
Abstract
The thermal degradation reactivities of cellulose and hemicellulose are substantially different in Japanese cedar (Cryptomeria japonica, a softwood) and Japanese beech (Fagus crenata, a hardwood). Uronic acid and its salts act as acid and base catalysts, respectively, and their specific placement in the cell walls has been considered a factor that influences degradation reactivity. In this study, the role of lignin in degradation reactivity was investigated using holocellulose prepared from cedar and beech woods. The thermal degradation reactivities of cellulose and hemicellulose in holocellulose were evaluated according to the recovery of hydrolyzable sugars from heat-treated samples and compared with those of wood samples. Results show that the reactivities of xylan and glucomannan in both woods became similar to those of the corresponding isolated samples when lignin was removed. By contrast, the cellulose in both woods became more reactive when lignin was removed, and the degradation could be separated into two modes depending on the reactivity. These results were analyzed in terms of the effect of lignin on the matrix of cell walls and the interaction between the matrix and surface molecules of cellulose microfibrils. Differential thermogravimetric curves of the holocellulose samples were obtained and explained in terms of the degradation of hemicellulose and cellulose. The reported findings will provide insights into the research fields of wood pyrolysis and cell wall ultrastructures.
Similar content being viewed by others
Introduction
The thermal degradation reactivities of component polymers in wood provide a fundamental basis for understanding the pyrolysis of wood and other lignocellulosic biomasses as well as the changes in the physical properties of wood due to heat treatment. Isolated polymers have been used to study thermal degradation reactivity and pathways. Moreover, our previous study [1] using Japanese cedar (Cryptomeria japonica, a softwood) and Japanese beech (Fagus crenata, a hardwood) showed that the thermal degradation reactivities of cellulose and hemicellulose differed substantially between the wood cell walls and isolated samples. Isolated xylan was more reactive than isolated glucomannan owing to the catalytic action of 4-O-methyl-d-glucuronic acid (4-O-MeGlcA) groups bound to the xylose chain [2], but the xylan reactivity was substantially reduced in the cell walls of both woods. The glucomannan reactivity in beech was improved compared with that of the isolated glucomannan, but this was not observed in the case of cedar.
In addition to the different hemicellulose reactivities, the thermal degradation behaviors of cellulose and hemicellulose were different in cedar and beech woods [1]. Cellulose and hemicellulose degraded synchronously in cedar wood, while the same components decomposed independently in different temperature ranges in beech wood. These observations reasonably explain the differential thermogravimetric (DTG) curves for cedar and beech woods. The DTG curve of cedar wood had one wide peak, while one shoulder was clearly visible on the lower temperature side of the peak in beech wood. This is a common difference between the softwood and hardwood DTG curves [2,3,4].
To understand the role of 4-O-MeGlcA as a catalyst, the location of 4-O-MeGlcA in the cell walls was evaluated for cedar and beech, based on the effect of demineralization on thermal degradation reactivity [5]. Because demineralization converts metal salts (base) into free acids, the components affected by demineralization should be near 4-O-MeGlcA. The results showed that 4-O-MeGlcA is located near xylan and glucomannan in beech but is located near cellulose and glucomannan in cedar. The latter arrangement cannot be explained by the ultrastructure that has been proposed for softwood cell walls, where glucomannan is tightly bound to the surface of cellulose microfibrils [6,7,8]. Such an arrangement would affect pyrolysis characteristics, which differ for cedar and beech woods.
Thus, hemicellulose and cellulose are located at specific positions in the cell wall, which determines their reactivities. The next question to be addressed is the influence of lignin on the thermal degradation reactivities of hemicellulose and cellulose in the cell walls. Although the state of lignin in cell walls (e.g., a network or particulate structure) is still controversial [9, 10], lignin makes the cell wall a hard material. Lignin–carbohydrate complex (LCC) linkages such as Cγ-ester with the 4-O-MeGlcA group, benzyl ether, and phenyl glycoside types [11,12,13,14,15,16] fix the location of hemicellulose within the cell wall. Furthermore, several researchers [17,18,19] have proposed covalent linkages between lignin and cellulose.
In this study, the influence of lignin on the thermal degradation reactivities of hemicellulose and cellulose was investigated using holocellulose samples prepared by removing lignin from cedar and beech woods. The pyrolytic reactivities of hemicellulose and cellulose were evaluated according to the recovery of hydrolyzable sugars from the heat-treated holocellulose. As pyrolysis experiments were performed at the same heating rate as that used in the thermogravimetric (TG) analysis, TG/DTG curves measured for holocellulose samples are used to discuss with the decomposition of hemicellulose and cellulose at each temperature.
Experimental
Materials
Holocellulose samples were prepared from Japanese cedar and Japanese beech according to the following procedure [20]. A wood sample (25 g, passed through an 80 mesh) was mixed with 1.5 L of 0.2 M acetic acid. Sodium chlorite (10 g) followed by glacial acetic acid (2 mL) was added under stirring, and the mixture was stirred for 1 h at 70 °C–80 °C. The same amounts of sodium chlorite and glacial acetic acid were added every 1 h (four times for softwood, five times for hardwood) to complete the reaction. After centrifugation, the suspended solids were collected and washed with distilled water until the solution became clear, and sodium chlorite was removed. The resulting solid product was washed with acetone to remove water and dried in an oven. Whatman CF-11 cellulose (Whatman PLC, Maidstone, UK) was used as isolated cellulose.
Inorganic contents of wood and holocellulose were analyzed by incinerating the samples in air at 600 °C for 2 h, followed by the elemental analysis with scanning electron microscope/energy dispersive X-ray spectroscopy (SEM–EDS). The ash content increased from 0.32 wt% to 0.96 wt% (cedar wood) and 0.38 wt% to 0.55 wt% (beech wood) by converting wood to holocellulose. In the holocellulose samples, sodium and chlorine were observed as the major inorganic components, as reported in literature [21, 22]. The originally existing cations would be replaced by sodium during the delignification stage, and chlorination of lignin aromatic rings may occur to some extent [22, 23]. It is well known that the inorganic components affect the wood pyrolysis behavior, but the influences observed for delignification were much greater than those of minerals [5].
Cellulose and hemicellulose in wood may be modified during the delignification stage [22, 23], but the degree is considered not to be significant, since the results of hydrolysable sugar determination were not so different between wood and holocellulose.
TG analysis
TG analysis (TGA-50, Shimadzu, Kyoto, Japan) was performed on holocellulose samples from Japanese cedar and beech. Each sample (1 mg) was placed in a platinum pan and heated from room temperature to 800 °C at a heating rate of 10 °C/min under a N2 flow of 10 mL/min (purity: 99.9998%, JAPAN FINE PRODUCTS, Mie, Japan). Before being supplied to the TG equipment, the N2 gas was passed through a deoxygenation column (Model 1000 O2 filter, GL Sciences, Tokyo, Japan) to remove oxygen.
Pyrolysis
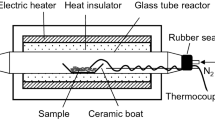
Figure 1 illustrates the experimental setup used in this study. An electric furnace (ARF-20KC, Asahi-Rika, Chiba, Japan) was used to heat the samples. For each experiment, a sample (20 mg) in a ceramic boat (As One, Osaka, Japan) was placed in a quartz glass tube (inner diameter: 15 mm; length: 400 mm; wall thickness: 1.5 mm). N2 then passed through the glass tube at a flow rate of 100 mL/min for 5 min to replace the air in the glass tube. The N2 flow rate was maintained by a mass flow controller (SEC-400MK3, Horiba, Kyoto, Japan). The sample was heated to a designated temperature (220 °C–380 °C at 20 °C intervals) at a heating rate of 10 °C/min, the same heating rate used for the TG analysis. The sample temperature was measured directly during the pyrolysis experiment by touching the tip of a fine thermocouple (0.25 mm in diameter) to the sample. When the sample temperature reached the designated temperature, the cover of the electric furnace was opened and the glass tube was immediately cooled to room temperature under an airflow.

Experimental setup
Hydrolyzable sugar analysis
Acid hydrolysis and methanolysis were separately conducted to convert cellulose and hemicellulose/pectin in the heat-treated samples into sugars and methyl glycosides, respectively. Hydrolysis was performed by treating each sample (before pyrolysis, 20 mg) together with the ceramic boat with 0.3 mL of an aqueous 72% H2SO4 solution at 30 °C in a sealed glass vial for 1 h in a water bath and shaking several times. Then, 8.4 mL of water was added and the mixture was heated in an autoclave at 120 °C for 1 h to complete the hydrolysis reaction. The mixture was filtered, and an aliquot of the filtrate (1 mL) was diluted 15 times with water; this was then neutralized with a Dionex OnGuard II A cartridge (Thermo Fisher Scientific, MA, USA). The glucose yield was determined via high-performance anion-exchange chromatography using a Prominence system (Shimadzu, Kyoto, Japan) equipped with an electrochemical detector (DECADE Elite, Antec Scientific, Zoeterwoude, Netherlands). A CarboPac PA1 column (4 mm × 250 mm) was used with an eluent of 85% distilled water/15% 0.2 M NaOH, flow rate of 1 mL/min, and column oven temperature of 35 °C.
Mild methanolysis [24,25,26,27] was conducted to determine the hydrolyzable sugars according to the methyl glycosides from the hemicellulose, pectin, and uronic acid groups. The ceramic boat and heat-treated sample were added to 4 mL of 2 M HCl in methanol solution (KOKUSAN CHEMICAL, Tokyo, Japan) in a sealed glass tube and heated at 60 °C for 16 h to complete the methanolysis reaction. After neutralization with pyridine (0.1 mL), a portion of the mixture (0.1 mL) was mixed with a glucitol/methanol solution (0.1 mL, 0.1 mg/mL) as an internal standard and dried under vacuum. Then, the resulting methyl glycoside mixture was trimethylsilylated with pyridine (0.1 mL), hexamethyldisilazane (0.15 mL), and trimethylchlorosilane (0.08 mL). The mixture was analyzed via gas chromatography–mass spectroscopy (GC–MS) using a QP2010 Ultra (Shimadzu, Kyoto, Japan). An Agilent CPSil 8CB column (length: 30 m; diameter: 0.25 mm) was used with an injector temperature of 260 °C, split ratio of 1:50, helium as the carrier gas, and a flow rate of 1.0 mL/min. The column temperature was kept at 100 °C for 2 min, increased at 4 °C/min to 220 °C, kept at 220 °C for 2 min, increased at 15 °C/min to 300 °C, and kept at 300 °C for 2 min. The signals originating from xylose, mannose, arabinose, galactose, and 4-O-MeGlcA were assigned based on the associated mass spectra and retention times in the literature [28, 29].
Results and discussion
TG/DTG profile in terms of component degradation
The TG/DTG curves measured for the holocellulose samples prepared from cedar and beech woods are illustrated in Fig. 2. Delignification lowered the temperature range in which weight loss occurred for both woods. In particular, the DTG peak temperatures were substantially lowered from 379 °C to 341 °C for cedar and from 381 °C to 353 °C for beech. This indicates that the cellulose in wood becomes very reactive when lignin is removed, because crystalline cellulose was more thermally stable than amorphous hemicellulose and degraded around the DTG peak temperature [1]. The shapes of the DTG curves for cedar and beech woods are different because of the different degradation behaviors of cellulose and hemicellulose [1]; these components degrade together in cedar but independently in beech. Removing lignin changed the shape of the DTG curve for cedar to that for beech: a shoulder can clearly be observed below the peak temperature.

TG/DTG curves of Japanese cedar and Japanese beech wood: untreated and after delignification. Dashed lines at 320 °C are inserted to facilitate comparison
To explain the TG/DTG profiles, the thermal degradation reactivities of hemicellulose and cellulose in holocellulose were determined according to the recovery rates of hydrolyzable sugars from heat-treated samples and compared with those of wood samples in previous reports [1, 2]. The reactivities of xylan and glucomannan were directly determined from the recovery rates of xylose and mannose, respectively, because these are the characteristic constituent sugars of these hemicelluloses. However, glucose is produced from cellulose and glucomannan, so the cellulose reactivity was determined according to the cellulose-derived glucose. This was estimated by subtracting the amount of glucose formed from glucomannan under the assumption that the mannose:glucose molar ratio in glucomannan is 3:1 [30, 31] and that both units have the same thermal degradation reactivity.
The amounts of cellulose, xylan, and glucomannan remaining in the pyrolyzed holocellulose were estimated based on the recovery data of the hydrolyzable sugars and their contents in each wood type; the corresponding results are plotted against the pyrolysis temperature in Fig. 3. By comparison with the DTG curves, the weight loss behavior during the heating process can be explained in terms of the degradation of cellulose and hemicellulose; this is because the same heating rate as that in the TG analysis was used with no heating time at a constant temperature.

DTG curves as compared with the recovery rates (wood basis) of cellulose ( ), glucomannan (
), glucomannan ( ), and xylan (
), and xylan ( ), in pyrolysis of untreated wood and holocelluose. Dashed lines at 320 °C are inserted to facilitate comparison. Results of untreated wood were taken from Wang et al. [1]
), in pyrolysis of untreated wood and holocelluose. Dashed lines at 320 °C are inserted to facilitate comparison. Results of untreated wood were taken from Wang et al. [1]
As indicated by the TG analysis, cellulose reactivity increased when lignin was removed. Although wood cellulose degraded continuously and gradually as the pyrolysis temperature was increased, the cellulose degradation in holocellulose was divided into two modes depending on the pyrolysis temperature. Degradation started at 260 °C–280 °C, and the reactivity increased sharply above 320 °C. In the low-temperature degradation mode (260 °C–280 °C), hemicellulose degraded together with cellulose. Consequently, the polysaccharide components that degraded at the DTG shoulder and peak temperatures could not be clearly separated into hemicellulose and cellulose. Most hemicellulose and 20%–25% of cellulose decomposed below the DTG shoulder temperature (around 320 °C) for both wood types, but the remaining cellulose degraded around the peak temperatures (above 320 °C) after the hemicellulose degraded.
Reactivities of isolated and wood polysaccharides
Figure 4 shows the recovery rates of hydrolyzable sugars plotted against the pyrolysis temperature, with holocellulose normalized as 100%. The results for isolated hemicellulose [1] and Whatman cellulose are also included for comparison. These plots can be used to discuss the cellulose and hemicellulose reactivities in holocellulose as compared with those of the original wood and isolated samples.

Comparison of the recovery rates of hydrolysable sugars, cellulose-derived glucose, mannose, and xylose, in pyrolysis of untreated wood (▲) and holocelllulose ( ) and Whatman cellulose, beech xylan, and konjac glucomannan (♦). Dashed lines at 320 °C are inserted to facilitate comparison
) and Whatman cellulose, beech xylan, and konjac glucomannan (♦). Dashed lines at 320 °C are inserted to facilitate comparison
Whatman cellulose is an example of pure cellulose; it withstood heating up to 320 °C and then degraded rapidly at higher temperatures. The low-temperature cellulose degradation mode as observed for holocellulose at 260 °C–320 °C was not detected. Accordingly, holocellulose is characterized by this low-temperature cellulose degradation, which may be caused by matrix degradation as discussed later. This phenomenon appears when lignin is removed from wood. It should be noted that the degradation behaviors of cellulose were different in cedar and beech woods but similar in their holocellulose samples.
As described in the previous paper [1], the hemicellulose reactivity in wood was different from those of isolated hemicelluloses, and the reactivity varied depending on the type of hemicellulose and wood. Xylan in wood was less reactive than isolated xylan, but glucomannan in beech was more reactive than isolated glucomannan. The high reactivity of glucomannan was explained by the presence of 4-O-MeGlcA near glucomannan in beech [5]. Removing lignin changed their reactivities to be similar to those of isolated xylan and glucomannan. A similar trend was observed for 4-O-MeGlcA, as shown in Fig. 5. Thus, lignin plays an important role in determining the thermal reactivities of hemicellulose and cellulose in wood; this role is probably due to the restraints of the specific locations of xylan, glucomannan, and 4-O-MeGlcA in the cell walls as discussed later. Removing lignin increases the mobility of these components in holocellulose.

Comparison of the recovery rates of 4-O-MeGlcA units in pyrolysis of untreated wood (▲), holocelllulose (
) and isolated xylan (♦). Dashed lines at 320 °C are inserted to facilitate comparison
The data in Figs. 4 and 5 are rearranged in Fig. 6 to understand the differences depending on the wood type. Although the temperature ranges at which cellulose and hemicellulose decomposed were similar, the shapes of the graphs differ for cedar and beech holocellulose. With cedar holocellulose, the recovery–temperature relationships of mannose and xylose show similar trends. Two reflection points can be observed at 260 °C and 300 °C; this indicates that the reactivity changed at these temperatures. This tendency is not observed for beech holocellulose. Thus, these results indicate that xylan and glucomannan degraded synchronously in cedar holocellulose, and the degradation can be divided into three types depending on the reactivity: < 260 °C, 260 °C–300 °C, and > 300 °C. The recovery–temperature relationship of cellulose-derived glucose indicates that the degradations of cellulose and hemicellulose occurred synchronously in cedar. The cellulose degradation started around 260 °C, at which point approximately half of the xylan and some glucomannan decomposed. After the rapid degradation of the remaining hemicellulose around 300 °C–320 °C, cellulose quickly degraded at 320 °C–340 °C. Therefore, the characteristic thermal degradation behaviors observed for cedar were maintained when lignin was removed. Unlike for beech holocellulose, the cellulose degradation is intimately related to the hemicellulose degradation in cedar holocellulose.

Influences of pyrolysis temperature on the recovery rates of hydrolysable sugars, cellulose-derived glucose (
), mannose (
), and xylose (
), in pyrolysis of cedar and beech holocellulose samples. Dashed lines at 320 °C are inserted to facilitate comparison
Role of lignin
On the basis of the present results, the role of lignin in wood pyrolysis is discussed using a schematic of a single cellulose microfibril surrounded by a matrix (Fig. 7), although further studies are necessary to confirm the following proposal. Xylan, glucomannan, and 4-O-MeGlcA are anchored in specific locations within the matrix [1, 5]. Meanwhile, 4-O-MeGlcA and its salts act as acid and base catalysts, respectively [2], which increase the thermal degradation reactivity of adjacent components. However, previous experimental results [1, 2] indicated that the catalytic activity is not effective in wood. For example, 4-O-MeGlcA is bound to xylan, but the xylan was very stable in both wood samples. Rather, 4-O-MeGlcA influences the glucomannan degradation in beech wood. All these features disappeared in holocellulose. These results can be reasonably explained by considering the role of lignin, which physically strengthens the ultrastructure formed between hemicellulose and the cellulose interface in the matrix. The formation of LCC linkages [11,12,13] may also be involved in this process.

A proposed role of lignification in determining thermal degradation reactivities of cellulose and hemicellulose in cedar (softwood) and beech (hardwood)
The thermal degradation of cellulose is known to occur nonuniformly at the crystallite level [32,33,34]; surface molecules preferentially tend to decompose as internal molecules are more stable owing to stabilization by filling the crystallites. Accordingly, the reactivity of surface molecules plays an important role in the thermal degradation of cellulose [35,36,37], and the matrix and its degradation are expected to affect the reactivity of cellulose microfibrils by affecting the surface cellulose molecules at the interface. This may induce cellulose degradation particularly in the low-temperature degradation mode of cellulose, which was observed in both types of holocellulose in the temperature range of 260 °C–320 °C (Fig. 4).
To understand the effect of low-temperature cellulose degradation (260 °C–320 °C) on the high-temperature cellulose degradation (> 320 °C), TG analysis was conducted for holocellulose samples at different heating rates of 1, 5, and 10 °C/min (Fig. 8). By decreasing the heating rate, TG and DTG curves shifted to lower temperature side. Although researchers try to explain these shifts with the time lag in temperature measurement [38], heating rates and sample weight (1 mg) used in the present TG analysis are very small to account for such a large shift (DTG peak temperature: 342 °C → 326 °C → 297 °C (10 °C/min → 5 °C/min → 1 °C/min) for cedar holocellulose, 357 °C → 338 °C → 307 °C (10 °C/min → 5 °C/min → 1 °C/min) for beech holocellulose).

TG/DTG curves measured for (a) cedar and (b) beech holocellulose samples at different heating rates of 1, 5, and 10 °C/min
By redrawing the TG/DTG curves in Fig. 8 with the unit of weight-loss rate changed from mg/min to mg/°C, the appearance of TG/DTG curves becomes very similar (Fig. 9a). This is confirmed by Fig. 9b, where TG/DTG curves are moved with respect to the temperature axis so that the peaks of the DTG curves are aligned. Surprisingly, these graphs match well. These results lead to a hypothesis; low- and high-temperature modes of cellulose degradation are closely related. Although the temperature range where thermal degradation of cellulose occurs is different depending on the heating rate, once thermal degradation of cellulose begins in low-temperature mode, this determines the subsequent cellulose degradation including high-temperature mode. This hypothesis gives insights in understanding cellulose pyrolysis, although further studied are necessary to confirm it.

TG/DTG curves of cedar and beech holocellulose samples redrawn with the unit of weight-loss rate changed from mg/min to mg/°C (a) and further moved with respect to the temperature axis so that the peaks of the DTG curves are aligned (b)
Conclusions
The thermal degradation reactivities of cellulose and hemicellulose in cedar and beech holocellulose were investigated via TG analysis and the recovery rates of hydrolyzable sugars. The following conclusions were obtained:
-
1.
The TG/DTG profiles of cedar and beech wood samples were different but became similar for holocellulose when lignin was removed.
-
2.
Removing lignin made the reactivity of hemicellulose similar to that of isolated hemicellulose while increasing the reactivity of cellulose. Additionally, the cellulose degradation can be divided into two modes: low temperature (260 °C–320 °C) and high temperature (> 320 °C).
-
3.
The TG analysis at different heating rates indicated that low- and high-temperature modes of cellulose thermal degradation are closely related.
-
4.
The TG/DTG profiles can be explained in terms of the degradation of hemicellulose and cellulose. For both types of holocellulose, 20%–25% of cellulose degraded at the DTG shoulder temperature (< 320 °C) with the degradation of hemicellulose, while the remaining cellulose degraded around the DTG peak temperature (> 320 °C).
-
5.
Cellulose degraded in response to the hemicellulose degradation in cedar holocellulose, which is in contrast to the results for beech holocellulose. This indicates that cellulose is intimately associated with hemicellulose in cedar and holocellulose.
-
6.
The hemicellulose in cedar holocellulose can be divided into three groups depending on the thermal degradation reactivity: > 260 °C, 260 °C–300 °C, and > 300 °C.
-
7.
Lignin is proposed to have a physical restraint role in determining the thermal degradation reactivities of cellulose and hemicellulose in cell walls.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- TG:
-
Thermogravimetric
- DTG:
-
Differential thermogravimetric
- 4-O-MeGlcA:
-
4-O-Methyl-d-glucuronic acid
- GC–MS:
-
Gas chromatography–mass spectroscopy
References
Wang J, Minami E, Kawamoto H (2020) Thermal reactivity of hemicellulose and cellulose in cedar and beech wood cell walls. J Wood Sci 66:41
Wang J, Asmadi M, Kawamoto H (2018) The effect of uronic acid moieties on xylan pyrolysis. J Anal Appl Pyrolysis 136:215–221
Yang H, Yan R, Chen H, Lee DH, Zheng C (2007) Characteristics of hemicellulose, cellulose and lignin pyrolysis. Fuel 86:1781–1788
Shen DK, Gu S, Bridgwater AV (2010) Study on the pyrolytic behaviour of xylan-based hemicellulose using TG-FTIR and Py-GC-FTIR. J Anal Appl Pyrolysis 87:199–206
Wang J, Minami E, Kawamoto H (2021) Location of uronic acid group in Japanese cedar and Japanese beech wood cell walls as evaluated by the influences of minerals on thermal reactivity. J Wood Sci 67:3
Åkerholm M, Salmén L (2001) Interactions between wood polymers studied by dynamic FT-IR spectroscopy. Polymer 42:963–969
Kumagai A, Endo T (2018) Comparison of the surface constitutions of hemicelluloses on lignocellulosic nanofibers prepared from softwood and hardwood. Cellulose 25:3885–3897
Terashima N, Kitano K, Kojima M, Yoshida M, Yamamoto H, Westermark U (2009) Nanostructural assembly of cellulose, hemicellulose, and lignin in the middle layer of secondary wall of ginkgo tracheid. J Wood Sci 55:409–416
Norgren M, Edlund H (2014) Lignin: recent advances and emerging applications. Curr Opin Colloid Interface Sci 19:409–416
Radotić K, Mićić M, Jeremić M (2005) New insights into the structural organization of the plant polymer lignin. Ann N Y Acad Sci 1048:215–229
Tarasov D, Leitch M, Fatehi P (2018) Lignin-carbohydrate complexes: properties, applications, analyses, and methods of extraction: a review. Biotechnol Biofuels 11:1–28
Du X, Pérez-Boada M, Fernández C, Rencoret J, del Río JC, Jiménez-Barbero J, Li J, Gutiérrez A, Martínez AT (2014) Analysis of lignin–carbohydrate and lignin–lignin linkages after hydrolase treatment of xylan–lignin, glucomannan–lignin and glucan–lignin complexes from spruce wood. Planta 239:1079–1090
Balakshin M, Capanema E, Gracz H, Chang H, Jameel H (2011) Quantification of lignin-carbohydrate linkages with high-resolution NMR spectroscopy. Planta 233:1097–1110
Yuan TQ, Sun SN, Xu F, Sun RC (2011) Characterization of lignin structures and lignin-carbohydrate complex (LCC) linkages by quantitative 13C and 2D HSQC NMR spectroscopy. J Agric Food Chem 59:10604–10614
Takahashi N, Koshijima T (1988) Ester linkages between lignin and glucuronoxylan in a lignin-carbohydrate complex from beech (Fagus crenata) wood. Wood Sci Technol 22:231–241
Balakshin M, Capanema E, Berlin A (2014) Isolation and analysis of lignin-carbohydrate complexes preparations with traditional and advanced methods: a review. Stud Nat Prod Chem 42:83–115
Nair SS, Yan N (2015) Effect of high residual lignin on the thermal stability of nanofibrils and its enhanced mechanical performance in aqueous environments. Cellulose 22:3137–3150
Zhang J, Choi YS, Yoo CG, Kim TH, Brown RC, Shanks BH (2015) Cellulose-hemicellulose and cellulose-lignin interactions during fast pyrolysis. ACS Sustain Chem Eng 3:293–301
Jin Z, Katsumata KS, Lam TBT, Iiyama K (2006) Covalent linkages between cellulose and lignin in cell walls of coniferous and nonconiferous woods. Biopolymers 83:103–110
Wise LE, Murphy M, D’Addieco AA (1946) Chlorite holocellulose, its fractionation and bearing on summative wood analysis and on studies on the hemicelluloses. Pap Trade J 122:35–43
Rabemanolontsoa H, Ayada S, Saka S (2011) Quantitative method applicable for various biomass species to determine their chemical composition. Biomass Bioenerg 35:4630–4635
Browning B, Bublitz L (1953) The isolation of holocellulose from wood. Tappi 36:452–458
Koeppen A, Cohen WE (1953) A study of “Chlorite” and “Chlorine” holocelluloses prepared from Eucalyptus regnans F. v. M Holzforschung 7:102–110
Bertaud F, Sundberg A, Holmbom B (2002) Evaluation of acid methanolysis for analysis of wood hemicelluloses and pectins. Carbohydr Polym 48:319–324
Bleton J, Mejanelle P, Sansoulet J, Goursaud S, Tchapla A (1996) Characterization of neutral sugars and uronic acids after methanolysis and trimethylsilylation for recognition of plant gums. J Chromatogr A 720:27–49
Li J, Kisara K, Danielsson S, Lindström ME, Gellerstedt G (2007) An improved methodology for the quantification of uronic acid units in xylans and other polysaccharides. Carbohydr Res 342:1442–1449
Asmadi M, Kawamoto H, Saka S (2017) Characteristics of softwood and hardwood pyrolysis in an ampoule reactor. J Anal Appl Pyrolysis 124:523–535
Ha YW, Thomas RL (1988) Simultaneous determination of neutral sugars and uronic acids in hydrocolloids. J Food Sci 53:574–577
Sundberg A, Sundberg K, Lillandt C, Holmbom B (1996) Determination of hemicelluloses and pectins in wood and pulp fibres by acid methanolysis and gas chromatography. Nord Pulp Pap Res J 11:216–219
Timell TE (1967) Recent progress in the chemistry of wood hemicelluloses. Wood Sci Technol 1:45–70
Tyminski A, Timell TE (1960) The constitution of a glucomannan from white spruce (Picea glauca). J Am Chem Soc 82:2823–2827
Kim DY, Nishiyama Y, Wada M, Kuga S, Okano T (2001) Thermal decomposition of cellulose crystallites in wood. Holzforschung 55:521–524
Kawamoto H, Saka S (2006) Heterogeneity in cellulose pyrolysis indicated from the pyrolysis in sulfolane. J Anal Appl Pyrolysis 76:280–284
Zickler GA, Wagermaier W, Funari SS, Burghammer M, Paris O (2007) In situ X-ray diffraction investigation of thermal decomposition of wood cellulose. J Anal Appl Pyrolysis 80:134–140
Kawamoto H (2016) Review of reactions and molecular mechanisms in cellulose pyrolysis. Curr Org Chem 20:1–15
Matsuoka S, Kawamoto H, Saka S (2014) What is active cellulose in pyrolysis? An approach based on reactivity of cellulose reducing end. J Anal Appl Pyrolysis 106:138–146
Nomura T, Kawamoto H, Saka S (2017) Pyrolysis of cellulose in aromatic solvents: reactivity, product yield, and char morphology. J Anal Appl Pyrolysis 126:209–217
Kan T, Strezov V, Evans T (2016) Effect of the heating rate on the thermochemical behavior and biofuel properties of sewage sludge pyrolysis. Energy Fuels 30:1564–1570
Acknowledgements
We thank Edanz Group (https://en-author-services.edanz.com/ac) for editing a draft of this manuscript.
Funding
This work was supported by the Japan Society for the Promotion of Science (Grant Numbers JP16H04954, JP19H03019) and the JST-Mirai Program (Grant Number JPMI20E3), Japan.
Author information
Authors and Affiliations
Contributions
The pyrolysis experiment, data analysis, and manuscript writing were conducted by JW under the supervision of HK and EM. Holocellulose samples and TG analysis were conducted by MA. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Wang, J., Minami, E., Asmadi, M. et al. Effect of delignification on thermal degradation reactivities of hemicellulose and cellulose in wood cell walls. J Wood Sci 67, 19 (2021). https://doi.org/10.1186/s10086-021-01952-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s10086-021-01952-0