
Abstract
Wood cell walls have complex ultra-structures, in which cellulose microfibrils are surrounded by a hemicellulose–lignin matrix. This is the first paper to discuss the effect of cell wall on the pyrolytic reactivity of hemicellulose and cellulose in wood by using Japanese cedar (Cryptomeria japonica, a softwood) and Japanese beech (Fagus crenata, a hardwood), along with isolated xylan and glucomannan. Pyrolysis was conducted by subjecting the samples to temperatures in the range of 220–380 °C, at a heating rate of 10 °C/min, under a N2 flow with the reactor quenched as soon as the temperature reached the set value. The remaining hemicellulose and cellulose in the heat-treated wood were evaluated by determining the yields of hydrolysable sugar. Isolated xylan containing the uronic acid group, probably acting as a catalyst, was more reactive than isolated glucomannan, whereas the xylan in both woods was remarkably stable and degraded across a similar temperature range to the glucomannan degradation. Thus, the majority of the hemicellulose fractions in cedar and beech unexpectedly exhibited similar reactivity, except for glucomannan in beech that degraded at lower temperatures. Differing thermogravimetric (TG) and derivative TG (DTG) profiles, measured for cedar and beech under similar heating conditions, were explained by the different cellulose reactivity, rather than the hemicellulose reactivity; cellulose decomposed with hemicellulose in cedar, while such decomposition was independent in beech. The observed reactivity is a new finding that is different from the currently understood ideas and may originate from the effects of the cell walls. The research herein provides important information on the kinetics and thermochemical conversion of lignocellulosic biomass.
Similar content being viewed by others
Explore related subjects
Find the latest articles, discoveries, and news in related topics.Introduction
The pyrolytic reactivity of wood polysaccharides has been studied using isolated cellulose and hemicelluloses [1,2,3,4,5]. Cellulose, a crystalline polysaccharide, exhibits a higher thermal stability than amorphous hemicelluloses and exhibits the maximum thermal degradation rate at ~ 350 °C, that yields volatile products and char [1, 2, 6]. Xylan and glucomannan are the major hemicellulose of hardwood and softwood, respectively, whose structures are illustrated in Fig. 1. The pyrolytic reactivity of hemicellulose has received a wealth of interdependent research by using isolated wood xylan [2,3,4,5, 7] and konjac glucomannan [8,9,10,11,12], and in addition, numerous studies have concluded that the thermal stability of xylan is lower than glucomannan [4, 5, 13]. With regard to xylan instability, Wang et al. [14] reported that the 4-O-methyl-d-glucuronic acid (4-O-MeGlcA) moiety and the Na salts in demineralized and original commercial beech xylan acted as acid and base catalysts, respectively, which promote the thermal degradation of the xylose units in xylan. The observed changes to the catalytic performance were also supported from the different compositions of the pyrolysis products; depolymerization/dehydration products such as furfural and 4-hydroxy-5,6-dihydro-2H-pyran-2-one were observed by acid catalysis, while fragmentation products were yielded by base catalysis.

Chemical structure of xylan and glucomannan
Conversely, the reactivity of hemicellulose and cellulose in wood cell walls has not been discussed in the literature. Only thermogravimetric (TG) and differential TG (DTG) profiles of wood samples have been discussed with the thermal reactivity of isolated hemicellulose and cellulose [1,2,3, 6, 15]. The DTG curve of hardwood typically exhibits a shoulder in the lower temperature region along with a peak, which has been reported to originate from xylan and cellulose, respectively [16,17,18]. This assignment appears to be reasonable because the shoulder is not observed for the typical DTG curve of softwood that contains only a smaller amount of xylan. Nevertheless, there are no reports to evidence this explanation. For these reasons, the work herein evaluates the pyrolytic reactivity of hemicelluloses and cellulose in wood by using Japanese cedar (Cryptomeria japonica, a softwood) and Japanese beech (Fagus crenata, a hardwood).
To evaluate the pyrolytic reactivity of wood polysaccharides in wood cell walls, hemicelluloses and cellulose remaining in heat-treated woods must be quantified during pyrolysis. Standard analysis methods for wood polysaccharides include hydrolysis [19] and methanolysis [20,21,22,23], which can be applied for this purpose. Herein, Japanese cedar and Japanese beech woods were pyrolyzed by heating to set temperatures from 220 to 380 °C (20 °C intervals and no holding period), and the thermal degradations of xylan, glucomannan and cellulose were evaluated from the remaining hydrolysable sugar content. Comparisons with isolated xylan and glucomannan provide insight into how the pyrolytic reactivity is influenced in wood cell wall. Finally, the obtained results allowed for the assignment of the TG/DTG curves.
Materials and methods
Materials
Beechwood xylan and konjac glucomannan were purchased from Megazyme (Wicklow, Ireland) and Carbosynth (Berkshire, United Kingdom), respectively, and used without any further purification. Extractive-free wood flour (80 mesh passed) was prepared from the sapwood of Japanese cedar and Japanese beech by extracting with acetone using a Soxhlet extractor.
TG analysis
TG analysis was conducted using a TGA-50 instrument (Shimadzu Corp., Kyoto, Japan). Oven-dried Japanese cedar or Japanese beech wood (1 mg) was placed into a platinum pan and heated from room temperature to 800 °C, at a heating rate of 10 °C/min under a N2 flow of 10 mL/min (purity: 99.9998%, JAPAN FINE PRODUCTS, Mie, Japan). N2 was first passed through a deoxygenation column (GL Sciences, Japan) to remove any oxygen contamination.
Pyrolysis experiment
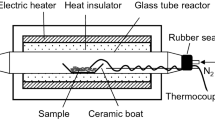
Figure 2 illustrates the experimental setup used in this study. A quartz glass tube (internal diameter 15 mm, length 400 mm, wall thickness 1.5 mm) was placed in an electric furnace (Asahi-Rika Seisakusho Co., Ltd., Chiba, Japan). For each experiment, the sample (20 mg) was placed into a ceramic boat (As One Corp. Osaka, Japan), and the ceramic boat placed within the glass tube. Thereafter, the air inside the glass tube was displaced by a N2 flow (100 mL/min) for 5 min using a SEC-400MK3 mass flow controller (Horiba, Kyoto, Japan), and the sample subjected to heat treatment at a set temperature (220–380 °C, at 20 °C intervals), at a heating rate of 10 °C/min, which was the same heating rate used for TG analysis. The sample temperature was directly measured during the pyrolysis experiment by contacting the tip of a fine thermocouple (0.25 mm in diameter, type K, Shinnetsu Co., Ltd., Ibaraki, Japan) to the sample whilst recording with a thermologger (AM-8000, Anritsu Corporation, Kanagawa, Japan). When the temperature reached the designated temperature, the cover of the electric furnace was opened and the glass tube was immediately cooled to room temperature under a flow of air.

Experimental setup for the pyrolysis experiment
Hydrolysable sugar analysis
Acid hydrolysis and methanolysis were conducted to determine the hydrolysable sugars from cellulose and hemicellulose/pectin in the heat-treated woods, respectively.
Hydrolysis was used to completely convert crystalline cellulose and was performed by treating the sample in a ceramic boat with 0.3 mL of an aqueous 72% H2SO4 solution at 30 °C in a glass tube for 1 h in a water bath. Thereafter, 8.4 mL of water was added to the mixture for dilution and the solution was heated in an autoclave at 120 °C for 1 h to complete the hydrolysis reactions. After quenching the system, the sample was filtered with filter paper (Kiriyama Glass Works Co., Tokyo, Japan). An aliquot of the filtrate was taken and diluted 15 times with water and neutralized with Dionex OnGuard II P cartridge (Thermo Fisher Scientific K.K., Tokyo, Japan). The glucose yield was determined by high-performance anion-exchange chromatography using a Dionex ICS-3000 instrument under the following conditions: column, CarboPac PA1 (4 mm × 250 mm); gas, N2; flow-rate, 1 mL/min; pressure, 1 atm; eluent, mixture of 0.2 M NaOH and distilled water; gradient-program, 15/85 (0–20 min); detector, electron capture; detector temperature, 35 °C. The eluent was degassed using an aspirator for 45 min prior to purging with N2 to prevent the absorption of CO2.
Milder methanolysis [20,21,22,23] was selected to determine the hydrolysable sugars from hemicellulose, pectin, and uronic acid groups. The ceramic boat containing heat-treated wood was placed into 4 mL of a 2 M solution of HCl in methanol in a glass tube comprising a screw cap and a Teflon-lined septum and heated at 60 °C for 16 h to complete the methanolysis reactions. After neutralization with 100 μL of pyridine, a portion of the mixture was mixed with a glucitol/methanol solution as an internal standard and dried under vacuum. Then, pyridine (100 μL), hexamethyldisilazane (150 μL) and trimethylchlorosilane (80 μL) were added and the mixture was heated at 60 °C for 30 min under stirring. The resulting trimethylsilyl derivatives were analyzed by GC–MS using a Shimadzu-2010 Plus gas chromatograph (Shimadzu Corporation) coupled to a Shimadzu QP 2010 Ultra mass spectrometer (Shimadzu Corporation). The instrumental conditions comprised: column, Agilent CPSil 8CB (length, 30 m; diameter, 0.25 mm); injector temperature, 260 °C; split ratio, 1:50; column temperature, 100 °C (2 min), 4 °C/min to 220 °C, 220 °C (2 min), 15 °C/min to 300 °C, 300 °C (2 min); carrier gas, helium; flow-rate, 1.0 mL/min. The MS scan parameters included a scan range of 35–600 m/z and a scan-interval of 0.3 s. The signals originating from methyl glycosides were assigned based on the associated mass spectra and retention times, as compared with literature data [24, 25].
Results and discussion
Hemicellulose and other minor saccharide reactivity in wood cell walls
Figure 3 compares the pictures of heat-treated Japanese beech and Japanese cedar woods subjected to temperatures of 220–380 °C (10 °C/min). Discoloration of the beech wood was initiated at 240 °C, with the color changing to brown and darkening further as the temperature increased. Conversely, discoloration of the cedar wood occurred at a higher temperature range of 320–340 °C. Thus, in terms of discoloration, beech wood is more reactive than cedar wood. These results may originate from the different pyrolytic reactivity of the wood components, which will be discussed in the following paragraphs.

Pictures of the appearance of: a Japanese cedar and b Japanese beech woods after being subjected to pyrolysis at various temperatures (10 °C/min, no holding period), under a nitrogen flow (100 mL/min)
Changes in the hydrolysable sugar content in the heat-treated woods are shown in Fig. 4, focusing on six sugar components: arabinose, glucose, galactose, mannose, xylose and 4-O-MeGlcA. The hydrolysable sugar content is shown as the percentage of sugar recovered against the yield from non-treated wood (normalized to 100%). Sugars other than glucose were determined as the corresponding methyl glycosides obtained by acidic methanolysis because of the instability of hemicellulose- and pectin-derived sugars when subjected to more severe hydrolysis conditions that hydrolyze stable crystalline cellulose.

Recovery rates of hydrolysable sugars from: a Japanese cedar and b Japanese beech woods after heating to various temperatures (10 °C/min, no holding period), under a nitrogen flow (100 mL/min). Black circle glucose (Glu), black pointing-up triangle xylose (Xyl), black square mannose (Man), white circle arabinose (Ara), white pointing-up triangle galactose (Gal), white square 4-O-methyl-d-glucuronic acid (4-O-MeGlcA)
Table 1 [26] shows sugar compositions determined from non-treated cedar and beech woods. The origins of these sugars should be discussed, based on literature [22, 27,28,29,30,31,32,33], prior to comparing the pyrolysis results. A significant amount of glucose originates from cellulose, however, glucose is also the constituent of glucomannan. Different xylose and mannose contents in cedar and beech woods are explained by the well-known compositional difference of hemicellulose in hardwood and softwood: xylan and trace amounts of glucomannan in hardwood, while softwood typically comprises glucomannan in major and xylan in minor amounts. Thus, changes in the yields of mannose and xylose directly indicate the degradation of glucomannan and xylan, respectively, during wood pyrolysis. The decrease in the recovery of glucose is related to cellulose degradation in beech, however, the contribution of glucomannan degradation must be considered for cedar.
Xylan in both cedar and beech woods contains 4-O-MeGlcA as an acidic sugar component that is expected to act as an acid and base (as metal uronate) catalyst [14], indicating that this unit may accelerate the degradation of wood cell wall components. The effectiveness of this acceleration effect in cell walls is particularly discussed in the present paper. Typically, the 4-O-MeGlcA content is greater in hardwood than softwood, as recognized for beech (20 g/kg) and cedar (9 g/kg), Table 1.
The origins of arabinose and galactose are more complex. Arabinose is a component of softwood xylan, but not of hardwood xylan. However, galactose is attached to the glucomannan chain of both types of wood species. For these minor sugars, the pectin content in the primary cell wall, which contains arabinose and galactose as the primary sugars [34,35,36,37,38], cannot be ignored. Accordingly, understanding the pyrolytic degradation of arabinose and galactose units is difficult with respect to the wood components.
In Fig. 4, degradation of glucose units is observed in the highest temperature region for both woods, which is consistent with the degradation of highly stable cellulose. Conversely, the temperature range where xylose and mannose units degrade is different; xylose and mannose units degrade at similar temperatures for cedar, however, in the pyrolysis of beech wood, the mannose units degrade at significantly lower temperatures than xylose. The xylose units in both woods are observed to degrade at similar temperatures, where mannose units in cedar also degrade. Consequently, hemicelluloses in the cell walls of both wood species are suggested to have similar reactivity, except for glucomannan in beech wood that is more reactive than other hemicelluloses.
The pyrolytic reactivity of the sugar units in hemicellulose in cedar and beech woods, as shown in Fig. 4, is compared with those of isolated xylan and glucomannan in Fig. 5, to elucidate the influence of the cell wall matrix. The results of the isolated hemicelluloses are shown in dashed lines. Commercial beech wood xylan, where the majority of the uronic acid moieties exist as Na salts, was used along with the demineralized (free carboxyl) sample as the isolated xylans, and their pyrolytic reactivities were evaluated by a similar procedure used in the present study [14]. Based on the analysis data of ten softwood and hardwood species in our previous paper [23], most of the free carboxylic 4-O-MeGlcA groups in xylan would form the salts with alkali and alkaline earth metal cations, although some are involved in the formation of ester linkages with lignin as discussed later.

Influence of pyrolysis temperature on recovery rates for: a xylose, b 4-O-MeGlcA, c mannose, d arabinose and e galactose in Japanese beech (black circle) and Japanese cedar (white circle) woods, compared with isolated xylan (white pointing-up triangle: Na+ salt, black pointing-up triangle: free carboxyl) and isolated glucomannan (x). Pyrolysis conditions: heating rate (10 °C/min)/nitrogen flow (100 mL/min)/no holding period
Glucomannan was isolated from Japanese cedar wood according to a previously reported procedure that includes the extraction of the residues obtained by the pre-extraction of xylan from holocellulose (delignified wood) using an aqueous solution of sodium hydroxide (24%) and boric acid (5%). However, that procedure concluded that the boric acid contaminant could not be removed from the isolated glucomannan even when using resins. For these reasons, konjac glucomannan was used as the isolated glucomannan herein.
The degradation temperature of the xylose units in both woods shifted to higher temperatures when compared with the isolated xylans, suggesting that xylan in the cell walls of both woods is significantly stabilized. Furthermore, the reactivity is observed to be similar to the glucomannan in cedar wood, as described above. The 4-O-MeGlcA units are also stabilized in the woods, however, the observed stability is different for cedar and beech. These results indicate that the 4-O-MeGlcA units attached to the xylan chain are restricted in the wood cell walls, where 4-O-MeGlcA fails to properly function as an acid and base catalyst for xylan degradation. These findings will provide new insight to research groups in the area of wood pyrolysis because xylan is currently believed to be more reactive than glucomannan in wood pyrolysis.
The reactivity of the mannose units exhibit the opposite trend for cedar and beech woods; mannose units in cedar wood degraded at slightly higher temperatures than konjac glucomannan, while the mannose units in beech wood degraded at significantly lower temperatures, although the glucomannan content is relatively low in beech. These results suggest that the environment where glucomannan exists is different in the cell wall matrix of cedar and beech woods. A possible explanation for the enhanced reactivity of beech wood glucomannan is that the 4-O-MeGlcA groups act as acid/base catalysts in the vicinity of glucomannan in the cell wall of beech wood, as discussed later.
Arabinose and galactose units in beech wood degrade at lower temperatures than those in cedar wood. The greater reactivity of the galactose units in beech can be explained by the reactivity of glucomannan, which was greater in beech than cedar, because galactose is the minor component of glucomannan in both woods. Nevertheless, the contribution of pectin must be considered for the reactivity of these minor sugar units. Discoloration of beech wood that initiated at a lower temperature of 240 °C, when compared with the cedar wood (Fig. 3), may be related to the greater reactivity of arabinose and galactose units along with glucomannan.
Cellulose reactivity and assignment of TG/DTG curves
Figure 6 illustrates the TG/DTG profiles measured for beech and cedar woods at the same heating rate (10 °C/min), used for the above pyrolysis experiments, along with the degradation behavior of cellulose and hemicelluloses, expressed as wt% based on the content in the wood, which were roughly estimated from the yields of hydrolysable sugars. The xylan and glucomannan contents in the pyrolyzed woods were calculated by multiplying the respective content in the original wood by the recovery rate of methyl xyloside and methyl mannoside obtained by methanolysis, respectively. The cellulose content was determined from the glucose yield obtained by hydrolysis by subtracting the yield from glucomannan, assuming that the reactivity of the glucose and mannose units in glucomannan is similar. Although the TG/DTG curves appear at slightly higher temperatures than the degradation of wood polysaccharides, the data set comparison is useful for TG/DTG curve assignment.

Thermogravimetric (TG)/Derivative TG profiles showing the recovery of cellulose, xylan and glucomannan from: a Japanese cedar and b Japanese beech woods (only cellulose-originated glucose was counted to determine the glucose yield)
A shoulder is clearly observed in the beech DTG curve along with a peak, while the DTG curve of cedar exhibits only one broad peak. Such a difference has previously been believed to originate from the greater reactivity of xylan that is more abundant in hardwood [29]. The present investigation, however, clarifies that the glucomannan in beech is significantly more reactive than xylan, which suggests that the shoulder in the beech DTG curve is not related to hemicellulose reactivity.
Contrary to this, the degradation behavior of cellulose is different for cedar and beech woods. Cellulose in beech is stable up to ~ 320 °C, where xylan and glucomannan almost degrade. Thus, thermal degradation of cellulose occurs independently of hemicellulose degradation in the beech wood cell wall. Conversely, cellulose in cedar wood typically degrades together with xylan and glucomannan degradation. Consequently, as indicated in Fig. 6, the overlapping temperature ranges for the degradation of cedar cellulose and hemicelluloses leads to one broad DTG peak.
Influence of the cell wall ultra-structure
Esterification with lignin may partially explain the ineffective catalytic activity of 4-O-MeGlcA in the wood cell wall. Three types of lignin–carbohydrate complex (LCC) linkages, Cγ-ester with 4-O-MeGlcA, benzyl ether, and phenyl glycoside, Fig. 7, are reported in softwood and hardwood cell walls [39,40,41,42,43]. This ester formation renders a portion of the 4-O-MeGlcA moieties inactive as acid/base catalysts for hemicelluloses and cellulose. Although the esterification rate is not clear presently, free 4-O-MeGlcA moieties are suggested to exist in the wood cell wall based on the cation-exchange ability [23, 44, 45] and distribution of alkali and alkaline earth metal cations within the cell walls [46]. Accordingly, some of the 4-O-MeGlcA moieties in the cell wall are ineffective, even without the formation of the ester linkages with lignin.

Three types of lignin–carbohydrate complex linkages
Acetyl groups are attached to xylan in beech and glucomannan in cedar, although the isolated hemicelluloses in Fig. 5 do not contain any acetyl groups. Such acetyl groups may affect the xylan and glucomannan reactivity in wood cell walls. However, this would not be important because xylans in both woods exhibited similar reactivities.
Xylan and glucomannan are normally involved in the formation of LCC linkages with lignin [39], indicating that hemicellulose and lignin exist in proximity by the formation of chemical bonds. These structures in the matrix of the wood cell wall may restrict the mobility of the 4-O-MeGlcA moieties, although this hypothesis needs to be confirmed by further investigations on the wood cell wall matrix and pyrolytic reactivity. The greater reactivity of glucomannan in beech wood can be explained by this hypothesis; glucomannan exists in the vicinity of 4-O-MeGlcA in the beech wood cell wall, whereas an attack of the xylose main chain in xylan by 4-O-MeGlcA is not possible. These results will be of significant interest to wood anatomists, as well as researchers in the field of pyrolysis.
The pyrolytic reactivity of cellulose is intrinsically determined by the crystalline nature. The molecules that comprise the nano-crystallites (tens of nm in cross-section) are stable, and hence, the thermal degradation is initiated from the surface molecules [47,48,49]. Prior to the decomposition, there is an “induction period” that is observed to activate cellulose, which led to the concept of “active cellulose” formation [50, 51]. The role of the reducing end during the activation of cellulose for thermal discoloration [52] and weight-loss behavior [53] is also suggested. Thus, the cellulose crystallite surface and hemicellulose–lignin matrix interface plays an important role when determining the cellulose reactivity, as illustrated in Fig. 8, which is suggested to be different for cedar and beech woods. Degradation of hemicellulose can activate the surface molecules of cedar cellulose, however, this is not observed for beech.

Role of the hemicellulose–lignin matrix and cellulose microfibril surface interface for cellulose reactivity during pyrolysis, which is expected to be different for Japanese cedar and Japanese beech woods
The assembly of cellulose and hemicelluloses in the wood cell walls has received significant attention in the field of wood anatomy, and different arrangements are proposed for softwood and hardwood cell walls, as illustrated in Fig. 9. Strong binding of glucomannan to cellulose in softwood cell walls has been reported [54,55,56,57]. Based on the results of dynamic mechanical analysis with FT-IR spectrometry, Åkerholm and Salmén [54] reported the close connection between cellulose and glucomannan in Norway spruce (Picea abies) wood fibers, although xylan showed no mechanical interaction with cellulose. Kumagai and Endo [55] utilized a quartz crystal microbalance for studying the action of cellulase during enzymatic hydrolysis of lignocellulose nanofibers prepared from Japanese cedar (a softwood) and eucalyptus (a hardwood). The report concluded that cellulose is covered by glucomannan in Japanese cedar, because the removal of glucomannan by the mannanase treatment was necessary for cellulase to bind to cellulose. Xylan and lignin are considered to exist between glucomannan-coated cellulose in softwood (Fig. 9a) [57].

Schematic view of the proposed cell wall arrangements of cellulose, hemicellulose and lignin for softwood and hardwood
Conversely, in the case of hardwood cell walls, Dammström [58] reported the dynamic FT-IR analysis data of aspen (Populus tremula), suggesting that xylan is strongly associated with cellulose, instead of glucomannan in the case of softwood. Association of xylan on cellulose is also used to explain the helicoidal array of cellulose microfibrils; negatively charged 4-O-MeGlcA moieties in xylan attached to the surface of cellulose microfibrils help to maintain the space between the microfibrils yielding a cholesteric mesophase [30, 59, 60]. However, there has been controversy because xylan in solution forms a threefold helical screw conformation, hindering xylan combining with cellulose having a twofold conformation [61, 62]. Simmons et al. [63] reported clear evidence for the binding of xylan to cellulose by solid-state NMR; xylan exhibiting a threefold helical screw in solution flattens into a twofold helicoidal screw to intimately bind to cellulose. These observations are also supported by theoretical calculations [61, 64,65,66]. These lines of information indicate that xylan binds to cellulose microfibrils instead of glucomannan in hardwood cell walls, as shown in Fig. 9b.
The cellulose reactivity of cedar and beech woods may be influenced differently by the variation in the assembly, although elucidation of the detailed mechanisms hitherto is not fully understood. Hence, further information with respect to the wood anatomy is necessary.
Conclusions
The thermal reactivity of xylan, glucomannan and cellulose in Japanese beech and Japanese cedar woods was investigated. The TG/DTG profiles were assigned from the results obtained herein. The results were also compared with those of isolated xylan and glucomannan, to understand the influence of the cell wall. Xylan is observed to be significantly stabilized in the cell walls of both woods, and the reactivity is similar to that of glucomannan in cedar. Glucomannan becomes unstable in beech. The 4-O-MeGlcA moieties and the corresponding salts do not act as acid/base catalysts in the cell walls, except for glucomannan in beech. This observation is partially explained by the ester formation with lignin. The observed differences in the cedar and beech TG/DTG curves do not arise from the differences in hemicellulose reactivity, but from the differences in cellulose reactivity; cellulose decomposes together with hemicellulose in cedar, however, the decomposition occurs independently in beech. The nature of the hemicellulose and cellulose microfibril assemblies for cedar and beech cell walls is considered as a reason for the different cellulose reactivity in cedar and beech woods.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- TG:
-
Thermogravimetric
- DTG:
-
Derivative thermogravimetric
- 4-O-MeGlcA:
-
4-O-Methyl-d-glucuronic acid
- LCC:
-
Lignin–carbohydrate complex
References
Yang H, Yan R, Chen H, Lee DH, Zheng C (2007) Characteristics of hemicellulose, cellulose and lignin pyrolysis. Fuel 86:1781–1788
Yang H, Yan R, Chen H, Zheng C, Lee DH, Liang DT (2006) In-depth investigation of biomass pyrolysis based on three major components: hemicellulose, cellulose and lignin. Energy Fuels 20:383–393
Shen DK, Gu S, Bridgwater AV (2010) The thermal performance of the polysaccharides extracted from hardwood: cellulose and hemicellulose. Carbohydr Polym 82:39–45
Shafizadeh F, McGinnis GD, Philpot CW (1972) Thermal degradation of xylan and related model compounds. Carbohydr Res 25:23–33
Wang S, Ru B, Lin H, Luo Z (2013) Degradation mechanism of monosaccharides and xylan under pyrolytic conditions with theoretic modeling on the energy profiles. Bioresour Technol 143:378–383
Zhou H, Long Y, Meng A, Chen S, Li Q, Zhang Y (2015) A novel method for kinetics analysis of pyrolysis of hemicellulose, cellulose, and lignin in TGA and macro-TGA. RSC Adv 5:26509–26516
Sanchez-Silva L, López-González D, Villaseñor J, Sánchez P, Valverde JL (2012) Thermogravimetric-mass spectrometric analysis of lignocellulosic and marine biomass pyrolysis. Bioresour Technol 109:163–172
Zhou X, Li W, Mabon R, Broadbelt LJ (2018) A mechanistic model of fast pyrolysis of hemicellulose. Energy Environ Sci 11:1240–1260
Räisänen U, Pitkänen I, Halttunen H, Hurtta M (2003) Formation of the main degradation compounds from arabinose, xylose, mannose and arabinitol during pyrolysis. J Therm Anal Calorim 72:481–488
Shen DK, Gu S, Bridgwater AV (2010) Study on the pyrolytic behaviour of xylan-based hemicellulose using TG-FTIR and Py-GC-FTIR. J Anal Appl Pyrolysis 87:199–206
Patwardhan PR, Brown RC, Shanks BH (2011) Product distribution from the fast pyrolysis of hemicellulose. Chemsuschem 4:1442–1449
Ohnishi A, Kato K, Takagi E (1977) Pyrolytic formation of 3-hydroxy-2-penteno-1,5-lactone from xylan, xylo-oligosaccharides, and methyl xylopyranosides. Carbohydr Res 58:387–395
Werner K, Pommer L, Broström M (2014) Thermal decomposition of hemicelluloses. J Anal Appl Pyrolysis 110:130–137
Wang J, Asmadi M, Kawamoto H (2018) The effect of uronic acid moieties on xylan pyrolysis. J Anal Appl Pyrolysis 136:215–221
Shen DK, Gu S (2009) The mechanism for thermal decomposition of cellulose and its main products. Bioresour Technol 100:6496–6504
Poletto M (2016) Thermal degradation and morphological aspects of four wood species used in lumber industry. Rev Árvore 40:941–948
Kim HS, Kim S, Kim HJ, Yang HS (2006) Thermal properties of bio-flour-filled polyolefin composites with different compatibilizing agent type and content. Thermochim Acta 451:181–188
Poletto M, Zattera AJ, Forte MMC, Santana RMC (2012) Thermal decomposition of wood: influence of wood components and cellulose crystallite size. Bioresour Technol 109:148–153
Harris JF. Baker AJ, Conner AH. Jeffries TW. Minor JL, Pettersen RC, Scott RW, Springer EL, Wegner TH, Zerbe JI (1985) Two-stage, dilute sulfuric acid hydrolysis of wood: an investigation of fundamentals. Gen. Tech. Rep. FPL-45. Madison, WI: U.S. Department of Agriculture, Forest Service, Forest Products Laboratory, p 73
Bertaud F, Sundberg A, Holmbom B (2002) Evaluation of acid methanolysis for analysis of wood hemicelluloses and pectins. Carbohydr Polym 48:319–324
Bleton J, Mejanelle P, Sansoulet J, Goursaud S, Tchapla A (1996) Characterization of neutral sugars and uronic acids after methanolysis and trimethylsilylation for recognition of plant gums. J Chromatogr A 720:27–49
Li J, Kisara K, Danielsson S, Mikael E, Lindström ME, Gellerstedt G (2007) An improved methodology for the quantification of uronic acid units in xylans and other polysaccharides. Carbohydr Res 342:1442–1449
Asmadi M, Kawamoto H, Saka S (2017) Characteristics of softwood and hardwood pyrolysis in an ampoule reactor. J Anal Appl Pyrolysis 124:523–535
Yw HA, Thomas RL (1988) Simultaneous determination of neutral sugars and uronic acids in hydrocolloids. J Food Sci 53:574–577
Sundberg A, Sundberg K, Lillandt C, Holmbom B (1996) Determination of hemicelluloses and pectins in wood and pulp fibres by acid methanolysis and gas chromatography. Nord Pulp Pap Res J 11:216–219
Rabemanolontsoa H, Saka S (2013) Comparative study on chemical composition of various biomass species. RSC Adv 3:3946–3956
Koshijima T (1985) Hemicellulose. In: Wood Chemistry (in Japanese), 3rd ed. Buneido-shuppan, pp 84–95
Scheller HV, Ulvskov P (2010) Hemicelluloses. Annu Rev Plant Biol 61:263–289
Zhou X, Li W, Mabon R, Broadbelt LJ (2017) A critical review on hemicellulose pyrolysis. Energy Technol 5:52–79
Reis D, Vian B (2004) Helicoidal pattern in secondary cell walls and possible role of xylans in their construction. Comptes Rendus Biol 327:785–790
Meier H (1961) Isolation and characterisation of an acetylated glucomannan from pine (Pinus sylvestris L.). Acta Chem Scand 15:1381–1385
Mian AJ, Timell TE (2011) Isolation and properties of a glucomannan from the wood of red maple (Acer rubrum L.). Can J Chem 38:1511–1517
Heinze T (2015) Cellulose: structure and properties. In: Rojas O (eds). Cellulose chemistry and properties: fibers, nanocelluloses and advanced materials. Adv Polym Sci, pp 1–52
Caffall KH, Mohnen D (2009) The structure, function, and biosynthesis of plant cell wall pectic polysaccharides. Carbohydr Res 344:1879–1900
Mohnen D (2008) Pectin structure and biosynthesis. Curr Opin Plant Biol 11:266–277
Ridley BL, O’Neill MA, Mohnen D (2001) Pectins: structure, biosynthesis, and oligogalacturonide-related signaling. Phytochemistry 57:929–967
Voragen AGJ, Coenen GJ, Verhoef RP, Schols HA (2009) Pectin, a versatile polysaccharide present in plant cell walls. Struct Chem 20:263–275
Thakur BR, Singh RK, Handa AK (1997) Chemistry and uses of pectin—a review. Crit Rev Food Sci Nutr 37:47–73
Tarasov D, Leitch M, Fatehi P (2018) Lignin-carbohydrate complexes: properties, applications, analyses, and methods of extraction: a review. Biotechnol Biofuels 11:1–28
Du X, Pérez-Boada M, Fernández C, Rencoret J, del Río JC, Jiménez-Barbero J, Li J, Gutiérrez A, Martínez AT (2014) Analysis of lignin–carbohydrate and lignin–lignin linkages after hydrolase treatment of xylan–lignin, glucomannan–lignin and glucan–lignin complexes from spruce wood. Planta 239:1079–1090
Balakshin M, Capanema E, Gracz H, Chang H, Jameel H (2011) Quantification of lignin-carbohydrate linkages with high-resolution NMR spectroscopy. Planta 6:1097–1110
Yuan TQ, Sun SN, Xu F, Sun RC (2011) Characterization of lignin structures and lignin-carbohydrate complex (LCC) linkages by quantitative 13C and 2D HSQC NMR spectroscopy. J Agric Food Chem 59:10604–10614
Takahashi N, Koshijima T (1988) Ester linkages between lignin and glucuronoxylan in a lignin-carbohydrate complex from beech (Fagus crenata) wood. Wood Sci Technol 22:231–241
DeGroot WF (1985) Preliminary investigation of the association of inorganic cations with carboxylic acid groups in wood. Carbohydr Res 142:172–178
Sjöström E, Janson T, Haglund P, Enström B (1965) The acidic groups in wood and pulp as measured by ion exchange. J Polym Sci Part C Polym Symp 11:221–241
Saka S, Mimori R (1994) The distribution of inorganic constituents in white birch wood as determined by SEM-EDXA. Mokuzai Gakkaishi 40:88–94
Kawamoto H, Saka S (2006) Heterogeneity in cellulose pyrolysis indicated from the pyrolysis in sulfolane. J Anal Appl Pyrolysis 76:280–284
Zickler GA, Wagermaier W, Funari SS, Burghammer M, Paris O (2007) In situ X-ray diffraction investigation of thermal decomposition of wood cellulose. J Anal Appl Pyrolysis 80:134–140
Kim DY, Nishiyama Y, Wada M, Kuga S, Okano T (2001) Thermal decomposition of cellulose crystallites in wood. Holzforschung 55:521–524
Bradbury AGW, Sakai Y, Shafizadeh F (1979) A kinetic model for pyrolysis of cellulose. J Appl Polym Sci 23:3271–3280
Kilzer FJ, Broido A (1965) Speculations on the nature of cellulose pyrolysis. Pyrodynamics 2:151–163
Matsuoka S, Kawamoto H, Saka S (2014) What is active cellulose in pyrolysis? An approach based on reactivity of cellulose reducing end. J Anal Appl Pyrolysis 106:138–146
Matsuoka S, Kawamoto H, Saka S (2011) Reducing end-group of cellulose as a reactive site for thermal discoloration. Polym Degrad Stab 7:1242–1247
Åkerholm M, Salmén L (2001) Interactions between wood polymers studied by dynamic FT-IR spectroscopy. Polymer 42:963–969
Kumagai A, Endo T (2018) Comparison of the surface constitutions of hemicelluloses on lignocellulosic nanofibers prepared from softwood and hardwood. Cellulose 25:3885–3897
Terashima N, Kitano K, Kojima M, Yoshida M, Yamamoto H, Westermark U (2009) Nanostructural assembly of cellulose, hemicellulose, and lignin in the middle layer of secondary wall of ginkgo tracheid. J Wood Sci 55:409–416
Salmén L, Burgert I (2009) Cell wall features with regard to mechanical performance. A review. COST Action E35 2004-2008: wood machining—micromechanics and fracture. Holzforschung 63:121–129
Dammström S, Lennart S, Gatenholm P (2009) On the interactions between cellulose and xylan, a biomimetic simulation of the hardwood cell wall. BioResources 4:3–14
Vian B, Roland JC, Reis D, Mosiniak M (2014) Distribution and possible morphogenetic role of the xylans within the secondary vessel wall of linden wood. IAWA J 13:269–282
Reis D, Vian B, Roland JC (1994) Cellulose-glucuronoxylans and plant cell wall structure. Micron 25:171–187
Busse-Wicher M, Gomes TCF, Tryfona T, Nikolovski N, Stott K, Gratham NJ, Bolam DN, Skaf MS, Dupree P (2014) The pattern of xylan acetylation suggests xylan may interact with cellulose microfibrils as a twofold helical screw in the secondary plant cell wall of Arabidopsis thaliana. Plant J 79:492–506
Nieduszynski I, Marchessault RH (1971) Structure of β-d-(1 → 4′)xylan hydrate. Nature 11:1335–1344
Simmons TJ, Mortimer JC, Bernardinelli OD, Pöppler AC, Brown SP, deAzevedo ER, Dupree R, Dupree P (2016) Folding of xylan onto cellulose fibrils in plant cell walls revealed by solid-state NMR. Nat Commun 7:1–9
Mikkelsen D, Flanagan BM, Wilson SM, Bacic A, Gidley MJ (2015) Interactions of arabinoxylan and (1,3)(1,4)-β-glucan with cellulose networks. Biomacromolecules 16:1232–1239
Pereira CS, Silveira RL, Dupree P, Skaf MS (2017) Effects of xylan side-chain substitutions on xylan-cellulose interactions and implications for thermal pretreatment of cellulosic biomass. Biomacromolecules 18:1311–1321
Busse-Wicher M, Li A, Silveira RL, Silveira CS, Tryfona T, Gomes TCF, Skaf MS, Dupree P (2016) Evolution of xylan substitution patterns in gymnosperms and angiosperms: implications for xylan interaction with cellulose. Plant Physiol 171:2418–2431
Acknowledgements
We are grateful to Dr. Mohd Asmadi, Faculty of Chemical and Energy Engineering, Universiti Teknologi Malaysia, for the preparation of wood samples and TG/DTG data. We thank Edanz Group (https://www.edanzediting.com/ac) for editing a draft of this manuscript.
Funding
This work was supported by JSPS KAKENHI Grant Numbers JP16H04954, JP19H03019.
Author information
Authors and Affiliations
Contributions
JW conducted the experiments, data analysis and wrote the manuscript under the supervision of HK and EM. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Wang, J., Minami, E. & Kawamoto, H. Thermal reactivity of hemicellulose and cellulose in cedar and beech wood cell walls. J Wood Sci 66, 41 (2020). https://doi.org/10.1186/s10086-020-01888-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s10086-020-01888-x