., Ltd (Shanghai, China). Genomic DNA was digested with the restriction endonuclease XmaI, a high fidelity restriction enzyme (New England Biolabs, USA), which recognizes an 6-nucleotide (nt) sequence (5'CCCGGG3'). The RAD library construction and DNA sequencing were done according to the protocol described by Baird et al. [29]. The DNA samples were performed in a single lane (library) of an Illumian Hiseq2000.
Map construction
Using the procedure of Ramsey et al [45], Ren et al. [41, 42] genotyped 153 SSR markers. The RAD-seq markers were sorted using the MSTMAP software [46]. MSTMAP v4.3 software was used to generate individual group tree for the 122 lines. SNP markers with >15 % missing value were removed. JoinMap 4.0 was used to calculate the 1894 SNP and 68 polymorphic SSR markers order and genetic distance using Kosambi’s mapping function [47, 48]. Markers were assigned to seven linkage groups using a test for independence LOD score of 7.0, and ordered using the regression mapping algorithm. Two morphological markers, row number (Vrs1) and naked caryopsis (Nud1), were also integrated into the map.
Phenotyping
After fully maturity, we randomly harvested four individual plants from each plot. Ten agronomic traits, spike number per plant (SP), main spike length (MSL), spikelet number per spike (SMS), spikelet number per plant (SLP), grain number per plant (GP), grain number per spikes (GS), grain weight per plant (GWP), grain weight per spikes (GWS), thousand grain weight (TGW) and spike density (SPD) were measured. Phenotypic data for those traits were measured as described by Ren et al. [42] except spike density (SPD) or spike (rachis) internodes length that was calculated using the number of fertile rachis nodes in a spike divided by length of main spike (cm). The mean values of twelve plants (four plants from each replicate x three replicates) sampled were subjected to statistical analysis. The phenotypic data of nine traits (except SPD) in 2009 and 2010 were from Ren et al. [42].
Phenotypic data analysis
Correlation and QTL analyses were performed for the data from each year. Homogeneity of variance and normality of distribution were tested before analysis of variance (ANOVA) using the general linear model (GLM). All analyses were performed using IBM SPSS Statistics 19 software (http://www.spss.com). P value less than 0.05 was considered as significance.
QTL analysis
The location of QTL and its genetic effect were detected through composite interval mapping (CIM) using QTL Cartographer version 2.5 [49]. After performing 1000 permutation test, a LOD (Likelihood of odd) threshold of 2.5 was used to declare putative QTLs [50]. Percentage of phenotypic variation explained and additive effect of each QTL were also calculated using QTL Cartographer 2.5. The confidence of interval was calculated using the two-LOD support interval, which was determined by finding the region on both sides of a QTL peak that corresponds to a decrease of 2 LOD score [51, 52]. The software MapChart 2.2 was used to draw QTL location on the map [53]. The putative QTLs were defined as ‘q’ + abbreviation trait name + detected QTL order on chromosome for CIM mapping. QTL in multiple environment interaction (QEI) mapping was conducted using the software IciMapping 4.0 (www.isbreeding.net). A LOD threshold ≥5.0 was set for declaring the QTL × environment interaction (QEI). The putative QTLs were defined as ‘q’ + ‘I’ + abbreviation trait name + detected QTL order on chromosome in multi-environment analysis using inclusive composite interval mapping (ICIM). QTLs linked to a target trait, which were stably identified from different years with clearly similar positions (overlapping intervals), were assumed to be the same one.
Results
Characteristics of agronomic traits
Table 1 showed 10 agronomic traits of the 122 individuals from the DH population and their parents in five consecutive years (year 2009–2013). The values of SP, MSL, GWP, GWS, and TGW in Huadamai 6 were higher than those in Huaai 11. The values of SMS, SLP, GS, GP and SPD were higher in Huaai 11 than those in Huadamai 6. The t-test showed that two parents were significant difference on all traits (p < 0.05) but GP. GP is determined by SP and GS. Because Huaai 11 had more grain number per spike and few spike number per plant, it could explain why GP in DH population was significant different although parents showed no significant different. Probabilistic of the distribution test showed nine traits (except SMS) displayed normal distribution with skewness and kurtosis of among -1 from 1 (Table 1), and SMS displayed bimodal distribution. Analysis of variance of the 122 DH lines and their parents showed highly significant genotype effect for the traits studied, and also a significant year effect for all traits except SPD and SMS (Additional file 1: Table S1). Genotype × environment interaction (GEI) was also significant for all traits. The variable coefficients ranged from 21.69–39.34 % in 2009, 20.46–34.78 % in 2010, 22.44–44.40 % in 2011, 21.40–40.57 % in 2012, and 16.06–39.49 % in 2013. Correlation coefficients among the 10 agronomic traits were given in (Additional file 2: Table S2). MSL, SMS, GP, GWP, SLP and GWS showed positive correlation with each other. SPD showed negative correlation with other traits. TGW showed significant negative correlation with SMS, SLP, GP and GS. SP showed significant positive correlation with GP and GWP, while GWS showed negative correlation with SP.
Marker genotyping
A total of 4992 SNP polymorphic markers and 153 polymorphic SSR markers were used for constructing a high density SNP map. Two morphological markers, row number (Vrs1) and naked caryopsis (Nud1), were also integrated into the map. Linkage analysis positioned morphological marker Vrs1 to the 0.72 cM marker interval flanked by SNP marker 2_522610509 and 2HL_34260490 (Fig. 1). The Nud1 was positioned to the 0.50 cM marker interval flanked by SNP marker M_96819_188 and 7HL_29967547 (Fig. 1). The final map was composed of 1894 SNP and 68 SSR markers (the co-segregation of markers and non-linked markers were excluded). The total length of genetic map was 1375.8 cM with an averaged inter-marker distance of 0.70 cM.

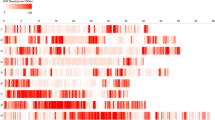
Chromosomes location of reliable QTL associated with 10 traits and two morphological traits (black). Genetic distance scale in centiMorgan (cM) is placed at left margin. Location of QTL is indicated for year 2009 (Cadmium Green Pale), 2010 (orange red), 2011 (green), 2012 (purple) and 2013 (pink)
Genome mapping of SNP markers
To compare SNP marker distribution in coding and noncoding regions of the barley genome, we searched the locations of all 4992 SNP markers on reference genome. The high-quality SNP sequences were blasted against the comprehensive barley gene index in the NCBI (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Using this database, 321 of 4992 (6.43 %) polymorphic SNP loci were aligned to multiple positions in gene index (Additional file 3: Table S3). High percentage of SNP sequences was positioned on candidate genes, suggesting that SNP markers were significantly rich in gene region.
QTL analysis
A total of 221 QTLs for 10 observed agronomic traits were detected in five consecutive years using CIM mapping (Additional file 4: Table S4). The numbers of QTLs detected were 50, 48, 41, 41 and 41 for the year 2009 to 2013, respectively (Table 2).
Main spike length and spikelet number on main spike
For MSL, a total of 26 QTLs were identified in single environment in five consecutive years, its LOD value ranged from 2.80 to 39.79, and individually accounted for 1.16–52.72 % of the phenotypic variation (Additional file 4: Table S4). Three reliable QTLs were detected in more than one year (Table 3 and Fig. 1). Of them, the major QTL qMSL2-7 was detected on chromosome 2H in four years, and accounted for 7.27–18.70 % of phenotypic variation with LOD value 7.24–17.32. qMSL7-4 and qMSL7-5 had positive additive effect on MSL and total explained 28.54–54.96 % of the phenotypic variation in three years (2009–2011) and four years (2009–2012), respectively. qMSL7-4 and qMSL7-5 were close to the SNP marker 7_542100185 and 7_249275418, respectively. The alleles from Huadamai6 increased MSL.
For SMS, a total of 27 QTLs were detected and dispersed on chromosome 1H, 2H, 4H and 7H. Each LOD value ranged from 2.58 to 87.42, and individually accounted for 1.07–82.22 % phenotypic variation (Additional file 4: Table S4). Six reliable QTLs were detected in more than one year (Table 3). qSMS2-1 (year 2012 and 2013), qSMS2-4 (year 2009 and 2013), qSMS4-4 (year 2009 and 2011) and qSMS4-5 (year 2012 and 2013) were detected in different two years and showed minor effects. qSMS2-7 had highest effect on SMS, and accounted for 72.76–82.22 % of phenotypic variation. qSMS2-7 was close to the SNP marker 2HL_22930294, and the favorable alleles came from Huaai11 increasing SMS. On chromosome 1H, qSMS1-1 had positive additive effect on SMS and explained 1.27–2.37 % of phenotypic variation in three years (year 2009, 2012 and 2013).
Spike density
A total of 24 QTLs were detected underlying SPD, whose LOD value ranged from 2.94 to 44.73, and individually accounted for 1.34–85.06 % of phenotypic variation (Additional file 4: Table S4). Four reliable QTLs were detected on chromosome 2H, 4H and 7H (Table 3). Two QTLs (qSPD2-4 and qSPD2-6) on chromosome 2H had positive additive effect on SPD. qSPD2-6 was detected in five years, and explained 16.85–23.94 % of phenotypic variation, while the qSPD2-4 was detected in year 2009 and 2012, explaining 4.38 % and 3.99 % of phenotypic variation. The qSPD4-1 on chromosome 4H was detected in four years (year 2009 to 2012), and had the effect on increasing SPD and explained 1.72–2.92 % of phenotypic variation. The qSPD7-3 on chromosome 7H had main effect on SPD, which was detected in three years (2009 to 2011), and accounted for 46.97–57.06 % of phenotypic variation.
Spike numbers per plant
For SP, a total of 18 QTLs were detected in five consecutive years, whose LOD value ranged from 2.55 to 19.22 and individually accounted for 3.50–51.97 % of phenotypic variation (Additional file 4: Table S4). Only one reliable QTL qSP5-1 on chromosome 5H was identified for SP in year 2011, 2012 and 2013, with effect on increasing spike number per plant and explained 6.18–20.32 % of phenotypic variation (Table 3).
Spikelet numbers per plant
A total of 18 QTLs influencing SLP were identified, individually explaining 3.50–51.97 % of phenotypic variation with LOD value 2.55–19.22 (Additional file 4: Table S4). Two reliable QTLs were detected in more than one year (Table 3). qSLP2-6 has highest effects on SLP, and was detected in four years (2009, 2011, 2012 and 2013), and accounted for 16.70–64.20 % of phenotypic variation. qSLP2-6 was close to the marker 2HL_34260490 with favorable alleles from Huaai11 for increasing SLP. qSLP4-2 on chromosome 4H had positive additive effect on SLP.
Thousand grain weight
For TGW, a total of 26 QTLs were detected in five consecutive years, whose LOD ranged from 2.69 to 49.64 and individually accounted for 1.39–61.78 % of phenotypic variation (Additional file 4: Table S4). Six reliable QTLs underlying TGW were detected in more than one year and were mapped on chromosome 2H, 5H and 7H (Table 3). Two QTLs (qTGW2-1 and qTGW2-2) were close to the Vrs 1 marker, and their additive effects were constancy in five years. qTGW2-1 was detected in year 2009 and 2013, and explained 44.03 % and 61.78 % of phenotypic variation, respectively, while qTgw2-2 was detected in year 2010, 2011 and 2012, explaining 43.45 % to 58.63 % of phenotypic variation. qTGW5-1 on chromosome 5H, was identified in year 2011 and 2013, with effect on decreasing thousand grain weight and explained 1.56 % and 1.39 % of phenotypic variation, respectively. Three QTLS (qTGW7-1, qTGW7-3 and qTGW7-4) on chromosome 7H had positive effect on TGW. qTGW7-1 was detected in three years, explaining 3.02–16.22 % of phenotypic variation. qTGW7-3 was detected in year 2009 and 2011, and explained 10.99 % and 3.00 % of phenotypic variation, respectively. qTGW7-4 was detected in year 2010 and 2013, and accounted for 6.64 % and 5.42 % of phenotypic variation, respectively.
Grain number per plant and grain number per spike
A total of 16 QTLs underlying GP were detected on chromosome 2H, 4H, 5H, 6H and 7H, individually explaining 2.54–57.24 % of phenotypic variation with LOD value 2.69–33.76 (Additional file 4: Table S4). One reliable QTL (qGP2-2) on chromosome 2H was identified for GP in three years, with effect on increasing grain number per plant. qGP2-2 explained 11.70–57.24 % of phenotypic variation (Table 3).
For GS, a total of 20 QTLs were detected in five consecutive years, whose LOD value ranged from 2.63 to 58.64 and individually accounted for 1.74–79.50 % of phenotypic variation (Additional file 4: Table S4). Two reliable QTLs were detected on chromosome 2H in more than one year (Table 3). qGS2-2 was detected in two years and accounted for 1.83 % and 4.73 % of phenotypic variation in 2010 and 2011, respectively, with favorable alleles from Huadamai 6. qGS2-4 was detected in four years and accounted for 41.14–79.50 % of phenotypic variation in 2010 to 2013, respectively, with favorable alleles from Huadamai6. qGS2-4, close to the marker 2HL_34260490, had main effect on decreasing GS.
Grain weight per plant and grain weight per spike
For GWP, a total of 18 QTLs were detected in five consecutive years, whose LOD value ranged from 2.52 to 21.59 and individually accounted for 3.09–40.82 % of phenotypic variation (Additional file 4: Table S4). Four reliable QTLs underlying GWP were detected in more than one year (Table 3). qGWP3-3 on chromosome 3H was identified in two years, explained 6.85 % and 6.01 % of phenotypic variation in 2010 and 2013, respectively. qGWP2-1 on chromosome 2H was identified in three years, with effect on increasing grain weights per plant, and explained 6.68 % to 11.72 % of phenotypic variation. Two QTLs (qGWP7-3 and qGWP7-4) on chromosome 7H had positive additive effect on grain weight per plant. qGWP7-3 accounted for 40.82 % and 11.85 % of phenotypic variation in 2010, and 2012, while qGWP7-4 accounted for 7.11 % and 28.85 % of phenotypic variation in 2012 and 2013, respectively.
For GWS, a total of 25 QTLs were detected in five consecutive years, whose LOD value ranged from 2.67 to 26.59 and individually accounted for 2.08–40.82 % of phenotypic variation (Additional file 4: Table S4). Two QTLs were detected in more than one year (Table 3). qGWS2-3 on chromosome 2H had effect on decreasing GWS, which was detected in three years and explained 12.44–16.83 % of phenotypic variation. qGWS7-8 on chromosome 7H was detected in two years, accounted for 4.08 % and 6.50 % of phenotypic variation in 2012 and 2013, respectively.
QTL × environment interaction (QEI) analysis
A total 71 of 111 significant QTLs for 10 agronomic traits were detected using both single and multiple environment analyses over five environments (Additional file 5: Table S5). Among them, 31 QTLs had major effects on their target traits, in which 12 QTLs were also major QTLs identified in single environment.
For MSL, 18 QTLs were detected across the five environments: one each on chromosome 3H and 6H,two each on chromosome 1H and 4H, four and eight on chromosome 7H and 2H, respectively. Eleven QTLs were detected in both single and multiple environments mapping while seven QTLs were only detected in multiple environments mapping analysis (Additional file 5: Table S5). Four QTLs (qIMSL3-1, qIMSL4-1, qIMSL4-2 and qIMSL6-1) were relatively stable, whose LODA value ranged from 4.81 to 6.02, and LODAE ranged from 0.54 to 1.97. Three QTLs (qIMSL2-8, qIMSL7-3 and qIMSL7-4) were detected in more than one year. The highest main-effect QTL underlying MSL identified in five years was qIMSL2-8, its LODA and LODAE was 88.78 and 42.64, respectively. This major QTL qIMSL2-8 was environment-specific, close to qMSL2-7 detected using CIM mapping.
A total 16 QTLs influencing SMS were identified across the five environments: one each on chromosome 1H and 3H,two each on chromosome 4H, four on chromosome 7H and eight on chromosome 2H. Eight QTLs were detected in both single and multiple environments mapping while other eight QTLs were only detected in multiple environments mapping analysis (Additional file 5: Table S5). Four QTLs (qISMS2-3, qISMS2-6, qISMS4-1 and qISMS4-2) were detected in more than one year. qISMS7-1 was relatively stable, its LODA and LODAE was 7.61 and 1.35, respectively. Three QTLs (qISMS2-1, qISMS2-2 and qISMS4-2) had strong QEI. LODA ranged from 1.07 to 1.73, and LODAE ranged from 5.02 to 5.17. The highest main-effect QTL underlying SMS identified in five years was qISMS2-6, its LODA and LODAE was 150.54 and 248.14, respectively. This major QTL qISMS2-6 was environment-specific, close to qSMS2-7 detected using CIM mapping.
For SPD, 15 QTLs were identified across the five environments: one each on chromosome 3H, 5H and 6H,three on chromosome 7H and nine on chromosome 2H. Ten QTLs were detected in both single and multiple environments mapping analyses, while five QTLs were only detected using multiple environments mapping (Additional file 5: Table S5). Three QTLs (qISPD2-6, qISPD2-9 and qISPD5-1) were detected in more than one year. Two QTLs (qISPD2-7 and qISPD7-1) were relatively stable, whose LODA ranged from 4.33 to 5.29, and LODAE ranged from 1.07 to 1.67. qISPD2-5 had strong QEI, its LODA and LODAE was 1.76 and 9.68, respectively. The highest main-effect QTL underlying SPD identified was qISPD2-9, its LODA and LODAE was 66.82 and 56.50, respectively. This major QTL qISPD2-9 was environment-specific, close to qSPD2-6 detected using CIM mapping.
For SP, 5 QTLs were identified across the five environments: one on chromosome 5H, two each on chromosome 2H and 7H. All QTLs were detected using both single and multiple environments mapping analyses (Additional file 5: Table S5). Only qISP5-1 was detected in more than one year. qISP2-1 had strong QEI, its LODA and LODAE was 1.24 and 4.83, respectively. Other four QTLs were identified with environment-specific effect. The highest main-effect QTL underlying SP identified was qISP7-2, its LODA and LODAE was 6.39 and 20.46, respectively.
For SLP, 4 QTLs were identified across the five environments: one each on chromosome 4H and 7H, two on chromosome 2H. All QTLs were detected in both single and multiple environments mapping analyses (Additional file 5: Table S5). Only qISLP2-2 was detected in more than one year. qISLP2-1 had strong QEI, its LODA and LODAE was 0.33 and 5.52, respectively. Other three QTLs were identified with environment-specific effect. The highest main-effect QTL underlying SP identified in four years was qISLP2-2, its LODA and LODAE was 26.16 and 61.52, respectively. This major QTL qISLP2-2 was environment-specific, close to qSLP2-6 detected using CIM mapping.
For TGW, 14 QTLs were identified across the five environments: one on chromosome 3H,two each on chromosome 1H, 2H, 4H and 5H, five on chromosome 7H. Ten QTLs were detected using both single and multiple environments mapping analyses, while four QTLs were only detected using multiple environments mapping (Additional file 5: Table S5). Six QTLs (qITGW2-1, qITGW2-2, qITGW5-1, qITGW7-1, qITGW7-4 and qITGW7-5) were detected in more than one year. qITGW3-1 was relatively stable, its LODA and LODAE was 4.14 and 1.15, respectively. qITGW4-1 had strong QEI, its LODA and LODAE was 0.50 and 6.96, respectively. The highest main-effect QTL underlying TGW identified was qITGW2-2, its LODA and LODAE was 91.64 and 89.10, respectively. This major QTL qITGW2-2 was environment-specific, close to qTGW2-2 detected using CIM mapping.
For GP, 5 QTLs were identified across the five environments: one on chromosome 2H,two each on chromosome 4H and 7H. Three QTLs were detected using both single and multiple environments mapping analyses, while two QTLs (qIGP4-1 and qIGP7-2) were only detected using multiple environments mapping (Additional file 5: Table S5). qIGP7-1 had strong QEI, its LODA and LODAE was 1.64 and 13.43, respectively. The highest main-effect QTL underlying GS identified in four years was qIGP2-1, its LODA and LODAE was 56.99 and 36.23, respectively. This major QTL qIGP2-1 was environment-specific, close to qGP2-1 detected using CIM mapping.
For GS, 11 QTLs were identified across the five environments: one on chromosome 3H, three each on chromosome 1H and 7H, four on chromosome 2H. Eight QTLs were detected using both single and multiple environments mapping analyses, while three QTLs were only detected using multiple environments mapping (Additional file 5: Table S5). Two QTLs (qIGS2-2 and qIGS2-3) were detected in more than one year. Three QTLs (qIGS7-1, qIGS7-2 and qIGS7-3) had strong QEI, whose LODA ranged from 0.91 to 1.71, and LODAE ranged from 5.69 to 9.35. The highest main-effect QTL underlying GS identified in four years was qIGS2-3, its LODA and LODAE was 86.98 and 110.08, respectively. This major QTL qIGS2-3 was environment-specific, close to qGS2-4 detected using CIM mapping.
For GWP, 8 QTLs were identified across the five environments: one on chromosome 6H, two each on chromosome 2H and 3H, three on chromosome 7H. Four QTLs were detected using both single and multiple environments mapping analyses, while four QTLs were only detected using multiple environments mapping (Additional file 5: Table S5). Only qIGWP2-1 was detected in more than one year. qIGWP7-2 had strong QEI, LODA and LODAE was 0.95 and 20.70, respectively. The highest main-effect QTL underlying GWP was qIGWP6-1, its LODA and LODAE was 12.25 and 28.30, respectively.
For GWS, 15 QTLs were identified across the five environments: one each on chromosome 3H, 5H and 6H, three each on chromosome 2H and 4H, seven on chromosome 7H. Eight QTLs were detected using both single and multiple environments mapping analyses, while seven QTLs were only detected using multiple environments mapping (Additional file 5: Table S5). Two QTLs (qIGWS2-3 and qIGWS7-6) were detected in more than one year. qIGWS4-3 was relatively stable, its LODA and LODAE was 4.59 and 1.20, respectively. The highest main-effect QTL underlying GWS was qIGWS7-2, its LODA and LODAE was 85.71 and 2.32, respectively.
Discussion
Advantages of mapping agronomic trait with SNP markers
Sequence-based genotyping method could provide high quality SNPs for constructing an ultra-high density genetic map. In the present study, a high density genetic linkage map containing 1894 SNP markers and 68 SSR markers covering 1375.8 cM were used to identify quantitative trait loci. In the previous study using 153 SSR markers to dissect agronomic traits, we detected 12 QTLs for eight agronomic traits, one for SP, two for MSL, one for SLP, two for GP,one for GS, two for GWP, two for GWS, and one for TGW in 2009 [42]. In present study, we detected 35 QTLs for those traits: three for SP, six for MSL, six for SLP, four for GS, five for GWP, five for GWS and six for TGW with SNP markers (Table 2). In 2010, we detected 6 QTLs for four agronomic traits: two for MSL, one for GWP, one for GWS, two for TGW [42]. In present study, we detected 38 QTLs for those traits: three for SP, six for MSL, six for SLP, three for GP, four for GS, five for GWP, five for GWS and six for TGW with SNP markers (Table 2). It demonstrated advantage of SNP in detecting power and resolution relative to SSR map. The SNP markers were distributed across all seven linkage groups, with polymorphic loci covering both coding and noncoding regions in the barley genome. The accuracy and quality of the SNP markers identified will provide more associated markers for marker-assistant breeding (MAS) [23, 28, 34, 54, 55].
QTL affecting grain yield and its related agronomic traits
In barley, main spike length (MSL) and spike density (SPD) are two of the most important spike morphological traits. They not only affect grain yield potential, but also the yield of malt extract [56]. In our study, three credible QTLs were detected for MSL using CIM mapping (Table 3). qMSL7-5 was detected in four years, indicating this QTL was not affected by environment. This QTL was also mapped on the same position with the dwarf gene btwd1 that we had previously studied, which was linked to the SSR marker Bmac0031 and Bmac0167 [41]. This QTL is likely same to the QTL Qel 7.1 on the chromosome 7H reported by Li et al. [57] , and different from the QTL reported by Sameri et al. [58]. On the terminal of chromosome 2HL, qMSL2-7 was close to the 2_598509820 marker, and is likely the same locus on 2HL reported by Xue et al. [59]. The QTL, qMSL1-2 (Additional file 4: Table S4), close to the marker 1H_10863328 and SSR markers Bmac90 and EBmac501 on chromosome 1H, is likely different from the Qsl-tera_1H reported by von Korff et al. [60]. The SSR marker Bmac90 was associated with days to heading on chromosome 1H [61]. QTLs for main MSL were reported to be located on all seven linkage groups [16, 22, 56–58, 62, 63].
QTLs conferring SPD were reported on 2H, 3H and 7H [57, 63–65]. On chromosome 2H, there were two significant QTLs (qSPD2-4 and qSPD2-6). qSPD2-6 was close to the SSR marker Ebmag793, which is near to the marker EBmac415 inferred from http://wheat.pw.usda.gov/GG3/, and is likely the same loci on chromosome 2HL as reported by Sameri et al. [57]. qSPD2-4 is likely new one for spike density (SPD). qSPD4-1 was detected in four years and close to the marker M_153521_1223, and is likely a new QTL. qSPD7-3 had main effect on spike density, is close to the marker 7_379313804 and SSR marker Bmac0031 on chromosome 7HS, and is likely the same loci reported by Sameri et al. and Shahinnia et al. [57, 65].
The spikelet number on main spike (SMS) and spikelet number per plant (SLP) are closely correlated. As expected, two QTL regions (qSMS2-7 and qSLP2-6, qSMS4-4 and qSLP4), located nearby to each other on the chromosome were detected (Table 3). QTLs conferring SMS were previously reported on chromosome 1H, 2H, 5H and 7H [19, 66]. The main effect QTL qSMS2-7 associated with the marker 2HL_22930294 was close to the morphological marker Vrs1 on chromosome 2H, explaining 72.76 % to 82.22 % of phenotypic variance. The Vrs1 locus, which primarily determines the row type of spike, has a pleiotropic effect on many agronomic characters such as the number of rachis nodes, spike length, stem length, thousand grain weight, fusarium head blight (FHB) resistance and heading date [18, 19, 60, 67–69]. qSMS4-4 and qSMS4-5 were close to the SSR marker HVM40, and are likely new loci underlying SMS. QTLs for SLP were detected on chromosome 2H and 4H, and QTLs for controlling spikelet number were rarely reported in barley.
For grain number per plant (GP) and grain numbers per spike (GS), there was a common QTL on chromosome 2H (Table 3). This QTL was close to the SNP marker 2HL_22930294, which was nearby the morphological marker Vrs1. QTLs for grain number per plant are reported to be located on chromosomes 1H [17, 22], 2H [56, 62, 63], 3H and 4H [70].
Only one credible QTL qSP5-1 for spike number per plant was detected and close to the SNP marker 5_6313908 and SSR marker GBM1176. The SSR marker GBM1176 tightly linked to SNP marker scssr07106 that was associated with a main QTL for abiotic stress on chromosome 5H [11]. QTLs underlying spike numbers per plant were previously reported on the 1H, 2H, 5H, 6H and 7H [22, 37, 66].
Thousand grain weight (TGW) is one of the major yield components having direct effect on final yield. As shown in Table 3, the SNP associated with TGW were previously reported on seven linkage groups [5, 17, 37, 59, 63, 71, 72]. The main effect QTL qTGW2-1 and qTGW2-2 associated with the marker 2HL_22930294 and 2HL_22930005, respectively, were close to the morphological marker Vrs1, explaining 43.45 % to 61.78 % of phenotypic variance. qTGW5-1 on the terminal of 5HS is likely new QTL and different from previously reported [17]. The qTGW7-1 on chromosome 7H was close to the marker 7HL_28107982, which is in the same region close to the morphological marker Nud1 on the chromosome 7H. This QTL seems to be a new one and different from the QTLs on chromosome 7H reported by Pillen et al. [17] and von Korff et al. [59].
QTLs conferring grain yield were reported to be located on all seven chromosomes [59, 62, 70, 73]. QTLs for GWP and GWS were mainly detected on chromosome 2H and 7H (Table 3). qGWP2-1 and qGWS2-3 were at the same region on chromosome. qGWP7-3 and qGWP7-4 are likely same ones reported by [17] on short arm of chromosome 7H.
QTL × environment interaction (QEI)
Because of genetic variation in complex traits including environment × genotype interaction effect, some QTLs were often detected with small effects and low stability across different environments. Thus, consistency of QTLs across different years and environments is a major concern for marker-assisted selection to improve complex traits, especially for yield traits. In this study, we used multi-environment analysis to detect QTLs with QEI effects and find stable QTLs across different environments for 10 agronomic traits related to yield. Nine QTLs (qIMSL3-1, qIMSL4-1, qIMSL4-2, qIMSL6-1, qISMS7-1, qISPD2-7, qISPD7-1, qITGW3-1 and qIGWS4-3) were detected with minimal QEI effects and stable across different years. These QTLs may be better ones for MAS-based breeding. Compared the QTLs detected using single analysis with CIM mapping and multi-environment analysis with ICIM mapping, we found that 71 QTLs identified using CIM mapping were also detected using ICIM mapping, especially for QTL detected in more than one environment. For instance, the main effect QTL (qMSL2-7) for MSL identified using CIM mapping located at 184.71 cM on the chromosome 2H, was also detected using ICIM mapping. However, some QTLs with special environment interaction detected using ICIM mapping were not detected using CIM.
Clustering of the detected QTLs on linkage groups
A number of QTLs underlying the agronomic traits were detected in this cross. Some were highly genetically correlated and mapped to similar positions (Fig. 1). There are 11 genomic regions controlling these traits studied here: one on chromosome 1H, three on the chromosome 2H, one on chromosome 3H, one on chromosome 4H, one on chromosome 5H and four on chromosome 7H. A cluster of QTL for SMS, GS, SLP, GP, GWP and TGW was found on chromosome 2H (Fig. 1). This cluster was close to the morphological marker Vrs1. Some QTLs for SPD and MSL were located on the same region, such as on long arm of chromosome 2H and on chromosome 7H, but some mapped independently. Other traits like SMS, SPD and SLP were also clustered on chromosome 4H.
Conclusions
A number of QTLs associated with multiple traits were found on certain chromosome regions, and some of these traits were significantly correlated with each other. The two parents used for mapping population construction are different row types (six-rowed dwarfing barley cultivar Huaai 11 and two-rowed barley cultivar Huadamai 6). A lot of QTLs detected were close to the morphological marker Vrs1 region, for example qSMS2-7, qSLP2-6, qTGW2-1, qTGW2-2, qGP2-2, qGS2-4 and qGWP2-1. On the terminal of chromosome 7H, the QTLs for MSL, SP and SPD were clustered together. The SSR maker GBM1149 and SNP marker 2_598509820 could be used for marker-assisted selection for these three traits.
Abbreviations
DH, Double haploid; RAD, Restriction site associated DNA; GP, Grain number per plant; GWP, Grain weight per plant; GS, Grain number per spike; TGW, Thousand grain weight; GWS, Grain weight per spike; SPD, pike density; ICIM, Inclusive composite interval mapping; LOD, Likelihood of odd; CIM, Composite interval mapping; QEI, QTL × environment interaction; MSL, Length of main spike; QTL, Quantitative trait locus; MAS, Marker assisted selection; SMS, Spikelet number on main spike; SLP, Spikelet number per plant; SNP, single nucleotide polymorphism; SP, Spike numbers per plant.SM, Spike morphology
References
Varshney RK, Graner A, Sorrells ME. Genomics-assisted breeding for crop improvement. Trends Plant Sci. 2005;10:621–30.
Toojinda T, Baird E, Booth A, Broers L, Hayes P, Powell W, et al. Introgression of quantitative trait loci (QTLs) determining stripe rust resistance in barley: an example of marker-assisted line development. Theor Appl Genet. 1998;96:123–31.
Yano M. Genetic and molecular dissection of naturally occurring variation. Curr Opin Plant Biol. 2001;4:130–5.
Kleinhofs A, Kilian A, Maroof MS, Biyashev RM, Hayes P, Chen F, et al. A molecular, isozyme and morphological map of the barley (Hordeum vulgare L.) genome. Theor Appl Genet. 1993;86:705–12.
Backes G, Graner A, Foroughi-Wehr B, Fischbeck G, Wenzel G, Jahoor A. Localization of quantitative trait loci (QTL) for agronomic important characters by the use of a RFLP map in barley (Hordeum vulgare L.). Theor Appl Genet. 1995;90:294–302.
Van Deynze AE, Nelson JC, Yglesias ES, Harrington SE, Braga DP, McCouch SR, et al. Comparative mapping in grasses. Wheat relationships. Mol Gen Genet. 1995;248:744–54.
Marquez-Cedillo L, Hayes P, Jones B, Kleinhofs A, Legge W, Rossnagel B, Sato K, et al. QTL analysis of malting quality in barley based on the doubled-haploid progeny of two elite North American varieties representing different germplasm groups. Theor Appl Genet. 2000;101:173–84.
Heun M, Kennedy A, Anderson J, Lapitan N, Sorrells M, Tanksley S. Construction of a restriction fragment length polymorphism map for barley (Hordeum vulgare L.). Genome. 1991;34:437–47.
Hayes PM, Blake T, Chen TH, Tragoonrung S, Chen F, Pan A, et al. Quantitative trait loci on barley (Hordeum vulgare L.) chromosome 7 associated with components of winterhardiness. Genome. 1993;36:66–71.
Qi X, Stam P, Lindhout P. Comparison and integration of four barley genetic maps. Genome. 1996;39:379–94.
Rostoks N, Mudie S, Cardle L, Russell J, Ramsay L, Booth A, et al. Genome-wide SNP discovery and linkage analysis in barley based on genes responsive to abiotic stress. Mol Genet Genomics. 2005;274:515–27.
Wenzl P, Li H, Carling J, Zhou M, Raman H, Paul E, et al. A high-density consensus map of barley linking DArT markers to SSR, RFLP and STS loci and agricultural traits. BMC Genomics. 2006;7:206.
Hearnden P, Eckermann P, McMichael G, Hayden M, Eglinton J, Chalmers K. A genetic map of 1,000 SSR and DArT markers in a wide barley cross. Theor Appl Genet. 2007;115:383–91.
Varshney RK, Marcel TC, Ramsay L, Russell J, Röder MS, Stein N, et al. A high density barley microsatellite consensus map with 775 SSR loci. Theor Appl Genet. 2007;114:1091–103.
Wang J, Yang J, Jia Q, Zhu J, Shang Y, Hua W, et al. A new QTL for plant height in barley (Hordeum vulgare L.) showing no negative effects on grain yield. PloS One. 2014;2:e90144.
Hori K, Kobayashi T, Shimizu A, Sato K, Takeda K, Kawasaki S. Efficient construction of high-density linkage map and its application to QTL analysis in barley. Theor Appl Genet. 2003;107:806–13.
Pillen K, Zacharias A, Léon J. Advanced backcross QTL analysis in barley (Hordeum vulgare L.). Theor Appl Genet. 2003;107:340–52.
Komatsuda T, Maxim P, Senthil N, Mano Y. High-density AFLP map of nonbrittle rachis 1 (btr1) and 2 (btr2) genes in barley (Hordeum vulgare L.). Theor Appl Genet. 2004;109:986–95.
Hori K, Kobayashi T, Sato K, Takeda K. QTL analysis of Fusarium head blight resistance using a high-density linkage map in barley. Theor Appl Genet. 2005;111:1661–72.
Szűcs P, Blake VC, Bhat PR, Chao S, Close TJ, Cuesta-Marcos A, et al. An integrated resource for barley linkage map and malting quality QTL alignment. Plant Genome. 2009;2:134–40.
Aghnoum R, Marcel TC, Johrde A, Pecchioni N, Schweizer P, Niks RE. Basal host resistance of barley to powdery mildew: connecting quantitative trait loci and candidate genes. Mol Plant Microbe. 2010;23:91–102.
Chutimanitsakun Y, Nipper RW, Cuesta-Marcos A, Cistué L, Corey A, Filichkina T, et al. Construction and application for QTL analysis of a Restriction Site Associated DNA (RAD) linkage map in barley. BMC Genomics. 2011;12:4.
Poland JA, Brown PJ, Sorrells ME, Jannink J-L. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PloS One. 2012;7:e32253.
Schuster SC. Next-generation sequencing transforms today′s biology. Nat Methods. 2008;5:16–8.
Mardis ER. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008;24:133–41.
Varshney RK, Nayak SN, May GD, Jackson SA. Next-generation sequencing technologies and their implications for crop genetics and breeding. Trends Biotechnol. 2009;27:522–30.
Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, Blaxter ML. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat Rev Genet. 2011;12:499–510.
Nielsen R, Paul JS, Albrechtsen A, Song YS. Genotype and SNP calling from next-generation sequencing data. Nat Rev Genet. 2011;12:443–51.
Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, et al. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PloS One. 2008;3:e3376.
Huang X, Feng Q, Qian Q, Zhao Q, Wang L, Wang A, et al. High-throughput genotyping by whole-genome resequencing. Genome Res. 2009;19:1068–76.
Andolfatto P, Davison D, Erezyilmaz D, Hu TT, Mast J, Sunayama-Morita T, et al. Multiplexed shotgun genotyping for rapid and efficient genetic mapping. Genome Res. 2011;21:610–7.
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PloS One. 2011;6:e19379.
Miller MR, Dunham JP, Amores A, Cresko WA, Johnson EA. Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Res. 2007;17:240–8.
Van Tassell CP, Smith TP, Matukumalli LK, Taylor JF, Schnabel RD, Lawley CT, et al. SNP discovery and allele frequency estimation by deep sequencing of reduced representation libraries. Nat Methods. 2008;5:247–52.
Pfender W, Saha M, Johnson E, Slabaugh M. Mapping with RAD (restriction-site associated DNA) markers to rapidly identify QTL for stem rust resistance in Lolium perenne. Theor Appl Genet. 2011;122:1467–80.
Barchi L, Lanteri S, Portis E, Valè G, Volante A, Pulcini L, et al. A RAD tag derived marker based eggplant linkage map and the location of QTLs determining anthocyanin pigmentation. PLoS One. 2012;7:e43740.
Peighambari SA, Samadi BY, Nabipour A, Charmet G, Sarrafi A. QTL analysis for agronomic traits in a barley doubled haploids population grown in Iran. Plant Sci. 2005;169:1008–13.
Bezant J, Laurie D, Pratchett N, Chojecki J, Kearsey M. Mapping QTL controlling yield and yield components in a spring barley (Hordeum vulgare L.) cross using marker regression. Mol Breed. 1997;3:29–38.
Lu CF, Shen LS, Tan Z, Xu Y, He P, Chen Y, et al. Comparative mapping of QTLs for agronomic traits of rice across environments using a doubled haploid population. Theor Appl Genet. 1996;93:1211–7.
Thomas WTB, Powell W, Waugh R, Chalmers KJ, Barua UM, Jack P, et al. Detection of quantitative trait loci for agronomic, yield, grain and disease characters in spring barley (Hordeum vulgare L.). Theor Appl Genet. 1995;91:1037–47.
Ren XF, Sun DF, Guan WW, Sun GL, Li CD. Inheritance and identification of molecular markers associated with a novel dwarfing gene in barley. BMC Genet. 2010;11:89.
Ren XF, Sun DF, Sun GL, Li CD, Dong WB. Molecular detection of QTL for agronomic and quality traits in a doubled haploid barley population. Aust J Crop Sci. 2013;7:878–86.
Liu LP, Sun GL, Ren XF, Li CD, Sun DF. Identification of QTL underlying physiological and morphological traits of flag leaf in barley. BMC Genet. 2015;16:1.
Davey JW, Cezard T, Fuentes‐Utrilla P, Eland C, Gharbi K, Blaxter ML. Special features of RAD sequencing data: implications for genotyping. Mol Ecol. 2013;22:3151–64.
Ramsay L, Macaulay M, Ivanissevich SD, MacLean K, Cardle L, Fuller J, et al. A simple sequence repeat-based linkage map of barley. Genetics. 2000;156:1997–2005.
Wu Y, Bhat PR, Close TJ, Lonardi S. Efficient and accurate construction of genetic linkage maps from noisy and missing genotyping data. PLos Genet. 2008;4:e1000212.
Van Os H, Andrzejewski S, Bakker E, Barrena I, Bryan GJ, Caromel B, et al. Construction of a 10,000-marker ultradense genetic recombination map of potato: providing a framework for accelerated gene isolation and a genomewide physical map. Genetics. 2006;173:1075–87.
Van Ooijen JW. JoinMap 4, Software for the calculation of genetic linkage maps in experimental populations. Wageningen, TheNetherlands: Kyazma BV; 2006.
Wang S, Basten CJ, Zeng ZB. Windows QTL Cartographer 2.5. Raleigh, NC: Department of Statistics, North Carolina State Univ; 2007.
Churchill G, Doerge R. Empirical threshold values for quantitative trait mapping. Genetics. 1994;138:963–71.
Lander ES, Botstein D. Mapping mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics. 1989;121:185–99.
Hackett CA. Statistical methods for QTL mapping in cereals. Plant Mol Biol. 2002;48:585–99.
Voorrips R. MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered. 2002;93:77–8.
Nicolae DL, Gamazon E, Zhang W, Duan S, Dolan ME, Cox NJ. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 2010;6:e1000888.
Close TJ, Bhat PR, Lonardi S, Wu Y, Rostoks N, Ramsay L, et al. Development and implementation of high-throughput SNP genotyping in barley. BMC Genomics. 2009;10:582.
Wang J, Yang J, McNeil DL, Zhou M. Identification and molecular mapping of a dwarfing gene in barley (Hordeum vulgare L.) and its correlation with other agronomic traits. Euphytica. 2010;175:331–42.
Li J, Huang X, Heinrichs F, Ganal M, Röder M. Analysis of QTLs for yield, yield components, and malting quality in a BC3-DH population of spring barley. Theor Appl Genet. 2005;110:356–63.
Sameri M, Takeda K, Komatsuda T. Quantitative trait loci controlling agronomic traits in recombinant inbred lines from a cross of oriental-and occidental-type barley cultivars. Breed Sci. 2006;56:243–52.
Xue DW, Zhou MX, Zhang XQ, Chen S, Wei K, Zeng FR, et al. Identification of QTLs for yield and yield components of barley under different growth conditions. J Zhejiang Univ Sci B. 2010;11:169–76.
Von Korff M, Grando S, Del Greco A, This D, Baum M, Ceccarelli S. Quantitative trait loci associated with adaptation to Mediterranean dryland conditions in barley. Theor Appl Genet. 2008;117:653–69.
Mesfin A, Smith K, Dill-Macky R, Evans C, Waugh R, Gustus C, et al. Quantitative trait loci for Fusarium head blight resistance in barley detected in a two-rowed by six-rowed population. Crop Sci. 2003;43:307–18.
Islamovic E, Obert DE, Oliver RE, Marshall JM, Miclaus KJ, Hang A, et al. A new genetic linkage map of barley (Hordeum vulgare L.) facilitates genetic dissection of height and spike length and angle. Field Crop Res. 2013;154:91–9.
Baghizadeh A, Taleei AR, Naghavi MR. QTL analysis for some agronomic traits in barley (Hordeum vulgare L.). Int J Agric Biol. 2007;9:372–4.
Taketa S, Yuo T, Sakurai Y, Miyake S, Ichii M. Molecular mapping of the short awn 2 (lks2) and dense spike 1 (dsp1) genes on barley chromosome 7H. Breed Sci. 2011;61:80–5.
Shahinnia F, Druka A, Franckowiak J, Morgante M, Waugh R, Stein N. High resolution mapping of Dense spike-ar (dsp. ar) to the genetic centromere of barley chromosome 7H. Theor Appl Genet. 2012;124:373–84.
Li J, Huang X, Heinrichs F, Ganal M, Röder M. Analysis of QTLs for yield components, agronomic traits, and disease resistance in an advanced backcross population of spring barley. Genome. 2006;49:454–66.
Baum M, Grando S, Backes G, Jahoor A, Sabbagh A, Ceccarelli S. QTLs for agronomic traits in the Mediterranean environment identified in recombinant inbred lines of the cross' Arta' × H. spontaneum 41-1. Theor Appl Genet. 2003;107:1215–25.
Pourkheirandish M, Wicker T, Stein N, Fujimura T, Komatsuda T. The barley six-rowed spike gene vrs1 reveals a breakdown of rice-barley micro collinearity by a transposition. Theor Appl Genet. 2007;114:s1357–1365.
Yoshida M, Kawada N, Tohnooka T. Effect of row type, flowering type and several other spike characters on resistance to Fusarium head blight in barley. Euphytica. 2005;141:217–27.
Teulat B, Merah O, Souyris I, This D. QTLs for agronomic traits from a Mediterranean barley progeny grown in several environments. Theor Appl Genet. 2001;103:774–87.
Kjaer B, Jensen J. Quantitative trait loci for grain yield and yield components in a cross between a six-rowed and a two-rowed barley. Euphytica. 1996;90:39–48.
Pasam RK, Sharma R, Malosetti M, van Eeuwijk FA, Haseneyer G, Kilian B, et al. Genome-wide association studies for agronomical traits in a world wide spring barley collection. BMC Plant Biol. 2012;12:1.
Marquez-Cedillo L, Hayes P, Kleinhofs A, Legge W, Rossnagel B, Sato K, et al. QTL analysis of agronomic traits in barley based on the doubled haploid progeny of two elite North American varieties representing different germplasm groups. Theor Appl Genet. 2001;103:625–37.
Funding
This study was supported in part by the National Natural Science Foundation of China (31301310 and 31228017) and the earmarked fund for China Agriculture Research System (CARS-5).
Availability of data and materials
All relevant datasets supporting the conclusions of this article are available within the article and its additional files.
Authors’ contributions
JW performed this study, statistical analysis and manuscript writing. XR,LL, QW and BD assisted in phenotyping and software analysis. DS and GS conceived this study, coordinated the experiments, and wrote the manuscript. CL produced the Huaai 11 and Huadamai 6 DH population. All authors have read and approved the manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Our research did not involve any human subjects, human material, or human data. All our studies have been performed in accordance with ethics committee polices. The field test in the Experimental Farm of Huazhong Agricultural had been allowed and not involved private interests.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Table S1.
Analysis of variance on 10 agronomic traits in DH lines. (XLSX 8 kb)
Additional file 2: Table S2:
Correlation coefficients among the 10 observed agronomic traits. (XLSX 9 kb)
Additional file 3: Table S3:
Genome mapping of SNP sequence markers on Hordeum gene index in the NCBI. (XLSX 37 kb)
Additional file 4: Table S4:
List of 221 QTLs detected for 10 agronomic traits, their position, likelihood of odd (LOD), additive effect, explained variance and 2-LOD confidence interval. (XLSX 27 kb)
Additional file 5: Table S5:
Putative QTLs for 10 agronomic traits through multiple environments QTL mapping. (XLSX 25 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wang, J., Sun, G., Ren, X. et al. QTL underlying some agronomic traits in barley detected by SNP markers. BMC Genet 17, 103 (2016). https://doi.org/10.1186/s12863-016-0409-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12863-016-0409-y