
Abstract
Parkinson’s disease (PD) starts at the molecular and cellular level long before motor symptoms appear, yet there are no early-stage molecular biomarkers for diagnosis, prognosis prediction, or monitoring therapeutic response. This lack of biomarkers greatly impedes patient care and translational research—l-DOPA remains the standard of care more than 50 years after its introduction. Here, we performed a large-scale, multi-tissue, and multi-platform proteomics study to identify new biomarkers for early diagnosis and disease monitoring in PD. We analyzed 4877 cerebrospinal fluid, blood plasma, and urine samples from participants across seven cohorts using three orthogonal proteomics methods: Olink proximity extension assay, SomaScan aptamer precipitation assay, and liquid chromatography–mass spectrometry proteomics. We discovered that hundreds of proteins were upregulated in the CSF, blood, or urine of PD patients, prodromal PD patients with DAT deficit and REM sleep behavior disorder or anosmia, and non-manifesting genetic carriers of LRRK2 and GBA mutations. We nominate multiple novel hits across our analyses as promising markers of early PD, including DOPA decarboxylase (DDC), also known as l-aromatic acid decarboxylase (AADC), sulfatase-modifying factor 1 (SUMF1), dipeptidyl peptidase 2/7 (DPP7), glutamyl aminopeptidase (ENPEP), WAP four-disulfide core domain 2 (WFDC2), and others. DDC, which catalyzes the final step in dopamine synthesis, particularly stands out as a novel hit with a compelling mechanistic link to PD pathogenesis. DDC is consistently upregulated in the CSF and urine of treatment-naïve PD, prodromal PD, and GBA or LRRK2 carrier participants by all three proteomics methods. We show that CSF DDC levels correlate with clinical symptom severity in treatment-naïve PD patients and can be used to accurately diagnose PD and prodromal PD. This suggests that urine and CSF DDC could be a promising diagnostic and prognostic marker with utility in both clinical care and translational research.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is characterized by selective neuronal degeneration of the substantia nigra and the accumulation of predominantly neuronal alpha-synuclein protein aggregates termed Lewy bodies [2]. The defining histopathologic feature of the disease is the loss of dopaminergic neurons in the substantia nigra but can also include noradrenergic neurons in the locus coeruleus [11] and serotonergic neurons in the dorsal raphe nuclei [16]. PD and other neurodegenerative diseases which present with parkinsonian-like symptoms have a high rate of misdiagnosis, particularly at initial symptom presentation and there are few molecular biomarkers to aid diagnosis and monitor patient care [65]. Newly developed alpha-synuclein seeding assays [36, 43, 47, 48, 55] (αSyn-SAA) can identify misfolded alpha-synuclein in cerebrospinal fluid (CSF) and can be used as a proxy for the presence of Lewy body pathology in the brain to assist diagnosis of PD and other synucleopathies. While a major advance, αSyn-SAAs has multiple limitations. The αSyn-SAA has limited utility for some patients with LRRK2 mutations and others who have nigrostriatal neurodegeneration without Lewy bodies at autopsy [52, 55]. Further, αSyn-SAA does not provide information about disease severity or progression which remains an important unmet need in PD research and care. It also remains unclear how sensitive and generalizable these tests will be for early detection of PD before emergence of motor symptoms [55]. Lastly, αSyn-SAA requires a lumbar puncture and a complex custom incubation assay which takes many days, posing challenges for widespread use in screening, clinical trials, and routine care.
Thus, there remains an urgent need for additional minimally invasive biomarkers which reflect the early underlying neurodegenerative processes in PD to augment diagnosis and to assess disease severity, forecast disease progression, and monitor response to therapeutics [37, 44, 56]. In particular, damage to dopaminergic neurons begins many years before the development of clinical motor symptoms, and biomarkers which can detect this damage in patients in the asymptomatic phase are urgently needed [58]. In Alzheimer’s disease (AD) research, the development of such quantitative CSF and blood plasma biomarkers which reflect early pathologic events in the brain and which correlate with symptoms and predict future decline enabled the development of new disease-modifying therapies [3, 9, 57]. Similarly, recent drug approvals for amyotrophic lateral sclerosis were based on a reduction in plasma neurofilament light [31], a biomarker of axonal injury and neurodegeneration.
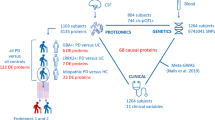
Quantitative proteomics has recently been used to develop disease-specific protein signatures as diagnostic biomarkers and holds great promise to enhance our current understanding of the molecular mechanisms underlying neurological diseases. In this study, we integrated CSF, plasma, and urine proteomics from three orthogonal proteomics assays in multiple independent human cohorts (Fig. 1) to identify disease-specific protein signatures of early PD that correlated with observed clinical severity and could distinguish PD participants from cognitively normal individuals and Alzheimer’s disease participants.

Illustration of the study design. Three proteomics platforms (Olink, SomaScan, LC–MS/MS) were used to analyze proteomics from three different biofluids (CSF, plasma, urine) in multiple cohorts (Stanford-5x, PPMI 1, and PPMI 2). CSF and plasma were collected from Stanford-5x and PPMI 1, while urine was collected from PPMI 2. Inset within each square are the number of samples from healthy controls (HC), Parkinson’s disease (PD), and AD-spectrum (AD) participants. The total sample numbers are summed across the rows and columns, providing information on the total number of samples run with each assay and biofluid
Methods
Stanford research cohorts
We included participants from five different studies of aging and neurodegeneration at Stanford University: (1) Biomarkers in PD Study (BPD), (2) the Pacific Udall Center (PUC), (3) Stanford Alzheimer’s Disease Research Center (ADRC), (4) Stanford Center for Memory Disorders Cohort Study (SCMD), and (5) Stanford Aging and Memory Study (SAMS). The combined cohort is, Hereafter, referred to as Stanford-5x. Data were collected between 2012 and 2018. Inclusion criteria for these analyses were (1) ages between 40 and 90 years (2) English or Spanish fluency for comprehensive neuropsychological testing, and (3) no contraindications to lumbar puncture. All participants provided written informed consent to participate in the parent studies following protocols approved by the Stanford Institutional Review Board.
A consensus panel consisting of one board-certified movement disorders neurologist or behavioral neurologist, one board-certified neuropsychologist, and other study personnel adjudicated the diagnosis for each participant. PD diagnosis was based on UK PD Society Brain Bank clinical diagnostic criteria [23]. We defined Early PD as participants with less than three years since diagnosis at the time of CSF collection. Participants on the AD spectrum (AD-s) included those with dementia or mild cognitive impairment likely due to AD based on the NIH Alzheimer’s Disease Diagnostic Guidelines [30, 40]. Participants with mild cognitive impairment who have decreased CSF Aβ-42 concentration, are more likely to have cognitive impairments due to AD [18]. To exclude participants without AD from the AD-s group, we excluded mild cognitive impairment participants who had CSF Aβ-42 concentration more than two standard deviations above the mean in AD [54, 62]. Participants with PD include those with no cognitive impairment, those with mild cognitive impairment [24] and those with dementia due to PD. Healthy controls (HC) were older individuals without a neurological diagnosis adjudicated as cognitively normal for age at the consensus meeting.
Some participants were excluded from the current study because of incomplete metadata on age, sex, or disease status. After filtering based on inclusion criteria, 201 CSF samples (71 PD, 78 HC, and 52 AD-s) and 250 blood plasma samples (68 PD, 105 HC, and 77 AD-s) were sent to Olink Proteomics AB (http://www.olink.com) for proteomics using a multiplex proximity extension assay [1], described in detail in a separate methods section 385 CSF samples (71 PD, 253 HC, 61 AD-s) and 1164 plasma samples (249 PD, 652 HC, 263 AD-s) were sent to SomaLogic Inc. (somalogic.com) for proteomics using a multiplex aptamer affinity assay, described in detail in a separate methods section.
Neurologic, motor and cognitive assessments
All participants completed a general neurological exam. PD participants completed the Movement Disorders Society-Unified Parkinson’s Disease Rating Scale (MDS-UPDRS III) [13] in the Off- and On-medication states, according to published criteria [34]. We calculated the Levodopa Equivalent Daily Dose (LEDD) using previously reported conversion factors [63, 66].
Global cognitive function was assessed using the Montreal Cognitive Assessment (MoCA) [33] in the ADRC, PUC and BPD, and the Mini-Mental State Exam [32] in the SAMS and SCMD studies. PD participants underwent neuropsychological testing in the on-medication state in order to assess cognitive function without interference by motor deficits.
CSF collection and assessment
A neurologist performed a lumbar puncture to collect CSF samples according to procedures standardized across all Stanford-5x cohorts [10]. Briefly, a 20–22 G spinal needle was inserted in the L4–L5 or L5–S1 interspace and CSF was collected in polypropylene tubes. The tubes were immediately frozen at − 80 °C in a centralized freezer in the Neuropathology Core of the Stanford ADRC.
PPMI cohort
Data used in the preparation of this article were obtained on August 8, 2023, from the Parkinson’s Progression Markers Initiative (PPMI) database (http://www.ppmi-info.org/access-dataspecimens/download-data), RRID:SCR 006431. For up-to-date information on the study, visit http://www.ppmi-info.org.
The Parkinson's Progression Markers Initiative (PPMI) is an ongoing observational, international study conducted in the United States, Europe, Israel, and Australia. The study has enrolled approximately 4000 participants to date which includes healthy adults (HC), de novo PD, prodromal (age 60 or older with DAT deficit and REM sleep behavior disorder (RBD) or hyposmia), and non-manifesting LRRK2 and GBA carrier participants. Participants undergo extensive clinical assessment, imaging, and molecular phenotyping. Here, we have used data from two PPMI sub-studies: project 190 and project 196. Both studies were performed by industry research groups in collaboration with PPMI and shared in the online PPMI portal as part of the PPMI data use agreement. While each study uses samples from PPMI participants, they have few overlapping participants (42 PD, 92 HC, and 5 genetic carriers) and no overlapping samples since they focused on different tissues and proteomics methods. We have labeled them as PPMI 1 (project 196) and PPMI 2 (project 190) in the main text for clarity.
PPMI patient nomenclature
PPMI has recruited multiple participant groups and a detailed description of groups is available at https://www.ppmi-info.org/study-design/study-cohorts/.
PD cohort: all participants of the PD cohort have a clinical diagnosis of PD and a positive dopamine transporter (DAT) SPECT. The PD cohort is comprised of several subgroups, which include the following key inclusion criteria:
De novo PD: people with untreated PD and within 2 years of diagnosis at enrollment. The initial phase of PPMI enrolled 423 untreated PD participants.
Genetic PD: people with PD and pathogenic genetic variant(s) in LRRK2 or GBA, within 7 years of diagnosis. Treatment with medication was allowed at enrollment, therefore, some of the participants were medicated and some were still de novo at baseline visit. The initial phase of PPMI enrolled 294 genetic PD participants (across variants).
Prodromal cohort: participants who are at risk of Parkinson’s based on clinical features, genetic variants, or other biomarkers.
Prodromal (RBD or anosmia): the initial phase of PPMI enrolled 67 prodromal volunteers age 60 or older with DAT deficit and REM sleep behavior disorder (RBD) or hyposmia.
Prodromal (non-manifesting genetic carriers): the initial phase of PPMI enrolled 445 prodromal volunteers with a genetic risk variant (SNCA, LRRK2, GBA).
Project 196 (PPMI 1)
A study document can be found at https://ida.loni.usc.edu/download/files/study/4bb082de-fa77-40ce-9dc5-494ec7fc0a1f/file/ppmi/PPMI_Project_196_Methods_Explore_20221212.pdf.
Briefly, project 196 is a longitudinal Olink proteomics study of de-novo PD and non-genetic prodromal participants age 60 or older with RBD or hyposmia and DAT deficit, with repeat sampling over 4 years. Data from study 196 were downloaded, totaling 924 CSF samples and 1160 plasma samples. The data are provided in Olink NPX units, a normalized arbitrary unit derived from sequence counts. The Olink NPX calculation and QC process are described in a separate section for clarity. Project 196 documents describe the data generation occurred in two separate experimental batches more than one year apart, so a batch effect analysis using principal component analysis (PCA) was performed. A batch effect corresponding to the experimental batch was observed via PCA. The study design utilized bridging samples to allow for correction of batch effects. We noted 89 bridging samples in both CSF and plasma datasets. We performed a batch correction based on PLATEID, using the removeBatchEffect function in the limma package for R 4.0.3. We confirmed via PCA that this removed the experimental batch effect. After batch correction, NPX values for replicate bridging samples were averaged to avoid duplicates in downstream analysis. The dataset was then further filtered to include only samples in which DDC was detected, since a fraction of samples were missing data from the Olink Cardiometabolic panel which contains DDC. After merging the filtered and QC’d data with participant level metadata, there were 765 CSF samples from 257 participants, consisting of 180 PD, 439 HC, and 146 prodromal samples. There were 859 plasma samples from 274 participants, consisting of 193 PD, 439 HC, and 227 prodromal samples. These data were used for all downstream CSF and plasma analysis in PPMI. For analysis of baseline samples, there were 243 CSF samples (69 de novo PD, 130 HC, 44 prodromal) and 262 plasma samples (74 de novo PD, 130 HC, 58 prodromal).
Project 190 (PPMI 2)
A study document can be found at the PPMI website, https://ida.loni.usc.edu/download/files/study/0a00d6e1-cd85-4340-9913-b2436a94acc1/file/ppmi/PPMI_190_Methods_Targeted_Untargeted_MS-based_proteomics_of_urine.pdf.
Briefly, project 190 is an LC–MS/MS proteomics study of urine samples from PD and non-manifesting GBA and LRRK2 mutation carriers, with a small amount of longitudinal data in LRRK2 carriers. Data from study 190 was provided in a normalized and QC’d form. After merging with patient-level metadata there were 1156 samples from 983 participants, consisting of 549 PD, 140 HC, and 467 non-manifesting carrier samples that were then used in this study.
Mass Spec proteomics (excerpted from project 190 study documents)
Proteins were extracted from neat urine samples and digested into peptides by using the MStern blotting sample preparation protocol. To determine urinary proteome profiles, purified peptides were loaded on Evosep Evotips, separated via an online-coupled Evosep One HPLC and analyzed on a Bruker timsTOF Pro mass spectrometer with a data-independent acquisition method and a gradient length of 44 min. Analysis of raw spectra was performed with DIA-NN.
Olink CSF and plasma proteomics
Plasma and CSF samples in the Stanford-5x and PPMI cohorts were sent to Olink Proteomics AB (Uppsala, Sweden. http://www.olink.com) for the quantification of up to 1536 proteins using a multiplex proximity extension assay [1]. This technology has been extensively vetted in biomarker studies and detailed methodology of the assay has been previously published [1]. Briefly, the proximity extension assay uses DNA oligonucleotide-labeled polyclonal antibodies which bind to each protein target. When two antibodies targeting different epitopes bind the same protein target, a proximity-dependent DNA ligation and elongation reaction can occur. The requirement for coincident binding leads to high specificity. The target protein levels can then be read out using quantitative PCR (qPCR) or next generation sequencing. This technology enables multiplex measurement of up to 96 or up to 384 protein targets in a single assay, depending on assay version. In the Stanford-5x cohorts, proteins from 13 different 96-protein panels were measured, resulting in quantification of 1196 proteins in both CSF and plasma samples. CSF and plasma protein levels were analyzed using the Cardiometabolic (v.3602), Cardiovascular II (v.5005), Cardiovascular III (v. 6112), Cell Regulation (v.3701), Development (v.3512), Immune Response (v.3201), Inflammation (v.3012), Metabolism (v.3402), Neuro Exploratory (v.3901), Neurology (v.8011), Oncology II (v.7002), Oncology III (v.4001) and Organ Damage (v.3301) 96-plex immunoassay Olink panels. In the PPMI cohort, four 384-protein panels from the Olink explore 1536 platform, the Cardiometabolic 384, Neurology 384, Oncology 384, and Inflammation 384, were run. Details on the exact assay version were not provided in study docs.
Olink data processing and quality control
A detailed description of the Olink data normalization and QC process can be found at https://olink.com/application/data-normalization-and-standardization/. Exact processing differs depending on assay version (qPCR-based readout or NGS-based readout).
In Stanford-5x, a qPCR-based readout was used. As previously described [1], eight control samples are run on each plate: two are external pooled plasma samples, which are used to assess potential intra-plate/run variation, three are Inter-Plate Controls (IPCs) and three are buffer blanks. The IPCs are formed from a pool of 92 antibodies. The median of the IPCs is used to normalize each assay and compensate for potential variation between runs and plates.
Briefly, protein expression data are reported in Normalized Protein eXpression (NPX), which is a normalized unit on a log2-scale. Calculation of NPX differs based on assay version, depending on if a qPCR readout or next generation sequencing readout was used. In Stanford-5x cohorts, a qPCR readout was used, and the NPX values are derived from the Ct or “threshold cycle”. This is the number of qPCR cycles needed for the signal to pass a fluorescence signal threshold. NPX is calculated from the Ct values using the following equations:
In the PPMI cohort, an NGS readout was used. Detailed information on normalization and the calculation of NPX from NGS reads can be found in the PPMI project 196 study documents and on the Olink website.
SomaScan proteomics
SomaScan assay
The SomaLogic SomaScan assay, which uses slow off-rate modified DNA aptamers (SOMAmers) to bind target proteins with high specificity, was used to quantify the relative concentration of 4979 protein targets in CSF and 7288 protein targets in plasma samples from Stanford-5x. The assay has been used in hundreds of studies and described in detail previously [15, 35]. Two versions of the SomaScan assay were used in this study. The v4 assay (4,979 protein targets) was used in CSF samples from Stanford-5x, and the v4.1 assay (7,288 protein targets) was used in plasma samples from Stanford-5x. All v4 probes are included in the v4.1 assay. Since protein levels across assay versions were not directly compared to each other, no bridging procedure was needed in the current study to harmonize values.
SomaScan normalization and QC
Standard Somalogic normalization, calibration, and quality control were performed on all samples [59,60,61, 69] by SomaLogic Inc. Detailed documentation can be found at https://somalogic.com/tech-notes/. Briefly, pooled reference standards and buffer standards are included on each plate to control for batch effects during assay quantification. Samples are normalized within and across plates using median signal intensities in reference standards to control for both within-plate and across-plate technical variation. Samples are further normalized to a pooled reference using an adaptive maximum likelihood procedure. Samples are additionally flagged by SomaLogic if signal intensities deviated significantly from the expected range and these samples were excluded from analysis. The resulting expression values are the provided data from Somalogic and are considered “raw” data. Raw data was then log10 transformed to reduce heteroscedasticity and increase power in downstream statistical modeling.
Statistical analyses
To examine demographic and clinical group differences, we used a non-parametric Wilcoxon sign-rank test or a non-parametric one-way ANOVA on ranks (Kruskal Wallis H-test).
When using longitudinal sample data, we ran differential expression analysis on protein levels using a multi-level linear-mixed effects model controlling for age, sex, race, ethnicity, and sample-relatedness. Sample-relatedness refers to longitudinally collected samples from a single individual, which we expect to be more correlated than samples from different individuals. When looking at samples from one timepoint only, we used a linear model controlling for age, sex, race, and ethnicity.
We used Benjamini–Hochberg false discovery rate control across the number of detected proteins to account for multiple testing. We studied the association between DDC levels and clinical measures of disease severity (MDS-UPDRS III, LEDD, MoCA) using linear regression analyses corrected for age and sex. We used principal component analysis to explore the relationship between global differences in protein expression profile and clinical/demographic variables. We tested correlations between principal components with a spearman correlation test with Bonferroni correction for multiple testing. All statistical analysis was done in R 4.0.3. We used the package lmerTest [19] and the dream function from the R package variancePartition [17] for mixed effects models. We used the glm function with a binomial link in R 4.0.3 to perform binary logistic regression. We used the pROC [46] and multiROC [68] packages in R 4.0.3 to generate and visualize receiver operator sensitivity–specificity curves and calculate area under the ROC curve.
Results
We utilized five independent Stanford research cohorts (Stanford-5x) and two independent cohorts from the Parkinson’s Progression Marker Initiative (PPMI) to identify biomarkers in this study (Fig. 1, Supplementary Table 1). We began by analyzing blood and CSF proteomics from Stanford-5x using two methods: proximity extension assay (Olink) and aptamer precipitation assay (SomaScan). We then replicated results in PPMI 1 using Olink and expanded the analysis into urine with LC–MS/MS proteomics from PPMI 2.
CSF and plasma proteomics identifies biomarkers of PD
We first compared the Olink CSF and plasma proteomes of people with PD, HC, and AD-s in our discovery cohorts (Stanford-5x). The CSF study population included 71 PD, 78 HC, and 52 AD-s samples. The plasma study population included 68 PD, 105 HC, and 77 AD-s samples. Analysis of the first 10 principal components for CSF and plasma proteins indicated that there was not a statistically significant global difference in protein expression between the disease groups. The first principal component in CSF did show a significant correlation with age (spearman p = 0.0005) (Supp Fig. 1).
To identify proteins whose expression differed between disease groups, we performed differential protein expression analysis on the Olink CSF and plasma proteomes using linear mixed-effects models while controlling for age, sex, education, ethnicity, and repeated longitudinal sampling (Fig. 2). When comparing people with PD to HC, there was 1 significant hit in CSF (Fig. 2a, Supplementary Table 2) and 10 significant hits in plasma (Fig. 2b, Supplementary Table 3) after proteome-wide multiple testing correction. Comparing people with PD to AD-s, there were three significant hits in CSF (Fig. 2c, Supplementary Table 4) and 9 significant hits in plasma (Fig. 2d, Supplementary Table 5). We also performed differential expression for age and sex (Supp Fig. 2), and replicated known top hits such as PTN and WFDC2 as significantly up in aging in plasma [21, 67], and CGA, CGB3, and PSPN as significantly differential between sexes [21, 64].

Differential expression analysis of CSF and plasma from Stanford-5x using Olink proximity extension assay proteomics identifies DDC (DDC) as a top hit in both tissues. Top horizontal dotted line indicates FDR significance threshold, bottom horizontal dotted line indicates raw p-value threshold. Dotted vertical lines indicate an arbitrary 0.25 effect size cutoff for moderate-effect proteins. Significant hits are shaded by effect size cutoff. a Results comparing Parkinson’s disease participants to healthy controls in CSF. b Results comparing Parkinson’s disease participants to Alzheimer’s disease participants in CSF. c Results comparing Parkinson’s disease participants to healthy controls in plasma d Results comparing Parkinson’s disease participants to Alzheimer’s disease participants in plasma. e CSF DDC levels plotted per disease group. f Plasma DDC levels plotted per disease group. Statistics from non-parametric one-way ANOVA test
The protein DOPA decarboxylase (DDC), also known as aromatic l-amino acid decarboxylase (AADC EC 4.1.1.28), was the top upregulated hit in PD when compared to both HC and AD-s, in both CSF and plasma (Fig. 2e). DDC was most elevated in PD patients, but it was also significantly upregulated in AD-s relative to HC. DDC remained significantly elevated after controlling for age, sex and levodopa equivalent daily dose [LEDD (F2,200 = 10.515, p < 0.001, partial η2 = 0.095]. We also looked specifically at individuals in our study with Early PD (≤ 2 years since diagnosis) compared to HC, and found significantly elevated concentrations in Early PD, F113 = 9.101, p = 2.7e−6 (Fig. 2f).
We used GTEx [7] to look at DDC expression levels in different tissues and verified that in the brain, DDC is highly expressed in the substantia nigra (Supp Fig. 3). DDC is directly involved in monoamine synthesis, most notably dopamine synthesis in dopaminergic neurons, though it plays a role in synthesis of serotonin and other trace amines as well. This mechanistic link to known PD pathogenesis makes it an appealing biomarker candidate for PD.
To expand our findings, we also analyzed 385 CSF samples (71 PD, 253 HC, 61 AD-s) and 1164 plasma samples (249 PD, 652 HC, 263 AD-s) using an orthogonal proteomics platform, the SomaScan assay, in Stanford-5x (Fig. 3). Unlike Olink, which utilizes a polyclonal antibody pool against each target, SomaScan uses a single high affinity aptamer (raised in-vitro) to bind each protein target. For this reason, Olink and SomaScan commonly have differential sensitivity to proteoform changes such as post-translational modification or degradation. We found that DDC was also upregulated in both CSF and plasma using the SomaScan assay (Fig. 3a, b), but the effects were smaller in magnitude than in the Olink assay. Interestingly, we did not find DDC was upregulated in AD-s participants using SomaScan, despite having more statistical power. These difference across assays may suggest that there are different DDC proteoforms in AD-s and PD, which could be investigated in future research to identify even more specific DDC-based biomarkers of PD, similar to phosphorylated Tau species in AD-s.

Differential expression analysis of CSF and plasma from Stanford-5x using SomaScan modified DNA aptamer proteomics replicates DDC as a hit and identifies additional top hits GAPDH and SUMF1. a DDC levels in CSF measured by SomaScan aptamer. b DDC levels in plasma measured by SomaScan aptamer. c, d Proteome-wide differential expression analysis. Top horizontal dotted line indicates FDR significance threshold, bottom horizontal dotted line indicates raw p-value threshold. Dotted vertical lines indicate an arbitrary 0.25 effect size cutoff for moderate-effect proteins. Significant hits are shaded by effect size cutoff. DDC is highlighted for comparison to Fig. 1. c Parkinson’s disease participants vs healthy participants in CSF. d Parkinson’s disease participants vs healthy participants in plasma. Statistics in A and B from non-parametric one-way ANOVA test
In a proteome-wide analysis of the SomaScan data, there were 29 significant hits in CSF PD vs HC and 7 significant hits in plasma PD vs HC after FDR correction (Fig. 3c, d, Supplementary Tables 6–7). GAPDH in CSF and SUMF1 in plasma stand out as significant proteins with large effect sizes. SUMF1 encodes sulfatase-modifying factor 1 (aka formylglycine-generating enzyme, FGE), which is responsible for generating the active site of all known sulfatases in the body [8, 50]. Although it has not been previously linked to PD, mutations in SUMF1 and its sulfatase targets cause a diversity of rare lysosomal storage diseases, many of which have large phenotypic overlap with PD [45, 53]. For example, mutations in the lysosomal storage disorder-causing gene GBA are a common risk factor for PD [53].
Averaged across CSF and plasma in both platforms, DDC was the most significant hit (p = 0.01), and given its plausible role in PD, we next tested if ADDC levels in CSF and plasma were associated with PD symptom severity and could thus have potential prognostic and clinical utility. We focused on the Olink assay for this analysis because of the larger effect size. We found that DDC levels in CSF were significantly associated with severity of motor symptoms assessed on the MDS-UPDRS III Off (β = 2.85, p = 0.022) (Fig. 4a), and On scores (β = 3.29, p = 0.0017) (Fig. 4a–c).

CSF DDC levels are correlated with motor function in PD participants. a Correlation between MDS-UPDRS III Off score and CSF DDC level. b Correlation between MDS-UPDRS III On score and CSF DDC level. c Effect size estimate and p-value of relationship between CSF DDC and UPDRS III scores after controlling for age and sex effects on scores. 95% confidence intervals are shown. MDS-UPDRS III Off p = 0.022. MDS-UPDRS III On p = 0.0017
DDC is elevated in prodromal PD and is associated with disease symptom severity
DDC’s direct involvement in dopamine synthesis raises questions about the role of dopaminergic medications in this finding. Although our analysis in Stanford-5x suggested that LEDD did not explain the relationship between DDC levels and symptom severity, the possibility that elevated DDC levels are driven by dopamine replacement therapy could not be fully excluded in Stanford-5x because nearly all PD participants were taking some form of dopamine replacement therapy at the time of blood and CSF collection.
To rule out this possibility, we turned to an unpublished Olink CSF and plasma proteomics study done in untreated, newly diagnosed PD participants (de novo PD) in the Parkinson’s Progression Marker Initiative (PPMI), a multicenter international prospective cohort study [28, 29]. Participants in the PPMI 1 de novo cohort are not taking any medications for PD at their baseline visit, so we first restricted analysis to baseline samples to rule out any medication influences. There were 243 baseline CSF samples (69 PD, 130 HC, 44 prodromal) and 262 baseline plasma samples (74 PD, 130 HC, 58 prodromal).
In treatment naïve, newly diagnosed, baseline PD participants, CSF DDC was significantly elevated compared to HC (Fig. 5b). In a proteome-wide analysis, DDC was the most significantly upregulated protein in CSF, and the only protein which passed the proteome-wide FDR significance threshold. (Fig. 5c, supplementary Table 8). DDC was even more strongly upregulated in prodromal participants with DAT deficit and RBD or hyposmia, where it remained the most significantly upregulated protein (Fig. 5d). This suggests that changes in DDC may happen early in the disease course before motor symptoms emerge. In prodromal participants, there were an additional 3 upregulated proteins and 56 downregulated proteins which passed proteome-wide FDR correction (Supplementary Table 9). There was no significant difference in DDC levels between prodromal participants with hyposmia and those with RBD, though those with hyposmia were trending higher (p = 0.19, Fig. 5e). CSF DDC was also significantly elevated in non-manifesting LRRK2 and GBA mutation carriers, though sample sizes were too small to draw a robust conclusion on this point (extended data Fig. 4).

Replication of DDC findings in CSF Olink proteomics from treatment-naïve and prodromal PD participants in the PPMI 1 cohort. a CSF DDC is significantly elevated in treatment-naïve PD (TN PD) and prodromal PD. b, c Proteome-wide differential expression analysis of TN PD vs healthy controls (B) and prodromal PD vs healthy controls (C) identifies DDC as the most differentially expressed protein. Top horizontal dotted line indicates FDR significance threshold, bottom horizontal dotted line indicates raw p-value threshold. Dotted vertical lines indicate an arbitrary 0.25 effect size cutoff for moderate-effect proteins. Significant hits are shaded by effect size cutoff. d CSF DDC is elevated in both hyposmic and RBD prodromal subtypes. Hyposmic participants trend higher but the effect is not significant (p = 0.19). e Correlation between MDS-UPDRS III score and CSF DDC level in treatment-naïve, baseline PD participants in PPMI. f Correlation between MDS-UPDRS total score and CSF DDC level in treatment-naïve, baseline PD participants in PPMI. g Effect size estimate and p value of relationship between CSF DDC and MDS-UPDRS III scores after controlling for age and sex effects on scores. 95% confidence intervals are shown. MDS-UPDRS III p = 0.00018, MDS-UPDRS total score p = 0.0063
Given these findings, we tested if DDC levels increase over time in longitudinally sampled PD or prodromal participants using a linear mixed effects model. This was difficult to resolve In PD participants, since after the baseline visit most participants begin dopaminergic medications to manage their PD. We found that DDC levels did increase significantly over time in PD participants, but this was confounded with LEDD. In prodromal participants, there was a trend towards increased DDC levels over time (B = 0.04, p = 0.07) but the effect size was small and only a small number of prodromal participants had multiple samples. Larger studies are needed to understand the relevance of this increase in prodromal PD.
Plasma DDC levels were not significantly elevated in de-novo PD participants at baseline or in prodromal PD participants (Extended data Fig. 5, Supplementary Table 10–11). This is in contrast to the findings in Stanford-5x, suggesting that medications may have an impact on DDC plasma levels. In both PD vs HC and Prodromal vs HC comparisons, no proteins in plasma passed proteome-wide FDR correction.
In PPMI 1, we replicated the finding that CSF DDC levels are significantly associated with symptom severity. In baseline samples from treatment-naïve PD participants, CSF DDC levels were significantly associated with MDS-UPDRS III score (β = 1.94, p = 0.0003) and MDS-UPDRS total symptom score (β = 2.07, p = 0.007) (Fig. 5F–H).
In contrast to these findings on DDC, CSF alpha-synuclein level was not significantly associated with any motor or nonmotor symptoms in any participant subset (Supp Fig. 6), suggesting that DDC levels could be an earlier and more quantitative correlate of clinical symptoms than CSF alpha-synuclein.
DDC and other proteins are elevated in PD urine samples
When considering the utility of clinical biomarkers for diagnostics and disease monitoring, an important feature is the invasiveness and ease of sample collection. Urine is an ideal biofluid for disease biomarkers because collection is simple and completely non-invasive. Therefore, to fully understand protein biomarkers of PD and early PD, we next examined an unpublished LC–MS/MS proteomics study conducted in a separate group of participants from the PPMI cohort (PPMI 2) with a focus on carriers of GBA or LRRK2 mutations (Fig. 7a). This study included 1156 samples (549 PD, 140 HC, and 467 non-manifesting carriers with high risk GBA or LRRK2 mutations) from 983 participants. 6487 proteins were detected in the study, but we restricted the analysis to 4222 proteins which were detected in at least 50 samples from every group to avoid bias from sparsely detected outliers.
We performed differential expression analysis in baseline PD samples (472 PD and 140 HC samples) and in non-manifesting carrier participant samples (140 HC, 363 non-manifesting carriers). In PD vs HC analysis, 254 proteins were significantly downregulated, and 165 proteins were significantly upregulated after proteome-wide FDR adjustment (Fig. 6b, Supplementary Table 12). We discovered, however, that due to a difference in recruitment for genetic carriers in PPMI 2, some PD baseline participants in this study were not treatment-naïve. We repeated differential expression analysis using only treatment-naïve baseline PD samples (298 de novo PD, 140 HC), and found that effects between the whole baseline population and the treatment-naïve only population correlated extremely well (r = 0.91, p < 2.2e−16) (Supplementary Fig. 7, Supplementary Table 13). This suggests the main effect of this difference was reduced statistical power in the treatment-naïve group due to smaller sample size. No proteins in the treatment-naïve only analysis passed proteome-wide significance, but top hits including DDC (effect size = 0.15, p = 0.014) remained the most statistically significant hits.

Liquid chromatography-mass spectrometry-mass spectrometry (LC–MS/MS) proteomics of urine samples in the PPMI 2 cohort. a, b Proteome-wide differential expression analysis of LC–MS/MS urine data in baseline PD vs healthy controls (A) and non-manifesting GBA and LRRK2 carriers vs healthy controls (B) identifies multiple strong hits including DDC. Top horizontal dotted line indicates FDR significance threshold, bottom horizontal dotted line indicates raw p-value threshold. Dotted vertical lines indicate an arbitrary 0.25 effect size cutoff for moderate-effect proteins. Significant hits are shaded by effect size cutoff. c Urine DDC levels are elevated in baseline PD (BL PD) and non-manifesting GBA and LRRK2 carriers. d Summary of differential expression effect sizes across all cohorts and measurement technologies for DDC (i) and DPP7 (ii). Error bars represent 95% confidence intervals. DDC is significantly elevated in all tissues by all measurement technologies, though effect strength varies across technologies and tissues. DDP7 is elevated in CSF and urine across all 3 technologies, with strongest effects in urine. e Urine DDC levels in non-manifesting carrier types. DDC is significantly elevated in both LRRK2 and GBA carriers, and is significantly more elevated in LRRK2 carriers. f Urine DDC levels go up by ~ 10% in LRRK2 carriers with repeated sampling at baseline and 24 months. Statistic is p-value of time covariate in a linear mixed-effects model which accounts for individual differences in baseline DDC levels, as well as age and sex. Bars are mean DDC levels at each timepoint in males of mean study population age
In PD vs non-manifesting carrier analysis, 329 proteins were significantly downregulated, and 259 proteins were significantly upregulated after proteome-wide FDR adjustment (Fig. 6c, Supplementary Table 14). Multiple top hits, including ENPEP, WFDC2, JPH3, GLB1, DPP7, and DDC had consistent and large effect sizes across baseline PD, de novo PD, and non-manifesting carrier groups.
DDC was a top upregulated protein in both baseline de novo PD and non-manifesting carrier urine samples (Fig. 6B–E, Supp Fig. 7), validating this biomarker across three different biofluids and three orthogonal proteomics platforms. Another notable large effect-size hit which was validated across all 3 proteomics platforms in CSF and urine in both baseline de novo PD and non-manifesting carriers was Dipeptidyl Peptidase 7 (DPP7), also known as Dipeptidyl Peptidase 2 (DPP2), a serine protease of unknown function. This protein was most significantly upregulated in urine of PD and non-manifesting carrier participants, where its effect was larger than DDC (Fig. 6e, f). DPP7 has a known role in immune cell quiescence, and increased DPP7 enzymatic activity has been repeatedly observed in the CSF of PD patients [25, 26]. Related DPP proteins have roles in synapse formation and maintenance, gut inflammation, and immune activation [20, 27, 41], suggesting that DPP7 may also play a role in these processes which have been implicated in PD pathogenesis.
The urine study allowed us to assess the relationship between DDC and genetic risk factors of PD. DDC was significantly elevated in non-manifesting GBA and LRRK2 mutation carriers compared to healthy participants without genetic risks (Fig. 6g). DDC was also significantly elevated in LRRK2 carriers compared to GBA carriers, but the expression between LRRK2 carriers and dual GBA/LRRK2 carriers was indistinguishable. Repeated sampling at baseline and a 2-year follow-up visit in 98 non-manifesting LRRK2 carriers allowed us to assess if urine DDC levels increased over time using a linear mixed effects model. Urine DDC levels increased significantly in LRRK2 carriers (B = 950, p = 0.0022) by an average of 10% over this time (Fig. 6h).
DDC discriminates PD and prodromal PD from AD and HC
Given that CSF DDC levels are consistently elevated in people with PD compared to HC and AD-s, we tested if DDC levels could accurately discriminate PD from HC and AD-s participants. We trained a logistic regression classifier of PD diagnosis against HC and AD-s diagnosis within a subset of the Stanford-5x cohorts (PUC/BPD/SCMD), and evaluated its performance in the remaining held-out cohorts (ADRC/SAMS) and on the independent PPMI 1 CSF and plasma datasets (Fig. 7a–e). We found that both CSF and plasma DDC levels were capable of discriminating PD in Stanford-5x with promising sensitivity and specificity (CSF ROC AUC = 0.88, plasma ROC AUC = 0.87), but CSF DDC outperformed plasma DDC in the larger fully independent test cohort PPMI (CSF ROC AUC = 0.80, plasma ROC AUC = 0.59). This makes sense given that plasma DDC levels were found to be medication-dependent while CSF DDC levels were not—classifiers trained on CSF DDC levels from patients taking dopaminergic medications in Stanford-5x were still able to diagnose PD in de novo PPMI 1 participants. We also applied the classifier to prodromal participants in PPMI 1 (Fig. 7f) and found CSF DDC levels did equivalently well at diagnosing prodromal PD (CSF ROC AUC = 0.79) without any adjustment of the algorithm. These data suggest CSF DDC may hold promise as a diagnostic biomarker of early PD even before the emergence of motor symptoms.

Development and testing of logistic regression classifiers to discriminate PD from HC and AD-s participants using DDC levels in CSF and plasma. Receiver operating characteristic (ROC) sensitivity–specificity curves of classifier performance are plotted. Area Under the Curve (AUC) is a measure of classifier performance, with an AUC of 1 being perfect classification with no false positives (perfect specificity) and no false negatives (perfect sensitivity). The diagonal (AUC = 0.5) represents a random guess. Classifier using CSF DDC levels is shown in blue. Classifier using plasma DDC levels is shown in red. Classifier using both CSF and plasma DDC levels shown in black. All classifiers include age and sex as additional covariates. a Performance on PD vs HC in the Stanford-5x sub-cohorts BPD, PUC, and SCMD, which were used to train the model. CSF AUC = 0.88. Plasma AUC = 0.87. Combined AUC = 0.92. b Performance on PD vs AD-s in the Stanford-5x sub-cohorts BPD, PUC, and SCMD. CSF AUC = 0.75. Plasma AUC = 0.81. Combined AUC = 0.79. c Performance on PD vs HC in the held-out test sub-cohorts from Stanford-5x ADRC and SAMS. CSF AUC = 0.80. Plasma AUC = 0.84. Combined AUC = 0.89. d Performance on PD vs AD-s in the held-out test sub-cohorts from Stanford-5x ADRC and SAMS. CSF AUC = 0.67. Plasma AUC = 0.78. Combined AUC = 0.73. e Performance on PD vs HC in the completely independent test cohort PPMI 1. CSF AUC = 0.80. Plasma AUC = 0.59. Combined AUC = 0.76. f Performance on prodromal PD vs HC in the completely independent test cohort PPMI 1. CSF AUC = 0.79. Plasma AUC = 0.61. Combined AUC = 0.74
Discussion
In the search for new biomarkers of Parkinson's disease (PD), we analyzed proteomics data from multiple large human studies of PD, AD, and prodromal PD, totaling approximately 5000 samples across three orthogonal proteomics platforms in CSF, plasma, and urine. We discovered multiple promising candidate biomarkers in each biofluid, including the proteins DDC, DPP7/2, SUMF1, ENPEP, WFDC2, and others. While an in-depth investigation of all the hits uncovered is beyond the scope of this work, this study serves as a powerful resource for the field to investigate candidate genes of interest in multiple tissues and demonstrates the utility of proteomics in biomarker discovery.
This study highlights the discovery of DDC as a multi-tissue biomarker candidate for both early diagnosis and disease monitoring of PD. We and others have recently suggested DDC as a biomarker of PD status in CSF based on findings from Olink proteomics [4, 38, 39, 49]. The findings reported here are a comprehensive and unbiased analysis of DDC as a biomarker across CSF, plasma, and urine using three proteomics platforms and considering multiple stages of PD. Our findings indicate that DDC levels are consistently elevated in CSF and urine of PD patients, regardless of dopamine replacement therapy, including pre-motor disease stages and newly diagnosed, treatment-naïve individuals. We demonstrated that CSF DDC levels correlate with clinical symptom severity and can be used to identify treatment-naïve PD and prodromal PD. These data strongly suggest that DDC, previously overlooked due to its association with L-DOPA therapy, could be a specific marker for monoaminergic neuron degeneration in PD.
The discovery of DDC elevated in the urine of de-novo PD and genetic carriers is particularly significant, since the impact of a simple, scalable, and non-invasive diagnostic test for early PD would be substantial. This finding, as well as the discovery of other promising early PD biomarkers in urine including DPP7/2, ENPEP, WFDC2, JPH3, GLB1, and others, should be replicated in additional cohorts with multiple detection approaches to understand the full utility of urine protein testing in PD. The finding that DDC is most strongly elevated in LRRK2 carriers is also of particular interest. Existing αSyn-SAA tests are unlikely to be helpful in many LRRK2-PD or Parkin-PD patients, who lack Lewy body pathology despite substantial nigrostriatal degeneration [52]. A recent αSyn-SAA study in PPMI found that αSyn-SAA was unable to identify LRRK2 carriers [55].
This study raises crucial questions about whether the elevation of DDC in CSF and urine are a direct consequence of neuronal loss, a compensatory upregulation due to neurodegeneration, or some other mechanism. Previous studies have suggested that the increasing loss of endogenous DDC in the brain leads to waning treatment responses to dopaminergic medication over time [6, 42, 51]. However, the changes in DDC early in the disease and in genetic carriers demonstrated in this study are not understood. Currently, phase II clinical trials are investigating increasing DDC enzymatic expression via gene therapy as a potential therapeutic intervention for PD motor fluctuations [5, 12]. Our data raise the need for further study into the mechanisms of elevated DDC levels. If elevated CSF levels of DDC are due to compensatory upregulation of DDC elsewhere in the brain as a response to nigrostriatal degeneration, this could suggest that the location of DDC in the brain, or other non-dopamine functions of nigrostriatal neurons, play a key role in disease symptoms and progression.
CSF DDC also shows promise as a quantitative biomarker for monitoring disease progression and motor symptom severity in PD. This capability is particularly important given the limitations of current clinical measures such as the UPDRS or MDS-UPDRS, which are subjective and can be affected by large placebo effects [14, 22], a significant challenge for exam-based outcome measures in clinical trials. The ability of CSF DDC to reflect underlying disease severity has the potential to facilitate the development of new treatments and provide more objective endpoints for clinical trials.
In conclusion, our research reports the differentially regulated levels of hundreds of proteins in CSF, blood plasma, and urine in multiple tissues in prodromal PD and PD. We highlight the discovery of CSF and urine DDC as promising novel biomarkers, which may serve as valuable adjuncts for both monitoring and diagnosing PD.
Data availability
Stanford-5x data used in this study will be made available on reasonable request to the Stanford-ADRC data release committee, https://web.stanford.edu/group/adrc/cgi-bin/web-proj/datareq.php. All Stanford-ADRC data will be made publicly available after an embargo period at this link: https://twc-stanford.shinyapps.io/adrc/. This analysis used data openly available from PPMI, see https://www.ppmi-info.org/ for more info.
References
Assarsson E, Lundberg M, Holmquist G, Björkesten J, Thorsen SB, Ekman D et al (2014) Homogenous 96-Plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS ONE 9:e95192. https://doi.org/10.1371/journal.pone.0095192
Braak H, Tredici KD, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24:197–211. https://doi.org/10.1016/S0197-4580(02)00065-9
Budd Haeberlein S, Aisen PS, Barkhof F, Chalkias S, Chen T, Cohen S et al (2022) Two randomized phase 3 studies of aducanumab in early Alzheimer’s disease. J Prev Alzheimers Dis 9:197–210. https://doi.org/10.14283/jpad.2022.30
del Campo M, Vermunt L, Peeters CFW, Sieben A, Hok-A-Hin YS, Lleó A et al (2023) CSF proteome profiling reveals biomarkers to discriminate dementia with Lewy bodies from Alzheimer´s disease. Nat Commun 14:5635. https://doi.org/10.1038/s41467-023-41122-y
Christine CW, Bankiewicz KS, Van Laar AD, Richardson RM, Ravina B, Kells AP et al (2019) Magnetic resonance imaging–guided phase 1 trial of putaminal AADC gene therapy for Parkinson’s disease. Ann Neurol 85:704–714. https://doi.org/10.1002/ana.25450
Ciesielska A, Samaranch L, Sebastian WS, Dickson DW, Goldman S, Forsayeth J et al (2017) Depletion of AADC activity in caudate nucleus and putamen of Parkinson’s disease patients; implications for ongoing AAV2-AADC gene therapy trial. PLoS ONE 12:e0169965. https://doi.org/10.1371/journal.pone.0169965
Consortium TGte (2020) The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369:1318–1330. https://doi.org/10.1126/science.aaz1776
Diez-Roux G, Ballabio A (2005) Sulfatases and human disease. Annu Rev Genom Hum Genet 6:355–379. https://doi.org/10.1146/annurev.genom.6.080604.162334
van Dyck CH, Swanson CJ, Aisen P, Bateman RJ, Chen C, Gee M et al (2023) Lecanemab in early Alzheimer’s disease. N Engl J Med 388:9–21. https://doi.org/10.1056/NEJMoa2212948
Engelborghs S, Niemantsverdriet E, Struyfs H, Blennow K, Brouns R, Comabella M et al (2017) Consensus guidelines for lumbar puncture in patients with neurological diseases. Alzheimers Dement Diagn Assess Dis Monit 8:111–126. https://doi.org/10.1016/j.dadm.2017.04.007
Gesi M, Soldani P, Giorgi FS, Santinami A, Bonaccorsi I, Fornai F (2000) The role of the locus coeruleus in the development of Parkinson’s disease. Neurosci Biobehav Rev 24:655–668. https://doi.org/10.1016/S0149-7634(00)00028-2
Ghilardi MF, Feigin AS, Battaglia F, Silvestri G, Mattis P, Eidelberg D et al (2007) l-Dopa infusion does not improve explicit sequence learning in Parkinson’s disease. Parkinsonism Relat Disord 13:146–151. https://doi.org/10.1016/j.parkreldis.2006.08.006
Goetz CG, Tilley BC, Shaftman SR, Stebbins GT, Fahn S, Martinez-Martin P et al (2008) Movement Disorder Society-sponsored revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS): Scale presentation and clinimetric testing results. Mov Disord 23:2129–2170. https://doi.org/10.1002/mds.22340
Goetz CG, Wuu J, McDermott MP, Adler CH, Fahn S, Freed CR et al (2008) Placebo response in Parkinson’s disease: comparisons among 11 trials covering medical and surgical interventions. Mov Disord 23:690–699. https://doi.org/10.1002/mds.21894
Gold L, Ayers D, Bertino J, Bock C, Bock A, Brody EN et al (2010) Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS ONE 5:e15004. https://doi.org/10.1371/journal.pone.0015004
Guttman M, Boileau I, Warsh J, Saint-Cyr JA, Ginovart N, McCluskey T et al (2007) Brain serotonin transporter binding in non-depressed patients with Parkinson’s disease. Eur J Neurol 14:523–528. https://doi.org/10.1111/j.1468-1331.2007.01727.x
Hoffman GE, Roussos P (2021) Dream: powerful differential expression analysis for repeated measures designs. Bioinformatics 37:192–201. https://doi.org/10.1093/bioinformatics/btaa687
Jack CR, Bennett DA, Blennow K, Carrillo MC, Feldman HH, Frisoni GB et al (2016) A/T/N: an unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 87:539–547. https://doi.org/10.1212/WNL.0000000000002923
Kuznetsova A, Brockhoff PB, Christensen RHB (2017) lmerTest Package: tests in linear mixed effects models. J Stat Softw 82:1–26. https://doi.org/10.18637/jss.v082.i13
Lambeir A-M, Durinx C, Scharpé S, Meester ID (2003) Dipeptidyl-peptidase IV from Bench to bedside: an update on structural properties, functions, and clinical aspects of the enzyme DPP IV. Crit Rev Clin Lab Sci. https://doi.org/10.1080/713609354
Lehallier B, Gate D, Schaum N, Nanasi T, Lee SE, Yousef H et al (2019) Undulating changes in human plasma proteome profiles across the lifespan. Nat Med 25:1843–1850. https://doi.org/10.1038/s41591-019-0673-2
Lidstone SC, Schulzer M, Dinelle K, Mak E, Sossi V, Ruth TJ et al (2010) Effects of expectation on placebo-induced dopamine release in Parkinson Disease. Arch Gen Psychiatry 67:857–865. https://doi.org/10.1001/archgenpsychiatry.2010.88
Litvan I, Bhatia KP, Burn DJ, Goetz CG, Lang AE, McKeith I et al (2003) SIC Task Force appraisal of clinical diagnostic criteria for parkinsonian disorders. Mov Disord 18:467–486. https://doi.org/10.1002/mds.10459
Litvan I, Goldman JG, Tröster AI, Schmand BA, Weintraub D, Petersen RC et al (2012) Diagnostic criteria for mild cognitive impairment in Parkinson’s disease: Movement Disorder Society Task Force guidelines. Mov Disord 27:349–356. https://doi.org/10.1002/mds.24893
Maes M-B, Martinet W, Schrijvers DM, Van der Veken P, De Meyer GRY, Augustyns K et al (2006) Dipeptidyl peptidase II and leukocyte cell death. Biochem Pharmacol 72:70–79. https://doi.org/10.1016/j.bcp.2006.04.009
Maes M-B, Scharpé S, De Meester I (2007) Dipeptidyl peptidase II (DPPII), a review. Clin Chim Acta 380:31–49. https://doi.org/10.1016/j.cca.2007.01.024
Malloy C, Ahern M, Lin L, Hoffman DA (2022) Neuronal roles of the multifunctional protein dipeptidyl peptidase-like 6 (DPP6). Int J Mol Sci. https://doi.org/10.3390/ijms23169184
Marek K, Chowdhury S, Siderowf A, Lasch S, Coffey CS, Caspell-Garcia C et al (2018) The Parkinson’s progression markers initiative (PPMI)—establishing a PD biomarker cohort. Ann Clin Transl Neurol 5:1460–1477. https://doi.org/10.1002/acn3.644
Marek K, Jennings D, Lasch S, Siderowf A, Tanner C, Simuni T et al (2011) The Parkinson Progression Marker Initiative (PPMI). Prog Neurobiol 95:629–635. https://doi.org/10.1016/j.pneurobio.2011.09.005
McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Kawas CH et al (2011) The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement J Alzheimers Assoc 7:263–269. https://doi.org/10.1016/j.jalz.2011.03.005
Miller TM, Cudkowicz ME, Genge A, Shaw PJ, Sobue G, Bucelli RC et al (2022) Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N Engl J Med 387:1099–1110. https://doi.org/10.1056/NEJMoa2204705
Molloy DW, Standish TIM (1997) A guide to the standardized mini-mental state examination. Int Psychogeriatr 9:87–94. https://doi.org/10.1017/S1041610297004754
Nasreddine ZS, Phillips NA, Bédirian V, Charbonneau S, Whitehead V, Collin I et al (2005) The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 53:695–699. https://doi.org/10.1111/j.1532-5415.2005.53221.x
Ng B, Varoquaux G, Poline JB, Thirion B, Greicius MD, Poston KL (2017) Distinct alterations in Parkinson’s medication-state and disease-state connectivity. NeuroImage Clin 16:575–585. https://doi.org/10.1016/j.nicl.2017.09.004
Oh HS-H, Rutledge J, Nachun D, Pálovics R, Abiose O, Moran-Losada P et al (2023) Organ aging signatures in the plasma proteome track health and disease. Nature 624:164–172. https://doi.org/10.1038/s41586-023-06802-1
Orrù CD, Ma TC, Hughson AG, Groveman BR, Srivastava A, Galasko D et al (2020) A rapid α-synuclein seed assay of Parkinson’s disease CSF panel shows high diagnostic accuracy. Ann Clin Transl Neurol 8:374–384. https://doi.org/10.1002/acn3.51280
Parnetti L, Gaetani L, Eusebi P, Paciotti S, Hansson O, El-Agnaf O et al (2019) CSF and blood biomarkers for Parkinson’s disease. Lancet Neurol 18:573–586. https://doi.org/10.1016/S1474-4422(19)30024-9
Paslawski W, Khosousi S, Hertz E, Markaki I, Boxer A, Svenningsson P (2023) Large-scale proximity extension assay reveals CSF midkine and DOPA decarboxylase as supportive diagnostic biomarkers for Parkinson’s disease. Transl Neurodegener 12:42. https://doi.org/10.1186/s40035-023-00374-w
Pereira JB, Kumar A, Hall S, Palmqvist S, Stomrud E, Bali D et al (2023) DOPA decarboxylase is an emerging biomarker for Parkinsonian disorders including preclinical Lewy body disease. Nat Aging 3:1201–1209. https://doi.org/10.1038/s43587-023-00478-y
Petersen RC (2004) Mild cognitive impairment as a diagnostic entity. J Intern Med 256:183–194. https://doi.org/10.1111/j.1365-2796.2004.01388.x
Pham CTN, Ley TJ (1999) Dipeptidyl peptidase I is required for the processing and activation of granzymes A and B in vivo. Proc Natl Acad Sci 96:8627–8632. https://doi.org/10.1073/pnas.96.15.8627
Poewe W (2009) Treatments for Parkinson disease—past achievements and current clinical needs. Neurology 72:S65–S73. https://doi.org/10.1212/WNL.0b013e31819908ce
Poggiolini I, Gupta V, Lawton M, Lee S, El-Turabi A, Querejeta-Coma A et al (2022) Diagnostic value of cerebrospinal fluid alpha-synuclein seed quantification in synucleinopathies. Brain 145:584–595. https://doi.org/10.1093/brain/awab431
Qiang JK, Wong YC, Siderowf A, Hurtig HI, Xie SX, Lee VM-Y et al (2013) Plasma apolipoprotein A1 as a biomarker for Parkinson disease. Ann Neurol 74:119–127. https://doi.org/10.1002/ana.23872
Robak LA, Jansen IE, van Rooij J, Uitterlinden AG, Kraaij R, Jankovic J et al (2017) Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 140:3191–3203. https://doi.org/10.1093/brain/awx285
Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez J-C et al (2011) pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform 12:77
Rossi M, Baiardi S, Teunissen CE, Quadalti C, van de Beek M, Mammana A et al (2021) Diagnostic value of the CSF α-synuclein real-time quaking-induced conversion assay at the prodromal MCI stage of dementia with Lewy bodies. Neurology 97:e930–e940. https://doi.org/10.1212/WNL.0000000000012438
Russo MJ, Orru CD, Concha-Marambio L, Giaisi S, Groveman BR, Farris CM et al (2021) High diagnostic performance of independent alpha-synuclein seed amplification assays for detection of early Parkinson’s disease. Acta Neuropathol Commun 9:179. https://doi.org/10.1186/s40478-021-01282-8
Rutledge J, Lehallier B, Zarifkar P, Losada PM, Ryman S, Yutsis M et al (2022) Aromatic l-amino acid decarboxylase is a novel fluid biomarker of Parkinson’s disease. 2022.11.09.22282149
Sabourdy F, Mourey L, Le Trionnaire E, Bednarek N, Caillaud C, Chaix Y et al (2015) Natural disease history and characterisation of SUMF1 molecular defects in ten unrelated patients with multiple sulfatase deficiency. Orphanet J Rare Dis 10:31. https://doi.org/10.1186/s13023-015-0244-7
Sánchez-Pernaute R, Harvey-White J, Cunningham J, Bankiewicz KS (2001) Functional effect of adeno-associated virus mediated gene transfer of aromatic l-amino acid decarboxylase into the striatum of 6-OHDA-Lesioned Rats. Mol Ther 4:324–330. https://doi.org/10.1006/mthe.2001.0466
Schneider SA, Alcalay RN (2017) Neuropathology of genetic synucleinopathies with Parkinsonism—review of the literature. Mov Disord Off J Mov Disord Soc 32:1504–1523. https://doi.org/10.1002/mds.27193
Shachar T, Bianco CL, Recchia A, Wiessner C, Raas-Rothschild A, Futerman AH (2011) Lysosomal storage disorders and Parkinson’s disease: Gaucher disease and beyond. Mov Disord 26:1593–1604. https://doi.org/10.1002/mds.23774
Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC et al (2009) Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann Neurol 65:403–413. https://doi.org/10.1002/ana.21610
Siderowf A, Concha-Marambio L, Lafontant D-E, Farris CM, Ma Y, Urenia PA et al (2023) Assessment of heterogeneity among participants in the Parkinson’s Progression Markers Initiative cohort using α-synuclein seed amplification: a cross-sectional study. Lancet Neurol 22:407–417. https://doi.org/10.1016/S1474-4422(23)00109-6
Sieber B-A, Landis S, Koroshetz W, Bateman R, Siderowf A, Galpern WR et al (2014) Prioritized research recommendations from the National Institute of Neurological Disorders and Stroke Parkinson’s Disease 2014 conference. Ann Neurol 76:469–472. https://doi.org/10.1002/ana.24261
Sims JR, Zimmer JA, Evans CD, Lu M, Ardayfio P, Sparks J et al (2023) Donanemab in early symptomatic Alzheimer disease: the TRAILBLAZER-ALZ 2 randomized clinical trial. JAMA 330:512–527. https://doi.org/10.1001/jama.2023.13239
Simuni T, Chahine L, Poston K, Brumm M, Buracchio T, Campbell M et al (2023) Biological definition of neuronal alpha-synuclein disease: towards an integrated staging system for research
SomaLogic (2020) SomaScan® v4 data standardization
SomaLogic SOMAscan® v4 data standardization and file specification technical note
SomaLogic technical specification: adaptive normalization using maximum likelihood
Sonnen JA, Montine KS, Quinn JF, Kaye JA, Breitner JCS, Montine TJ (2008) Biomarkers for cognitive impairment and dementia in elderly people. Lancet Neurol 7:704–714. https://doi.org/10.1016/S1474-4422(08)70162-5
Su PC, Ma Y, Fukuda M, Mentis MJ, Tseng H-M, Yen R-F et al (2001) Metabolic changes following subthalamotomy for advanced Parkinson’s disease. Ann Neurol 50:514–520. https://doi.org/10.1002/ana.1232
Tanaka T, Biancotto A, Moaddel R, Moore AZ, Gonzalez-Freire M, Aon MA et al (2018) Plasma proteomic signature of age in healthy humans. Aging Cell 17:e12799. https://doi.org/10.1111/acel.12799
Tolosa E, Garrido A, Scholz SW, Poewe W (2021) Challenges in the diagnosis of Parkinson’s disease. Lancet Neurol 20:385–397. https://doi.org/10.1016/S1474-4422(21)00030-2
Tomlinson CL, Stowe R, Patel S, Rick C, Gray R, Clarke CE (2010) Systematic review of levodopa dose equivalency reporting in Parkinson’s disease. Mov Disord 25:2649–2653. https://doi.org/10.1002/mds.23429
Walker KA, Chen J, Zhang J, Fornage M, Yang Y, Zhou L et al (2021) Large-scale plasma proteomic analysis identifies proteins and pathways associated with dementia risk. Nat Aging 1:473–489. https://doi.org/10.1038/s43587-021-00064-0
Wei R, Wang J (2018) multiROC: calculating and visualizing ROC and PR curves across multi-class classifications
Williams SA, Kivimaki M, Langenberg C, Hingorani AD, Casas JP, Bouchard C et al (2019) Plasma protein patterns as comprehensive indicators of health. Nat Med 25:1851–1857. https://doi.org/10.1038/s41591-019-0665-2
Acknowledgements
We thank Dr. Jacob Hall, Dr. Veronica Santini, Dr. Sharon Sha and Dr. Laurice Yang for performing lumbar punctures in these cohorts. We thank Michelle Fenesy and Anisa Marshall for their assistance with neuropsychological testing and scoring. We thank Jeffrey Bernstein, Nicole Corso, Wanjia Guo, Marc Harrison, Madison Hunt, Manasi Jayakumar, Anna Khazenzon, Celia Litovsky, Natalie Tanner, and Monica Thieu for assistance with SAMS data collection. We thank the participants and family members from the Stanford Alzheimer’s Disease Research Center (ADRC) and the Pacific Udall Center (PUC), and we thank Stanford ADRC and PUC investigators for their contributions to these data. Stanford ADRC investigators are Victor W. Henderson (principal investigator); Administrative Core—Nusha Askari, Katrin I. Andreasson, Victor W. Henderson (leader), Frank M. Longo, Tony Wyss-Coray, and Jerome A. Yesavage; Clinical Core—Carolyn A. Fredericks, Jacob N. Hall, Victor W. Henderson (leader), Kathleen L. Poston, Veronica Rameriz, Allyson C. Rosen, Veronica E. Santini, Sharon J. Sha, Christina Wyss-Coray, Laurice Yang, and Maya V. Yutsis; Biostatistics and Data Management Core—Janet Hwang and Lu Tian (leader); Neuropathology and Biospecimens Core—Donald E. Born, Divya Channappa, Thomas J. Montine (leader), Ahmad Salehi, O. Hannes Vogel, and Tony Wyss-Coray; Outreach, Recruitment, and Education Core—Allyson C. Rosen, Vyjeyanthi S. Periyakoil (leader); Imaging Core—Michael D. Greicius (leader), Elizabeth C. Mormino; Stanford University, Stanford, CA, and the Veterans Affairs Palo Alto Health Care System, Palo Alto, CA. PPMI – a public-private partnership – is funded by the Michael J. Fox Foundation for Parkinson’s Research and funding partners, including 4D Pharma, Abbvie, AcureX, Allergan, Amathus Therapeutics, Aligning Science Across Parkinson's, AskBio, Avid Radiopharmaceuticals, BIAL, BioArctic, Biogen, Biohaven, BioLegend, BlueRock Therapeutics, BristolMyers Squibb, Calico Labs, Capsida Biotherapeutics, Celgene, Cerevel Therapeutics, Coave Therapeutics, DaCapo Brainscience, Denali, Edmond J. Safra Foundation, Eli Lilly, Gain Therapeutics, GE HealthCare, Genentech, GSK, Golub Capital, Handl Therapeutics, Insitro, Janssen Neuroscience, Jazz Pharmaceuticals, Lundbeck, Merck, Meso Scale Discovery, Mission Therapeutics, Neurocrine Biosciences, Neuropore, Pfizer, Piramal, Prevail Therapeutics, Roche, Sanofi, Servier, Sun Pharma Advanced Research Company, Takeda, Teva, UCB, Vanqua Bio, Verily, Voyager Therapeutics, the Weston Family Foundation and Yumanity Therapeutics. Some figure components created with BioRender.com.
Funding
This research was supported by grants from the NIH (NS075097, KP; NS115114, KP; AG048076, AW; P50 AG047366 and P30 AG066515, VWH, KP, LT, TJM, and TWC; NS062684 TJM, LT, and KP), Michael J. Fox Foundation for Parkinson’s disease Research (KP, TWC), Alzheimer’s Association and McKnight Foundation (GK), The Knight Initiative for Brain Resilience (KP and TWC).
Author information
Authors and Affiliations
Contributions
JR, KP, TW–C, BL, and PZ conceptualized the study. JR performed all the analysis and statistics. JR and KP wrote the manuscript, which was approved by all authors. BL and PZ assisted in analysis on early PD in Stanford-5x. JR and BL contributed code for binary classifiers. JR, DW, PG, and CC performed mendelian randomization and polygenic risk score analyses which were not included in the final manuscript due to null results. All data was processed and normalized by JR, with assistance from PML and MS. All authors except DW, PG, and CC contributed to data collection in the 5 Stanford cohorts analyzed in this study.
Corresponding authors
Ethics declarations
Conflict of interest
J.R., T.W-C, B.L., and K.P. have filed a patent application related to this work. J.R. and T.W-C. are advisors of Teal Omics, where they have an equity stake. K.P. is on the Scientific Advisory Board for Curasen, where she receives consulting fees and stock options. K.P. is on the Scientific Advisory Board for Amprion, where she receives stock options.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rutledge, J., Lehallier, B., Zarifkar, P. et al. Comprehensive proteomics of CSF, plasma, and urine identify DDC and other biomarkers of early Parkinson’s disease. Acta Neuropathol 147, 52 (2024). https://doi.org/10.1007/s00401-024-02706-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00401-024-02706-0