
Abstract
Background
Population-based studies have highlighted a close relationship between migraine and stroke. Migraine, especially with aura, is a risk factor for both ischemic and hemorrhagic stroke. Interestingly, stroke risk is highest for migraineurs who are young and otherwise healthy.
Main body
Preclinical models have provided us with possible mechanisms to explain the increased vulnerability of migraineurs’ brains towards ischemia and suggest a key role for enhanced cerebral excitability and increased incidence of microembolic events. Spreading depolarization (SD), a slowly propagating wave of neuronal depolarization, is the electrophysiologic event underlying migraine aura and a known headache trigger. Increased SD susceptibility has been demonstrated in migraine animal models, including transgenic mice carrying human mutations for the migraine-associated syndrome CADASIL and familial hemiplegic migraine (type 1 and 2). Upon experimentally induced SD, these mice develop aura-like neurological symptoms, akin to patients with the respective mutations. Migraine mutant mice also exhibit an increased frequency of ischemia-triggered SDs upon experimental stroke, associated with accelerated infarct growth and worse outcomes. The severe stroke phenotype can be explained by SD-related downstream events that exacerbate the metabolic mismatch, including pericyte contraction and neuroglial inflammation. Pharmacological suppression of the genetically enhanced SD susceptibility normalizes the stroke phenotype in familial hemiplegic migraine mutant mice. Recent epidemiologic and imaging studies suggest that these preclinical findings can be extrapolated to migraine patients. Migraine patients are at risk for particularly cardioembolic stroke. At the same time, studies suggest an increased incidence of coagulopathy, atrial fibrillation and patent foramen ovale among migraineurs, providing a possible path for microembolic induction of SD and, in rare instances, stroke in hyperexcitable brains. Indeed, recent imaging studies document an accelerated infarct progression with only little potentially salvageable brain tissue in acute stroke patients with a migraine history, suggesting an increased vulnerability towards cerebral ischemia.
Conclusion
Preclinical models suggest a key role for enhanced SD susceptibility and microembolization to explain both the occurrence of migraine attacks and the increased stroke risk in migraineurs. Therapeutic targeting of SD and microembolic events, or potential causes thereof, will be promising for treatment of aura and may also prevent ischemic infarction in vulnerable brains.
Similar content being viewed by others


Background
Migraine and Aura
Migraine is a chronic or episodic neurological disorder that is typically characterized by throbbing or pulsatile unilateral headaches lasting for 4–72 h. The high prevalence of migraine and the resulting disability places migraine among the top diseases culminating in a high socioeconomic burden. Thirty percent of migraineurs develop transient neurological symptoms in the setting of an attack, the so-called migraine aura [1]. Aura symptoms characteristically occur up to 1 h prior to the headache, but sometimes can overlap with the headache phase. At least two migraine attacks preceded by an aura are needed to establish the diagnosis of migraine with aura (MA). The clinical characteristics of migraine aura included in the formal migraine classification [2] International Classification of Headache Disorders (ICHD-3) are visual, sensory, language, or motor symptoms as well as brainstem symptoms [1]. Visual symptoms are the most commonly encountered aura feature. Brainstem aura symptoms are rare but particularly common in familial hemiplegic migraine [1, 3].
Since the initial description by Leão in 1944 [4], spreading depolarization (SD) has been recognized as the electrophysiological correlate of migraine aura. SD is characterized by prompt, self-propagating neuronal depolarization waves that spread at a speed of 3–5 mm/min [5, 6]. Strongest evidence for a key role of SD in migraine aura comes from a functional MRI study that showed retinotopic congruence between the visual aura perception and SD-typical BOLD signal changes traversing the occipital cortex [7]. Preclinical studies in transgenic mice for familial hemiplegic migraine (FHM) further underscore a key role for SD in migraine pathophysiology. FHM is an autosomal dominant severe migraine subtype, with associated hemiplegic aura preceding some attacks. FHM has a prevalence of 5/100000. Three FHM mutations have been identified so far; the mutations are found in voltage dependent, P/Q type calcium channel alpha 1A subunit CACNA1A for FHM1; ATPase, Na+/K+ transporting, alpha 2 polypeptide ATP1A2 for FHM2; and sodium channel, voltage gated, type 1 alpha subunit SCN1A for FHM3 [8, 9]. In transgenic mice carrying human mutations for FHM type 1, experimental induction of SD produces migraine-aura like symptoms. Mice with the R192Q mutation develop transient hemiplegia, whereas mice with the severe S218 L mutation also develop seizures, similar to the clinical phenotype in patients with the respective mutation [10]. These severe aura symptoms were associated with a facilitated subcortical spread of SD [11]. Upon exposure to an SD trigger (KCl or electrical stimulation), FHM1 mice show a reduced threshold for SD induction and develop a higher number of SDs, with S218 L mice carrying the stronger gain-of-function mutation exhibiting a more severe SD phenotype when compared to R192Q mice [10]. This enhanced SD susceptibility seems to be related to stronger cortical synapses, as indicated by larger axonal boutons and an increased percentage of highly excitable mushroom type dendritic spines with a high number of excitatory NMDA receptors [12]. Interestingly, SD susceptibility is further increased in female mice compared to male FHM1 mice, consistent with an increased migraine incidence in females compared to males. Hormonal ablation in FHM1 mice successfully abrogated the gender difference in SD susceptibility [13], underscoring the importance of sex hormones in further modulating the genetically enhanced SD susceptibility. An increased SD susceptibility has also been demonstrated in transgenic mice for FHM type 2 [14], as well as familial migraine and advanced sleep phase [15]. Importantly, SD can be induced by microembolic events [16], and even occlusion of a single cortical arteriole is sufficient to trigger SD [17], providing a candidate mechanism for SD induction in the migraine-susceptible brain. SD also plays a major role in other diseases, and worsens outcomes in ischemic stroke, intracranial hemorrhage, traumatic brain injury and subarachnoid hemorrhage [18, 19].
Migraine and stroke
The World Health Organization lists stroke as the second leading cause of death and the third leading cause of serious long-lasting disability [20]. Recent reports have highlighted the fact that 90% of strokes are preventable, which reflects an opportunity to decrease stroke related mortality and morbidity [21]. Strategies primarily target modifiable vascular risk factors such as hypertension, hyperlipidemia, diabetes and smoking [22]. Over the years, data accumulating from experimental and clinical studies have pointed out an important role for migraine as another potentially modifiable risk factor contributing to the stroke burden [23,24,25,26]. The association between migraine and stroke, both of which are considered as multifaceted neurovascular disorders, is especially pronounced in young female patients with no other stroke risk factors; overall, a history of migraine doubles the risk of stroke [23,24,25,26]. Importantly, the stroke risk is higher in migraineurs with aura compared to those without aura.
The relationship between migraine and stroke has been studied for years, both experimentally and clinically. Numerous theories have been proposed, involving a shared genetic basis, vascular dysfunction, patent foramen ovale (PFO), atrial fibrillation, increased inflammation and excitotoxicity, as well as abnormally increased coagulation [18, 27,28,29,30,31,32,33,34,35,36]. Twin studies suggest a contribution of familial factors to underlie the migraine stroke association [37], and a genome-wide analysis of common variants has identified a shared genetic susceptibility to migraine and ischemic stroke [34]. Preclinical studies have confirmed an increased cerebral vulnerability to ischemia in transgenic mice carrying human migraine mutations. In FHM1 transgenic mice, occlusion of the middle cerebral artery causes an increased number of ischemia-triggered SDs with facilitated initiation of anoxic depolarization, known to exacerbate the metabolic mismatch and worsen infarcts. Accordingly, diffusion weighted MRI documents an accelerated expansion of the infarct core in migraine mutant mice, with only a small amount of potentially salvageable brain tissue, the so-called penumbra [8]. In fact, high-frequent ischemic depolarizations have been shown to adversely affect tissue and neurological outcomes in the setting of cerebral ischemia even in wild-type mice [38]. Accordingly, migraine prophylactic drugs that suppress the genetically increased SD susceptibility in FHM mutant mice reduce the number of ischemia-triggered SDs upon experimental middle cerebral artery occlusion, and improve, even normalize, the severe stroke phenotype [39]. Another autosomal dominant rare migraine-associated disease is cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). CADASIL is caused by mutations in the NOTCH3 gene and is characterized by vasculopathy in perforator cerebral arteries secondary to fibrosis and accumulation of osmiophilic substances [40]. Migraine with aura is generally the first symptom and found in 30–40% of CADASIL patients. At later stages, ischemic stroke develops in some patients, while migraine attacks lessen or even stop [2, 40]. Patients typically show a reduced vascular smooth muscle cell function/relaxation [41]. Similarly, transgenic mice expressing the human Notch 3 R90C mutation as well as Notch 3 knockout mice develop arterial pathological hallmarks of CADASIL as well as cerebrovascular dysfunction, and show an enhanced susceptibility to SD [9, 11]. Upon experimental middle cerebral artery occlusion, stroke sizes are enlarged with an increased frequency of ischemia-triggered spreading depolarizations, and neurological outcomes are worse when compared to wild-type littermates [42]. These experimental data suggest enhanced SD susceptibility to be a key factor for the increased stroke risk in migraine-susceptible brains. Along the same line, factors increasing the likelihood of SD occurrence, or “endogenous” SD trigger factors, seem increased in migraineurs. For example, genetic and epidemiologic studies document an increased incidence of hypercoagulability [43], persistent foramen ovale [44], and atrial fibrillation [36] among migraineurs, which facilitate microembolic events in cerebral vasculature as triggers for SD, migraine and possibly stroke.
Mechanisms underlying the migraine, SD and Stroke Association
Consistent with the accelerated infarct growth in migraine mutant mice, acute stroke patients with a history of migraine also show rapid infarct expansion. There is only a small amount of potentially salvageable brain tissue/mismatch in migraineurs, when penumbra is determined by cerebral blood volume (CBV) / mean transit time (MTT) mismatch on CT perfusion [45], or diffusion weighted imaging (DWI) / MTT mismatch on MR perfusion [46]. Additionally, the amount of penumbra that could be salvaged was smaller among MA patients in comparison to MO and non-migraineous cases, highlighting increased brain tissue vulnerability in migraineurs [45]. Preclinical data support a key role for SD susceptibility in mediating stroke risk in migraineurs. In fact, as outlined below in more detail, there is evidence in migraine-susceptible brains for an increased incidence of 1) SD triggering factors such as microemboli, 2) a reduced threshold for SD induction, 3) an increased frequency of ischemia-triggered SD, and 4) worse consequences of individual SDs on the metabolic mismatch. However, considering that migraineurs also have an increased risk of myocardial infarction, venous thromboembolism and atrial fibrillation, a systemic dysfunction not limited to the cerebral vasculature could also contribute to the increased stroke risk in migraineurs [36, 47, 48].
Increased SD susceptibility and its consequences
Preclinical and imaging studies highlight enhanced SD susceptibility as a candidate mechanism increasing brain vulnerability to ischemia and thereby contributing to the stroke risk in migraineurs. These findings have paved the way for studies focusing on the consequences of SD, and thereby migraine, on ischemic stroke. Following SD, cerebral blood flow is reduced for hours, after a brief initial functional hyperemia, with an associated decrease in induced neuronal and glial calcium responses [49, 50]. Even a single episode of SD in rats is associated with a long-lasting rise in the cerebral metabolic rate of oxygen, a reduction in cerebral blood flow and impaired neurovascular coupling [49]. Migraineurs’ brains might even be more vulnerable to the negative and sometimes long-lasting effects of SD on microvasculature, leading to an accumulation of pathological cellular changes secondary to repetitive ischemic events in the long run [5, 6, 51]. In fact, SD-related changes in neuronal calcium levels and transient hypoxia are more severe in FHM transgenic mice when compared to wild-type mice, suggesting that the consequences of SD are more pronounced in migraine-susceptible brains [12, 50]. Therefore, cerebral hypoperfusion that remains unnoticed in the non-migraneur’s brain might cause profound ischemic lesions, and in rare instances even clinically manifest stroke in a migraine-susceptible brain. Indeed, white matter abnormalities, infarct-like lesions as well as volumetric changes in gray and white matter were found to be more frequent in migraineurs, particularly with aura, when compared to controls [52,53,54].
SD-typical dynamic changes in cerebral blood flow are attributed to an impaired vascular reactivity of cortical vessels [55]. In fact, SD impairs vascular smooth muscle function as evidenced by a reduced vasodilatory response of isolated rat middle cerebral artery to extraluminal acidosis, and an increased vascular reaction to extraluminal K+ [55]. Similar observations have been made in migraine patients [56]. Migraineurs exhibit an increased cerebrovascular reactivity to hypocapnia and decreased vascular reactivity to vasodilatory agents such as acetazolamide or L-arginine, pointing to a reduced tone of cerebral vessels and/or endothelial dysfunction [57, 58]. These findings suggest a reduced vasomotor reserve in cerebral microvasculature in migraineurs, and recent studies suggest a key role for pericytes to underlie this altered cerebrovascular reactivity.
Role for Pericytes and autoregulation in mediating the effects of SD
Pericytes are contractile mural cells in the cerebral and retinal vasculature that express alpha-smooth muscle actin and cover capillaries [59,60,61]. As a critical component of the neurovascular unit, pericytes have an important role in regulating microcirculation via constriction and relaxation, thereby controlling local cerebral blood flow in physiological and disease states such as stroke [62,63,64,65,66,67,68]. In the setting of cerebral ischemia, prolonged constriction of pericytes might lead to microvascular occlusion and unfavorable outcomes [69, 70]. Recently, it has been shown that capillary pericytes also have an active role in the regulation of cortical blood vessels during and after SD [71]. In fact, SD-induced prolonged vasoconstriction is strongest in first order capillaries with a persistent increase in pericyte calcium. Following SD, somatosensory stimulation fails to evoke further changes in capillary diameter and pericyte calcium, suggesting a key role for pericytes in mediating long-lasting oligemia following SD [71]. Recent studies in Notch3 transgenic mice indeed revealed a loss of pericytes with reduced coverage of capillaries and Notch3 aggregations around the few remaining pericytes which might explain microcirculatory dysfunction and ischemia in CADASIL mutant mice and patients [72,73,74,75]. In fact, CADASIL mutant mice show blood brain barrier leakage, reduced vasomotor reactivity to CO2, and narrowing or occlusion of microvessels, which may result from reduced pericyte function [74, 75]. Similarly, in patients with CADASIL, resting cerebral blood flow and vasodilatory response are reduced while there is an increase in oxygen extraction fraction and endothelial dysfunction [76,77,78,79]. And just very recently, structural changes involving pericytes and endothelial cells of microvessels have also been identified in FHM patients [80]. Therefore, prevention or treatment of pericyte constriction may become a therapeutic target in MA and migraine-related cerebral ischemia [71, 81].
The potent vasodilator calcitonin gene-related peptide (CGRP), and pharmacological suppression thereof, may further modulate the altered cerebrovascular autoregulation and the increased vulnerability to cerebral ischemia in migraineurs. CGRP receptors are present in both the nervous and cardiovascular system, underscoring a significant role for CGRP in regulating vascular resistance and regional blood flow in cerebral health and disease [82]. In fact, endogenous CGRP is protective against neuronal damage in the setting of acute or chronic stroke, as suggested by experiments using CGRP knockout mice. CGRP has been shown to reduce infarct size [83], and CGRP is protective against cerebral vasospasm in the setting of subarachnoid hemorrhage [84]. CGRP might also be protective in individuals with chronic bilateral carotid stenosis by reducing subsequent neuronal injury and cognitive impairment [83]. The protective role of CGRP in the setting of cerebral ischemia may be particularly relevant in patients with migraine for the following reasons. During migraine headache, activation of the trigeminovascular system triggers the release of CGRP from trigeminal sensory nerves [85,86,87], and stimulation of sensory fibers has been shown to increase CGRP with consecutive dilatation of cerebral and dural vessels [88]. Accordingly, CGRP was found to be elevated in external jugular venous blood samples of migraine patients during migraine attacks [89]. In turn, intravenous infusion of CGRP has been shown to cause attacks in some migraine patients [90]. Because CGRP may act as a vasodilatory safeguard during cerebral ischemic events in migraine patients, pharmacological CGRP blockade, efficacious as acute and preventive treatment of migraine [86, 91], may exacerbate the increased stroke risk in migraineurs. Antibodies against CGRP or its receptor may further enhance vulnerability to cerebral ischemia in migraineurs, with the risk of transient mild ischemic events progressing to an ischemic stroke [92]. However, no safety issues suggesting a cerebrovascular risk for anti-CGRP drugs have emerged from clinical trials so far, with possible long-term effects still not sufficiently investigated [93].
Role for Neuroinflammation in mediating the effects of SD
Neuroinflammation contributes to ischemic complications related to migraine. Inflammatory cascades are involved in the detrimental effects of SD in migraine and stroke. Animal studies showed that SD induces neuronal and glial release of inflammatory mediators, dural mast cell degranulation as well as activation the trigeminovascular system [94,95,96,97]. Within minutes after SD, the neuronal hemichannel pannexin 1 opens and forms a pore complex with the ligand-gated cation channel P2X7, allowing the release of excitatory neurotransmitters to sustain SD and activate neuroinflammation [96]. Inhibition of SD-induced opening of the neuronal Pannexin1 megachannel suppresses SD and reduces SD-induced inflammatory downstream cascades that might lead to headache [97], including upregulation of interleukin-1 beta (IL-1beta), inducible nitric oxide synthase and cyclooxygenase-2 in the cortex. IL-1beta as a pro-inflammatory cytokine in rat trigeminal ganglia cells causes the release of prostaglandin E2/CGRP and induces the activation of meningeal nociceptors, mechanically sensitizing and activating nociceptors that innervate the intracranial meninges and possibly involved in initiating throbbing headache [98,99,100,101]. Accordingly, pore-inhibitors also suppress surrogates for trigeminovascular activation, including the expression of calcitonin gene-related peptide in the trigeminal ganglion and c-Fos in the trigeminal nucleus caudalis [97]. Therefore, inhibition of neuroinflammation might be protective in both migraine and stroke by suppressing SD and direct cellular damage in the setting of ischemia [96, 97].
Increased SD triggers in Migraineurs
Cerebral microembolization may induce SD and thereby trigger a migraine attack. In mice, different types of microemboli injected through the carotid artery, mimicking embolization in humans, were found to induce SD [16]. Embolic occlusion of even a single penetrating artery imitating cerebral microembolism has been shown to induce SD, leading to selective neuronal death and small infarction [17]. Similarly, thrombotic occlusion of a single ascending cortical vein can also induce SD, albeit less frequently [102,103,104]. These microembolic events might contribute to long-lasting hypoperfusion if they are repetitive and if there is a predisposing condition like MA, with increased vulnerability to cerebral ischemia [40]. Microembolic small vessel occlusion in the setting of persistent foramen ovale or silent vessel dissections that remain completely unnoticed in non-migrainous brains might therefore lead to SD and ischemic complications or even infarct in migraine susceptible brains. This hypothesis is consistent with an enhanced likelihood of clot formation in migraineurs, who reportedly show an increased incidence of hypercoagulability [43] and atrial fibrillation [36]. As a potential path for cerebral microembolism, an increased incidence of persistent foramen ovale has been reported in migraineurs [105], and paradoxical air microembolism as well as cardiac catheterization with presumably associated microembolic events have been shown to induce headache in migraineurs [106,107,108]. Along the same line, the increased stroke risk in migraineurs is highest for stroke of cardioembolic etiology, and in the peri-operative emboli-prone setting [109, 110].
Conclusions
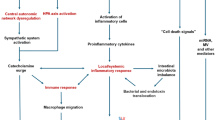
Studies suggest a double-hit hypothesis linking migraine with stroke (Fig. 1). In the setting of a genetically enhanced cerebral excitability, microembolization and/or inflammatory mediators among other factors might trigger SD, which causes or exacerbates focal ischemia; these insults remain transient or asymptomatic in most instances. However, in the setting of a vulnerable/hyperexcitable brain, these perturbations might be more severe or prolonged, escalating into further downstream events such as silent ischemic lesions or even ischemic stroke.

The interplay between migraine and stroke. Shared genetic factors and associated clinical features commonly observed in migraine patients contribute to the link between migraine and stroke. The underlying mechanism involves facilitation of spreading depolarization (SD), the electrophysiological correlate of aura, via increased SD trigger factors and/or reduced threshold for SD induction. SD then causes migraine as well as neuroinflammation and vascular dysfunction, increasing the brain’s vulnerability to ischemia. This cascade may result in clinically silent ischemic lesions that are frequently observed in migraineurs, or even cause ischemic stroke
Availability of data and materials
Not applicable.
Abbreviations
- CADASIL:
-
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy
- GWAS:
-
Genome-wide association study
- ICHD:
-
International Classification of Headache Disorders
- KCl:
-
Potassium chloride
- MA:
-
Migraine with aura
- MO:
-
Migraine without aura
- MRI:
-
Magnetic resonance imaging
- PFO:
-
Patent foramen ovale
- SD:
-
Spreading depolarization
References
Russell MB, Olesen J (1995) Increased familial risk and evidence of genetic factor in migraine. BMJ 311(7004):541–544
Headache Classification Committee of the International Headache Society (IHS) (2018) The international classification of headache disorders, 3rd edition. Cephalalgia 38(1):1–211
Thomsen LL, Eriksen MK, Roemer SF, Andersen I, Olesen J, Russell MB (2002) A population-based study of familial hemiplegic migraine suggests revised diagnostic criteria. Brain. 125:1379–1391
Leão AAP (1944) Spreading depression of activity in the cerebral cortex. J Neurophysiol 7:359–390
Somjen GG (2001) Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol Rev 81:1065–1096
Dreier JP (2011) The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med 17:439–447
Hadjikhani N, Sanchez Del Rio M, Wu O, Schwartz D, Bakker D, Fischl B, Kwong KK, Cutrer FM, Rosen BR, Tootell RB, Sorensen AG, Moskowitz MA (2001) Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc Natl Acad Sci U S A 98:4687–4692
Eikermann-Haerter K, Lee JH, Yuzawa I, Liu CH, Zhou Z, Shin HK, Zheng Y, Qin T, Kurth T, Waeber C, Ferrari MD, van den Maagdenberg AM, Moskowitz MA, Ayata C (2012) Migraine mutations increase stroke vulnerability by facilitating ischemic depolarizations. Circulation. 125(2):335–345
Chen SP, Tolner EA, Eikermann-Haerter K (2016) Animal models of monogenic migraine. Cephalalgia. 36(7):704–721
Eikermann-Haerter K, Dileköz E, Kudo C, Savitz SI, Waeber C, Baum MJ, Ferrari MD, van den Maagdenberg AM, Moskowitz MA, Ayata C (2009) Genetic and hormonal factors modulate spreading depression and transient hemiparesis in mouse models of familial hemiplegic migraine type 1. J Clin Invest 119:99–109
Eikermann-Haerter K, Yuzawa I, Qin T, Wang Y, Baek K, Kim YR, Hoffmann U, Dilekoz E, Waeber C, Ferrari MD, van den Maagdenberg AM, Moskowitz MA, Ayata C (2011) Enhanced subcortical spreading depression in familial hemiplegic migraine type 1 mutant mice. J Neurosci 31:5755–5763
Eikermann-Haerter K, Arbel-Ornath M, Yalcin N, Yu ES, Kuchibhotla KV, Yuzawa I, Hudry E, Willard CR, Climov M, Keles F, Belcher AM, Sengul B, Negro A, Rosen IA, Arreguin A, Ferrari MD, van den Maagdenberg AM, Bacskai BJ, Ayata C (2015) Abnormal synaptic Ca (2+) homeostasis and morphology in cortical neurons of familial hemiplegic migraine type 1 mutant mice. Ann Neurol 78(2):193–210
Eikermann-Haerter K, Baum MJ, Ferrari MD, van den Maagdenberg AM, Moskowitz MA, Ayata C (2009) Androgenic suppression of spreading depression in familial hemiplegic migraine type 1 mutant mice. Ann Neurol 66(4):564–568
Kros L, Lykke-Hartmann K, Khodakhah K (2018) Increased susceptibility to cortical spreading depression and epileptiform activity in a mouse model for FHM2. Sci Rep 8:16959
Brennan KC, Bates EA, Shapiro RE, Zyuzin J, Hallows WC, Huang Y, Lee HY, Jones CR, Fu YH, Charles AC, Ptáček LJ (2013) Casein kinase iδ mutations in familial migraine and advanced sleep phase. Sci Transl Med 5(183):183ra56 1-11
Nozari A, Dilekoz E, Sukhotinsky I, Stein T, Eikermann-Haerter K, Liu C, Wang Y, Frosch MP, Waeber C, Ayata C, Moskowitz MA (2010) Microemboli may link spreading depression, migraine aura, and patent foramen ovale. Ann Neurol 67:221–229
Donmez-Demir B, Yemisci M, Kilic K, Gursoy-Ozdemir Y, Soylemezoglu F, Moskowitz M, Dalkara T (2018) Microembolism of single cortical arterioles can induce spreading depression and ischemic injury; a potential trigger for migraine and related MRI lesions. Brain Res 1679:84–90
Ayata C, Lauritzen M (2015) Spreading depression, spreading depolarizations, and the cerebral vasculature. Physiol Rev 95:953–993
Lauritzen M, Strong AJ (2017) Spreading depression of Leão' and its emerging relevance to acute brain injury in humans. J Cereb Blood Flow Metab 37(5):1553–1570
Johnson W, Onuma O, Owolabi M, Sachdev S (2016) Stroke: a global response is needed. Bull World Health Organ 9:633–708
O'Donnell MJ, Xavier D, Liu L, Zhang H, Chin SL, Rao-Melacini P, Rangarajan S, Islam S, Pais P, MJ MQ, Mondo C, Damasceno A, Lopez-Jaramillo P, Hankey GJ, Dans AL, Yusoff K, Truelsen T, Diener HC, Sacco RL, Ryglewicz D, Czlonkowska A, Weimar C, Wang X, Yusuf S, INTERSTROKE investigators (2010) Risk factors for ischaemic and intracerebral haemorrhagic stroke in 22 countries (the INTERSTROKE study): a case-control study. Lancet 376(9735):112–123
Johnston SC, Mendis S, Mathers CD (2009) Global variation in stroke burden and mortality: estimates from monitoring, surveillance, and modelling. Lancet Neurol 8(4):345–354
Chang CL, Donaghy M, Poulter N (1999) Migraine and stroke in young women: case-control study. The World Health Organisation collaborative study of cardiovascular disease and steroid hormone contraception. BMJ. 318:13–18
Kurth T, Schurks M, Logroscino G, Buring JE (2009) Migraine frequency and risk of cardiovascular disease in women. Neurology. 73:581–588
Spector JT, Kahn SR, Jones MR, Jayakumar M, Dalal D, Nazarian S (2010) Migraine headache and ischemic stroke risk: an updated meta-analysis. Am J Med 123:612–624
Kurth T, Chabriat H, Bousser MG (2012) Migraine and stroke: a complex association with clinical implications. Lancet Neurol 11:92–100
Tietjen GE (2007) Migraine and ischaemic heart disease and stroke: potential mechanisms and treatment implications. Cephalalgia. 27:981–987
Tietjen GE (2007) Migraine as a systemic disorder. Neurology. 68:1555–1556
Stam AH, Haan J, van den Maagdenberg AM, Ferrari MD, Terwindt GM (2009) Migraine and genetic and acquired vasculopathies. Cephalalgia. 29:1006–1017
Eikermann-Haerter K, Ayata C (2010) Cortical spreading depression and migraine. Curr Neurol Neurosci Rep. 10:167–173
Eikermann-Haerter K (2014) Spreading depolarization may link migraine and stroke. Headache. 54:1146–1157
Kazemi H, Speckmann EJ, Gorji A (2014) Familial hemiplegic migraine and spreading depression. Iran J Child Neurol 8:6–11
Sacco S, Kurth T (2014) Migraine and the risk for stroke and cardiovascular disease. Curr Cardiol Rep 16:524
Malik R, Freilinger T, Bendik S, Winsvold BS, Anttila V, Vander Heiden J et al (2015) Shared genetic basis for migraine and ischemic stroke. A genome-wide analysis of common variants. Neurology. 84:2132–2145
Eikermann-Haerter K, Yuzawa I, Dilekoz E, Joutel A, Moskowitz MA, Ayata C (2011) Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy syndrome mutations increase susceptibility to spreading depression. Ann Neurol 69(2):413–418
Sen S, Androulakis XM, Duda V, Alonso A, Chen LY, Soliman EZ, Magnani J, Trivedi T, Merchant AT, Gottesman RF, Rosamond WD (2018) Migraine with visual aura is a risk factor for incident atrial fibrillation: A cohort study. Neurology 91(24):e2202–e2210
Lantz M, Sieurin J, Sjölander A, Waldenlind E, Sjöstrand C, Wirdefeldt K (2017) Migraine and risk of stroke: a national population-based twin study. Brain. 140(10):2653–2662
Shin HK, Dunn AK, Jones PB, Boas DA, Moskowitz MA, Ayata C (2006) Vasoconstrictive neurovascular coupling during focal ischemic depolarizations. J Cereb Blood Flow Metab 26:1018–1030
Eikermann-Haerter K, Lee JH, Yalcin N, Yu ES, Daneshmand A, Wei Y, Zheng Y, Can A, Sengul B, Ferrari MD, van den Maagdenberg AM, Ayata C (2015) Migraine prophylaxis, ischemic depolarizations, and stroke outcomes in mice. Stroke. 46:229–236
Gladstone JP, Dodick DW (2005) Migraine and cerebral white matter lesions: when to suspect cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). Neurologist. 11(1):19–29
de Boer I, Stam AH, Buntinx L, Zielman R, van der Steen I, van den Maagdenberg AMJM, de Koning EJP, Ferrari MD, de Hoon JN, Terwindt GM (2018) RVCL-S and CADASIL display distinct impaired vascular function. Neurology. 91(10):e956–e963
Lee JH, Eikermann-Haerter K, Joutel A, Moskowitz MA, Ayata C (2009) Enlarged infarcts in mice expressing the archetypal Notch3 R90C CADASIL mutation. J Cereb Blood Flow Metab 29:S253-S254
Tietjen GE, Collins SA (2018) Hypercoagulability and migraine. Headache. 58(1):173–183
Wilmshurst PT (2018) Migraine with aura and persistent foramen ovale. Eye (Lond) 32:184–188
Pezzini A, Busto G, Zedde M, Gamba M, Zini A, Poli L, Caria F, De Giuli V, Simone AM, Pascarella R, Padovani A, Padroni M, Gasparotti R, Colagrande S, Fainardi E (2018) Vulnerability to infarction during cerebral ischemia in migraine sufferers. Stroke. 49(3):573–578
Mawet J, Eikermann-Haerter K, Park KY, Helenius J, Daneshmand A, Pearlman L, Avery R, Negro A, Velioglu M, Arsava EM, Ay H, Ayata C (2015) Sensitivity to acute cerebral ischemic injury in migraineurs: a retrospective case-control study. Neurology. 85(22):1945–1949
Bigal ME, Kurth T, Santanello N, Buse D, Golden W, Robbins M, Lipton RB (2010) Migraine and cardiovascular disease: a population-based study. Neurology. 74(8):628–635
Adelborg K, Szépligeti SK, Holland-Bill L, Ehrenstein V, Horváth-Puhó E, Henderson VW, Sørensen HT (2018) Migraine and risk of cardiovascular diseases: Danish population based matched cohort study. BMJ. 360:k96
Piilgaard H, Lauritzen M (2009) Persistent increase in oxygen consumption and impaired neurovascular coupling after spreading depression in rat neocortex. J Cereb Blood Flow Metab 29(9):1517–1527
Khennouf L, Gesslein B, Lind BL, van den Maagdenberg AM, Lauritzen M (2016) Activity-dependent calcium, oxygen, and vascular responses in a mouse model of familial hemiplegic migraine type 1. Ann Neurol 80:219–232
Gryglas A, Smigiel R (2017) Migraine and stroke: What's the link? What to do? Curr Neurol Neurosci Rep 17(3):22
Kruit MC, van Buchem MA, Hofman PA, Bakkers JT, Terwindt GM, Ferrari MD, Launer J (2004) Migraine as a risk factor for subclinical brain lesions. JAMA. 28:427–434
Bashir A, Lipton RB, Ashina S, Ashina M (2013) Migraine and structural changes in the brain: a systematic review and meta-analysis. Neurology. 81:1260–1268
Chong CD, Schwedt TJ, Hougaard A (2019) Brain functional connectivity in headache disorders: A narrative review of MRI investigations. J Cereb Blood Flow Metab 39(4):650-669
Seitz I, Dirnagl U, Lindauer U (2004) Impaired vascular reactivity of isolated rat middle cerebral artery after cortical spreading depression in vivo. J Cereb Blood Flow Metab 24(5):526–530
Lauritzen M (1994) Pathophysiology of the migraine aura. The spreading depression theory. Brain. 117:199–210
Thomsen LL, Iversen HK, Olesen J (1995) Increased cerebrovascular pCO2 reactivity in migraine with aura - a transcranial Doppler study during hyperventilation. Cephalalgia. 15:211–215
Perko D, Pretnar-Oblak J, Sabovic M, Zvan B, Zaletel M (2011) Cerebrovascular reactivity to L-arginine in the anterior and posterior cerebral circulation in migraine patients. Acta Neurol Scand 124(4):269–274
Armulik A, Genové G, Betsholtz C (2011) Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell 21(2):193–215
Hartmann DA, Underly RG, Grant RI, Watson AN, Lindner V, Shih AY (2015) Pericyte structure and distribution in the cerebral cortex revealed by high-resolution imaging of transgenic mice. Neurophotonics. 2(4):041402
Alarcon-Martinez L, Yilmaz-Ozcan S, Yemisci M, Schallek J, Kılıç K, Can A, Di Polo A, Dalkara T (2018) Capillary pericytes express α-smooth muscle actin, which requires prevention of filamentous-actin depolymerization for detection. Elife 21:7
Peppiatt CM, Howarth C, Mobbs P, Attwell D (2006) Bidirectional control of CNS capillary diameter by pericytes. Nature. 443(7112):700–704
Gursoy-Ozdemir Y, Yemisci M, Dalkara T (2012) Microvascular protection is essential for successful neuroprotection in stroke. J Neurochem 123(Suppl 2):2–11
Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z, Toga AW, Jacobs RE, Liu CY, Amezcua L, Harrington MG, Chui HC, Law M, Zlokovic BV (2015) Blood-brain barrier breakdown in the aging human hippocampus. Neuron. 85(2):296–302
Fernández-Klett F, Priller J (2015) Diverse functions of pericytes in cerebral blood flow regulation and ischemia. J Cereb Blood Flow Metab 35(6):883–887
Attwell D, Mishra A, Hall CN, O'Farrell FM, Dalkara T (2016) What is a pericyte? J Cereb Blood Flow Metab 36(2):451–455
Kisler K, Nelson AR, Rege SV, Ramanathan A, Wang Y, Ahuja A, Lazic D, Tsai PS, Zhao Z, Zhou Y, Boas DA, Sakadžić S, Zlokovic BV (2017) Pericyte degeneration leads to neurovascular uncoupling and limits oxygen supply to brain. Nat Neurosci 20(3):406–416
Dalkara T, Alarcon-Martinez L, Yemisci M (2016) Role of Pericytes in Neurovascular Unit and Stroke. In: Chen J, Zhang JH, Hu X (eds) Non-Neuronal Mechanisms of Brain Damage and Repair After Stroke. Springer International Publishing Springer Series in Translational Stroke Research, Switzerland, pp 25–43
Yemisci M, Gursoy-Ozdemir Y, Vural A, Can A, Topalkara K, Dalkara T (2009) Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat Med 15:1031–1037
Hall CN, Reynell C, Gesslein B, Hamilton NB, Mishra A, Sutherland BA, O'Farrell FM, Buchan AM, Lauritzen M, Attwell D (2014) Capillary pericytes regulate cerebral blood flow in health and disease. Nature 508(7494):55–60
Khennouf L, Gesslein B, Brazhe A, Octeau JC, Kutuzov N, Khakh BS, Lauritzen M (2018) Active role of capillary pericytes during stimulation-induced activity and spreading depolarization. Brain. 141(7):2032–2046
Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P, Alamowitch S, Domenga V, Cécillion M, Marechal E, Maciazek J, Vayssiere C, Cruaud C, Cabanis EA, Ruchoux MM, Weissenbach J, Bach JF, Bousser MG, Tournier-Lasserve E (1996) Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature. 383(6602):707–710
Dziewulska D, Lewandowska E (2012) Pericytes as a new target for pathological processes in CADASIL. Neuropathology. 32(5):515–521
Gu X, Liu XY, Fagan A, Gonzalez-Toledo ME, Zhao LR (2012) Ultrastructural changes in cerebral capillary pericytes in aged Notch3 mutant transgenic mice. Ultrastruct Pathol 36:48–55
Ghosh M, Balbi M, Hellal F, Dichgans M, Lindauer U, Plesnila N (2015) Pericytes are involved in the pathogenesis of CADASIL. Ann Neurol 78(6):887–900
Chabriat H, Pappata S, Ostergaard L, Clark CA, Pachot-Clouard M, Vahedi K, Jobert A, Le Bihan D, Bousser MG (2000) Cerebral hemodynamics in cadasil before and after acetazolamide challenge assessed with MRI bolus tracking. Stroke. 31:1904–1912
Bruening R, Dichgans M, Berchtenbreiter C, Yousry T, Seelos KC, Wu RH, Mayer M, Brix G, Reiser M (2001) Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy: decrease in regional cerebral blood volume in hyperintense subcortical lesions inversely correlates with disability and cognitive performance. AJNR Am J Neuroradiol 22:1268–1274
Pfefferkorn T, von Stuckrad-Barre S, Herzog J, Gasser T, Hamann GF, Dichgans M (2001) Reduced cerebrovascular CO2 reactivity in CADASIL: a transcranial doppler sonography study. Stroke. 32:17–21
Ayata C (2010) CADASIL: experimental insights from animal models. Stroke. 41:S129–S134
Dziewulska D, Kierdaszuk B (2018) Ultrastructural changes in microvessels in familial hemiplegic migraine with CACNA1A mutation. Clin Neuropathol 37(6):283–287
Cheng J, Korte N, Nortley R, Sethi H, Tang Y, Attwell D (2018) Targeting pericytes for therapeutic approaches to neurological disorders. Acta Neuropathol 136(4):507–523
Shin HK, Hong KW (2004) Importance of calcitonin gene-related peptide, adenosine and reactive oxygen species in cerebral autoregulation under normal and diseased conditions. Clin Exp Pharmacol Physiol 31(1–2):1–7
Zhai L, Sakurai T, Kamiyoshi A, Ichikawa-Shindo Y, Kawate H, Tanaka M, Xian X, Hirabayashi K, Dai K, Cui N, Tanimura K, Liu T, Wei Y, Tanaka M, Tomiyama H, Yamauchi A, Igarashi K, Shindo T (2018) Endogenous calcitonin gene-related peptide suppresses ischemic brain injuries and progression of cognitive decline. J Hypertens 36(4):876–891
Johansson SE, Abdolalizadeh B, Sheykhzade M, Edvinsson L, Sams A (2019) Vascular pathology of large cerebral arteries in experimental subarachnoid hemorrhage: vasoconstriction, functional CGRP depletion and maintained CGRP sensitivity. Eur J Pharmacol 846:109–118
Moskowitz MA, Reinhard JF Jr, Romero J, Melamed E, Pettibone DJ (1979) Neurotransmitters and the fifth cranial nerve: is there a relation to the headache phase of migraine? Lancet. 2(8148):883–885
Edvinsson L, Villalón CM, MaassenVanDenBrink A (2012) Basic mechanisms of migraine and its acute treatment. Pharmacol Ther 136(3):319–333
Goadsby PJ, Edvinsson L (1993) The trigeminovascular system and migraine: studies characterizing cerebrovascular and neuropeptide changes seen in humans and cats. Ann Neurol 33:48–56
Goadsby PJ, Edvinsson L, Ekman R (1988) Release of vasoactive peptides in the extracerebral circulation of humans and the cat during activation of the trigeminovascular system. Ann Neurol 23:193–196
Goadsby PJ, Edvinsson L, Ekman R (1990) Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann Neurol 22:183–187
Lassen LH, Haderslev PA, Jacobsen VB, Iversen HK, Sperling B, Olesen J (2002) CGRP may play a causative role in migraine. Cephalalgia. 22(1):54–61
Deen M, Correnti E, Kamm K, Kelderman T, Papetti L, Rubio-Beltrán E, Vigneri S, Edvinsson L, MaassenVanDenBrink A (2017) European Headache Federation School of Advanced Studies (EHF-SAS). Blocking CGRP in migraine patients - a review of pros and cons. J Headache Pain 18(1):96
MaassenVanDenBrink A, Meijer J, Villalón CM, Ferrari MD (2016) Wiping out CGRP: potential cardiovascular risks. Trends Pharmacol Sci 37(9):779–788
Favoni V, Giani L, Al-Hassany L, Asioli GM, Butera C, de Boer I, Guglielmetti M, Koniari C, Mavridis T, Vaikjärv M, Verhagen I, Verzina A, Zick B, Martelletti P, Sacco S, European Headache Federation School of Advanced Studies (EHF-SAS) (2019) CGRP and migraine from a cardiovascular point of view: what do we expect from blocking CGRP? J Headache Pain 20(1):27
Moskowitz MA, Nozaki K, Kraig RP (1993) Neocortical spreading depression provokes the expression of c-fos protein-like immunoreactivity within trigeminal nucleus caudalis via trigeminovascular mechanisms. J Neurosci 13:1167–1177
Bolay H, Reuter U, Dunn AK, Huang Z, Boas DA, Moskowitz MA (2002) Intrinsic brain activity triggers trigeminal meningeal afferents in a migraine model. Nat Med 8:136–142
Karatas H, Erdener SE, Gursoy-Ozdemir Y, Lule S, Eren-Kocak E, Sen ZD, Dalkara T (2013) Spreading depression triggers headache by activating neuronal Panx1 channels. Science. 339:1092–1095
Chen SP, Qin T, Seidel JL, Zheng Y, Eikermann M, Ferrari MD, van den Maagdenberg AMJM, Moskowitz MA, Ayata C, Eikermann-Haerter K (2017) Inhibition of the P2X7-PANX1 complex suppresses spreading depolarization and neuroinflammation. Brain. 140(6):1643–1656
Yamasaki Y, Matsuura N, Shozuhara H, Onodera H, Itoyama Y, Kogure K (1995) Interleukin-1 as a pathogenetic mediator of ischemic brain damage in rats. Stroke. 26:676–680 discussion 681
Neeb L, Hellen P, Boehnke C, Hoffmann J, Schuh-Hofer S, Dirnagl U et al (2011) IL-1beta stimulates COX-2 dependent PGE (2) synthesis and CGRP release in rat trigeminal ganglia cells. PLoS One 6:e17360
Lambertsen KL, Biber K, Finsen B (2012) Inflammatory cytokines in experimental and human stroke. J Cereb Blood Flow Metab 32:1677–1698
Zhang X, Burstein R, Levy D (2012) Local action of the proinflammatory cytokines IL-1beta and IL-6 on intracranial meningeal nociceptors. Cephalalgia. 32:66–72
Orr SL, Dos Santos MP, Jurencak R, Michaud J, Miller E, Doja A (2014) Central nervous system venulitis presenting as migraine. Headache. 54:541–544
Sakadzic S, Mandeville ET, Gagnon L, Musacchia JJ, Yaseen MA, Yucel MA, Lefebvre J, Lesage F, Dale AM, Eikermann-Haerter K, Ayata C, Srinivasan VJ, Lo EH, Devor A, Boas DA (2014) Large arteriolar component of oxygen delivery implies a safe margin of oxygen supply to cerebral tissue. Nat Commun 5:5734
Dönmez-Demir B, Yemisci M, Dalkara T (2018) Data of ascending cortical vein occlusion induced spreading depression. Data Brief 18:1462–1465
Snijder RJ, Luermans JG, de Heij AH, Thijs V, Schonewille WJ, Van De Bruaene A, Swaans MJ, Budts WI, Post MC (2016) Patent Foramen Ovale With Atrial Septal Aneurysm Is Strongly Associated With Migraine With Aura: A Large Observational Study. J Am Heart Assoc 5(12):e003771
Noheria A, Roshan J, Kapa S, Srivathsan K, Packer DL, Asirvatham SJ (2011) Migraine headaches following catheter ablation for atrial fibrillation. J Interv Card Electrophysiol 30(3):227–232
Sevgi EB, Erdener SE, Demirci M, Topcuoglu MA, Dalkara T (2012) Paradoxical air microembolism induces cerebral bioelectrical abnormalities and occasionally headache in patent foramen ovale patients with migraine. J Am Heart Assoc 1(6):e001735
Buture A, Khalil M, Ahmed F (2017) Iatrogenic visual aura: a case report and a brief review of the literature. Ther Clin Risk Manag 13:643–646
Androulakis XM, Kodumuri N, Giamberardino LD, Rosamond WD, Gottesman RF, Yim E, Sen S (2016) Ischemic stroke subtypes and migraine with visual aura in the ARIC study. Neurology. 87(24):2527–2532
Timm FP, Houle TT, Grabitz SD, Lihn A, Stokholm JB, Eikermann-Haerter K, Nozari A, Kurth T, Eikermann M (2017) Migraine and risk of perioperative ischemic stroke and hospital readmission: hospital based registry study. BMJ. 356:i6635
Acknowledgements
None.
Funding
The article-processing charges for the article have been sponsored by the European Headache Federation.
Author information
Authors and Affiliations
Contributions
MY and KEH were responsible for writing and critically revising the manuscript. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declared that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Yemisci, M., Eikermann-Haerter, K. Aura and Stroke: relationship and what we have learnt from preclinical models. J Headache Pain 20, 63 (2019). https://doi.org/10.1186/s10194-019-1016-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s10194-019-1016-x