
Abstract
Serum or plasma is frequently utilized in biomedical research; however, its application is impeded by the requirement for invasive sample collection. The non-invasive nature of urine collection makes it an attractive alternative for disease characterization and biomarker discovery. Mass spectrometry-based protein profiling of urine has led to the discovery of several disease-associated biomarkers. Proteomic analysis of urine has not only been applied to disorders of the kidney and urinary bladder but also to conditions affecting distant organs because proteins excreted in the urine originate from multiple organs. This review provides a progress update on urinary proteomics carried out over the past decade. Studies summarized in this review have expanded the catalog of proteins detected in the urine in a variety of clinical conditions. The wide range of applications of urine analysis—from characterizing diseases to discovering predictive, diagnostic and prognostic markers—continues to drive investigations of the urinary proteome.
Similar content being viewed by others

Blood and its cellular constituents (e.g., lymphocytes) are the most commonly used specimens for laboratory testing and diagnosis. However, collecting blood samples is an invasive procedure and the development of more non-invasive methods of clinical testing and diagnosis is important for healthcare delivery [1]. The non-invasive nature of urine collection and its deployment for research as well as clinical testing offers a distinct advantage over other body fluids or tissue specimens. A simple urinalysis is often an essential component of managing different disorders including diabetes, kidney diseases and urinary tract infection [2, 3]. Notably, detecting the presence of human chorionic gonadotropin (hCG) hormone in urine to detect pregnancy is perhaps the most popular point-of-care test in medical diagnostics.
In contrast to serum or blood, which are commonly used but require invasive methods for collection, urine can be collected in a simple and non-invasive fashion and in significant quantities [4, 5]. Indeed, urine has also become a valuable specimen in the field of biomedical research due to its widespread availability among patients and the straightforward collection process. Various urine collection methods such as first-morning, 24-h or random spot collection can facilitate easy follow-up and time-based studies [6,7,8]. From an analytical standpoint, the composition of urine proteome is less complex than that of serum or plasma making it more feasible for proteomic analyses where abundant proteins can obscure signals derived from less abundant proteins [9, 10]. The glomeruli of a healthy kidney filter 150–180 L of urine each day of which 99% is reabsorbed [4]. Among the proteins observed in urine of healthy individuals, 70% originate in the urinary tract, while the remaining are proteins filtered from plasma [11]. Liver and kidney are major organs that maintain homeostasis in blood and urine reflects any alterations. It should be noted that the earliest described urinary biomarkers are from the field of nephrology [12, 13]. Thus, analysis of urinary proteins as biomarkers is of great importance not only for diseases of the urinary tract, but also for diseases affecting more distant organs [14]. Several analytical techniques have been employed to study urine proteome; however, mass spectrometry has emerged as a sensitive method for global urinary protein analysis. One of the earliest mass spectrometry-based proteomics analysis of urine identified 124 proteins [15] while current studies using high resolution mass spectrometry routinely identify between 1000 and 3000 proteins depending on the methods and instrumentation [10, 16].
Numerous factors such as sample collection, protein normalization, intra- and inter-individual differences in protein amount are to be considered when carrying out urine proteomics. Although urine is a less complex biospecimen, these inter- and intra-individual variations leads to the disparity in the protein content of urine. Moreover, urine collected at different times during the course of a day can also exhibit variation in the protein content. Usually, the second urine in the morning or 24-h urine is considered to be a “gold standard” for proteome analysis. In 24-h collection, the average information of urinary proteins excreted in a day can be obtained while a morning urine (usually second urine) collection avoids the diurnal variation if samples were collected at different time-points during the day [17, 18]. However, the 24-h collection of urine might not be practical due to its dependence of patient compliance and errors that often arise during collection [19]. Other factors such as protein intake, posture, circadian rhythm and normal physical activities can also affect the proteome [20, 21]. First voided or early morning (second) or spot (random) collection have been deployed to minimize the errors caused during 24-h collection [22, 23]. In such cases, the protein amount is usually normalized with respect to total creatinine by considering the protein/creatinine ratio, which is a current practice for quantifying urinary proteins [24, 25].
Analytical methods for urinary proteomics
Urine proteome has been studied using several analytical techniques that can be unbiased, e.g., mass spectrometry-based approaches or biased, e.g. proximity extension assay-based (Olink) or aptamer based (Somalogic) approaches [26,27,28,29]. In this review, we will restrict ourselves to mass-spectrometry-based approaches that have been employed to profile proteins that make up the urine proteome in both healthy and disease states.
-
1.
Matrix-assisted laser desorption ionization- time-of-flight mass spectrometry (MALDI-TOF–MS): MALDI-TOF mass spectrometry has also been used in the study of urine proteomics [30, 31]. This approach makes use of protein or peptide profiling in samples that have been crystallized using a MALDI matrix, often 2,5-Dihydroxybenzoic acid. MALDI is a soft-ionization technique that can analyze hundreds of samples or analytes in a short period of time. MALDI-TOF/TOF-based analyses permit accurate identification of peptides from crude mixtures such as the urinary proteome.
-
2.
Capillary electrophoresis coupled with mass spectrometry (CE-MS): This method is based on use of a mass spectrometer with capillary electrophoresis (CE) at the front end [32, 33]. Based on how proteins move across a gel when subjected to an electrical field, CE separates proteins in a single step with excellent resolution. CE-MS offers the following benefits: 1. It provides fast separation [34]; 2. It is robust and uses cost-effective capillaries rather than costly LC columns [35]; 3. It is compatible with a broad range of buffers and analytes [36] and 4. It provides a stable constant flow, without the need for elution gradients that might otherwise interfere with MS detection [37]. As high molecular weight proteins tend to precipitate at low pH used for CE, analysis of such proteins with CE is not efficient. In addition, the volume of sample that can be introduced into a capillary for separation by CE is small, potentially limiting the detection sensitivity of CE-MS [38,39,40]. Despite efficient separation and low sample requirements, CE-MS alone cannot provide definitive identification (sequence of peptides and proteins) being analyzed. Identification of peptides by their molecular weight alone can lead to false positives. For confident identification and quantification, fragmentation of the precursor ions followed by detection of fragment ions is carried out using tandem mass spectrometry (MS/MS) approaches. Studies have been reported in which CE-MS is performed in parallel with LC–MS/MS to map the precursor ions identified in CE-MS data to peptide sequences using fragmentation information in LC–MS/MS data, which can be a tedious process [41, 42]. However, several studies have utilized ESI for ionization of analytes that are separated using CE prior to performing MS/MS. Such analyses, termed CE-ESI–MS/MS, also allow for definitive peptide identification [43,44,45]. However, it is important to note that CE-ESI–MS/MS favors basic and hydrophilic peptides with low molecular masses, thus leading to an underrepresentation of peptides with relatively larger molecular weight [45].
-
3.
Liquid chromatography coupled with tandem mass spectrometry (LC–MS/MS): The majority of LC–MS-based methods involve trypsin digestion of proteins followed by LC-based separation of the resulting tryptic peptides prior to tandem mass spectrometry [10, 46]. This technique has been applied to identify and quantify thousands of urine proteins including low abundance proteins [10, 16, 47]. One of the major advantages of this approach is the high resolution of separation of the proteins by LC columns based upon different properties of the peptides including hydrophobicity, size or affinity prior to mass spectrometry analysis. This separation of the peptides provides an excellent coverage of proteins identifying the high as well as low abundance proteins in urine [48]. Moreover, this technique can be automated and used to separate large amounts of analytes (protein/peptides) on an high performance liquid chromatography (HPLC) column or tiny amounts of analytes (peptides) on a capillary LC column [49]. Despite providing high-resolution separation, this method is sensitive to interfering compounds in the urine (e.g., salts), which have to be removed during sample processing. The time taken for data acquisition makes it somewhat challenging to analyze very larger sample sets [40].
Urinary proteomics
Urinary proteomics is a burgeoning field of research that focuses on the comprehensive analysis of proteins present in urine (Fig. 1) providing valuable insights into various physiological and pathological processes. Due to its potential in the early diagnosis, prognosis and monitoring of a wide range of diseases non-invasively, urinary proteomics has gained prominence in biomarker discovery. Through advancements in mass spectrometry and high-throughput technologies, unraveling the intricate proteomic signatures within urine has now become feasible, shedding light on the normal [10, 16] as well as disease mechanisms and facilitating personalized medicine in several diseases such as urothelial cancer [50], prostate cancer [51], diabetes [52] and chronic kidney disease (CKD) [53]. Numerous mass spectrometry-based proteomics studies have been performed to profile and quantify the urinary proteins (Table 1). In mass spectrometry-based urine proteome investigations, LC–MS/MS is the most often used technology [10, 54]. More recently, a number of targeted mass spectrometry approaches as well as data dependent acquisition (DDA) [55] and data independent acquisition (DIA) [56, 57] approaches have been used as quantification techniques in urine proteomics studies. Specific biomarker-based investigations for urine proteomics have used targeted mass spectrometry methods such as multiple reaction monitoring (MRM) [58, 59] parallel reaction monitoring (PRM) [60, 61] and a newly developed SureQuant approach for absolute quantification and validation of the targeted proteins [62]. Following sections provide a brief overview of cataloging studies or interesting applications ranging from normal proteome profiling to biomarker discovery for numerous disorders including cancer that were reported over the past decade. A pictorial representation for the same is depicted in Fig. 2.

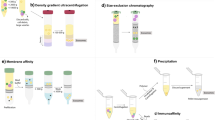
A schematic of the various analytical approaches for urine proteomics. The workflow of mass spectrometry-based approaches to enrich and study urinary proteins, glycoproteins and peptides is shown

A timeline of key studies in the field of urinary proteomics: Highlights of major studies and applications of urinary proteomics over the past decade
Cataloging studies
A number of studies aiming to profile urinary proteins in apparently healthy individuals have contributed to our understanding of normal human urinary proteins and established the baseline for urinary proteomics in several other disorders. Profiling the proteome of urine using mass spectrometry started in the early 2000s. The first LC–MS/MS analysis reported 124 proteins in the normal human urine [15]. Although this work was not expressly intended to identify biomarkers, it did demonstrate the untapped potential of urinary proteome and its usefulness for biomarker discovery. Subsequently, a proteomic study of normal human urine profiled 226 proteins separated using three different approaches. This study employed three different methods of protein and peptide separation in parallel, i.e., GeLC-MS/MS, LC–MS/MS and 2D—LC–MS/MS to achieve such depth [63] with more than 100 proteins identified, including proteins with low molecular mass. Additionally, 171 urinary proteins were newly identified along with 4 male-specific proteins in this study [63]. Shortly thereafter, an LC–MS/MS study was performed identifying more than 1500 proteins in human urine [64]. A proteomic analysis of non-prefractionated urine samples revealed around 1300 proteins including phosphoproteins in the normal human urine proteome for the first time demonstrating the utility of mass spectrometry-based proteomics to identify proteins that are post-translationally modified [65]. In 2011, in a study by Marimuthu et al. 1823 proteins were detected in a comprehensive analysis of human urine proteome [10] identifying around 600 proteins which were not reported to constitute human urine proteome. Many more studies have subsequently been carried out focussing on expanding the protein catalog of urinary proteome. A combination of different analytical approaches to increase the sensitivity and reproducibility of human urine proteome analysis by mass spectrometry yielded 3429 proteins [16]. By utilizing various separation strategies, including direct one-dimensional liquid chromatography-tandem mass spectrometry (LC–MS/MS), two-dimensional LC–MS/MS and gel-eluted liquid fraction entrapment electrophoresis/liquid-phase isoelectric focusing followed by two-dimensional LC–MS/MS, a total of 6085 proteins were found in the human urine proteome representing the largest database of the urine proteome till date [47].
Urinary proteomics in bladder cancer
Proteomics of urine has been widely adopted in multiple diseases especially in cancer for biomarker discovery in a non-invasive fashion. Although urine proteomics has often been performed in the context of renal disorders, some cancers such as lung [66] and prostate [67] cancer have been studied using urine proteomics approach. Over the past decade, nearly 400 studies have been carried out in cancer patients using urine samples. In bladder cancer, the urine is in direct contact with the tumor and its analysis often yields key proteomic alterations [12]. Some of the studies performed on bladder cancer urine proteomics since 2011 are discussed below.
Selevsek et al. reported a mass spectrometry-based targeted proteomics approach to quantify urinary proteins and peptides in bladder cancer. Stable isotope-labeled peptides were used for absolute quantification of nanogram amounts of endogenous peptides in urine using selected reaction monitoring (SRM) [68]. This study showed the potential of targeted mass spectrometry approaches in detection of peptides and proteins in the urine. An increased level of alpha-1-antitrypsin (A1AT) glycoprotein in urine from bladder cancer cohort was identified in a LC–MS/MS-based urine proteomics [69]. In this study, ~ 200 urinary glycoproteins were enriched and subsequently analyzed using a label-free quantitative approach for identification of novel non-invasive biomarkers to diagnose bladder cancer. An overexpression of ADAM28 protein in urine was found to be associated with bladder cancer invasion by two different groups [70, 71]. A combination of different proteomic approaches including western blot and LC–MS/MS was reported in these studies. Increased levels of several other proteins such as apolipoprotein E, alpha-1-antitrypsin and fibrinogen were also found and believed to be associated with recurrence in an LC–MS/MS analysis of urine from non-muscle invasive bladder cancer [72]. A phosphoproteomic study showed a downregulation of a profilin-1 in urine suggesting its association with bladder cancer invasion [73]. This study applied immobilized metal affinity chromatography (IMAC)-based phosphopeptide enrichment to urine samples. A high-resolution mass spectrometry-based analysis revealed a multiplex protein panel of five urine proteins including coronin-1A, apolipoprotein A4, semenogelin-2, gamma synuclein and DJ-1/PARK7 that could serve as diagnostic biomarkers in transitional bladder cancer [74]. Apart from discovery-style proteomics, numerous targeted mass spectrometry approaches have been deployed to validate the protein expression in urine from bladder cancer patients [75, 76]. Not only secreted proteins but extracellular vesicles in urine could also be effectively used as a sample for bladder cancer biomarker discovery [77,78,79,80,81]. Extracellular vesicles are secreted by the cells from various tissues into biological fluids and have roles in intercellular communication and signaling [82] and hence could be used in biomarker research. Recently, Carvalho et al. reported proteomic signatures that can indicate recurrence of bladder cancer [83]. LC–MS/MS-based monitoring of urine proteome at different stages of the disease have shown the utility of these proteins to be used as biomarkers for recurrence of bladder cancer [83]. These studies prove the ability of urine proteomics in discovering non-invasive biomarkers for bladder cancer and could be applied to other cancers.
Urinary proteomics in diabetic nephropathy
In addition to cancer, urinary proteomics has been used to characterize a number of other illnesses, including rheumatoid arthritis [84, 85], urinary tract infections [86], mucopolysaccharidosis [31], COVID-19 [87,88,89,90], neurological diseases [91,92,93,94] and diabetes [95]. A discovery study of urine from type-1 diabetes patients resulted in the identification of proteins that are dysregulated in type-1 diabetes and understanding the molecular mechanisms associated with the complications in type-1 diabetes [96]. According to a study by Fisher et al., patients with diabetic nephropathy had lower levels of urine proteins (e.g., uteroglobin) that are linked to the glomerular filtration rate. This was discovered using immunodepletion of high abundance urinary proteins, followed by fractionation and MALDI-MS analysis [97]. In 2013, Manwaring et al. showed how targeted mass spectrometry-based proteomics might be used to find new diagnostic biomarkers in pediatric patients with Fabry disease and type-1 diabetes [98]. Several other studies have been conducted to elucidate the proteomic alterations in urine from diabetes and associated complications such as nephropathy and retinopathy making it feasible to identify biomarkers for these conditions [99,100,101]. Lysosomal proteins were detected abundantly in the urine from young individuals with type-1 diabetes suggesting its correlation with inflammation in the kidney [102]. A similar study by another group showed the variations in lysosomal function are associated with alterations at proteome level in urine from type-1 diabetes [103]. In another study, alterations in the kallikrein-kinin system were shown to be involved in type-1 diabetes [104]. Urinary chronic kidney disease 3 (CKD3) classifier may be used to predict mortality in people with type 2 diabetes and microalbuminuria as well as to serve as a progressive marker for diabetic nephropathy [105]. Potential prospects for use in pathology and treatment include elevated levels of vascular cell adhesion molecule 1 (VCAM-1) and neprilysin in patients with baseline diabetic nephropathy and diabetic nephropathy-treated patients with persisting albuminuria, respectively [106]. Recently, a data independent analysis-mass spectrometry (DIA-MS) approach was applied to the urinary exosomal proteomics to uncover non-invasive protein-based urinary biomarkers in diagnosing diabetic nephropathy [107].
Urine glycoproteomics
A large number of proteins excreted in the urine are glycoproteins with either N- and/or O-linked glycosylation and may be of potential diagnostic value [108, 109]. Prostate specific antigen (PSA) is an N-linked glycoprotein with a key role in the diagnosis of prostate cancer. Urinary levels of PSA have been shown to be valuable in the differential diagnosis of prostate cancer and benign prostatic hyperplasia when serum levels of PSA are inconclusive [110]. A number of studies have reported on the N-linked glycoproteome of urine and urinary exosomes (Table 2). Methods and technologies used for analyzing the urinary glycoproteome have evolved over the years for characterization at the level of released glycans, deglycosylated peptides and intact glycopeptides with site-specific glycosylation information. For instance, Blaschke et al. have characterized n-glycans released enzymatically from urinary proteins along with prostatic secretions by using MALDI-imaging mass spectrometry [108]. They report that a majority of n-glycans from urinary glycoproteins are biantennary, fucosylated and sialylated [108]. However, the identification of glycoproteins and their glycosylation sites requires alternate methods of sample preparation and mass spectrometry analysis. Huo et al. reported a method to identify soluble urinary glycoproteins and glycoproteins from urinary extracellular vesicles (EVs) along with their glycosylation sites for potential applications towards comprehensive screening [111]. They enriched glycopeptides using hydrophilic interaction liquid chromatography (HILIC) and enzymatically deglycosylated them using PNGase F to identify over 600 n-glycoprotein groups in urine. However, deglycosylated peptide analysis does not provide information on the composition of glycans attached to the identified site, which is critical for biomarker discovery and screening. This information can be obtained by analyzing intact glycopeptides. Halim et al. captured urine-derived glycoproteins containing sialic acid using hydrazide chemistry, digested the captured proteins and analyzed the attached glycopeptides after releasing them by desialylation [54]. LC–MS/MS-based analysis of intact glycopeptides from complex samples has evolved considerably over the past few years [112]. Saraswat et al. enriched glycopeptides derived from urinary exosomes using both lectin affinity and size exclusion chromatography methods and analyzed intact n-glycopeptides by LC–MS/MS [113]. Belczacka et al. performed MS analysis using different technologies on intact urinary glycopeptides to identify both N- and O-linked glycopeptides [114].
Some of the above-mentioned technologies have been applied to study urinary glycoproteomic alterations in various cancers and disorders of the urinary tract and other organ systems. Sathe et al. profiled the urinary n-glycoproteome by analysis of subtypes of bladder cancer by quantitative analysis of deglycosylated n-glycopeptides to identify altered glycosylation levels between the subtypes [115]. Wang et al. performed an exploratory study of enzymatically deglycosylated peptides from prostate cancer patient urine-derived proteins to identify 1044 n-glycosylation sites [116]. Dong et al. developed targeted assays for a three-signature panel of urinary glycoproteins relevant to prostate cancer. They developed a PRM assay to detect peptides from urinary proteins after enzymatic deglycosylation [61]. More recently, Li et al. profiled intact N-linked glycopeptides from urinary extracellular vesicle (EV)-derived glycoproteins in hepatocellular carcinoma (HCC). They identified 756 intact glycopeptides with quantitative information and describe significant changes in the glycoproteome of urinary EVs in HCC [117]. The potential for quantitative analysis of N- and O-linked glycopeptides from urinary glycoproteins is immense in areas of disease characterization and biomarker development. However, it should be noted that available assays for measuring n-glycans and n-glycopeptides in urine can be laborious with multiple sample processing steps [108]. Therefore, there is also a need to optimize these methods to simpler formats before large-scale deployment as diagnostic markers.
Urinary peptidomics
Peptidomics is an emerging field of study that focuses on the comprehensive characterization and quantification of endogenous generated peptides in biological samples including tissues, body fluids and cells. These endogenous peptides play crucial roles in diverse biological and metabolic processes such as communication and signaling, immune response and enzymatic regulation by serving as peptide hormones [118], neuropeptides [119], cytokines [120] and enzyme inhibitors [121]. Numerous applications of peptidomics have led to its adoption across multiple research areas including clinical diagnostics, biomarker discovery and pharmacology. Researchers employ various analytical techniques such as mass spectrometry, liquid chromatography and electrophoresis to study the peptidome from complex biological matrices [122,123,124]. Within the realm of human peptidomics, numerous investigations have been carried out to explore the peptidome of different tissues and diseases. For instance, in a study by Nongonierma and Fitzgerald, the authors characterized peptides derived from dietary proteins to determine their inhibitory effects on dipeptidyl peptidase IV in the human gastrointestinal tract. Their findings highlighted the potential of dietary protein-derived peptides as inhibitors of dipeptidyl peptidase IV with potential implications for managing conditions like diabetes [125]. Another study by Li et al. delved into the peptidomics of human infantile hemangioma tissue. Using LC–MS/MS, the researchers identified and compared endogenous peptides in normal skin and hemangioma tissue. This study provides insights into the role of endogenous peptides in hemangioma development and may contribute to the development of targeted treatments for this condition [126]. Notably, peptidomics has been carried out on several body fluids such as urine, serum [127, 128] and cerebrospinal fluid (CSF) [129, 130] where such endogenously generated peptides are likely to be abundant.
Urinary peptidomics focuses on the study of peptides that can be detected in urine (Fig. 1). Urine peptidomics has gained significant attention in recent years due to its potential for biomarker discovery and disease diagnosis [4, 131,132,133]. Studies have been conducted to explore the peptidome of urine and its clinical applications in cancer [134] and numerous other disorders including chronic kidney disease [135], celiac disease [136] and diabetic nephropathy [137]. Table 3 provides a brief list of notable applications of urinary peptidomics in various physiological conditions. Various facets of urinary peptidomics under both normal and diseased conditions, including cancer, are elaborated upon in the following sections.
Cataloging studies
Detecting and quantifying the endogenous peptides in urine, which span a wide concentration range, has posed a challenge for a single technique. Mullen et al. couple a reflectron time-of-flight analyzer with a capillary electrophoresis (CE) system or a nanoflow high-performance liquid chromatography (HPLC) to generate a catalog of ~ 4500 urinary peptides [138]. Despite the challenges of low peptide abundance and high concentrations of salts and metabolites in urine, a study by Yang et al. used highly ordered mesoporous silica particles to extract peptides [139]. A total of 193 peptides were identified many of which were derived from proteins not commonly found in urine proteome databases. Urinary peptidomics has also been applied to assess age-related peptidome changes in healthy and diseased individuals ranging from 20 to 86 years old [140]. A peptidomic analysis of urinary exosomes led to identification of 3115 unique endogenous peptide fragments corresponding to 942 proteins [141]. A comprehensive analysis of urine proteome and peptidome was performed by Di Meo, et al. using an integrated analytical protocol developed with ultrafiltration [142]. The analysis of urine samples from healthy individuals led to the identification of 1754 proteins in addition to 4543 endogenous peptides derived from 566 proteins in the peptidomic analysis [142]. More recently, a comparative peptidomic analysis of plasma and urine was carried out to explore the origin of peptides in urine. This study identified 561 plasma and 1461 urinary endogenous peptides with only 90 peptides detected in both urine and plasma suggesting that most plasma peptides are not present in urine potentially due to tubular reabsorption [41]. In addition to the above-mentioned efforts, several other peptidomic studies have also been conducted in past decade [143,144,145,146].
Urine peptidomics for biomarkers in renal cancer
The field of urinary peptidomics has shown great promise in identifying biomarkers linked to renal cancer, in particular renal cell carcinoma (RCC). RCC is asymptomatic at early stage and at the time of clinical presentation, the tumor has frequently progressed to an advanced stage [147, 148]. This emphasizes the necessity for early detection and accurate diagnostic methods [149]. A peptidomic analysis was conducted to explore urine peptide signatures that could differentiate malignant kidney tumors from benign masses and controls [150]. Using a MALDI-TOF profiling approach with urine pre-purification, two distinct peptide clusters were identified with higher abundance in patients’ urine and could be linked to proteins associated with tumorigenesis and progression, including meprin 1α (MEP1A), probable G-protein coupled receptor 162 (GPCR162), osteopontin (OPN), phosphorylase b kinase regulatory subunit alpha (PHKA1) and secreted and transmembrane protein 1 (SECTM1) [150]. This study demonstrated the potential of urinary peptidomics in detecting peptide-based biosignatures that can distinguish malignant RCC from benign tumors and healthy controls with potential applications for biomarker discovery for better outcome and disease management. An attempt of urinary peptidomics for discovering a peptide-based biomarker for progression and aggressiveness of RCC resulted in identification of numerous urine peptides in correlation with grading and staging of tumor [151]. Using MALDI-TOF profiling, this study identified 15, 26 and 5 peptides in urine which showed significant alteration of their urinary concentration in concordance with tumour size, stage and grade, respectively [151]. With the potential to enhance early diagnosis and treatment approaches, these findings offer a strong foundation for future studies to evaluate the usefulness of these peptides as possible biomarkers for managing RCC patients. Early detection and prognostic evaluation of RCC are critical for effective management, especially in cases of small renal masses (SRMs) [152]. A quantitative peptidomic approach was deployed to identify potential diagnostic and prognostic biomarkers for early-stage RCC-SRMs revealed the 9 endogenous urinary peptides significantly overexpressed in RCC-SRM [153]. Furthermore, two urinary peptides showed significantly higher expression in progressive clear cell RCC-SRMs compared to nonprogressive cases highlighting the utility of peptide biomarker in escalating clinical diagnosis and disease management [153]. These works illustrate the application of urinary peptidomics in renal cancer research and highlight the potential benefits for better RCC diagnosis and treatment.
Urine peptidomics in chronic kidney diseases (CKD)
Urine peptidomics has been exploited in non-cancer disorders to delve into possible biomarkers and molecular signatures connected to different ailments. This covers a broad spectrum including chronic kidney disease (CKD) [42], cardiovascular diseases [154], diabetes [137, 155], autoimmune disorders, infectious diseases [156] and neurological conditions [157, 158]. The urinary peptidome is primarily derived from the filtration and reabsorption processes within the kidneys. As CKD is a condition characterized by impaired kidney function, studying urinary peptides provides valuable insights into kidney health and dysfunction. A CE-MS-based peptidomic study of urine was conducted to generate a database consisting of 5,010 unique urinary peptides presenting a diverse pool of potential biomarkers for diagnosing and monitoring various diseases including CKD [42]. Another study aimed at screening a high-risk population for developing CKD utilized CE-MS-based urinary peptidomics for identifying a new classifier i.e., CKD273 classifier attributed to different stages of CKD for early risk assessment and intervention [159]. Around 5000 urinary peptides were identified in a mass spectrometry analysis from apparently healthy individuals as well as patients with CKD of which 63 collagen-derived peptides were significantly upregulated and found to be associated with glomerular filtration rate (GFR) [160]. These results point to a substantial correlation between kidney function loss and collagen peptides suggesting that decreased collagen breakdown rather than increased production may be the cause of fibrosis in other organs as well [160]. In 2022, a peptidomic analysis was performed to compare different mass spectrometry approaches including CE-MS, LC–MS and MALDI-MS for profiling samples from diabetic nephropathy patients [161]. The study concluded that a front-end separation plays a crucial role in reliable peptide identification at the MS level providing a better technological approach to be deployed for urine peptidomics [161]. Recently, a link between glycosylation and CKD was established using CE-MS-based urine peptidome analysis which reflected 17 O-linked glycopeptides primarily derived from insulin-like growth factor-II (IGF2). One of these 17 glycopeptides showed a strong negative correlation with age-related estimated glomerular filtration rate (eGFR) suggesting its utility as a peptide biosignature in diagnosis of CKD [162]. In summary, urine-based peptidomics presents a promising approach for better understanding the molecular mechanisms driving the course of CKD and for identifying new biomarkers for early diagnosis and prognosis.
Promises and pitfalls
The field of urinary proteomics holds promise for identifying biomarkers using non-invasive sampling techniques [4, 163]. Understanding the mechanisms behind disease can be greatly enhanced by analyzing the urine proteome not only for kidney-related disorders but also for the conditions affecting distant organs [66, 163,164,165]. Nonetheless, proteomic analysis of urine presents unique challenges such as variability [166, 167] including variation in protein abundance and contaminants [168, 169]. Thus, an appropriate proteomics experiment involving urine necessitates standardized procedures to minimize such variables. It is also important to note that the path from discovery of molecular biomarkers to clinical translation can be quite arduous and not always successful. Studies towards biomarker discovery efforts are best begun with a clear clinical question accompanied by a sound experimental design. Reliable identification and validation of biomarkers can be facilitated by high-throughput and sensitive platforms. Despite all these limitations, urinary proteomics shows significant promise in the development of non-invasive diagnostics eliminating the need for invasive procedures and offering a more patient-friendly approach to healthcare [163, 164]. Box 1 summarizes the different aspects for urinary proteomics including applications and challenges in the field.
Conclusions
MS-based analysis of urinary proteins as well as post-translational modifications such as glycosylation has led to cataloging of a number of proteins originating from both urinary tract and distant organs. Promising biomarkers have been identified in urine as well as extracellular vesicles harvested from urine. Research in this area has evolved significantly through the use of analytical technologies such as nanoflow LC–MS/MS and could be further enhanced with newer methods like ion mobility spectrometry. Overall, such developments in characterizing the urinary proteome could have a high impact on how diseases are diagnosed and treated non-invasively in the future.
Data availability
No datasets were generated or analysed during the current study.
References
Leuzy A, Mattsson-Carlgren N, Palmqvist S, Janelidze S, Dage JL, Hansson O. Blood-based biomarkers for Alzheimer’s disease. EMBO Mol Med. 2022;14(1): e14408.
Marsden J, Pickering D. Urine testing for diabetic analysis. Commun Eye Health. 2015;28(92):77.
Schmiemann G, Kniehl E, Gebhardt K, Matejczyk MM, Hummers-Pradier E. The diagnosis of urinary tract infection: a systematic review. Dtsch Arztebl Int. 2010;107(21):361–7.
Decramer S, de Peredo AG, Breuil B, Mischak H, Monsarrat B, Bascands JL, et al. Urine in clinical proteomics. Mol Cell Proteom. 2008;7(10):1850–62.
Thongboonkerd V. Practical points in urinary proteomics. J Proteome Res. 2007;6(10):3881–90.
Fan G, Gong T, Lin Y, Wang J, Sun L, Wei H, et al. Urine proteomics identifies biomarkers for diabetic kidney disease at different stages. Clin Proteom. 2021;18(1):32.
Chen Z, Kim J. Urinary proteomics and metabolomics studies to monitor bladder health and urological diseases. BMC Urol. 2016;16:11.
Pejcic M, Stojnev S, Stefanovic V. Urinary proteomics–a tool for biomarker discovery. Ren Fail. 2010;32(2):259–68.
Kalantari S, Jafari A, Moradpoor R, Ghasemi E, Khalkhal E. Human urine proteomics: analytical techniques and clinical applications in renal diseases. Int J Proteom. 2015;2015:782798.
Marimuthu A, O’Meally RN, Chaerkady R, Subbannayya Y, Nanjappa V, Kumar P, et al. A comprehensive map of the human urinary proteome. J Proteome Res. 2011;10(6):2734–43.
Thongboonkerd V, McLeish KR, Arthur JM, Klein JB. Proteomic analysis of normal human urinary proteins isolated by acetone precipitation or ultracentrifugation. Kidney Int. 2002;62(4):1461–9.
Ahn JH, Kang CK, Kim EM, Kim AR, Kim A. Proteomics for early detection of non-muscle-invasive bladder cancer: clinically useful urine protein biomarkers. Life. 2022;12(3):395.
Gao Y. Urine-an untapped goldmine for biomarker discovery? Sci China Life Sci. 2013;56(12):1145–6.
Thongboonkerd V, Malasit P. Renal and urinary proteomics: current applications and challenges. Proteomics. 2005;5(4):1033–42.
Spahr CS, Davis MT, McGinley MD, Robinson JH, Bures EJ, Beierle J, et al. Towards defining the urinary proteome using liquid chromatography-tandem mass spectrometry. I. Profiling an unfractionated tryptic digest. Proteomics. 2001;1(1):93–107.
Santucci L, Candiano G, Petretto A, Bruschi M, Lavarello C, Inglese E, et al. From hundreds to thousands: widening the normal human urinome (1). J Proteom. 2015;112:53–62.
Wang HB, Li R, Liu R, Cui XF, Pan WJ, Sun A. Second morning ACR could be the alternative to first morning ACR to assess albuminuria in elderly population. J Clin Lab Anal. 2016;30(2):175–9.
Kawamura M, Ohmoto A, Hashimoto T, Yagami F, Owada M, Sugawara T. Second morning urine method is superior to the casual urine method for estimating daily salt intake in patients with hypertension. Hypertens Res. 2012;35(6):611–6.
Bottini PV, Ribeiro Alves MA, Garlipp CR. Electrophoretic pattern of concentrated urine: comparison between 24-hour collection and random samples. Am J Kidney Dis. 2002;39(1):E2.
Koopman MG, Koomen GC, van Acker BA, Arisz L. Circadian rhythm in glomerular transport of macromolecules through large pores and shunt pathway. Kidney Int. 1996;49(5):1242–9.
Greenhill A, Gruskin AB. Laboratory evaluation of renal function. Pediatr Clin North Am. 1976;23(4):661–79.
Witte EC, Lambers Heerspink HJ, de Zeeuw D, Bakker SJ, de Jong PE, Gansevoort R. First morning voids are more reliable than spot urine samples to assess microalbuminuria. J Am Soc Nephrol. 2009;20(2):436–43.
Ginsberg JM, Chang BS, Matarese RA, Garella S. Use of single voided urine samples to estimate quantitative proteinuria. N Engl J Med. 1983;309(25):1543–6.
Kaminska J, Dymicka-Piekarska V, Tomaszewska J, Matowicka-Karna J, Koper-Lenkiewicz OM. Diagnostic utility of protein to creatinine ratio (P/C ratio) in spot urine sample within routine clinical practice. Crit Rev Clin Lab Sci. 2020;57(5):345–64.
Schwab SJ, Christensen RL, Dougherty K, Klahr S. Quantitation of proteinuria by the use of protein-to-creatinine ratios in single urine samples. Arch Intern Med. 1987;147(5):943–4.
Daza J, Salome B, Okhawere K, Bane O, Meilika KN, Korn TG, et al. Urine supernatant reveals a signature that predicts survival in clear-cell renal cell carcinoma. BJU Int. 2023;132(1):75–83.
Daniels JR, Ma JZ, Cao Z, Beger RD, Sun J, Schnackenberg L, et al. Discovery of novel proteomic biomarkers for the prediction of kidney recovery from dialysis-dependent AKI patients. Kidney360. 2021;2(11):1716–27.
Ferreira JP, Rossignol P, Bakris G, Mehta C, White WB, Zannad F. Blood and urine biomarkers predicting worsening kidney function in patients with type 2 diabetes post-acute coronary syndrome: an analysis from the EXAMINE trial. Am J Nephrol. 2021;52(12):969–76.
Dong L, Watson J, Cao S, Arregui S, Saxena V, Ketz J, et al. Aptamer based proteomic pilot study reveals a urine signature indicative of pediatric urinary tract infections. PLoS ONE. 2020;15(7): e0235328.
Masood A, Benabdelkamel H, Jammah AA, Ekhzaimy AA, Alfadda AA. Identification of protein changes in the urine of hypothyroid patients treated with thyroxine using proteomics approach. ACS Omega. 2021;6(3):2367–78.
Yuan X, Meng Y, Chen C, Liang S, Ma Y, Jiang W, et al. Proteomic approaches in the discovery of potential urinary biomarkers of mucopolysaccharidosis type II. Clin Chim Acta. 2019;499:34–40.
Kammeijer GSM, Nouta J, de la Rosette J, de Reijke TM, Wuhrer M. An in-depth glycosylation assay for urinary prostate-specific antigen. Anal Chem. 2018;90(7):4414–21.
Frantzi M, Metzger J, Banks RE, Husi H, Klein J, Dakna M, et al. Discovery and validation of urinary biomarkers for detection of renal cell carcinoma. J Proteom. 2014;98:44–58.
Johannesson N, Wetterhall M, Markides KE, Bergquist J. Monomer surface modifications for rapid peptide analysis by capillary electrophoresis and capillary electrochromatography coupled to electrospray ionization-mass spectrometry. Electrophoresis. 2004;25(6):809–16.
Mischak H, Coon JJ, Novak J, Weissinger EM, Schanstra JP, Dominiczak AF. Capillary electrophoresis-mass spectrometry as a powerful tool in biomarker discovery and clinical diagnosis: an update of recent developments. Mass Spectrom Rev. 2009;28(5):703–24.
Hernandez-Borges J, Neususs C, Cifuentes A, Pelzing M. On-line capillary electrophoresis-mass spectrometry for the analysis of biomolecules. Electrophoresis. 2004;25(14):2257–81.
Neususs C, Pelzing M, Macht M. A robust approach for the analysis of peptides in the low femtomole range by capillary electrophoresis-tandem mass spectrometry. Electrophoresis. 2002;23(18):3149–59.
Ibanez C, Simo C, Garcia-Canas V, Cifuentes A, Castro-Puyana M. Metabolomics, peptidomics and proteomics applications of capillary electrophoresis-mass spectrometry in foodomics: a review. Anal Chim Acta. 2013;802:1–13.
Albalat A, Mischak H, Mullen W. Urine proteomics in clinical applications: technologies, principal considerations and clinical implementation. Prilozi. 2011;32(1):13–44.
Fliser D, Novak J, Thongboonkerd V, Argiles A, Jankowski V, Girolami MA, et al. Advances in urinary proteome analysis and biomarker discovery. J Am Soc Nephrol. 2007;18(4):1057–71.
Magalhaes P, Pontillo C, Pejchinovski M, Siwy J, Krochmal M, Makridakis M, et al. Comparison of urine and plasma peptidome indicates selectivity in renal peptide handling. Proteom Clin Appl. 2018;12(5): e1700163.
Good DM, Zurbig P, Argiles A, Bauer HW, Behrens G, Coon JJ, et al. Naturally occurring human urinary peptides for use in diagnosis of chronic kidney disease. Mol Cell Proteom. 2010;9(11):2424–37.
Zhang Z, Hebert AS, Westphall MS, Coon JJ, Dovichi NJ. Single-shot capillary zone electrophoresis-tandem mass spectrometry produces over 4400 phosphopeptide identifications from a 220 ng sample. J Proteome Res. 2019;18(8):3166–73.
Klein J, Papadopoulos T, Mischak H, Mullen W. Comparison of CE-MS/MS and LC-MS/MS sequencing demonstrates significant complementarity in natural peptide identification in human urine. Electrophoresis. 2014;35(7):1060–4.
Li Y, Champion MM, Sun L, Champion PA, Wojcik R, Dovichi NJ. Capillary zone electrophoresis-electrospray ionization-tandem mass spectrometry as an alternative proteomics platform to ultraperformance liquid chromatography-electrospray ionization-tandem mass spectrometry for samples of intermediate complexity. Anal Chem. 2012;84(3):1617–22.
Chen CL, Lin TS, Tsai CH, Wu CC, Chung T, Chien KY, et al. Identification of potential bladder cancer markers in urine by abundant-protein depletion coupled with quantitative proteomics. J Proteom. 2013;85:28–43.
Zhao M, Li M, Yang Y, Guo Z, Sun Y, Shao C, et al. A comprehensive analysis and annotation of human normal urinary proteome. Sci Rep. 2017;7(1):3024.
Neverova I, Van Eyk JE. Role of chromatographic techniques in proteomic analysis. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;815(1–2):51–63.
Niwa T. Biomarker discovery for kidney diseases by mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;870(2):148–53.
Lin SY, Chang CH, Wu HC, Lin CC, Chang KP, Yang CR, et al. Proteome profiling of urinary exosomes identifies alpha 1-antitrypsin and H2B1K as diagnostic and prognostic biomarkers for urothelial carcinoma. Sci Rep. 2016;6:34446.
Fujita K, Kume H, Matsuzaki K, Kawashima A, Ujike T, Nagahara A, et al. Proteomic analysis of urinary extracellular vesicles from high Gleason score prostate cancer. Sci Rep. 2017;7:42961.
Limonte CP, Valo E, Drel V, Natarajan L, Darshi M, Forsblom C, et al. Urinary proteomics identifies cathepsin D as a biomarker of rapid eGFR decline in type 1 diabetes. Diabetes Care. 2022;45(6):1416–27.
Chebotareva N, Vinogradov A, McDonnell V, Zakharova NV, Indeykina MI, Moiseev S, et al. Urinary protein and peptide markers in chronic kidney disease. Int J Mol Sci. 2021;22(22):12123.
Halim A, Nilsson J, Ruetschi U, Hesse C, Larson G. Human urinary glycoproteomics; attachment site specific analysis of N- and O-linked glycosylations by CID and ECD. Mol Cell Proteom. 2012;11(4):M111.013649.
Shao C, Zhao M, Chen X, Sun H, Yang Y, Xiao X, et al. Comprehensive analysis of individual variation in the urinary proteome revealed significant gender differences. Mol Cell Proteom. 2019;18(6):1110–22.
Meng W, Huan Y, Gao Y. Urinary proteome profiling for children with autism using data-independent acquisition proteomics. Transl Pediatr. 2021;10(7):1765–78.
Muntel J, Xuan Y, Berger ST, Reiter L, Bachur R, Kentsis A, et al. Advancing urinary protein biomarker discovery by data-independent acquisition on a quadrupole-orbitrap mass spectrometer. J Proteome Res. 2015;14(11):4752–62.
van Duijl TT, Ruhaak LR, Smit NPM, Pieterse MM, Romijn F, Dolezal N, et al. Development and provisional validation of a multiplex LC-MRM-MS test for timely kidney injury detection in urine. J Proteome Res. 2021;20(12):5304–14.
Anania VG, Yu K, Pingitore F, Li Q, Rose CM, Liu P, et al. Discovery and qualification of candidate urinary biomarkers of disease activity in lupus nephritis. J Proteome Res. 2019;18(3):1264–77.
Sun Y, Guo Z, Liu X, Yang L, Jing Z, Cai M, et al. Noninvasive urinary protein signatures associated with colorectal cancer diagnosis and metastasis. Nat Commun. 2022;13(1):2757.
Dong M, Lih TM, Hoti N, Chen SY, Ponce S, Partin A, et al. Development of parallel reaction monitoring assays for the detection of aggressive prostate cancer using urinary glycoproteins. J Proteome Res. 2021;20(7):3590–9.
Joshi N, Bhat F, Bellad A, Sathe G, Jain A, Chavan S, et al. Urinary proteomics for discovery of gastric cancer biomarkers to enable precision clinical oncology. OMICS. 2023;27(8):361–71.
Sun W, Li F, Wu S, Wang X, Zheng D, Wang J, et al. Human urine proteome analysis by three separation approaches. Proteomics. 2005;5(18):4994–5001.
Adachi J, Kumar C, Zhang Y, Olsen JV, Mann M. The human urinary proteome contains more than 1500 proteins, including a large proportion of membrane proteins. Genome Biol. 2006;7(9):R80.
Li QR, Fan KX, Li RX, Dai J, Wu CC, Zhao SL, et al. A comprehensive and non-prefractionation on the protein level approach for the human urinary proteome: touching phosphorylation in urine. Rapid Commun Mass Spectrom. 2010;24(6):823–32.
Zhang C, Leng W, Sun C, Lu T, Chen Z, Men X, et al. Urine proteome profiling predicts lung cancer from control cases and other tumors. EBioMedicine. 2018;30:120–8.
Swensen AC, He J, Fang AC, Ye Y, Nicora CD, Shi T, et al. A comprehensive urine proteome database generated from patients with various renal conditions and prostate cancer. Front Med. 2021;8:548212.
Selevsek N, Matondo M, Sanchez Carbayo M, Aebersold R, Domon B. Systematic quantification of peptides/proteins in urine using selected reaction monitoring. Proteomics. 2011;11(6):1135–47.
Yang N, Feng S, Shedden K, Xie X, Liu Y, Rosser CJ, et al. Urinary glycoprotein biomarker discovery for bladder cancer detection using LC/MS-MS and label-free quantification. Clin Cancer Res. 2011;17(10):3349–59.
Tyan YC, Yang MH, Chen SC, Jong SB, Chen WC, Yang YH, et al. Urinary protein profiling by liquid chromatography/tandem mass spectrometry: ADAM28 is overexpressed in bladder transitional cell carcinoma. Rapid Commun Mass Spectrom. 2011;25(19):2851–62.
Yang MH, Chu PY, Chen SC, Chung TW, Chen WC, Tan LB, et al. Characterization of ADAM28 as a biomarker of bladder transitional cell carcinomas by urinary proteome analysis. Biochem Biophys Res Commun. 2011;411(4):714–20.
Linden M, Lind SB, Mayrhofer C, Segersten U, Wester K, Lyutvinskiy Y, et al. Proteomic analysis of urinary biomarker candidates for nonmuscle invasive bladder cancer. Proteomics. 2012;12(1):135–44.
Zoidakis J, Makridakis M, Zerefos PG, Bitsika V, Esteban S, Frantzi M, et al. Profilin 1 is a potential biomarker for bladder cancer aggressiveness. Mol Cell Proteom. 2012;11(4):M111.009449.
Kumar P, Nandi S, Tan TZ, Ler SG, Chia KS, Lim WY, et al. Highly sensitive and specific novel biomarkers for the diagnosis of transitional bladder carcinoma. Oncotarget. 2015;6(15):13539–49.
Chen CL, Lai YF, Tang P, Chien KY, Yu JS, Tsai CH, et al. Comparative and targeted proteomic analyses of urinary microparticles from bladder cancer and hernia patients. J Proteome Res. 2012;11(12):5611–29.
Chen YT, Chen HW, Domanski D, Smith DS, Liang KH, Wu CC, et al. Multiplexed quantification of 63 proteins in human urine by multiple reaction monitoring-based mass spectrometry for discovery of potential bladder cancer biomarkers. J Proteom. 2012;75(12):3529–45.
Tomiyama E, Fujita K, Matsuzaki K, Narumi R, Yamamoto A, Uemura T, et al. EphA2 on urinary extracellular vesicles as a novel biomarker for bladder cancer diagnosis and its effect on the invasiveness of bladder cancer. Br J Cancer. 2022;127(7):1312–23.
Igami K, Uchiumi T, Shiota M, Ueda S, Tsukahara S, Akimoto M, et al. Extracellular vesicles expressing CEACAM proteins in the urine of bladder cancer patients. Cancer Sci. 2022;113(9):3120–33.
Tomiyama E, Matsuzaki K, Fujita K, Shiromizu T, Narumi R, Jingushi K, et al. Proteomic analysis of urinary and tissue-exudative extracellular vesicles to discover novel bladder cancer biomarkers. Cancer Sci. 2021;112(5):2033–45.
Lee J, McKinney KQ, Pavlopoulos AJ, Niu M, Kang JW, Oh JW, et al. Altered proteome of extracellular vesicles derived from bladder cancer patients urine. Mol Cells. 2018;41(3):179–87.
Nawaz M, Camussi G, Valadi H, Nazarenko I, Ekstrom K, Wang X, et al. The emerging role of extracellular vesicles as biomarkers for urogenital cancers. Nat Rev Urol. 2014;11(12):688–701.
Oeyen E, Hoekx L, De Wachter S, Baldewijns M, Ameye F, Mertens I. Bladder cancer diagnosis and follow-up: the current status and possible role of extracellular vesicles. Int J Mol Sci. 2019;20(4):821.
Carvalho LB, Capelo JL, Lodeiro C, Dhir R, Pinheiro LC, Lopez-Fernandez H, et al. Pathway-guided monitoring of the disease course in bladder cancer with longitudinal urine proteomics. Commun Med. 2023;3(1):8.
Siebert S, Porter D, Paterson C, Hampson R, Gaya D, Latosinska A, et al. Urinary proteomics can define distinct diagnostic inflammatory arthritis subgroups. Sci Rep. 2017;7:40473.
Kang MJ, Park YJ, You S, Yoo SA, Choi S, Kim DH, et al. Urinary proteome profile predictive of disease activity in rheumatoid arthritis. J Proteome Res. 2014;13(11):5206–17.
Vitko D, Cho PS, Kostel SA, DiMartino SE, Cabour LD, Migliozzi MA, et al. Characterizing patients with recurrent urinary tract infections in vesicoureteral reflux: a pilot study of the urinary proteome. Mol Cell Proteom. 2020;19(3):456–66.
Staessen JA, Wendt R, Yu YL, Kalbitz S, Thijs L, Siwy J, et al. Predictive performance and clinical application of COV50, a urinary proteomic biomarker in early COVID-19 infection: a prospective multicentre cohort study. Lancet Digit Health. 2022;4(10):e727–37.
Bi X, Liu W, Ding X, Liang S, Zheng Y, Zhu X, et al. Proteomic and metabolomic profiling of urine uncovers immune responses in patients with COVID-19. Cell Rep. 2022;38(3):110271.
Chen Y, Zhang N, Zhang J, Guo J, Dong S, Sun H, et al. Immune response pattern across the asymptomatic, symptomatic and convalescent periods of COVID-19. Biochim Biophys Acta Proteins Proteom. 2022;1870(2):140736.
Chavan S, Mangalaparthi KK, Singh S, Renuse S, Vanderboom PM, Madugundu AK, et al. Mass spectrometric analysis of urine from COVID-19 patients for detection of SARS-CoV-2 Viral antigen and to study host response. J Proteome Res. 2021;20(7):3404–13.
Wang Y, Zhang J, Song W, Tian X, Liu Y, Wang Y, et al. A proteomic analysis of urine biomarkers in autism spectrum disorder. J Proteom. 2021;242:104259.
Iwan K, Clayton R, Mills P, Csanyi B, Gissen P, Mole SE, et al. Urine proteomics analysis of patients with neuronal ceroid lipofuscinoses. iScience. 2021;24(2):102020.
Wang S, Kojima K, Mobley JA, West AB. Proteomic analysis of urinary extracellular vesicles reveal biomarkers for neurologic disease. EBioMedicine. 2019;45:351–61.
Suganya V, Geetha A, Sujatha S. Urine proteome analysis to evaluate protein biomarkers in children with autism. Clin Chim Acta. 2015;450:210–9.
Rossing K, Mischak H, Dakna M, Zurbig P, Novak J, Julian BA, et al. Urinary proteomics in diabetes and CKD. J Am Soc Nephrol. 2008;19(7):1283–90.
Soggiu A, Piras C, Bonizzi L, Hussein HA, Pisanu S, Roncada P. A discovery-phase urine proteomics investigation in type 1 diabetes. Acta Diabetol. 2012;49(6):453–64.
Fisher WG, Lucas JE, Mehdi UF, Qunibi DW, Garner HR, Rosenblatt KP, et al. A method for isolation and identification of urinary biomarkers in patients with diabetic nephropathy. Proteom Clin Appl. 2011;5(11–12):603–12.
Manwaring V, Heywood WE, Clayton R, Lachmann RH, Keutzer J, Hindmarsh P, et al. The identification of new biomarkers for identifying and monitoring kidney disease and their translation into a rapid mass spectrometry-based test: evidence of presymptomatic kidney disease in pediatric Fabry and type-I diabetic patients. J Proteome Res. 2013;12(5):2013–21.
Marikanty RK, Gupta MK, Cherukuvada SV, Kompella SS, Prayaga AK, Konda S, et al. Identification of urinary proteins potentially associated with diabetic kidney disease. Indian J Nephrol. 2016;26(6):434–45.
Zubiri I, Posada-Ayala M, Sanz-Maroto A, Calvo E, Martin-Lorenzo M, Gonzalez-Calero L, et al. Diabetic nephropathy induces changes in the proteome of human urinary exosomes as revealed by label-free comparative analysis. J Proteom. 2014;96:92–102.
Caseiro A, Barros A, Ferreira R, Padrao A, Aroso M, Quintaneiro C, et al. Pursuing type 1 diabetes mellitus and related complications through urinary proteomics. Transl Res. 2014;163(3):188–99.
Suh MJ, Tovchigrechko A, Thovarai V, Rolfe MA, Torralba MG, Wang J, et al. Quantitative differences in the urinary proteome of siblings discordant for type 1 diabetes include lysosomal enzymes. J Proteome Res. 2015;14(8):3123–35.
Singh H, Yu Y, Suh MJ, Torralba MG, Stenzel RD, Tovchigrechko A, et al. Type 1 diabetes: urinary proteomics and protein network analysis support perturbation of lysosomal function. Theranostics. 2017;7(10):2704–17.
Vitova L, Tuma Z, Moravec J, Kvapil M, Matejovic M, Mares J. Early urinary biomarkers of diabetic nephropathy in type 1 diabetes mellitus show involvement of kallikrein-kinin system. BMC Nephrol. 2017;18(1):112.
Currie GE, von Scholten BJ, Mary S, Flores Guerrero JL, Lindhardt M, Reinhard H, et al. Urinary proteomics for prediction of mortality in patients with type 2 diabetes and microalbuminuria. Cardiovasc Diabetol. 2018;17(1):50.
Guillen-Gomez E, Bardaji-de-Quixano B, Ferrer S, Brotons C, Knepper MA, Carrascal M, et al. Urinary proteome analysis identified neprilysin and VCAM as proteins involved in diabetic nephropathy. J Diabetes Res. 2018;2018:6165303.
Ding X, Zhang D, Ren Q, Hu Y, Wang J, Hao J, et al. Identification of a non-invasive urinary exosomal biomarker for diabetic nephropathy using data-independent acquisition proteomics. Int J Mol Sci. 2023;24(17):13560.
Blaschke CRK, Hartig JP, Grimsley G, Liu L, Semmes OJ, Wu JD, et al. Direct n-glycosylation profiling of urine and prostatic fluid glycoproteins and extracellular vesicles. Front Chem. 2021;9:734280.
Kawahara R, Saad J, Angeli CB, Palmisano G. Site-specific characterization of N-linked glycosylation in human urinary glycoproteins and endogenous glycopeptides. Glycoconj J. 2016;33(6):937–51.
Bolduc S, Lacombe L, Naud A, Gregoire M, Fradet Y, Tremblay RR. Urinary PSA: a potential useful marker when serum PSA is between 2.5 ng/mL and 10 ng/mL. Can Urol Assoc J. 2007;1(4):377–81.
Huo B, Chen M, Chen J, Li Y, Zhang W, Wang J, et al. A sequential separation strategy for facile isolation and comprehensive analysis of human urine n-glycoproteome. Anal Bioanal Chem. 2018;410(28):7305–12.
Bagdonaite I, Malaker SA, Polasky DA, Riley NM, Schjoldager K, Vakhrushev SY, et al. Glycoproteomics. Nat Rev Method Prime. 2022;2(1):1–29.
Saraswat M, Joenvaara S, Musante L, Peltoniemi H, Holthofer H, Renkonen R. N-linked (N-) glycoproteomics of urinary exosomes. [Corrected]. Mol Cell Proteom. 2015;14(2):263–76.
Belczacka I, Pejchinovski M, Krochmal M, Magalhaes P, Frantzi M, Mullen W, et al. Urinary glycopeptide analysis for the investigation of novel biomarkers. Proteom Clin Appl. 2019;13(3): e1800111.
Sathe G, George IA, Deb B, Jain AP, Patel K, Nayak B, et al. Urinary glycoproteomic profiling of non-muscle invasive and muscle invasive bladder carcinoma patients reveals distinct n-glycosylation pattern of CD44, MGAM, and GINM1. Oncotarget. 2020;11(34):3244–55.
Wang Y, Lih TM, Hoti N, Sokoll LJ, Chesnut G, Petrovics G, et al. Differentially expressed glycoproteins in pre- and post-digital rectal examination urine samples for detecting aggressive prostate cancer. Proteomics. 2023;23(7–8): e2200023.
Li D, Jia S, Wang S, Hu L. Glycoproteomic analysis of urinary extracellular vesicles for biomarkers of hepatocellular carcinoma. Molecules. 2023;28(3):1293.
Boschmann M, König W. Peptide and protein hormones: structure, regulation, activity: a reference manual. Nahrung. 1994;38:225.
Schaffer M, Beiter T, Becker HD, Hunt TK. Neuropeptides: mediators of inflammation and tissue repair? Arch Surg. 1998;133(10):1107–16.
Schroder JM. Peptides and cytokines. Arch Dermatol Res. 1992;284(Suppl 1):S22–6.
Oshima G, Shimabukuro H, Nagasawa K. Peptide inhibitors of angiotensin I-converting enzyme in digests of gelatin by bacterial collagenase. Biochim Biophys Acta. 1979;566(1):128–37.
Steiner C, Ducret A, Tille JC, Thomas M, McKee TA, Rubbia-Brandt L, et al. Applications of mass spectrometry for quantitative protein analysis in formalin-fixed paraffin-embedded tissues. Proteomics. 2014;14(4–5):441–51.
Domon B, Aebersold R. Mass spectrometry and protein analysis. Science. 2006;312(5771):212–7.
Baggerman G, Vierstraete E, De Loof A, Schoofs L. Gel-based versus gel-free proteomics: a review. Comb Chem High Throughput Screen. 2005;8(8):669–77.
Nongonierma AB, FitzGerald RJ. Features of dipeptidyl peptidase IV (DPP-IV) inhibitory peptides from dietary proteins. J Food Biochem. 2019;43(1): e12451.
Li Q, Li J, Chen L, Gao Y, Li J. Endogenous peptides profiles of human infantile hemangioma tissue and their clinical significance for treatment. J Cell Biochem. 2018;119(6):4636–43.
Zhang W, Li D, Xu B, Xu L, Lyu Q, Liu X, et al. Serum peptidome profiles immune response of COVID-19 vaccine administration. Front Immunol. 2022;13:956369.
Zhao Y, Tong D, Wang M, Xu C, Gong X, Wang Z, et al. Peptidomics analysis reveals serum biomarkers in spinal cord injury patients. Crit Rev Eukaryot Gene Expr. 2022;32(2):1–9.
Holtta M, Minthon L, Hansson O, Holmen-Larsson J, Pike I, Ward M, et al. An integrated workflow for multiplex CSF proteomics and peptidomics-identification of candidate cerebrospinal fluid biomarkers of Alzheimer’s disease. J Proteome Res. 2015;14(2):654–63.
Westman-Brinkmalm A, Ruetschi U, Portelius E, Andreasson U, Brinkmalm G, Karlsson G, et al. Proteomics/peptidomics tools to find CSF biomarkers for neurodegenerative diseases. Front Biosci. 2009;14(5):1793–806.
Beasley-Green A. Urine proteomics in the era of mass spectrometry. Int Neurourol J. 2016;20(Suppl 2):S70–5.
Bauca JM, Martinez-Morillo E, Diamandis EP. Peptidomics of urine and other biofluids for cancer diagnostics. Clin Chem. 2014;60(8):1052–61.
Ling XB, Mellins ED, Sylvester KG, Cohen HJ. Urine peptidomics for clinical biomarker discovery. Adv Clin Chem. 2010;51:181–213.
Krochmal M, van Kessel KEM, Zwarthoff EC, Belczacka I, Pejchinovski M, Vlahou A, et al. Urinary peptide panel for prognostic assessment of bladder cancer relapse. Sci Rep. 2019;9(1):7635.
Schanstra JP, Zurbig P, Alkhalaf A, Argiles A, Bakker SJ, Beige J, et al. Diagnosis and prediction of CKD progression by assessment of urinary peptides. J Am Soc Nephrol. 2015;26(8):1999–2010.
Palanski BA, Weng N, Zhang L, Hilmer AJ, Fall LA, Swaminathan K, et al. An efficient urine peptidomics workflow identifies chemically defined dietary gluten peptides from patients with celiac disease. Nat Commun. 2022;13(1):888.
Siwy J, Schanstra JP, Argiles A, Bakker SJ, Beige J, Boucek P, et al. Multicentre prospective validation of a urinary peptidome-based classifier for the diagnosis of type 2 diabetic nephropathy. Nephrol Dial Transplant. 2014;29(8):1563–70.
Mullen W, Albalat A, Gonzalez J, Zerefos P, Siwy J, Franke J, et al. Performance of different separation methods interfaced in the same MS-reflection TOF detector: a comparison of performance between CE versus HPLC for biomarker analysis. Electrophoresis. 2012;33(4):567–74.
Yang X, Hu L, Ye M, Zou H. Analysis of the human urine endogenous peptides by nanoparticle extraction and mass spectrometry identification. Anal Chim Acta. 2014;829:40–7.
Nkuipou-Kenfack E, Bhat A, Klein J, Jankowski V, Mullen W, Vlahou A, et al. Identification of ageing-associated naturally occurring peptides in human urine. Oncotarget. 2015;6(33):34106–17.
Liu X, Chinello C, Musante L, Cazzaniga M, Tataruch D, Calzaferri G, et al. Intraluminal proteome and peptidome of human urinary extracellular vesicles. Proteom Clin Appl. 2015;9(5–6):568–73.
Di Meo A, Batruch I, Yousef AG, Pasic MD, Diamandis EP, Yousef GM. An integrated proteomic and peptidomic assessment of the normal human urinome. Clin Chem Lab Med. 2017;55(2):237–47.
Piovesana S, Capriotti AL, Cerrato A, Crescenzi C, La Barbera G, Lagana A, et al. Graphitized carbon black enrichment and UHPLC-MS/MS allow to meet the challenge of small chain peptidomics in urine. Anal Chem. 2019;91(17):11474–81.
Cuervo D, Loli C, Fernandez-Alvarez M, Munoz G, Carreras D. Determination of doping peptides via solid-phase microelution and accurate-mass quadrupole time-of-flight LC-MS. J Chromatogr B Analyt Technol Biomed Life Sci. 2017;1065–1066:134–44.
Thomas A, Gorgens C, Guddat S, Thieme D, Dellanna F, Schanzer W, et al. Simplifying and expanding the screening for peptides <2 kDa by direct urine injection, liquid chromatography, and ion mobility mass spectrometry. J Sep Sci. 2016;39(2):333–41.
Padoan A, Basso D, La Malfa M, Zambon CF, Aiyetan P, Zhang H, et al. Reproducibility in urine peptidome profiling using MALDI-TOF. Proteomics. 2015;15(9):1476–85.
Bahadoram S, Davoodi M, Hassanzadeh S, Bahadoram M, Barahman M, Mafakher L. Renal cell carcinoma: an overview of the epidemiology, diagnosis, and treatment. G Ital Nefrol. 2022;39(3):1.
Gray RE, Harris GT. Renal cell carcinoma: diagnosis and management. Am Fam Phys. 2019;99(3):179–84.
Maher ER. Hereditary renal cell carcinoma syndromes: diagnosis, surveillance and management. World J Urol. 2018;36(12):1891–8.
Chinello C, Cazzaniga M, De Sio G, Smith AJ, Gianazza E, Grasso A, et al. Urinary signatures of renal cell carcinoma investigated by peptidomic approaches. PLoS ONE. 2014;9(9): e106684.
Chinello C, Cazzaniga M, De Sio G, Smith AJ, Grasso A, Rocco B, et al. Tumor size, stage and grade alterations of urinary peptidome in RCC. J Transl Med. 2015;13:332.
Sanchez A, Feldman AS, Hakimi AA. Current management of small renal masses, including patient selection, renal tumor biopsy, active surveillance, and thermal ablation. J Clin Oncol. 2018;36(36):3591–600.
Di Meo A, Batruch I, Brown MD, Yang C, Finelli A, Jewett MAS, et al. Identification of prognostic biomarkers in the urinary peptidome of the small renal mass. Am J Pathol. 2019;189(12):2366–76.
Zhang ZY, Ravassa S, Nkuipou-Kenfack E, Yang WY, Kerr SM, Koeck T, et al. Novel urinary peptidomic classifier predicts incident heart failure. J Am Heart Assoc. 2017;6(8):e005432.
Brewer HB Jr, Zech LA, Gregg RE, Schwartz D, Schaefer EJ. NIH conference: type III hyperlipoproteinemia: diagnosis, molecular defects, pathology, and treatment. Ann Intern Med. 1983;98(5 Pt 1):623–40.
Xiao D, Meng FL, He LH, Gu YX, Zhang JZ. Analysis of the urinary peptidome associated with Helicobacter pylori infection. World J Gastroenterol. 2011;17(5):618–24.
Patel PD, Stafflinger JE, Marwitz JH, Niemeier JP, Ottens AK. Secreted peptides for diagnostic trajectory assessments in brain injury rehabilitation. Neurorehabil Neural Repair. 2021;35(2):169–84.
Shan D, Wang H, Khatri P, Niu Y, Song W, Zhao S, et al. The urinary peptidome as a noninvasive biomarker development strategy for prenatal screening of down’s syndrome. OMICS. 2019;23(9):439–47.
Rodriguez-Ortiz ME, Pontillo C, Rodriguez M, Zurbig P, Mischak H, Ortiz A. Novel urinary biomarkers for improved prediction of progressive egfr loss in early chronic kidney disease stages and in high risk individuals without chronic kidney disease. Sci Rep. 2018;8(1):15940.
Mavrogeorgis E, Mischak H, Latosinska A, Vlahou A, Schanstra JP, Siwy J, et al. Collagen-derived peptides in CKD: a link to fibrosis. Toxins. 2021;14(1):10.
Jiang J, Zhan L, Dai L, Yao X, Qin Y, Zhu Z, et al. Evaluation of the reliability of MS1-based approach to profile naturally occurring peptides with clinical relevance in urine samples. Rapid Commun Mass Spectrom. 2022. https://doi.org/10.1002/rcm.9369.
Lohia S, Latosinska A, Zoidakis J, Makridakis M, Mischak H, Glorieux G, et al. Glycosylation analysis of urinary peptidome highlights IGF2 glycopeptides in association with CKD. Int J Mol Sci. 2023;24(6):5402.
Voss J, Goo YA, Cain K, Woods N, Jarrett M, Smith L, et al. Searching for the noninvasive biomarker holy grail: are urine proteomics the answer? Biol Res Nurs. 2011;13(3):235–42.
Shama A, Soni T, Jawanda IK, Upadhyay G, Sharma A, Prabha V. The latest developments in using proteomic biomarkers from urine and serum for non-invasive disease diagnosis and prognosis. Biomark Insights. 2023;18:11772719231190218.
Catanese L, Siwy J, Mischak H, Wendt R, Beige J, Rupprecht H. Recent advances in urinary peptide and proteomic biomarkers in chronic kidney disease: a systematic review. Int J Mol Sci. 2023;24(11):9156.
Nagaraj N, Mann M. Quantitative analysis of the intra- and inter-individual variability of the normal urinary proteome. J Proteome Res. 2011;10(2):637–45.
Zurbig P, Schiffer E, Mischak H. Capillary electrophoresis coupled to mass spectrometry for proteomic profiling of human urine and biomarker discovery. Methods Mol Biol. 2009;564:105–21.
Su SB, Poon TC, Thongboonkerd V. Human body fluid. Biomed Res Int. 2013;2013:918793.
Mischak H, Thongboonkerd V, Schanstra JP, Vlahou A. Renal and urinary proteomics. Proteom Clin Appl. 2011;5(5–6):211–3.
Castagna A, Olivieri O, Milli A, Dal Bosco M, Timperio AM, Zolla L, et al. Female urinary proteomics: new insight into exogenous and physiological hormone-dependent changes. Proteom Clin Appl. 2011;5(5–6):343–53.
He W, Huang C, Luo G, Dal Pra I, Feng J, Chen W, et al. A stable panel comprising 18 urinary proteins in the human healthy population. Proteomics. 2012;12(7):1059–72.
Zerefos PG, Aivaliotis M, Baumann M, Vlahou A. Analysis of the urine proteome via a combination of multi-dimensional approaches. Proteomics. 2012;12(3):391–400.
Raj DA, Fiume I, Capasso G, Pocsfalvi G. A multiplex quantitative proteomics strategy for protein biomarker studies in urinary exosomes. Kidney Int. 2012;81(12):1263–72.
Tyan YC, Yang MH, Chung TW, Lu CY, Tsai WC, Jong SB. Assessing human urinary proteome using a mass spectrometry-based profiling system combined with magnetic nanoparticles. Clin Chim Acta. 2013;420:54–61.
Zheng J, Liu L, Wang J, Jin Q. Urinary proteomic and non-prefractionation quantitative phosphoproteomic analysis during pregnancy and non-pregnancy. BMC Genom. 2013;14:777.
Bakun M, Senatorski G, Rubel T, Lukasik A, Zielenkiewicz P, Dadlez M, et al. Urine proteomes of healthy aging humans reveal extracellular matrix (ECM) alterations and immune system dysfunction. Age. 2014;36(1):299–311.
Gu YM, Thijs L, Liu YP, Zhang Z, Jacobs L, Koeck T, et al. The urinary proteome as correlate and predictor of renal function in a population study. Nephrol Dial Transplant. 2014;29(12):2260–8.
Hogan MC, Johnson KL, Zenka RM, Charlesworth MC, Madden BJ, Mahoney DW, et al. Subfractionation, characterization, and in-depth proteomic analysis of glomerular membrane vesicles in human urine. Kidney Int. 2014;85(5):1225–37.
Haniff AN, Gam LH. Identification of urinary protein biomarkers for tobacco smoking. Biotechnol Appl Biochem. 2016;63(2):266–72.
Hildonen S, Skarpen E, Halvorsen TG, Reubsaet L. Isolation and mass spectrometry analysis of urinary extraexosomal proteins. Sci Rep. 2016;6:36331.
Zhao M, Liu X, Sun H, Guo Z, Liu X, Sun W. Evaluation of urinary proteome library generation methods on data-independent acquisition ms analysis and its application in normal urinary proteome analysis. Proteom Clin Appl. 2019;13(5): e1800152.
Roux-Dalvai F, Gotti C, Leclercq M, Helie MC, Boissinot M, Arrey TN, et al. Fast and accurate bacterial species identification in urine specimens using LC-MS/MS mass spectrometry and machine learning. Mol Cell Proteom. 2019;18(12):2492–505.
Tang X, Xiao X, Sun H, Zheng S, Xiao X, Guo Z, et al. 96DRA-Urine: a high throughput sample preparation method for urinary proteome analysis. J Proteom. 2022;257:104529.
Xiao X, Sun H, Liu X, Guo Z, Zheng S, Xu J, et al. Qualitative and quantitative proteomic and metaproteomic analyses of healthy human urine sediment. Proteom Clin Appl. 2022;16(2): e2100007.
Zhou L, Lu X, Wang X, Huang Z, Wu Y, Zhou L, et al. A pilot urinary proteome study reveals widespread influences of circadian rhythm disruption by sleep deprivation. Appl Biochem Biotechnol. 2023. https://doi.org/10.1007/s12010-023-04666-9.
Husi H, Stephens N, Cronshaw A, MacDonald A, Gallagher I, Greig C, et al. Proteomic analysis of urinary upper gastrointestinal cancer markers. Proteom Clin Appl. 2011;5(5–6):289–99.
Li F, Chen DN, He CW, Zhou Y, Olkkonen VM, He N, et al. Identification of urinary Gc-globulin as a novel biomarker for bladder cancer by two-dimensional fluorescent differential gel electrophoresis (2D-DIGE). J Proteom. 2012;77:225–36.
Schiffer E, Bick C, Grizelj B, Pietzker S, Schofer W. Urinary proteome analysis for prostate cancer diagnosis: cost-effective application in routine clinical practice in Germany. Int J Urol. 2012;19(2):118–25.
Lei T, Zhao X, Jin S, Meng Q, Zhou H, Zhang M. Discovery of potential bladder cancer biomarkers by comparative urine proteomics and analysis. Clin Genitourin Cancer. 2013;11(1):56–62.
Metzger J, Negm AA, Plentz RR, Weismuller TJ, Wedemeyer J, Karlsen TH, et al. Urine proteomic analysis differentiates cholangiocarcinoma from primary sclerosing cholangitis and other benign biliary disorders. Gut. 2013;62(1):122–30.
Rainczuk A, Condina M, Pelzing M, Dolman S, Rao J, Fairweather N, et al. The utility of isotope-coded protein labeling for prioritization of proteins found in ovarian cancer patient urine. J Proteome Res. 2013;12(9):4074–88.
Kuo CJ, et al. Shotgun proteomics analysis of differentially expressed urinary proteins involved in the hepatocellular carcinoma. J Proteom Bioinform. 2014;7:34–40.
Li C, Li H, Zhang T, Li J, Liu L, Chang J. Discovery of Apo-A1 as a potential bladder cancer biomarker by urine proteomics and analysis. Biochem Biophys Res Commun. 2014;446(4):1047–52.
Haj-Ahmad TA, Abdalla MA, Haj-Ahmad Y. Potential urinary protein biomarker candidates for the accurate detection of prostate cancer among benign prostatic hyperplasia patients. J Cancer. 2014;5(2):103–14.
Adeola HA, Soares NC, Paccez JD, Kaestner L, Blackburn JM, Zerbini LF. Discovery of novel candidate urinary protein biomarkers for prostate cancer in a multiethnic cohort of South African patients via label-free mass spectrometry. Proteom Clin Appl. 2015;9(5–6):597–609.
Halder S, Dey RK, Chowdhury AR, Bhattacharyya P, Chakrabarti A. Differential regulation of urine proteins in urothelial neoplasm. J Proteom. 2015;127(Pt A):185–92.
Huang CH, Kuo CJ, Liang SS, Chi SW, Hsi E, Chen CC, et al. Onco-proteogenomics identifies urinary S100A9 and GRN as potential combinatorial biomarkers for early diagnosis of hepatocellular carcinoma. BBA Clin. 2015;3:205–13.
Husi H, Skipworth RJ, Cronshaw A, Stephens NA, Wackerhage H, Greig C, et al. Programmed cell death 6 interacting protein (PDCD6IP) and Rabenosyn-5 (ZFYVE20) are potential urinary biomarkers for upper gastrointestinal cancer. Proteom Clin Appl. 2015;9(5–6):586–96.
Li C, Zang T, Wrobel K, Huang JT, Nabi G. Quantitative urinary proteomics using stable isotope labelling by peptide dimethylation in patients with prostate cancer. Anal Bioanal Chem. 2015;407(12):3393–404.
Overbye A, Skotland T, Koehler CJ, Thiede B, Seierstad T, Berge V, et al. Identification of prostate cancer biomarkers in urinary exosomes. Oncotarget. 2015;6(30):30357–76.
Radon TP, Massat NJ, Jones R, Alrawashdeh W, Dumartin L, Ennis D, et al. Identification of a three-biomarker panel in urine for early detection of pancreatic adenocarcinoma. Clin Cancer Res. 2015;21(15):3512–21.
Beretov J, Wasinger VC, Millar EK, Schwartz P, Graham PH, Li Y. Proteomic analysis of urine to identify breast cancer biomarker candidates using a label-free LC-MS/MS approach. PLoS ONE. 2015;10(11): e0141876.
Chiang CY, Pan CC, Chang HY, Lai MD, Tzai TS, Tsai YS, et al. SH3BGRL3 protein as a potential prognostic biomarker for urothelial carcinoma: a novel binding partner of epidermal growth factor receptor. Clin Cancer Res. 2015;21(24):5601–11.
Sandim V, Pereira Dde A, Kalume DE, Oliveira-Carvalho AL, Ornellas AA, Soares MR, et al. Proteomic analysis reveals differentially secreted proteins in the urine from patients with clear cell renal cell carcinoma. Urol Oncol. 2016;34(1):5.e11-25.
Guo J, Ren Y, Hou G, Wen B, Xian F, Chen Z, et al. A comprehensive investigation toward the indicative proteins of bladder cancer in urine: from surveying cell secretomes to verifying urine proteins. J Proteome Res. 2016;15(7):2164–77.
Duriez E, Masselon CD, Mesmin C, Court M, Demeure K, Allory Y, et al. Large-scale SRM screen of urothelial bladder cancer candidate biomarkers in urine. J Proteome Res. 2017;16(4):1617–31.
Wang W, Wang S, Zhang M. Identification of urine biomarkers associated with lung adenocarcinoma. Oncotarget. 2017;8(24):38517–29.
Lee H, Kim K, Woo J, Park J, Kim H, Lee KE, et al. Quantitative proteomic analysis identifies AHNAK (neuroblast differentiation-associated protein AHNAK) as a novel candidate biomarker for bladder urothelial carcinoma diagnosis by liquid-based cytology. Mol Cell Proteom. 2018;17(9):1788–802.
Pang L, Li Q, Li Y, Liu Y, Duan N, Li H. Urine proteomics of primary membranous nephropathy using nanoscale liquid chromatography tandem mass spectrometry analysis. Clin Proteom. 2018;15:5.
Jayapalan JJ, Lee CS, Lee CC, Ng KL, Junit SM, Hashim OH. iTRAQ analysis of urinary proteins: potential use of gelsolin and osteopontin to distinguish benign thyroid goiter from papillary thyroid carcinoma. Clin Biochem. 2018;53:127–31.
Sandow JJ, Rainczuk A, Infusini G, Makanji M, Bilandzic M, Wilson AL, et al. Discovery and validation of novel protein biomarkers in ovarian cancer patient urine. Proteom Clin Appl. 2018;12(3): e1700135.
Chokchaichamnankit D, Watcharatanyatip K, Subhasitanont P, Weeraphan C, Keeratichamroen S, Sritana N, et al. Urinary biomarkers for the diagnosis of cervical cancer by quantitative label-free mass spectrometry analysis. Oncol Lett. 2019;17(6):5453–68.
Frantzi M, Gomez Gomez E, Blanca Pedregosa A, Valero Rosa J, Latosinska A, Culig Z, et al. CE-MS-based urinary biomarkers to distinguish non-significant from significant prostate cancer. Br J Cancer. 2019;120(12):1120–8.
Ortiz MV, Ahmed S, Burns M, Henssen AG, Hollmann TJ, MacArthur I, et al. Prohibitin is a prognostic marker and therapeutic target to block chemotherapy resistance in Wilms’ tumor. JCI Insight. 2019;4(15):e127098.
Zhao H, Zhao X, Lei T, Zhang M. Screening, identification of prostate cancer urinary biomarkers and verification of important spots. Invest New Drugs. 2019;37(5):935–47.
Hiltbrunner S, Mints M, Eldh M, Rosenblatt R, Holmstrom B, Alamdari F, et al. Urinary exosomes from bladder cancer patients show a residual cancer phenotype despite complete pathological downstaging. Sci Rep. 2020;10(1):5960.
Shimura T, Dayde D, Wang H, Okuda Y, Iwasaki H, Ebi M, et al. Novel urinary protein biomarker panel for early diagnosis of gastric cancer. Br J Cancer. 2020;123(11):1656–64.
Zhan Z, Guan Y, Mew K, Zeng W, Peng M, Hu P, et al. Urine alpha-fetoprotein and orosomucoid 1 as biomarkers of hepatitis B virus-associated hepatocellular carcinoma. Am J Physiol Gastrointest Liver Physiol. 2020;318(2):G305–12.
van Huizen NA, van Rosmalen J, Dekker LJM, van den Coebergh Braak RRJC, Verhoef C, IJzermans JNM, et al. Identification of a collagen marker in urine improves the detection of colorectal liver metastases. J Proteome Res. 2020;19(1):153–60.
Ni M, Zhou J, Zhu Z, Yuan J, Gong W, Zhu J, et al. A novel classifier based on urinary proteomics for distinguishing between benign and malignant ovarian tumors. Front Cell Dev Biol. 2021;9:712196.
Chen CJ, Chou CY, Shu KH, Chen HC, Wang MC, Chang CC, et al. Discovery of novel protein biomarkers in urine for diagnosis of urothelial cancer using iTRAQ proteomics. J Proteome Res. 2021;20(5):2953–63.
Zhou M, Kong Y, Wang X, Li W, Chen S, Wang L, et al. LC-MS/MS-based quantitative proteomics analysis of different stages of non-small-cell lung cancer. Biomed Res Int. 2021;2021:5561569.
Fan H, Li X, Li ZW, Zheng NR, Cao LH, Liu ZC, et al. Urine proteomic signatures predicting the progression from premalignancy to malignant gastric cancer. EBioMedicine. 2022;86:104340.
Feng Y, Liu S, Zha R, Sun X, Li K, Wu D, et al. Prostate cancer-associated urinary proteomes differ before and after prostatectomy. Ther Adv Med Oncol. 2022;14:17588359221131532.
Njoku K, Pierce A, Geary B, Campbell AE, Kelsall J, Reed R, et al. Quantitative SWATH-based proteomic profiling of urine for the identification of endometrial cancer biomarkers in symptomatic women. Br J Cancer. 2023;128(9):1723–32.
Prestagiacomo LE, Tradigo G, Aracri F, Gabriele C, Rota MA, Alba S, et al. Data-independent acquisition mass spectrometry of EPS-urine coupled to machine learning: a predictive model for prostate cancer. ACS Omega. 2023;8(7):6244–52.
Johnston O, Cassidy H, O’Connell S, O’Riordan A, Gallagher W, Maguire PB, et al. Identification of beta2-microglobulin as a urinary biomarker for chronic allograft nephropathy using proteomic methods. Proteom Clin Appl. 2011;5(7–8):422–31.
Lee SM, Park JS, Norwitz ER, Kim SM, Kim BJ, Park CW, et al. Characterization of discriminatory urinary proteomic biomarkers for severe preeclampsia using SELDI-TOF mass spectrometry. J Perinat Med. 2011;39(4):391–6.
Wu J, Chen YD, Yu JK, Shi XL, Gu W. Analysis of urinary proteomic patterns for type 2 diabetic nephropathy by ProteinChip. Diabetes Res Clin Pract. 2011;91(2):213–9.
Bandin F, Siwy J, Breuil B, Mischak H, Bascands JL, Decramer S, et al. Urinary proteome analysis at 5-year followup of patients with nonoperated ureteropelvic junction obstruction suggests ongoing kidney remodeling. J Urol. 2012;187(3):1006–11.
Bellei E, Cuoghi A, Monari E, Bergamini S, Fantoni LI, Zappaterra M, et al. Proteomic analysis of urine in medication-overuse headache patients: possible relation with renal damages. J Headache Pain. 2012;13(1):45–52.
Molin L, Seraglia R, Lapolla A, Ragazzi E, Gonzalez J, Vlahou A, et al. A comparison between MALDI-MS and CE-MS data for biomarker assessment in chronic kidney diseases. J Proteom. 2012;75(18):5888–97.
Kalantari S, Rutishauser D, Samavat S, Nafar M, Mahmudieh L, Rezaei-Tavirani M, et al. Urinary prognostic biomarkers and classification of IgA nephropathy by high resolution mass spectrometry coupled with liquid chromatography. PLoS ONE. 2013;8(12): e80830.
Su L, Zhou R, Liu C, Wen B, Xiao K, Kong W, et al. Urinary proteomics analysis for sepsis biomarkers with iTRAQ labeling and two-dimensional liquid chromatography-tandem mass spectrometry. J Trauma Acute Care Surg. 2013;74(3):940–5.
Thaivalappil S, Bauman N, Saieg A, Movius E, Brown KJ, Preciado D. Propranolol-mediated attenuation of MMP-9 excretion in infants with hemangiomas. JAMA Otolaryngol Head Neck Surg. 2013;139(10):1026–31.
Su L, Cao L, Zhou R, Jiang Z, Xiao K, Kong W, et al. Identification of novel biomarkers for sepsis prognosis via urinary proteomic analysis using iTRAQ labeling and 2D-LC-MS/MS. PLoS ONE. 2013;8(1): e54237.
Aregger F, Uehlinger DE, Witowski J, Brunisholz RA, Hunziker P, Frey FJ, et al. Identification of IGFBP-7 by urinary proteomics as a novel prognostic marker in early acute kidney injury. Kidney Int. 2014;85(4):909–19.
Matafora V, Zagato L, Ferrandi M, Molinari I, Zerbini G, Casamassima N, et al. Quantitative proteomics reveals novel therapeutic and diagnostic markers in hypertension. BBA Clin. 2014;2:79–87.
Mucha K, Bakun M, Jazwiec R, Dadlez M, Florczak M, Bajor M, et al. Complement components, proteolysis-related, and cell communication-related proteins detected in urine proteomics are associated with IgA nephropathy. Pol Arch Med Wewn. 2014;124(7–8):380–6.
Sedic M, Gethings LA, Vissers JP, Shockcor JP, McDonald S, Vasieva O, et al. Label-free mass spectrometric profiling of urinary proteins and metabolites from paediatric idiopathic nephrotic syndrome. Biochem Biophys Res Commun. 2014;452(1):21–6.
Sigdel TK, Ng YW, Lee S, Nicora CD, Qian WJ, Smith RD, et al. Perturbations in the urinary exosome in transplant rejection. Front Med. 2014;1:57.
Sylvester KG, Ling XB, Liu GY, Kastenberg ZJ, Ji J, Hu Z, et al. Urine protein biomarkers for the diagnosis and prognosis of necrotizing enterocolitis in infants. J Pediatr. 2014;164(3):607-12.e1-7.
Young BL, Mlamla Z, Gqamana PP, Smit S, Roberts T, Peter J, et al. The identification of tuberculosis biomarkers in human urine samples. Eur Respir J. 2014;43(6):1719–29.
Bourderioux M, Nguyen-Khoa T, Chhuon C, Jeanson L, Tondelier D, Walczak M, et al. A new workflow for proteomic analysis of urinary exosomes and assessment in cystinuria patients. J Proteome Res. 2015;14(1):567–77.
Hall AM, Vilasi A, Garcia-Perez I, Lapsley M, Alston CL, Pitceathly RD, et al. The urinary proteome and metabonome differ from normal in adults with mitochondrial disease. Kidney Int. 2015;87(3):610–22.
Lee MY, Huang CH, Kuo CJ, Lin CL, Lai WT, Chiou SH. Clinical proteomics identifies urinary CD14 as a potential biomarker for diagnosis of stable coronary artery disease. PLoS ONE. 2015;10(2): e0117169.
Lewandowicz A, Bakun M, Kohutnicki R, Fabijanska A, Kistowski M, Imiela J, et al. Changes in urine proteome accompanying diabetic nephropathy progression. Pol Arch Med Wewn. 2015;125(1–2):27–38.
Ovrehus MA, Zurbig P, Vikse BE, Hallan SI. Urinary proteomics in chronic kidney disease: diagnosis and risk of progression beyond albuminuria. Clin Proteom. 2015;12(1):21.
Seetho IW, Ramirez-Torres A, Albalat A, Mullen W, Mischak H, Parker RJ, et al. Urinary proteomic profiling in severe obesity and obstructive sleep apnoea with CPAP treatment. Sleep Sci. 2015;8(2):58–67.
Singh V, Stingl C, Stoop MP, Zeneyedpour L, Neuteboom RF, Smitt PS, et al. Proteomics urine analysis of pregnant women suffering from multiple sclerosis. J Proteome Res. 2015;14(5):2065–73.
Yu Y, Pieper R. Urinary pellet sample preparation for shotgun proteomic analysis of microbial infection and host-pathogen interactions. Methods Mol Biol. 2015;1295:65–74.
Cantley LG, Colangelo CM, Stone KL, Chung L, Belcher J, Abbott T, et al. Development of a targeted urine proteome assay for kidney diseases. Proteom Clin Appl. 2016;10(1):58–74.
Dwivedi RC, Navarrete M, Choi N, Spicer V, Rigatto C, Arora RC, et al. A proteomic evaluation of urinary changes associated with cardiopulmonary bypass. Clin Proteom. 2016;13:17.
Gonzalez-Calero L, Martin-Lorenzo M, Ramos-Barron A, Ruiz-Criado J, Maroto AS, Ortiz A, et al. Urinary Kininogen-1 and Retinol binding protein-4 respond to Acute Kidney Injury: predictors of patient prognosis? Sci Rep. 2016;6:19667.
Konvalinka A, Batruch I, Tokar T, Dimitromanolakis A, Reid S, Song X, et al. Quantification of angiotensin II-regulated proteins in urine of patients with polycystic and other chronic kidney diseases by selected reaction monitoring. Clin Proteom. 2016;13:16.
Ramachandrarao SP, Hamlin AA, Awdishu L, Overcash R, Zhou M, Proudfoot J, et al. Proteomic analyses of urine exosomes reveal new biomarkers of diabetes in pregnancy. Madridge J Diabetes. 2016;1(1):11–22.
Sigdel TK, Gao Y, He J, Wang A, Nicora CD, Fillmore TL, et al. Mining the human urine proteome for monitoring renal transplant injury. Kidney Int. 2016;89(6):1244–52.
Starodubtseva NL, Kononikhin AS, Bugrova AE, Chagovets V, Indeykina M, Krokhina KN, et al. Investigation of urine proteome of preterm newborns with respiratory pathologies. J Proteom. 2016;149:31–7.
Aggarwal A, Gupta R, Negi VS, Rajasekhar L, Misra R, Singh P, et al. Urinary haptoglobin, alpha-1 anti-chymotrypsin and retinol binding protein identified by proteomics as potential biomarkers for lupus nephritis. Clin Exp Immunol. 2017;188(2):254–62.
Fabris A, Bruschi M, Santucci L, Candiano G, Granata S, Dalla Gassa A, et al. Proteomic-based research strategy identified laminin subunit alpha 2 as a potential urinary-specific biomarker for the medullary sponge kidney disease. Kidney Int. 2017;91(2):459–68.
Pollock N, Dhiman R, Daifalla N, Farhat M, Campos-Neto A. Discovery of a unique Mycobacterium tuberculosis protein through proteomic analysis of urine from patients with active tuberculosis. Microb Infect. 2018;20(4):228–35.
Rauniyar N, Yu X, Cantley J, Voss EZ, Belcher J, Colangelo CM, et al. Quantification of urinary protein biomarkers of autosomal dominant polycystic kidney disease by parallel reaction monitoring. Proteom Clin Appl. 2018;12(5): e1700157.
Zou L, Wang X, Guo Z, Sun H, Shao C, Yang Y, et al. Differential urinary proteomics analysis of myocardial infarction using iTRAQ quantification. Mol Med Rep. 2019;19(5):3972–88.
Ding W, Qiu B, Cram DS, Chen X, Li S, Zhou X, et al. Isobaric tag for relative and absolute quantitation based quantitative proteomics reveals unique urinary protein profiles in patients with preeclampsia. J Cell Mol Med. 2019;23(8):5822–6.
Doykov ID, Heywood WE, Nikolaenko V, Spiewak J, Hallqvist J, Clayton PT, et al. Rapid, proteomic urine assay for monitoring progressive organ disease in Fabry disease. J Med Genet. 2020;57(1):38–47.
Fang X, Wu H, Lu M, Cao Y, Wang R, Wang M, et al. Urinary proteomics of Henoch-Schonlein purpura nephritis in children using liquid chromatography-tandem mass spectrometry. Clin Proteom. 2020;17:10.
Koziolek M, Mueller GA, Dihazi GH, Jung K, Altubar C, Wallbach M, et al. Urine E-cadherin: a marker for early detection of kidney injury in diabetic patients. J Clin Med. 2020;9(3):639.
Sun Y, Wang F, Zhou Z, Teng J, Su Y, Chi H, et al. Urinary proteomics identifying novel biomarkers for the diagnosis of adult-onset Still’s disease. Front Immunol. 2020;11:2112.
Bellei E, Bergamini S, Rustichelli C, Monari E, Dal Porto M, Fiorini A, et al. Urinary proteomics reveals promising biomarkers in menstrually related and post-menopause migraine. J Clin Med. 2021;10(9):1854.
Rudnicki M, Siwy J, Wendt R, Lipphardt M, Koziolek MJ, Maixnerova D, et al. Urine proteomics for prediction of disease progression in patients with IgA nephropathy. Nephrol Dial Transplant. 2021;37(1):42–52.
Virreira Winter S, Karayel O, Strauss MT, Padmanabhan S, Surface M, Merchant K, et al. Urinary proteome profiling for stratifying patients with familial Parkinson’s disease. EMBO Mol Med. 2021;13(3): e13257.
Zhou Z, You Y, Wang F, Sun Y, Teng J, Liu H, et al. Urine proteomics differentiate primary thrombotic antiphospholipid syndrome from obstetric antiphospholipid syndrome. Front Immunol. 2021;12:702425.
Mejia-Vilet JM, Shapiro JP, Zhang XL, Cruz C, Zimmerman G, Mendez-Perez RA, et al. Association between urinary epidermal growth factor and renal prognosis in lupus nephritis. Arthritis Rheumatol. 2021;73(2):244–54.
Liu L, Deng J, Yang Q, Wei C, Liu B, Zhang H, et al. Urinary proteomic analysis to identify a potential protein biomarker panel for the diagnosis of tuberculosis. IUBMB Life. 2021;73(8):1073–83.
Ahmed S, Odumade OA, van Zalm P, Smolen KK, Fujimura K, Muntel J, et al. Urine proteomics for noninvasive monitoring of biomarkers in bronchopulmonary dysplasia. Neonatology. 2022;119(2):193–203.
Kaushik A, Bandyopadhyay S, Porwal C, Srinivasan A, Rukmangadachar LA, Hariprasad G, et al. 2D-DIGE based urinary proteomics and functional enrichment studies to reveal novel Mycobacterium tuberculosis and human protein biomarker candidates for pulmonary tuberculosis. Biochem Biophys Res Commun. 2022;619:15–21.
Lima T, Barros AS, Trindade F, Ferreira R, Leite-Moreira A, Barros-Silva D, et al. Application of proteogenomics to urine analysis towards the identification of novel biomarkers of prostate cancer: an exploratory study. Cancers. 2022;14(8):2001.
Wei D, Melgarejo JD, Thijs L, Temmerman X, Vanassche T, Van Aelst L, et al. Urinary proteomic profile of arterial stiffness is associated with mortality and cardiovascular outcomes. J Am Heart Assoc. 2022;11(8): e024769.
Hadisurya M, Li L, Kuwaranancharoen K, Wu X, Lee ZC, Alcalay RN, et al. Quantitative proteomics and phosphoproteomics of urinary extracellular vesicles define putative diagnostic biosignatures for Parkinson’s disease. Commun Med. 2023;3(1):64.
Hallqvist J, Pinto RC, Heywood WE, Cordey J, Foulkes AJM, Slattery CF, et al. A multiplexed urinary biomarker panel has potential for Alzheimer’s disease diagnosis using targeted proteomics and machine learning. Int J Mol Sci. 2023;24(18):13758.
Kononikhin AS, Brzhozovskiy AG, Bugrova AE, Chebotareva NV, Zakharova NV, Semenov S, et al. Targeted MRM quantification of urinary proteins in chronic kidney disease caused by glomerulopathies. Molecules. 2023;28(8):3323.
Morales M, Alayi TD, Tawalbeh SM, Sydenstricker AV, Spathis R, Kim H, et al. Urine proteomics by mass spectrometry identifies proteins involved in key pathogenic pathways in patients with juvenile dermatomyositis. Rheumatology. 2023;62(9):3161–8.
Wei D, Melgarejo JD, Van Aelst L, Vanassche T, Verhamme P, Janssens S, et al. Prediction of coronary artery disease using urinary proteomics. Eur J Prev Cardiol. 2023;30(14):1537–46.
Watanabe Y, Hirao Y, Kasuga K, Kitamura K, Nakamura K, Yamamoto T. Urinary proteome profiles associated with cognitive decline in community elderly residents—a pilot study. Front Neurol. 2023;14:1134976.
Zhao X, Zheng S, Li Y, Huang J, Zhang W, Xie Y, et al. An Integrated mass spectroscopy data processing strategy for fast identification, in-depth, and reproducible quantification of protein o-glycosylation in a large cohort of human urine samples. Anal Chem. 2020;92(1):690–8.
Chen SY, Dong M, Yang G, Zhou Y, Clark DJ, Lih TM, et al. Glycans, glycosite, and intact glycopeptide analysis of N-linked glycoproteins using liquid handling systems. Anal Chem. 2020;92(2):1680–6.
Shen Y, Xiao K, Tian Z. Site- and structure-specific characterization of the human urinary n-glycoproteome with site-determining and structure-diagnostic product ions. Rapid Commun Mass Spectrom. 2021;35(1): e8952.
Jia X, Chen J, Sun S, Yang W, Yang S, Shah P, et al. Detection of aggressive prostate cancer associated glycoproteins in urine using glycoproteomics and mass spectrometry. Proteomics. 2016;16(23):2989–96.
Zhang Y, Zhao W, Zhao Y, Mao Y, Su T, Zhong Y, et al. Comparative glycoproteomic profiling of human body fluid between healthy controls and patients with papillary thyroid carcinoma. J Proteome Res. 2020;19(7):2539–52.
Dong M, Lih TM, Chen SY, Cho KC, Eguez RV, Hoti N, et al. Urinary glycoproteins associated with aggressive prostate cancer. Theranostics. 2020;10(26):11892–907.
Su Z, Wang X, Gao X, Liu Y, Pan C, Hu H, et al. Excessive activation of the alternative complement pathway in autosomal dominant polycystic kidney disease. J Intern Med. 2014;276(5):470–85.
Guo Z, Liu X, Li M, Shao C, Tao J, Sun W, et al. Differential urinary glycoproteome analysis of type 2 diabetic nephropathy using 2D-LC-MS/MS and iTRAQ quantification. J Transl Med. 2015;13:371.
Xu M, Jin H, Ge W, Zhao L, Liu Z, Guo Z, et al. Mass spectrometric analysis of urinary n-glycosylation changes in patients with Parkinson’s disease. ACS Chem Neurosci. 2023;14(18):3507–17.
Sauvage FL, Gastinel LN, Marquet P. Untargeted screening of urinary peptides with liquid chromatography coupled to hybrid linear-ion trap tandem mass spectrometry. J Chromatogr A. 2012;1259:138–47.
Fedou C, Breuil B, Golovko I, Decramer S, Magalhaes P, Muller F, et al. Comparison of the amniotic fluid and fetal urine peptidome for biomarker discovery in renal developmental disease. Sci Rep. 2020;10(1):21706.
Martens DS, Thijs L, Latosinska A, Trenson S, Siwy J, Zhang ZY, et al. Urinary peptidomic profiles to address age-related disabilities: a prospective population study. Lancet Healthy Longev. 2021;2(11):e690–703.
Bryan RT, Wei W, Shimwell NJ, Collins SI, Hussain SA, Billingham LJ, et al. Assessment of high-throughput high-resolution MALDI-TOF-MS of urinary peptides for the detection of muscle-invasive bladder cancer. Proteom Clin Appl. 2011;5(9–10):493–503.
Smith CR, Batruch I, Bauca JM, Kosanam H, Ridley J, Bernardini MQ, et al. Deciphering the peptidome of urine from ovarian cancer patients and healthy controls. Clin Proteom. 2014;11(1):23.
Nakayama K, Inoue T, Sekiya S, Terada N, Miyazaki Y, Goto T, et al. The C-terminal fragment of prostate-specific antigen, a 2331 Da peptide, as a new urinary pathognomonic biomarker candidate for diagnosing prostate cancer. PLoS ONE. 2014;9(9): e107234.
Padoan A, Basso D, Zambon CF, Prayer-Galetti T, Arrigoni G, Bozzato D, et al. MALDI-TOF peptidomic analysis of serum and post-prostatic massage urine specimens to identify prostate cancer biomarkers. Clin Proteom. 2018;15:23.
Ling XB, Sigdel TK, Lau K, Ying L, Lau I, Schilling J, et al. Integrative urinary peptidomics in renal transplantation identifies biomarkers for acute rejection. J Am Soc Nephrol. 2010;21(4):646–53.
Perez V, Sanchez A, Bayes B, Navarro-Munoz M, Lauzurica R, Pastor MC, et al. Effect of paricalcitol on the urinary peptidome of kidney transplant patients. Transplant Proc. 2010;42(8):2924–7.
Sylvester KG, Ling XB, Liu GY, Kastenberg ZJ, Ji J, Hu Z, et al. A novel urine peptide biomarker-based algorithm for the prognosis of necrotising enterocolitis in human infants. Gut. 2014;63(8):1284–92.
Sigdel TK, Nicora CD, Hsieh SC, Dai H, Qian WJ, Camp DG 2nd, et al. Optimization for peptide sample preparation for urine peptidomics. Clin Proteom. 2014;11(1):7.
Wang Y, Chen J, Chen L, Zheng P, Xu HB, Lu J, et al. Urinary peptidomics identifies potential biomarkers for major depressive disorder. Psychiatr Res. 2014;217(1–2):25–33.
Kononikhin AS, Starodubtseva NL, Bugrova AE, Shirokova VA, Chagovets VV, Indeykina MI, et al. An untargeted approach for the analysis of the urine peptidome of women with preeclampsia. J Proteom. 2016;149:38–43.
Fu G, Du Y, Chu L, Zhang M. Discovery and verification of urinary peptides in type 2 diabetes mellitus with kidney injury. Exp Biol Med. 2016;241(11):1186–94.
Wei R, Gao B, Shih F, Ranger A, Dearth A, Mischak H, et al. Alterations in urinary collagen peptides in lupus nephritis subjects correlate with renal dysfunction and renal histopathology. Nephrol Dial Transplant. 2017;32(9):1468–77.
Markoska K, Pejchinovski M, Pontillo C, Zurbig P, Jacobs L, Smith A, et al. Urinary peptide biomarker panel associated with an improvement in estimated glomerular filtration rate in chronic kidney disease patients. Nephrol Dial Transplant. 2018;33(5):751–9.
Van JAD, Clotet-Freixas S, Zhou J, Batruch I, Sun C, Glogauer M, et al. Peptidomic analysis of urine from youths with early type 1 diabetes reveals novel bioactivity of uromodulin peptides in vitro. Mol Cell Proteom. 2020;19(3):501–17.
Starodubtseva N, Nizyaeva N, Baev O, Bugrova A, Gapaeva M, Muminova K, et al. SERPINA1 peptides in urine as a potential marker of preeclampsia severity. Int J Mol Sci. 2020;21(3):914.
Wendt R, Thijs L, Kalbitz S, Mischak H, Siwy J, Raad J, et al. A urinary peptidomic profile predicts outcome in SARS-CoV-2-infected patients. EClinicalMedicine. 2021;36:100883.
Siwy J, Wendt R, Albalat A, He T, Mischak H, Mullen W, et al. CD99 and polymeric immunoglobulin receptor peptides deregulation in critical COVID-19: a potential link to molecular pathophysiology? Proteomics. 2021;21(20): e2100133.
Brogna C, Cristoni S, Petrillo M, Querci M, Piazza O, Van den Eede G. Toxin-like peptides in plasma, urine and faecal samples from COVID-19 patients. F1000Res. 2021;10:550.
Petra E, Siwy J, Vlahou A, Jankowski J. Urine peptidome in combination with transcriptomics analysis highlights MMP7, MMP14 and PCSK5 for further investigation in chronic kidney disease. PLoS ONE. 2022;17(1): e0262667.
18–23 October 1987. Annual meeting, Optical Society of America. Rochester, New York. Abstracts. J Opt Soc Am A. 1987;4(13):P1–144.
Mavrogeorgis E, He T, Mischak H, Latosinska A, Vlahou A, Schanstra JP, et al. Urinary peptidomic liquid biopsy for non-invasive differential diagnosis of chronic kidney disease. Nephrol Dial Transplant. 2023. https://doi.org/10.1093/ndt/gfad200.
Marx D, Anglicheau D, Caillard S, Moulin B, Kochman A, Mischak H, et al. Urinary collagen peptides: Source of markers for bone metabolic processes in kidney transplant recipients. Proteom Clin Appl. 2023;17(4): e2200118.
Funding
This work was supported in part by Grants from NCI to A.P. (U01CA271410 and P30CA15083).
Author information
Authors and Affiliations
Contributions
Conceptualization, NJ and AP; data curation, NJ, KG, and VG; funding acquisition, AP; Investigation, NJ, KG and VG; methodology, NJ, and AP; project administration, AP; resources, NJ, VG and AP; supervision, AP; visualization, NJ, RKK and AP; writing—original draft, NJ; writing—review and editing, NJ, KG, VG, RKK and AP. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Consent for publication
Not applicable.
Competing interests
The authors declare no competing of interests.
Ethics approval
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Joshi, N., Garapati, K., Ghose, V. et al. Recent progress in mass spectrometry-based urinary proteomics. Clin Proteom 21, 14 (2024). https://doi.org/10.1186/s12014-024-09462-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12014-024-09462-z