
Abstract
The thermal degradation reactivities of hemicellulose and cellulose in wood cell walls are significantly different from the thermal degradation behavior of the respective isolated components. Furthermore, the degradation of Japanese cedar (Cryptomeria japonica, a softwood) is distinct from that of Japanese beech (Fagus crenata, a hardwood). Lignin and uronic acid are believed to play crucial roles in governing this behavior. In this study, the effects of ball milling for various durations of time on the degradation reactivities of cedar and beech woods were evaluated based on the recovery rates of hydrolyzable sugars from pyrolyzed wood samples. The applied ball-milling treatment cleaved the lignin β-ether bonds and reduced the crystallinity of cellulose, as determined by X-ray diffraction. Both xylan and glucomannan degraded in a similar temperature range, although the isolated components exhibited different reactivities because of the catalytic effect of uronic acid bound to the xylose chains. These observations can be explained by the more homogeneous distribution of uronic acid in the matrix of cell walls as a result of ball milling. As observed for holocelluloses, cellulose in the ball-milled woods degraded in two temperature ranges (below 320 °C and above); a significant amount of cellulose degraded in the lower temperature range, which significantly changed the shapes of the thermogravimetric curves. This report compares the results obtained for cedar and beech woods, and discusses them in terms of the thermal degradation of the matrix and cellulose microfibrils in wood cell walls and role of lignin. Such information is crucial for understanding the pyrolysis and heat treatment of wood.
Similar content being viewed by others
Introduction
Pyrolysis-based technologies have received significant attention in recent years owing to their potential for converting wood biomass into biofuels and biochemicals [1, 2]. Cellulose, hemicellulose, and lignin are the major components of wood, and their thermal degradation reactivities provide the fundamental basis for pyrolysis-based technologies. Cellulose microfibrils are embedded in the hemicellulose–lignin matrix within the nanoscale cell wall structure, whose properties can also affect the thermal reactivity of the wood. However, this relationship has not been fully elucidated because the thermal reactivities of wood components have primarily been investigated using isolated components. Thermogravimetric (TG) analysis is frequently used for such studies, but to our knowledge, the results have not yet been discussed in terms of the degradation of wood components at each temperature.
Our group [3] previously evaluated the thermal degradation reactivities of hemicellulose and cellulose in wood by assessing the quantity of hydrolyzable sugars that remained after heat-treating Japanese cedar (Cryptomeria japonica, a softwood) and Japanese beech (Fagus crenata, a hardwood). The treatment involved conditions similar to those used for TG analysis, no heating time at constant temperatures. Employing this method, the results of TG analysis can be explained by the thermal degradation of wood polysaccharides at each temperature. Isolated xylan degraded at a lower temperature than isolated glucomannan because of the catalytic action of 4-O-methyl-d-glucuronic acid (4-O-MeGlcA) and its salt (base), which were bound to the xylose chains [4]. However, the xylans in cedar and beech woods were stable and degraded in the temperature range similar to that of isolated glucomannan [3]. In contrast, the glucomannan in beech wood was very reactive [3]. Therefore, the hemicellulose reactivity in wood is quite different from that of isolated xylan or isolated glucomannan. The thermal reactivity of cellulose in cedar and beech were also different; cellulose degraded together with hemicellulose in cedar wood, but independently in beech wood [3].
The distinct thermal degradation reactivities of cedar and beech woods may be attributed to the specific placement of cell wall components. The location of 4-O-MeGlcA was evaluated via demineralization, which converts metal salts to free 4-O-MeGlcA. The results indicated that it is located near cellulose and glucomannan in cedar wood, but near xylan and glucomannan in beech wood [5].
Lignin is believed to play an important role in determining the thermal reactivity of hemicellulose in wood, because the reactivities of xylan and glucomannan in wood became similar to those of isolated xylan and glucomannan by removing lignin [6]. The different thermal degradation behaviors of cedar and beech woods largely disappeared, and their differential thermogravimetric (DTG) curves adopted similar shapes, although the tendency for hemicellulose/cellulose co-degradation in cedar was maintained. Based on these results, lignification during the biosynthesis of cell walls is considered to (i) introduce enhanced physical restraint and (ii) fix the specific placement of hemicellulose in cell walls, particularly for 4-O-MeGlcA.
The thermal degradation of cellulose in cedar and beech woods was improved after delignification (relative to pure cellulose). The thermal degradation of cellulose in holocellulose occurred in two stages depending on the pyrolysis temperature (with some cellulose degrading below 320 °C and the rest degrading at higher temperatures) [6]. The improved reactivity may be related to the formation of pores in the matrix when the lignin was removed; however, the details of this activation mechanism are not entirely understood.
Ball milling can disrupt the matrix in wood cell walls, but this process does not remove any components from the wood. Therefore, assessing the effects of ball milling on the hemicellulose and cellulose reactivities in wood can improve our understanding of the thermal degradation of these components in wood cell walls. Ball milling is a commonly used method for pretreating wood and other lignocellulosic biomass to improve enzyme reactivity [7,8,9,10] and to separate lignin [11,12,13,14]. This process is also known to reduce the crystallinity of cellulose [15,16,17] and cleave the lignin β-ether linkages [12, 14], i.e., the dominant type of linkage. Although several research groups have reported on the TG analysis of ball-milled wood, to our knowledge, this method’s impact on the thermal reactivities of cellulose and hemicellulose in wood have not been investigated.
In the present study, cedar and beech wood samples were ball milled for various amounts of time (ranging from 10 min to 48 h), and the thermal degradation reactivities of the resulting wood samples were investigated using TG analysis. The amount of hydrolyzable sugar remaining in pyrolyzed ball-milled wood was also evaluated after treating samples at a heating rate similar to TG analysis (with no time spent heating at a constant temperature). The effects of the ball-milling process on the thermal degradation reactivities of hemicellulose and cellulose are discussed in terms of the cleavage of lignin β-ether linkages, the loss of cellulose crystallinity, and the cell wall ultrastructure.
Experimental
Preparation and characterization of ball-milled samples
Extractive-free Japanese cedar (Cryptomeria japonica) and Japanese beech (Fagus crenata) (80 mesh passed), and isolated cellulose (Whatman CF-11, Whatman plc, Maidstone, UK) powders were ball-milled using a vibratory ball mill (VS-1, Chuo Kakohki, Aichi, Japan). Approximately, 150 g of each powder was placed in a stainless steel jar (inner diameter = 110 mm, height = 120 mm) with about 450 stainless steel balls (diameter = 12.7 mm), which occupied about 80% of the jar’s inner volume. The jar was sealed with a lid and vibrated for a predetermined, designated time (between 10 min and 48 h). During the ball-milling process, the jar was cooled by an outer layer water-cooled jacket. Ball-milling process was conducted in air, but no extensive oxidation was expected to occur, since the literature [18, 19] reports that any differences were not observed in the FT-IR spectra of ball-milled wood samples in nitrogen and in air.
It has been reported that the ball-milling process decreases the cellulose crystallinity [15,16,17] and the degree of polymerization of lignin due to cleavage of the ether linkages [12, 14]. To study the effect of these changes on pyrolysis, the unmilled and ball-milled samples were analyzed by X-ray diffraction (XRD; RINT 2000V, Rigaku, Tokyo, Japan). The cellulose crystallinity index (defined as Xc) was determined from the obtained XRD pattern using the following equation [20]:
where I002 is the peak intensity of the 002 lattice diffraction of cellulose, and Iam is that of the nearby amorphous region.
Thioacidolysis is an acid-catalyzed solvolysis technique that employs ethanethiol, and the yield of thioacidolysis products (trithioethyl monolignols) represents an index of the number of β-ether linkages in lignin. Thioacidolysis was performed according to a published procedure [21], and the thioacidolysis products were quantified using gas chromatography–mass spectrometry (GC–MS; QP2010 Ultra, Shimadzu, Kyoto, Japan) after trimethylsilyl derivatization. The GC–MS used a CP-Sil 8CB column (Agilent Technologies, CA, USA; length = 30 m, diameter = 0.25 mm, thickness = 0.25 μm) where the injector temperature = 260 °C; split ratio = 1:50; column temperature = 130 °C (5 min), + 5 °C/min until 260 °C, 260 °C (5 min); carrier gas = H2. The quantitative determination of G and S monomers was performed based on the peak areas of total-ion chromatograms by comparing those of tetracosane (C24) and hexacosane (C26) used as internal standards [9, 21]. A typical example of the total-ion chromatogram is illustrated in Additional file 1: Fig. S2.
TG analysis
TG analysis was conducted using a TG analyzer (TGA-50; Shimadzu, Kyoto, Japan). Ball-milled wood samples from Japanese cedar and Japanese beech wood (1 mg) were heated from room temperature up to 800 °C in a platinum pan at a heating rate of 10 °C/min under a N2 flow of 10 mL/min (purity = 99.9998%, JAPAN FINE PRODUCTS, Mieken, Japan). A deoxygenation column (GL Sciences, Japan) was used to remove any O2 contamination in the N2.
Pyrolysis experiments
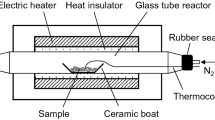
Figure 1 illustrates the pyrolysis experimental setup. An electric furnace (ARF-20KC, Asahi-Rika, Chiba, Japan) was used to heat the samples. In each pyrolysis experiment, the sample was placed in a ceramic boat (AS ONE, Osaka, Japan), which was inserted into a quartz glass tube (inner diameter = 15 mm, length = 400 mm, wall thickness = 1.5 mm). N2 gas was supplied into the glass tube at a flow rate of 100 mL/min using a mass flow controller (SEC-400MK3, Horiba, Kyoto, Japan). The sample was heated from room temperature to a designated temperatures (i.e., 20 °C intervals between 220 and 380 °C) at the same heating rate as in the TG analysis (10 °C/min) to allow for accurate comparisons. The sample temperature was measured by placing a fine thermocouple (0.25 mm in diameter; SHINNETSU, Ibaraki, Japan) in contact with the sample. When the sample temperature reached the designated temperature, the cover of the electric furnace was opened and the glass tube was immediately cooled to room temperature under a flow of air.

Experimental setup
Hydrolyzable sugar analysis
Acid hydrolysis and methanolysis of the pyrolyzed samples were performed separately to convert cellulose into glucose and hemicellulose/pectin into methyl glycosides, respectively. For the hydrolysis experiments, each sample was treated with 0.3 mL of aqueous 72% H2SO4 solution in a ceramic boat at 30 °C for 1 h in a sealed glass vial. Then, 8.4 mL of distilled water was added to the mixture and heated in an autoclave at 120 °C for 1 h to complete the hydrolysis reaction. After the hydrolysate solution was filtered, diluted, and neutralized, the glucose yield was determined via high-performance anion-exchange chromatography using a Prominence system (Shimadzu) equipped with an electrochemical detector (DECADE Elite, Antec Scientific, Zoeterwoude, Netherlands) under the following conditions: column = CarboPac PA1 (4 mm × 250 mm); eluent = 85% distilled water/15% 0.2 M NaOH; flow rate = 1 mL/min; column oven temperature = 35 °C.
The milder methanolysis experiments [22,23,24,25] was performed to determine the yields of hydrolyzable sugars, such as methyl glycosides, derived from hemicellulose, pectin, and uronic acid groups. Each pyrolyzed sample was placed in a sealed glass vial along with the ceramic boat, and 4 mL of 2 M HCl solution in methanol was added. The vial was then heated at 60 °C for 16 h to complete the methanolysis reaction. After neutralization, the addition of internal standard (glucitol) and trimethylsilyl derivatization, the resulting products were analyzed by GC–MS under the aforementioned conditions, except the temperature program was changed to the following: 100 °C (2 min), 4 °C/min up to 220 °C, 220 °C (2 min), 15 °C/min up to 300 °C, 300 °C (2 min). The peaks originating from hemicellulose/pectin and 4-O-MeGlcA were assigned based on the mass spectra, and the retention times were compared with published data [26, 27]. Typical examples of chromatograms from sugar analysis are illustrated in Additional file 1: Fig. S3.
In the present study, the hydrolysable sugar analysis was conducted three times and the average value was used for discussion. In addition, the pyrolysis experiments were repeated more than twice to confirm the reproducibility of the results.
Results and discussion
Characterization and TG analysis of ball-milled wood
The intensity of the XRD signals originating from cellulose crystallites decreased with increasing milling time for both cedar and beech wood samples (Additional file 1: Fig. S1); this phenomenon has also been reported previously [15,16,17, 28]. The crystallinity index calculated from the X-ray diffractograms using the 002 lattice diffraction decreased from 68 to 5% after ball milling for 1–2 h (Fig. 2).

Influence of ball-milling time on the crystallinity index evaluated by XRD analysis for cedar ( ) and beech (
) and beech ( ) wood
) wood
Although there is less information regarding the effect of ball milling on lignin than on cellulose, some literature reports [12, 14] determined that ball milling reduced the degree of polymerization of lignin and increased the phenolic structure by cleaving the ether linkages. Therefore, the β-ether structures remaining in the ball-milled wood samples were quantified using the thioacidolysis method to evaluate the effect of ball-milling time on the cleavage of the β-ether bonds (the most abundant type of linkage in lignin). Thioacidolysis involves ethanethiol-assisted, acid-catalyzed solvolysis and leads to the formation of trithioethyl monomeric products through the cleavage of β-ether bonds. The derivatization followed by reductive cleavage (DFRC) method has also been used for quantitative analysis of the β-ether linkages, but Holtman et al. [29] reported that thioacidolysis was a better strategy for evaluating the β-ether linkages in ball-milled wood.
The relative yields of thioacidolysis products, namely guaiacyl (G)-type in cedar and G- and syringyl (S)-types in beech, are plotted as a function of the milling time in Fig. 3. These yields (normalized relative to 100% for unmilled wood) decreased as the milling time increased, and were about 20% after 4 h for both woods. This indicates that the β-ether linkages were cleaved during the ball-milling process, and that the efficiency was similar for cedar and beech woods, but slightly higher for cedar. No significant difference was observed between the S and G types in beech wood.

Influence of ball-milling time on the yields of thioacidolysis products from a cedar and b beech wood
The TG and DTG profiles obtained for the ball-milled cedar and beech woods are presented in Figs. 4 and 5, respectively. The TG/DTG curves of the ball-milled woods for the 10 min and 4 h experiments are shown in Fig. 6 to represent typical examples and for comparison with the unmilled wood. As the ball-milling time increased, the TG curve shifted toward lower temperatures and tended to level off after 4 h of milling. The characteristics of the DTG curves of cedar and beech were largely unchanged; the beech DTG curve has a distinct shoulder, but cedar has one wide peak.

Influence of milling time on the TG curves of ball-milled a cedar and b beech wood

Influence of milling time on the DTG curves of ball-milled a cedar and b beech wood

TG/DTG curves of ball-milled a cedar and b beech wood for 10 min and 4 h
After a very short (10 min) milling time, the temperature ranges in which the TG curves changed were different for cedar versus beech samples; the change occurred near the DTG shoulder temperature for beech, but near the DTG peak temperature for cedar. Accordingly, the DTG peak tended to shift more for cedar, whereas the DTG shoulder shifted for beech. Specifically, the DTG peak temperature shifted from 374 to 365 °C (cedar), or 367 to 362 °C (beech), and the DTG shoulder temperature of beech shifted from 320 to 302 °C. These observations indicate that some heat-resistant cellulose in the cedar wood became reactive after short-term ball milling, while the hemicellulose was influenced to a greater extent in the beech wood.
When the milling time was extended to 4 h, the TG curves of both woods shifted toward lower temperatures, within the wider temperature range of 250–400 °C. The DTG peak temperatures decreased more significantly and became similar (cedar = 354 °C and beech = 355 °C). This result indicated that their cellulose reactivities became similar after 4 h of milling, although unmilled cedar cellulose was more stable. The ball-milled wood also exhibited improved weight-loss rates in the temperature range near the DTG shoulder in beech, thus changing the shape of DTG curve.
Polysaccharide reactivity
The amount of unreacted hemicellulose and cellulose remaining in 4-h-ball-milled wood after pyrolysis was evaluated based on the hydrolyzable sugars, and the results are plotted against the pyrolysis temperature in Fig. 7. The recovery rates are shown as normalized values relative to the contents in unmilled wood [30]. The quantities of xylan and glucomannan were determined based on the yields of methyl xyloside and methyl mannoside, respectively, obtained after methanolysis. The amount of cellulose-derived glucose in the hydrolysate was calculated by subtracting the yield of glucose derived from glucomannan, which was obtained from the methyl mannoside yield by assuming that (i) the mannose:glucose ratio in glucomannan was 3:1 [31, 32] and (ii) their thermal reactivities were the same.

Thermal degradation behaviors of cellulose ( ), glucomannan (
), glucomannan ( ) and xylan (
) and xylan ( ) in 4-h-ball-milled cedar and beech wood (4 h), compared with the DTG curves. Results of unmilled wood were obtained from Wang et al. [3]
) in 4-h-ball-milled cedar and beech wood (4 h), compared with the DTG curves. Results of unmilled wood were obtained from Wang et al. [3]
The recovery data are compared with the DTG profiles in Fig. 7. Because the heating conditions applied in the pyrolysis experiments were similar to those used for TG analysis, the results from these two types of experiments can be directly compared. Previously reported results for the unmilled woods [3] are also included for comparison.
In the 4-h ball-milled samples, some of the cellulose degraded at temperatures lower than 320 °C, whereas the cellulose in unmilled cedar and beech woods was relatively more stable in this temperature range. Therefore, the cellulose in ball-milled wood tended to degrade in two different temperature ranges. A similar trend was reported for holocellulose [6], but the degradation in the lower temperature range was greater for ball-milled wood. In this work, it was determined that 39% and 38% of the cellulose in ball-milled cedar and beech woods, respectively, degraded below 320 °C; the remaining cellulose degraded at higher temperatures around the DTG peaks. This result is consistent with the characteristics of the DTG curve of ball-milled beech wood, i.e., the shoulder intensity increased significantly. Although the DTG shoulder was not clearly observed for ball-milled cedar wood, the weight-loss rate at temperatures below 320 °C increased.
It is interesting to note that ball milling and delignification have similar effects on cellulose reactivity in wood, although the ball-milling process does not remove any components from the wood. About 80% of the β-ether linkages were cleaved after ball milling for 4 h (Fig. 3), suggesting that the enhanced thermal reactivity of cellulose may be related to the cleavage of lignin ether linkages, rather than the formation of pores in the cell wall matrices due to lignin removal. Loosening the cell wall structure by cleaving lignin chains is considered a potential reason for this observation, and this relationship is discussed further below.
The recovery rates of hydrolyzable sugars derived from cellulose, xylan, and glucomannan in ball-milled wood (after 10 min and 4 h) were compared with those of pure cellulose (Whatman CF-11), isolated xylan, and isolated glucomannan, respectively, to better understand the polysaccharide reactivity in ball-milled wood (Fig. 8). The results of 4-h-ball-milled pure cellulose are also included for comparison. The recovery data are summarized for each ball-milled wood in Fig. 9.

Recovery rates of hydrolyzable sugars (cellulose-derived glucose, mannose and xylose) after pyrolysis of ball-milled a cedar and b beech wood for 10 min ( ) and 4 h (
) and 4 h ( ), compared with unmilled wood (
), compared with unmilled wood ( ), Whatman CF-11 cellulose (
), Whatman CF-11 cellulose ( : unmilled,
: unmilled,  : ball-milled (4 h) and isolated hemicellulose (
)
: ball-milled (4 h) and isolated hemicellulose (
)

Recovery rates of hydrolyzable sugars, cellulose-derived glucose ( ), mannose (
), mannose ( ) and xylose (
) and xylose ( ), following pyrolysis of ball-milled cedar and beech wood for 10 min and 4 h
), following pyrolysis of ball-milled cedar and beech wood for 10 min and 4 h
In the case of cedar wood, even after a short milling time of 10 min, the recovery rates of hydrolyzable sugars from xylan and glucomannan decreased greatly, although their contributions to the weight loss were small (Fig. 6). Interestingly, clear discontinuities were observed at 300 °C for mannose and xylose in the cedar wood, indicating that xylan and glucomannan in ball-milled cedar wood are divided into two parts with different reactivities; 62% of xylan and 57% of glucomannan degraded at temperatures < 300 °C, and the remaining portions degraded at higher temperatures. A similar trend was observed for the thermal degradation of cellulose in ball-milled cedar wood (4 h), as described earlier, although the discontinuous temperature observed for xylan and glucomannan (300 °C) was slightly lower than that of cellulose (320 °C). This trend is clearly visible in Fig. 9.
The aforementioned features were only observed for cedar samples, so they may be related to the thermal degradation characteristics specific to cedar wood, in which cellulose degrades together with hemicellulose. The close assembly of xylan, glucomannan, and cellulose microfibrils (surface molecules) was demonstrated based on the conversion of metal salts to free 4-O-MeGlcA [5]. In general, the thermal degradation behaviors of cellulose and hemicellulose presented in the current study support such assembly.
The crystallinity of cellulose in cedar wood decreased from 68 to 51% following 10 min of milling, and some of the lignin β-ether linkages were cleaved (Figs. 2 and 3). Such modifications improved the mobility of the assembled xylan, glucomannan, and cellulose in cedar wood, resulting in enhanced thermal degradation and weight reduction at temperatures around the DTG peak where cellulose degraded (~ 370 °C, Fig. 6). The recovery data presented in Fig. 8 confirm the improved cellulose reactivity in this temperature range.
The recovery data for the 10-min-ball-milled beech wood (Fig. 8) indicate that the reactivities of xylan and cellulose increased. This is consistent with the changes in the TG/DTG profiles shown in Fig. 6, in which the TG/DTG curves shifted in the wide temperature range between 250 and 400 °C. In contrast, the glucomannan reactivity decreased (Fig. 8). This is also reasonably explained by the improved mobility of the matrix. The high glucomannan reactivity observed in beech wood was explained based on the 4-O-MeGlcA located near glucomannan [5], although it is bound to the xylose chain in xylan. The improved mobility of the cell wall matrix may diminish this effect.
When the milling time was increased to 4 h, the reactivity of xylan in beech wood increased, and xylan and glucomannan in both woods degraded in a similar temperature range, which was much lower than that of isolated glucomannan (Figs. 8 and 9). This is interesting, because isolated xylan bearing 4-O-MeGlcA moieties (salts or free carboxyls) was more reactive than isolated glucomannan because of the base or acid catalysis of 4-O-MeGlcA [4]. Therefore, ball milling should make the matrix components more homogeneous, thus allowing 4-O-MeGlcA to influence most of the hemicellulose components equally. This is an aspect of ball-milled wood that was not observed for holocellulose pyrolysis, where the thermal degradation reactivities of xylan and glucomannan were similar to those of isolated xylan and isolated glucomannan, respectively. This is likely because the delignification process did not allow the matrix components to mix efficiently.
The recovery rate of 4-O-MeGlcA is shown in Fig. 10. The thermal degradation reactivity of 4-O-MeGlcA in holocellulose was improved to a level similar to that of isolated xylan [6]; however, the improvement was not very significant in ball-milled wood, particularly for beech. This may be related to the ester linkages formed with lignin, which may be resistant to the ball-milling process, but further investigations are necessary to better explain this enhanced stability.

Recovery rate of 4-O-MeGlcA as a methanolysis product after the pyrolysis of ball-milled a cedar and b beech wood for 10 min (
) and 4 h (
), compared with unmilled wood (
) and isolated hemicellulose (
)
As discussed earlier, cellulose in wood degraded in two stages (some at temperatures below 320 °C and some at higher temperatures). In contrast, ball-milled Whatman cellulose degraded only in one mode corresponding to the high-temperature degradation of ball-milled wood. Therefore, the low-temperature degradation is characteristic of cellulose in wood. The-low-temperature-mode degradation would be explained by matrix-induced degradation as discussed later.
Role of ball milling on thermal reactivity of wood polysaccharides
The impact of ball milling on the polysaccharide reactivity of wood cell walls is discussed here using a schematic image of the interface between cellulose microfibril and hemicellulose–lignin matrix depicted in Fig. 11. Cellulose microfibrils play an important role owing to their crystalline nature. Cellulose molecules inside the crystallites are more stable than the surface molecules because of the packing in the crystallites [33], so the reactivity of the surface molecules is a critical factor for initiating the thermal degradation of bulk cellulose [34,35,36]. The difference between cellulose in wood and pure cellulose lies in this feature. Therefore, the reactivity of the matrix and its influence on the surface cellulose molecules must be considered to comprehensively understand the thermal degradation of cellulose in wood cell walls.

Schematic images of the hemicellulose–lignin matrix and the interface with cellulose microfibril and their modifications expected to be caused by ball-milling process
Our group’s previous work [3] indicated that the xylan and glucomannan reactivities are significantly influenced by the matrix construction. The arrangement of these molecules (including 4-O-MeGlcA, which has a catalytic effect) is precisely determined in wood cell walls, and it is different in cedar (softwood) versus beech (hardwood) [5]. Because these characteristic reactivities disappeared following the removal of lignin, the physical restraining effect of lignification during cell wall biosynthesis is considered to be the main reason for these distinctions [6]. Overall, the matrix is considered to be rigid and tightly associated with cellulose microfibrils in unmilled wood cell walls. Due to this tightly coagulated structure, hemicellulose and surface cellulose molecules in wood are stable for thermal degradation. The mobility must be sufficient to rearrange these polysaccharides into the transition state of thermal degradation reaction.
After the ball-milling process, the crystallinity index of cellulose in wood decreased significantly, indicating that the matrix and the interface with the cellulose microfibrils was disturbed in such a way to improve the mobility. Cleavage of the lignin β-ether linkages diminishes the physical restraining effect and should help further improve the mobility of the matrix components. These modifications would improve the thermal degradation reactivity of xylan and glucomannan. In addition to the improved mobility, the ball-milling process created a more homogeneous distribution of 4-O-MeGlcA, which improves the reactivity of glucomannan to similar levels of xylan degradation due to the catalytic effect of 4-O-MeGlcA. This feature is not the case for holocellulose, in which xylan and glucomannan have the same reactivity as isolated xylan and glucomannan. Therefore, xylan and glucomannan in both cedar and beech holocelluloses exist without affecting each other.
Unlike beech wood, short (10-min) ball milling significantly improved the reactivity of xylan and glucomannan, and the reactivity of cellulose was also improved. These results may correlate with the characteristic thermal degradation of cedar wood; cellulose, xylan and glucomannan degrade together like one ingredient [3]. Xylan and glucomannan may strongly coagulate with the surface molecules of cellulose microfibrils. This is also supported by the fact that delignification and the subsequent removal of xylan are necessary to isolate glucomannan from softwood [37]. Due to the rigid nature of crystalline cellulose microfibrils, ball milling disrupts the interface more efficiently.
Through loosening the matrix and the interface by ball milling, the surface cellulose molecules of cellulose microfibrils may become reactive and degrade in the low-temperature range in two modes of cellulose degradation that is characteristic of cellulose in holocellulose and ball-milled wood. The 4-O-MeGlcA groups and other matrix components and their thermal degradation products may induce the cellulose degradation in this temperature range through the action on the surface cellulose molecules. This proposed mechanism provides a reasonable explanation for why the thermal degradation of cellulose proceeds in two stages.
Conclusions
The thermal reactivities of hemicellulose and cellulose in ball-milled cedar and beech woods were evaluated to understand the effect of ball milling on wood pyrolysis. The main findings are as follows:
-
1.
Lignin β-ether linkages were cleaved during ball milling, along with the decrease in the cellulose crystallinity. These modifications would improve the mobility of matrix and the interface with cellulose microfibrils.
-
2.
Short-term ball milling (10 min) significantly improved the thermal degradation reactivities of xylan and glucomannan in cedar wood, but did not for beech wood. These components degraded together in two temperature ranges (below 300 °C and above), depending on the reactivity; this was not the case for beech wood. It was suggested that xylan, glucomannan and cellulose are more intimately coagulated in cedar (softwood) than beech (hardwood).
-
3.
Long-term ball milling (4 h) increased the thermal degradation reactivities of xylan and glucomannan (except for the glucomannan in beech wood, whose reactivity decreased after ball milling), due to the improved mobility. All these hemicelluloses degraded in a similar temperature range, likely because of the influence of catalytic 4-O-MeGlcA, which was more homogeneously distributed throughout the matrix after ball milling.
-
4.
Long-term ball milling (4 h) increased the thermal degradation reactivity of cellulose, which degraded in two different temperature ranges (below 320 °C and above). Improved mobility of surface molecules of cellulose microfibrils and the degradation induced by the matrix and its thermal degradation were considered for the reasons.
-
5.
The effects of ball milling on the thermal degradation reactivities of cellulose and hemicellulose were explainable in terms of the disturbance of the cell wall ultrastructure and cleavage of lignin ether linkages. Tightly coagulated matrix and interface structures due to lignification would make wood polysaccharides very stable against heat.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- TG:
-
Thermogravimetric
- DTG:
-
Derivative thermogravimetric
- 4-O-MeGlcA:
-
4-O-Methyl-d-glucuronic acid
- XRD:
-
X-Ray diffraction
- GC–MS:
-
Gas chromatography–mass spectrometry
References
Wang S, Dai G, Yang H, Luo Z (2017) Lignocellulosic biomass pyrolysis mechanism: a state-of-the-art review. Prog Energy Combust Sci 62:33–86
Bridgwater AV (2012) Review of fast pyrolysis of biomass and product upgrading. Biomass Bioenergy 38:68–94
Wang J, Minami E, Kawamoto H (2020) Thermal reactivity of hemicellulose and cellulose in cedar and beech wood cell walls. J Wood Sci 66:41
Wang J, Asmadi M, Kawamoto H (2018) The effect of uronic acid moieties on xylan pyrolysis. J Anal Appl Pyrolysis 136:215–221
Wang J, Minami E, Kawamoto H (2021) Location of uronic acid group in Japanese cedar and Japanese beech wood cell walls as evaluated by the influences of minerals on thermal reactivity. J Wood Sci 67:3
Wang J, Minami E, Asmadi M, Kawamoto H (2021) Effect of delignification on thermal degradation reactivities of hemicellulose and cellulose in wood cell walls. J Wood Sci 67:19
Piras CC, Fernández-Prieto S, De Borggraeve WM (2019) Ball milling: a green technology for the preparation and functionalisation of nanocellulose derivatives. Nanoscale Adv 1:937–947
Sun Y, Cheng J (2002) Hydrolysis of lignocellulosic materials for ethanol production: a review. Bioresour Technol 83:1–11
Sipponen MH, Laakso S, Baumberger S (2014) Impact of ball milling on maize (Zea mays L.) stem structural components and on enzymatic hydrolysis of carbohydrates. Ind Crops Prod 61:130–136
Hideno A, Kawashima A, Anzoua KG, Yamada T (2013) Comparison of the enzymatic digestibility of physically and chemically pretreated selected line of diploid-Miscanthus sinensis Shiozuka and triploid-M.×giganteus. Bioresour Technol 146:393–399
Crestini C, Melone F, Sette M, Saladino R (2011) Milled wood lignin: a linear oligomer. Biomacromol 12:3928–3935
Ikeda T, Holtman K, Kadla JF, Chang H, Jameel H (2002) Studies on the effect of ball milling on lignin structure using a modified DFRC method. J Agric Food Chem 50:129–135
Obst JR, Kirk TK (1988) Isolation of lignin. Methods Enzymol 161:3–12
Fujimoto A, Matsumoto Y, Chang HM, Meshitsuka G (2005) Quantitative evaluation of milling effects on lignin structure during the isolation process of milled wood lignin. J Wood Sci 51:89–91
Hideno A (2016) Comparison of the thermal degradation properties of crystalline and amorphous cellulose, as well as treated lignocellulosic biomass. BioResources 11:6309–6319
Mattonai M, Pawcenis D, del Seppia S, Łojewska J, Ribechini E (2018) Effect of ball-milling on crystallinity index, degree of polymerization and thermal stability of cellulose. Bioresour Technol 270:270–277
Ling Z, Wang T, Makarem M, Cintrón MS, Cheng HN, Kang X, Bacher M, Potthast A, Rosenau T, King H, Delhom C, Nam S, Edwards JV, Kim SH, Xu F, Frech Ad (2019) Effects of ball milling on the structure of cotton cellulose. Cellulose 26:305–328
Forziati FH, Stone WK, Rowen JW, Appel WD (1950) Cotton powder for infrared transmission measurements. J Res Natl Bur Stand 45:109
Schwanninger M, Rodrigues JC, Pereira H, Hinterstoisser B (2004) Effects of short-time vibratory ball milling on the shape of FT-IR spectra of wood and cellulose. Vib Spectrosc 36:23–40
Segal L, Creely JJ, Martin AE, Conrad CM (1959) An empirical method for estimating the degree of crystallinity of native cellulose using the X-ray diffractometer. Text Res J 29:786–794
Rolando C, Monties B, Lapierre C (1992) Thioacidolysis. In: Lin SY, Dence CW (eds) Methods in lignin chemistry. Springer, Heidelberg, pp 334–349
Bertaud F, Sundberg A, Holmbom B (2002) Evaluation of acid methanolysis for analysis of wood hemicelluloses and pectins. Carbohydr Polym 48:319–324
Bleton J, Mejanelle P, Sansoulet J, Goursaud S, Tchapla A (1996) Characterization of neutral sugars and uronic acids after methanolysis and trimethylsilylation for recognition of plant gums. J Chromatogr A 720:27–49
Li J, Kisara K, Danielsson S, Lindström ME, Gellerstedt G (2007) An improved methodology for the quantification of uronic acid units in xylans and other polysaccharides. Carbohydr Res 342:1442–1449
Asmadi M, Kawamoto H, Saka S (2017) Characteristics of softwood and hardwood pyrolysis in an ampoule reactor. J Anal Appl Pyrolysis 124:523–535
Yw HA, Thomas RL (1988) Simultaneous determination of neutral sugars and uronic acids in hydrocolloids. J Food Sci 53:574–577
Sundberg A, Sundberg K, Lillandt C, Holmbom B (1996) Determination of hemicelluloses and pectins in wood and pulp fibres by acid methanolysis and gas chromatography. Nord Pulp Pap Res J 11:216–219
Jiang J, Wang J, Zhang X, Wolcott M (2017) Assessing multi-scale deconstruction of wood cell wall subjected to mechanical milling for enhancing enzymatic hydrolysis. Ind Crops Prod 109:498–508
Holtman KM, Chang HM, Jameel H, Kadla JF (2003) Elucidation of lignin structure through degradative methods: comparison of modified DFRC and thioacidolysis. J Agric Food Chem 51:3535–3540
Rabemanolontsoa H, Saka S (2013) Comparative study on chemical composition of various biomass species. RSC Adv 3:3946–3956
Timell TE (1967) Recent progress in the chemistry of wood hemicelluloses. Wood Sci Technol 1:45–70
Tyminski A, Timell TE (1960) The constitution of a glucomannan from white spruce (Picea glauca). J Am Chem Soc 82:2823–2827
Kawamoto H (2016) Review of reactions and molecular mechanisms in cellulose pyrolysis. Curr Org Chem 20:2444–2457
Kawamoto H, Saka S (2006) Heterogeneity in cellulose pyrolysis indicated from the pyrolysis in sulfolane. J Anal Appl Pyrolysis 76:280–284
Zickler GA, Wagermaier W, Funari SS, Burghammer M, Paris P (2007) In situ X-ray diffraction investigation of thermal decomposition of wood cellulose. J Anal Appl Pyrolysis 80:134–140
Kim DY, Nishiyama Y, Wada M, Kuga S, Okano T (2001) Thermal decomposition of cellulose crystallites in wood. Holzforschung 55:521–524
Timell TE (1961) Isolation of galactoglucomannans from the wood of gymnosperms. Tappi 44:88–96
Acknowledgements
We thank Suzanne Adam, PhD, from Edanz Group (https://en-author-services.edanz.com/ac) for editing a draft of this manuscript.
Funding
This work was supported by the Japan Society for the Promotion of Science (Grant Numbers JP16H04954, JP19H03019) and the JST-Mirai Program (Grant Number JPMJMI20E3), Japan.
Author information
Authors and Affiliations
Contributions
The pyrolysis experiments, hydrolysis, methanolysis, thioacidolysis, data analysis, and manuscript writing were conducted by JW. Ball-milled sample preparation and thermogravimetric analysis were conducted by MA. This study was conducted under the supervision of HK and EM. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
Effect of ball-milling time on the cellulose crystallinity in Japanese (a) cedar and (b) beech wood. Fig. S2. A typical example of chromatogram of thioacidolysis mixture obtained from 4-hour-ball-milled beech wood (IS1: tetracosane (C24) IS2: hexacosane (C26)). Fig. S3. Typical examples of chromatograms of reaction mixtures obtained by methanolysis (a) and hydrolysis (b) of 4-hour-ball-milled beech wood. (IS: glucitol).
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Wang, J., Minami, E., Asmadi, M. et al. Thermal degradation of hemicellulose and cellulose in ball-milled cedar and beech wood. J Wood Sci 67, 32 (2021). https://doi.org/10.1186/s10086-021-01962-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s10086-021-01962-y