
Abstract
The last decade has been marked by an exponential increase in the number of publications on the physiological role of the normal human gut microbiota. The idea of a symbiotic relationship between the human organism and normal microbiota of its gastrointestinal tract has been firmly established as an integral part of the current biomedical paradigm. However, the type of this symbiosis varies from mutualism to parasitism and depends on the functional state of the host organism. Damage caused to the organism by external agents can lead to the emergence of conditionally pathogenic properties in the normal gut microbiota, mediated by humoral factors and affecting the outcome of exogenous exposure. Among the substances produced by symbiotic microbiota, there are an indefinite number of compounds with systemic toxicity. Some occur in the intestinal chyme in potentially lethal amounts in the case they enter the bloodstream quickly. The quick entry of potential toxicants is prevented by the intestinal barrier (IB), a set of structural elements separating the intestinal chyme from the blood. Hypothetically, severe damage to the IB caused by exogenous toxicants can trigger a leakage and subsequent systemic redistribution of toxic substances of bacterial origin. Until recently, the impact of such a redistribution on the outcome of acute exogenous poisoning remained outside the view of toxicology. The present review addresses causal relationships between the secondary dysfunction of the IB and complications of acute poisoning. We characterize acute systemic toxicity of such waste products of the normal gut microflora as ammonia and endotoxins, and demonstrate their involvement in the formation of such complications of acute poisoning as shock, sepsis, cerebral insufficiency and secondary lung injuries. The principles of assessing the functional state of the IB and the approaches to its protection in acute poisoning are briefly considered.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
The legal analysis of the causes of a patient’s death implies a choice out of four possibilities: untimely, incorrect, inadequate medical care, or lack of its connection with the outcome of the disease [1]. One of the sources of error in this choice is the unpredictability of complications of acute diseases, so vividly manifested during the recent COVID-19 pandemic [2]. Unpredictability is equally inherent to complications of acute poisoning: acute circulatory disorders, sepsis, secondary acute lung injuries and acute cerebral insufficiency develop not in all patients. The reasons for such an individual variability require further detailization.
As we have shown previously, the probability of fatal complications in acute poisoning depends on the functional state of the vascular endothelium [3]. Some of its damaging substances are produced by the normal gut microbiota [4]. Based on the concept that the human body is an ecosystem with the gut microbiota being its integral constituent [5, 6], these substances are considered hereinafter as endogenous. In healthy people, the ability of such substances to cross the enterohematic or intestinal barrier (IB), a system of structural elements separating the intestinal chyme from blood, is insignificant, and their latent leakage leads to a number of chronic diseases [4, 6]. In mild exogenous poisoning, the effects of endogenous substances of intestinal origin can be overshadowed by the direct toxic effect of the xenobiotic. However, their quick and massive entry into the bloodstream in severe exogenous poisoning can aggravate the state of the organism and be involved in the pathogenesis of complications.
Direct or indirect damage to the IB by a xenobiotic, hereinafter referred to as its secondary dysfunction, can provoke the entry of metabolites and cellular bacterial components into the bloodstream in doses affecting the outcome of acute exogenous poisoning. The composition of the mixture of these substances a priori depends on that of the gut microbiota, which is individually variable [5] and, according to some estimates [7], even unique. This variability may underlie the stochasticity of complications of acute poisoning.
There is a large body of data characterizing the effect of xenobiotics on the IB. There are also numerous data on the involvement of toxicants of intestinal origin in the pathogenesis of critical states of the organism. This allows tracking for the first time the relationship between secondary dysfunction of the IB and some complications of acute poisoning. Such a relationship is the subject of the present review.
The aim of the review was to reveal the ways to prevent complications of acute poisoning through the identification of the role of secondary dysfunction of the IB in their pathogenesis.
SOURCE SELECTION ALGORITHM
The search for data characterizing the relationship between complications of acute poisoning and secondary IB dysfunction revealed an obvious novelty of this subject matter. Therefore, this review only provides basic information, formulates the concept, but is in no way systematic. It mainly cites the publications of fundamental importance that came out over the last 10 years or earlier. The main source of information was the PubMed database. The following queries were used: “acute poisoning AND (gut microbiota OR intestinal microflora OR gut microflora OR metabolome OR human gut microbiome OR gut barrier OR leaky gut syndrome OR intestinal barrier OR gut–brain axis OR gut–liver axis)”. No conference abstracts were used for analysis.
STRUCTURE AND FUNCTION OF THE NORMAL INTESTINAL BARRIER
The IB comprises of mucin, the intestinal mucosal epithelium [8], symbiotic epithelium-associated microorganisms [9], and the endothelium of submucosal blood and lymphatic capillaries [10]. Substances that escaped absorption by blood and lymphatic capillary plexuses in the intestinal wall have to overcome additional structural elements of the IB on their way to blood: the layer of smooth muscle cells, the mesothelium of visceral and parietal peritoneal sheets with the fluid-filled peritoneal cavity in between, and the endothelium of blood and lymphatic capillaries of the parietal peritoneum.
The mucin layer, 150 µm thick, consists of hydrated glycoproteins and separates epithelial cells from the aggressive environment of the intestinal luminal space. Mucin production is impaired in mucosal ischemia, which is most dangerous for the colon with its high density of bacterial colonization. Normally, symbiontic aerobic bacteria oust the pathogenic microflora from the apical surface of entero- and colonocytes, while providing them with essential substances [5]. The main function of the 20-µm epithelial cell monolayer is the selective absorption of substances from the chyme.
Due to the presence of small-intestinal villi and large-intestinal crypts, the absorptive surface area of the gastrointestinal mucosa reaches 200 m2 [8]. Normally, substances are mainly absorbed from the intestinal chyme transcellularly. The proportion of paracellular transport is presumably proportional to that of intercellular contacts on the luminal surface of the intestine, which is estimated at 0.1% [11]. Intercellular contacts are of two types, tight and adherens junctions, and consist of actin; they provide mechanical strength to the epithelium by connecting the plasma membranes of neighboring cells to the intracellular cytoskeleton [8]. Intercellular contacts serve as an object of damaging effect of a number of xenobiotics [12], against which the proportion of paracellular transport increases.
A part of the chyme substances that overcome the intestinal epithelium, enter the submucosal network of blood and lymphatic capillaries and then proceed further to the basins of the portal vein and thoracic ducts, which is the main pathway under normal conditions. The other part, having passed through the visceral and parietal peritoneal sheets, as well as a 0.25-mm fluid-filled peritoneal cavity between them, enters lymphatic vessels of the thoracic duct basin or the blood capillary network of the inferior vena cava basin. This process, transperitoneal diffusion, is facilitated by the absence of the large-intestinal diffusion barrier in the form of a continuous longitudinal muscular layer [13]. Transperitoneal diffusion of ammonia [14] and endotoxin has been shown experimentally [15]. The substances involved in transperitoneal diffusion avoid presystemic metabolism in the liver on their way to the general blood flow. In portal hypertension, the role of transperitoneal diffusion may increase due to the delayed delivery of substances of intestinal origin to the portal vein basin [16]. Transperitoneal diffusion serves as a target for detoxification therapy with a peritoneal dialysis [17].
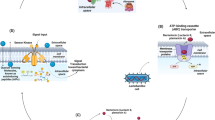
Normally, the IB is impermeable to intestinal bacteria: DNA of Escherichia coli and Bacteroides was detected in blood plasma only against the IB injury [18]. Substances to be removed from the organism penetrate from the chyme into blood via passive diffusion [19]. Their absorption is approximated [20] by the equation of the Fick’s first law of diffusion for membranes: J = D – (Ci – Co), where J is a diffusion flux density (mol × m–2 × s–1), D—membrane permeability coefficient (m × s–1), Ci and Co—concentrations of a substance on the epithelial and endothelial sides of the membrane, respectively (mol × m–3). Bioavailability of toxic products of the gut microbiota increases with values of the multipliers (Ci – Co) and D. The former is limited by the content of these substances in the chyme, the latter by the state of the IB. Presumably, absorption increases with increasing hydraulic pressure of the chyme due to smooth muscle spasm and (or) the intensification of gas production [21]. The routes of diffusion of substances from the intestinal chyme into lymph and blood are depicted schematically in Fig. 1.

Diffusion of substances from the colonal chyme into the blood and lymph. The arrows show the endpoints of diffusion.
NORMAL GUT MICROBIOTA
Prokaryotes of more than four hundred species inhabit the gastrointestinal tract (GIT) of a healthy human. The total number of their cells exceeds the number of cells of the host organism, while their mass is estimated at 0.3 % of the body weight [22]. The density of bacterial colonization in the lower (distal) GIT is higher than in its upper (proximal) division (Table 1).
Samples of gastric contents taken in fasting people are practically sterile. In hypochlorhydria, there are more bacteria in the gastric lumen, and they are mainly represented by obligate anaerobes, streptococci, lactobacilli, neisseria and staphylococci. At pH > 5.0, the microbial composition of gastric contents is indistinguishable from that of the small intestine. In 2/3 people aged 51–60 years, the ammonia-producing Gram-negative bacterium Helicobacter pylori is detected in the mucin layer of the gastric mucosa. In H. pylori-infected individuals, the pH of gastric contents increases, which adversely changes the composition of distal gut microbiota [23].
In the duodenum and jejunum, bacterial vegetation is counterbalanced by their rapid removal due to secretion, motility and bactericidal effect of bile, with Gram-positive cocci (Streptococci, Peptococci) and bacilli (Lactobacilli, Bifidobacteria) being predominant. The microbiota of the terminal ileum is similar to that of the cecum due to reflux from the latter [5]. In the colonic chyme, bacteria account for an average of 27% of its dry weight [22]. Anaerobic bacteria (predominated by Escherichia coli, Bacterioides fragilis, Lactobacilli and Bifidobacteria) in the colon are 1000 times as numerous as aerobic bacteria [5]. Anaerobes are represented by Gram-positive (Bifidobacteria, Eubacteria, Propionibacterium) and Gram-negative (Fusobacterium, Enterobacteriaceae) bacteria. The colonic mucosa is inhabited not only by symbiotic bacteria, but also by conditionally pathogenic urease-expressing bacteria from the family Enterobacteriaceae [24].
The composition of the normal gut microbiota depends on diet [25], age, antibacterial drug intake, and a number of uncontrollable conditions [5]. The production of toxic substances in the intestine is promoted by the predominance of Proteobacteria and Fusobacteria over Bacterioidetes while suppressing Lactobacilli and Bifidobacteria [26]. The number of microorganisms in the GIT depends on its motility. The normal gastrointestinal transit time is 10–48 min for the stomach, 2.5–4.0 h for the small intestine [27], and 25–40 h for the large intestine [22]. The duration of chyme passage through the large intestine determines the greatest contribution of its microbiota to the production of toxic substances [5].
TOXIC PRODUCTS OF NORMAL GUT MICROBIOTA
The normal intestinal microbiota produces substances both essential and toxic for the host [28]. The validity of the hypothesis formulated by I.I. Mechnikov more than a century ago on the ability of substances produced by the gut microbiota from nutrients to cause systemic pathological processes under certain conditions [29] is now fully proven. From 2011 to 2021, the annual number of publications available under the keywords “gut barrier”, “gut–brain axis”, “gut–liver axis”, “gut microbiota”, “intestinal microflora”, “gut microbiota AND metabolome”, “intestinal barrier”, “leaky gut syndrome” and “human gut microbiome” has grown exponentially. On the PubMed.gov Web site, it increased by 7, 16, 17, 18, 23, 23, 30, 34, 37, 40, and 44 times, respectively, during this time, whereas in previous years, the growth was linear.
Toxic substances of bacterial origin with systemic toxicity include bacterial exotoxins, endotoxin, and products of bacterial transformation of food proteins, amino acids, amino alcohols and phospholipids (ammonia, amines, phenols, heterocycles). Some amines, heterocycles and phenols are converted in the liver into secondary toxicants (trimethylamine-N-oxide, indoxyl sulfate, p-cresyl sulfate) involved in the pathogenesis of cardiovascular and other chronic diseases. Ammonia and endotoxin, asymptomatically circulating in the blood of a healthy individual in trace amounts, can exhibit acute systemic toxicity at higher concentrations, as discussed below.
Ammonia. No less than 2/3 of the ammonia produced in the organism is of intestinal origin. In enterocytes, the main mechanism of its formation is a glutaminase reaction, while in the colon, it is derived due to the metabolic activity of bacteria: deamination of amino acids and nitrogenous bases, as well as hydrolysis of the urea diffusing from blood to the luminal surface of the mucosa [30]. The ureolytic activity of microorganisms associated with the colonic mucosa accounts for the formation of half of the ammonia of intestinal origin [24].
From the intestinal chyme, ammonia enters the general blood flow through both the portal vein and the liver, as well as via transperitoneal diffusion [14]. In a healthy individual, about 4 g of ammonia is delivered into blood (mainly portal) from the intestine daily [31]. Its level in the blood of hepatic veins depends positively on that in the portal blood, while the latter on the ammonia content in the chyme [32]. Due to presystemic metabolism of ammonia, its content in hepatic venous blood is 2–3 times lower than in portal blood [33] and an order of magnitude lower than in mesenteric venous blood of the large intestine [5].
In arterial blood plasma, the normal concentration of ammonia ([NH3] + [NH4 +]) is 30 µM [34]. Its elevation is associated with neurotoxicity, first described in the laboratory of I.P. Pavlov [35]. At ([NH3] + [NH4 +]) values in blood plasma up to 100 µM, hyperammonemia is asymptomatic; at 100–200 µM, vomiting, ataxia, irritability and hyperactivity are noted, while at values more than 200 µM, there ensue convulsions and coma [36]. At an average colonic chyme volume of 0.4 L [22] and an ammonia content of 5.7–39.0 mmol/L [37], the colonic ammonia pool is 2.3–15.6 mmol/L. If this amount were evenly distributed in 5 L of blood, 2 L of lymph, and 0.4 L of chyme at the same time instant, the plasma ammonia concentration would reach 311–2108 µM, i.e., the values far above the threshold of coma.
Ammonia enters cells in an unionized form, as NH3. Considering the ammonia basicity constant (pKa = 9.15 at t = 37°C), the proportion of ammonia present as NH3 in blood plasma with a pH of 7.36 is 1.6%. The pH of the cytoplasm is lower, making NH3 to diffuse into cells even when the total blood level of both forms of ammonia is normal [30]. In metabolic acidosis or gaseous alkalosis, an increased difference between pH values in blood plasma and the cytoplasm intensifies ammonia influx into cells along the NH3 concentration gradient [38]. This explains the absorption of ammonia from blood by the brain, the accumulation of osmotically active glutamine therein, and brain edema with increasing ammonia levels in brain tissue [39]. In this case, neurotoxic effects of ammonia are possible even in the absence of hyperammonemia.
In addition to neurotoxicity, ammonia has endothelial toxicity, as demonstrated both in vitro and in vivo. In cultured brain capillary endothelial cells, it induces oxidative stress, the accumulation of nitrogen oxide (NO) and arachidonic acid peroxidation products [40, 41], destroys extracellular matrix, increases plasma membrane permeability to a fluorescein-isothiocyanate dextran derivative, causes the expression of the transmembrane carrier of arginine, the NO synthase substrate [40]. Endothelial cell growth medium treated with ammonium salt causes swelling of cultured astrocytes, which shows the involvement of the vascular endothelium in the development of brain edema during hyperammonemia [42]. In phagocytes, ammonia activates Toll-like type receptors 4, which enhances the inflammatory response to endotoxin and formation of free oxygen and nitrogen radicals, causes swelling and increased permeability of the cerebrovascular endothelium [42].
Hyperammonemia causes endothelium-dependent NO-mediated dilation of cerebral arterioles, which is involved in the development of brain edema and increased intracranial pressure in acute hepatic failure [38]. Intravenous administration of ammonium acetate to rabbits increased the permeability of their blood–brain barrier (BBB) to polyethylene glycol-400 [43]. An increased BBB permeability was also noted in patients with chronic hepatic encephalopathy [44]. Thus, the intensification of the ammonia entry into the brain induces oxidative stress, inflammatory injury, increased BBB permeability, brain edema, cerebral blood flow dysregulation, and impaired neurological functions.
Endotoxin. The content of lipopolysaccharides of Gram-negative bacteria, cumulatively known as “endotoxin”, in the colonic chyme of an adult individual is close to 2.5 g/L [45]. In blood plasma it is nine orders of magnitude lower, 9 ng/L, which is the threshold of inflammatory activation of macrophages and endotheliocytes. Moderate endotoxemia is observed in periodontitis, whereas in diabetes mellitus, liver cirrhosis, Alzheimer’s disease, and sepsis, plasma endotoxin levels can reach 500 ng/L [46]. In blood, endotoxin is present in the active free form or as complexes with proteins, usually undetectable in laboratory tests [17].
Endotoxin has a pronounced endotoxicity, which is manifested by oxidative stress, glycocalyx destruction, leukocyte adhesion, vasospasm, thrombosis [47] and increased BBB permeability [48]. Endotoxin content in blood, typical for sepsis, entails a “cytokine storm” and septic shock [49]. Lipopolysaccharides of Escherichia coli have a more pronounced proinflammatory effect compared to those of Bacteroides dorei and Bacteroides vulgatus [50]. Endotoxemia leads to thromboxane-dependent pulmonary vasoconstriction and pulmonary hypertension [51] combined with systemic vasodilation [52, 53]. The effect of endotoxin on endothelial permeability manifests itself in an increased permeability of the BBB both for endotoxin itself and for other substances [4, 54], noncardiogenic pulmonary edema [55], liver [56, 57] and renal [58] dysfunction. In endotoxemia, the ability of the liver to incorporate ammonia into urea is impaired [59]. The systemic toxicity profiles of ammonia and endotoxin are presented in Table 2.
EVALUATION OF INTESTINAL BARRIER PERMEABILITY
For an integral evaluation of the IB permeability, the levels of substances injected into the stomach are determined in blood or urine. Ideal permeability markers are biochemically inert substances that overcome the IB via passive diffusion. Of the substances determined in urine, preference is given to those that are not reabsorbed in the renal tubules. In the case of increased excretion of these substances with urine, increased IP permeability is postulated for other, similar in size, molecules [11]. Molecular weight, hydrophilic-lipophilic (log Poctanol/water), and the number of hydrogen bonds formed by the molecule are a priori indicators of the predominance of paracellular or transcellular transport of the test substance. The latter route is promoted by moderate hydrophilicity (log Poctanol/water is within the range from 0 to 5) and the ability of the molecule to form no more than five donor or acceptor hydrogen bonds. The molecular weight and effective molecular diameter of the substance are used to evaluate the pore size in the IB (Table 3).
The evaluation of IB permeability using these markers proceeds from the assumption of their preferential absorption in a certain segment of the GIT. Sucrose is used to assess the permeability of the gastric wall, and sucralose is used to assess that of the colonic wall. However, it is possible that the site of absorption changes under pathological conditions or the influence of therapy. For example, lactulose and mannitol are absorbed mainly in the small intestine, because in the colon they are quickly broken down by microorganisms [65]. However, against the suppression of the latter by antibiotics, these markers can accumulate in the chyme of the large intestine and be absorbed therein.
For the selective evaluation of mucosal epithelial permeability, isolated loops of the intestinal segment are consecutively filled with a marker solution, ligated and incubated in a buffer solution. The marker accumulation rate is considered to be an indicator of epithelial permeability, since circulation and hence the vascular endothelium, do not function under these conditions [61]. The barrier function of GIT vascular endothelium can be selectively evaluated using dyes (Evans blue, fluorescein) or compounds labeled with radioactive isotopes [66]. Intravital microscopy [67] and ex vivo microscopy of GIT tissues provide a valuable supplement to a direct investigation of the transport of substances across the IB [68].
INTESTINAL BARRIER DAMAGE IN ACUTE POISONING
Direct damage to the IB by xenobiotics. Many xenobiotics and their metabolites damage the IB directly. Local action of irritants and cauterizing substances triggers a cascade of pathological processes, including protein denaturation, tissue necrosis, release of inflammatory mediators, and blood plasma transudation [69]. Ethanol [70], arsenic, salts of heavy metals [69, 71], opioids [72], some mycotoxins [73] exhibit direct enterotoxicity. Its intensity in non-steroidal anti-inflammatory drugs made them a means for experimental modeling of acute gastroenteritis [74]. Dextran sulfate and 2,4,6-trinitrobenzene sulfonate are used to experimentally simulate acute colitis [75]. Some mycotoxins cause inflammatory damage to the IB by enhancing the effect of endotoxin thereupon [76]. T-2 mycotoxins and deoxynivalenol increase IB permeability to polyethylene glycol-4000 [77].
Direct enterotoxicity is possible not only by ingestion, but also through other routes of entry of toxicants into the organism. The small intestinal and, to a lesser extent, gastric and colonic epithelium is a typical cell renewal system with a high proliferative activity. This makes it sensitive to substances that cause reproductive or interphase death of enterocytes. Differences in enterotoxic effects of such substances are determined only by the level of target cell differentiation. Adriamycin induces apoptosis of enterocyte at position 4–5 from the stem cell located at the base of the intervillar crypt, isopropyl methanesulfonate, nitrogen mustard and actinomycin D at position 6–7; fluorouracil, mileran, cyclophosphamide and cycloheximide at position 7–9, vincristine and hydroxyurea at position 10–11 [68]. At a sufficient dose, all these toxicants cause denudation of the small intestinal epithelium. In rats with acute cyclophosphamide intoxication, IB permeability to methylene blue, mannitol and lactulose increases [63]. Enterotoxicity of cytostatic drugs in hematopoietic stem cell transplantation is one of the factors that limit patient survival [78].
An increased IB permeability against the background of its chemical damage has been demonstrated experimentally. In rats administered ammonium acetate through the gastric probe, intraperitoneal injection of cyclophosphamide accelerated ammonia and glutamine accumulation in blood and the brain [79] and provoked acute neurological disorders, such as opisthotonus and apnea, characteristic of the effect of ammonium salts at much higher doses [80, 81]. At the same time, isolated administration of the ammonium salt did not affect animal behavior significantly (Fig. 2).
In the above examples, direct damage to the IB by cyclophosphamide metabolites (aldocyclophosphamide, acrolein, phosphoramide mustard, etc.) promoted the entry of ammonia from the chyme into blood at a rate at which hyperammonemia exceeded the seizure threshold.
IB damage in acute systemic hypoxia. In critical states of the organism, acute intestinal hypoxia can be caused by a disruption of external respiration and circulation. In acute severe poisoning, such states include exotoxic shock, while in the absence of respiratory support—respiratory depression, neuromuscular blockade, and bronchoobstructive syndrome. Due to “centralization” of blood circulation in such patients, the GIT experiences deeper hypoxia than the “vital organs”.
Aerobic type of energy metabolism of enterocytes predisposes them to the breakdown of ATP resynthesis in acute hypoxia [82]. This is evidenced by intestinal damage during high-altitude hypoxia, as manifested in inflammation, ulceration and bleeding that exacerbated altitude sickness [83]. Normobaric hypoxia provoked an increase in IB permeability in rats during treadmill running [84]. IB permeability increases in hemolytic anemia [85] and acute blood loss [86]. Uncoupling of oxidative phosphorylation in colonocytes impairs their resistance to Escherichia coli [87]. Mice with more active oxidative phosphorylation in the colonic mucosa are more resistant to the local toxic effects of dextran sulfate or trinitrobenzene sulfonate [88]. Hypoxia reduces mucin production in the intestine, which entails inflammatory alterations in the mucosa. The submucosal capillary plexus in the small intestine is more developed than in the large intestine, which accounts for a higher sensitivity of the latter to ischemia [89].
IB damage due to smooth muscle spasms. Gastrointestinal smooth muscle spasm is a characteristic symptom of poisoning with cholinesterase inhibitors and serotonergic drugs, but can also be a reaction to circulatory “centralization”, or a result of the effect of serotonin produced by Enterobacteria on smooth muscles [90]. Serotonergic stimulation of the colon is accompanied, in addition to spasms of its own smooth muscles, by arterial spasm and local circulatory hypoxia [91]. Microcirculatory disorders in the intestine can damage it in the same way as it occurs under systemic hypoxia. The hypothesis of the “muscle spasm–ischemia–pain” triad as a factor of increased IB permeability in irritable bowel syndrome has been put forward [90]. In acute poisoning, the IB reaction to such changes may be identical.
IB damage in gastrointestinal stasis. Gastrointestinal stasis is a potentially fatal complication in intensive care unit (ICU) patients [92]. It is one of the specific manifestations of opioid (cholinergic antagonist and serotonin agonist) toxicity [11]. It has been observed in a rat barbiturate coma model [93], as well as in acute poisoning with alkylating toxicants. Figure 3 shows the position of radiological barium sulfate shadows in rats that received barium sulfate suspension through the gastric probe immediately after intraperitoneal (i.p.), subcutaneous (s.c.) or intragastric (gavage) administration of cyclophosphamide at a dose of 1000 mg/kg [94]. A sharp suppression of propulsive activity is quite evident in all GIT divisions, being most pronounced after intragastric infusion of a toxicant.
In gastrointestinal stasis, bacterial vegetation is not compensated by their removal, due to which the type of host–normal gut microbiota relationships becomes no longer mutualistic and approaches parasitism. Not only the total luminal concentration of ammonia produced by bacteria ([NH3] + [NH4 +]) but also the pH of the chyme, and hence the proportion of ammonia represented in a highly penetrant NH3 form, increase [37]. The cytotoxic effect of NH3 reduces the life cycle of colonocytes [95] and mucin production [96], which can damage the IB and increase the bioavailability of substances produced by colonic microflora.
Considerable gas production, characteristic of gastrointestinal stasis, can intensify the diffusion of substances through the IB. It increases the barometric pressure in the intestinal lumen and, according to Dalton’s first law, the partial vapor pressure of volatile substances. According to Dalton’s second law, it increases the tension of the corresponding gases dissolved in the chyme. Damage to the IB, as well as an increased tension of volatile substances in the chyme, are potential factors that increase their bioavailability in gastrointestinal stasis.
Inflammatory damage to the IB. Damage to the EGB by an exogenous toxicant promotes endotoxin entry into blood. It stimulates the secretion of proinflammatory cytokines (TNF-α, IFN-γ and IL-6) by phagocytes, leading to both systemic and local inflammation [97]. Cyclophosphamide administration to pygmy pigs at a dose of 50 mg/kg increased TNF-α and IL-6 serum levels and IB permeability [98]. Intestinal mucositis leads to bacteremia in hematopoietic stem cell recipients during cytostatic therapy [99]. Submucosal inflammation and leukocytoclastic vasculitis accompanied by mucosal ulceration have been reported in rats administered with mercuric chloride [100]. Acute inflammation of the colon provokes inflammation in other organs as well [101], which may promote the entry of toxic substances of intestinal origin from blood.
Thus, in acute severe poisonings, there are prerequisites for an increase in the IB permeability for metabolites and cellular components of the intestinal microflora. The mechanisms of secondary IB dysfunction in acute poisoning include direct damaging effects of xenobiotics or their metabolites, gastrointestinal stasis, smooth muscle spasm, gastrointestinal inflammation and hypoxia. Along with secondary IB dysfunction, the delivery of toxic substances from the intestinal chyme into the general blood flow in acute poisoning can be promoted by the excessive growth of the intestinal microbiota and increased gas production. All these synergistic factors lead to endotoxemia.

Symptoms of acute intoxication with ammonium acetate and/or cyclophosphamide in rats. Animals were photographed 30 min after the intragastric (gavage) administration of ammonium acetate (AA) at a dose of 12 mmol/kg and/or cyclophosphamide (CY) at a dose of 1000 mg/kg intraperitoneally (Rejniuk, Schäfer, and Ivnitsky 2010).

A diagram showing the position of the X-ray barium sulfate shadow in the rat gastrointestinal tract after the administration of cyclophosphamide and infusion of barium sulfate suspension. The tone of the circles is proportional to the percentage of the area occupied by a shadow in the corresponding division of the gastrointestinal tract. Black circles: 75–100% of the whole X-ray barium sulfate shadow. White circles: no barium sulfate shadow (Ivnitsky et al. 2012).
ENDOTOXEMIA IN ACUTE POISONING
Endotoxemia is an elevated blood level of biologically active substances produced in the organism. Proceeding from the ideas of the organism as an ecosystem with the intestinal microbiota as its constituent [5, 6], the compounds produced by the latter are classified as endogenous substances.
The blood level of medium molecular weight (MMW) substances is used as an integral index of endotoxemia [102]. In patients with uncomplicated poisoning with psychopharmacological drugs, blood levels of such substances were by 32% higher than in healthy donors, and those of endotoxin—10 times higher [103]. After intestinal lavage, the blood level of MMW substances decreased by 18 %, endotoxin—by half, while without lavage, these indices of endotoxemia continued to grow. Thus, in poisoning with psychopharmacological drugs, the colonic chyme is a source of endotoxemia.
The issue of which specific substances are involved in endotoxemia formation under acute poisoning is of interest. One of them is endotoxin, whose increased blood level has been shown in severe acute poisoning with colchicine [97], verapamil [104], and ethanol [105]. Manifest endotoxemia accompanies patients’ preparation for hematopoietic stem cell transplantation, during which cytostatic drugs are administered at potentially lethal doses. Endotoxin accumulation in blood in such a treatment leads to a systemic inflammatory response [106, 107].
In acute exogenous poisonings, endotoxemia is paralleled with hyperammonemia [108, 109]. Listed below are xenobiotics, in acute poisonings with which it has been documented (to exclude liver injuries from the causes of hyperammonemia, specific hepatotoxic agents are not omitted):
– acetazolamide [110];
– calcium hopantenate [111];
– clozapine [112];
– cocaine [113];
– cyclophosphan [79];
– cyclopropane [114];
– cyanides [115];
– ethanol [116];
– diethyl ether [117];
– fluoroacetate [118];
– 5-fluorouracil [119];
– homocysteine thiolactone [120];
– lead salts [121];
– methanol [122];
– oxygen [123];
– sodium thiopental [93];
– trimethoprim [124];
– valproates [125].
Thus, endotoxemia in acute poisoning involves endotoxin and ammonia, while changes in blood levels of other components of the intestinal metabolome require further investigation. The role of endotoxin and ammonia in the pathogenesis of acute complications is characterized by the information provided below on endotoxemia in the corresponding syndromes of various etiologies.
ENDOTOXEMIA IN CRITICAL STATES OF AN ORGANISM
Critical states of an organism are endotoxemia patterns, a constellation of clinical manifestations of endotoxemia. The role of endotoxin and ammonia in the pathogenesis of various critical states is discussed below, depending on whether the symptoms of these states are reproduced by endotoxin or ammonia administration.
Shock is an acute state of inadequate tissue blood supply, with hyperammonemia being one of its biochemical markers. Plasma and brain tissue ammonia levels are manifold elevated in rabbits and dogs when modeling heat or insulin shock [126]. In patients with septic shock, venous blood ammonia levels were 80% higher, while in those died eventually of septic shock, 2.2 times higher compared to patients with sepsis but without shock [127]. In patients with hemorrhagic shock, ammonia levels in arterial blood plasma were twice as high as in patients with hemorrhage but without shock [128]. In severe trauma, venous blood ammonia levels were twice as high as in controls, while in deceased patients, it was by 70% higher than in survivors [129]. Plasma ammonia levels in patients admitted to the ICU because of cardiac arrest was 4.8 times higher than that of patients with spontaneous circulation [130]. Hyperammonemia not only accompanies shock but is also involved in its pathogenesis. The administration of ammonium salts to rabbits reproduced key manifestations of heat and insulin shock [126].
In experiments, shock is also reproduced by endotoxin administration [131]. Endotoxin accumulation in blood is characteristic of septic [132], traumatic and hemorrhagic [133] shock.
IB damage is one of the mechanisms of endotoxemia in shock. When modeling hemorrhagic shock in rats, the animals were losing claudin-3, a protein of intestinal epithelium tight junctions, with urine [134]. Using MMW substances as endotoxemia markers, it has been shown that, in hemorrhagic shock, they accumulate first in intestinal tissues, then in portal blood, and finally in carotid artery blood [135].
Thus, hyperammonemia and endotoxinemia are characteristic of endotoxemia that accompanies shock and is involved in the development of its different types.
Sepsis is a systemic inflammatory response to endotoxemia and bacteremia, which most frequently complicate acute poisoning in alcohol addicts [105]. Sepsis developed in 6.4% of the methanol poisoning victims [136]. Endotoxemia constantly accompanies sepsis [137], while bacteremia has only been observed in 67% of such patients [138]. Endotoxin accumulation in blood occurs in sepsis caused by Gram-negative, as well as Gram-positive bacteria and fungi, due to IB damage [139].
Clinical manifestations of sepsis are reproduced by endotoxin administration to animals [140]. In endotoxemia characteristic of sepsis, the blood coagulation cascade is activated, resulting in disseminated microvascular thrombosis [141], which leads to kidney [10], heart and liver [142] damages, multiple organ failure [132], and pulmonary edema [143].
Ammonia is also involved in the pathogenesis of multiple organ failure in sepsis. Its plasma level in patients with this diagnosis, measured on admission to the emergency department, correlated positively with the likelihood of developing multiple organ failure within the following 28 days [96].
Secondary acute lung injuries in acute poisoning include non-cardiogenic pulmonary edema, shock and wet lung syndromes. The involvement of endotoxin in the pathogenesis of these conditions follows from endotoxemia that accompanies all of them [144] and endotoxin-induced damage to alveolar–capillary (air–blood) barrier cells both in vitro [145] and in vivo [146]. Endotoxin administration to rats caused acute lung injury resembling such in COVID-19 and manifesting itself as an alveolar cavity hemorrhage, cytokine storm, coagulopathy, and pulmonary hypertension [147]. Escherichia coli endotoxin caused hemodynamic disorders in the pig lungs: its administration into the right atrial cavity increased blood pressure in the pulmonary artery [53].
In portocaval anastomosis, hyperammonemia is the predictor of pulmonary hypertension [148]. In patients with severe acute pancreatitis, the development of a lethal complication, pulmonary edema, is caused by excessive bacterial colonization of the small intestine [149].
Thus, endotoxemia accompanies secondary acute lung injuries under acute poisoning and is involved in the development of pulmonary hypertension. Ammonia is another marker of endotoxemia, promising for the prediction of secondary acute lung injuries in acute poisoning.
Acute cerebral insufficiency is a syndrome resulting from diffuse brain damage. It is observed in severe poisoning with substances having different mechanisms of action, and manifests itself in impaired consciousness and mental confusion, motor disorders, accelerated catabolism, acute respiratory and circulatory disorders [150].
Some manifestations of acute cerebral insufficiency may be associated with increased ammonia and/or endotoxin delivery to the brain. Hyperammonemia is characteristic of acute liver failure complicated by brain edema and increased intracranial pressure [151]. Intracranial pressure, neurological disorders, and mortality are positively related to serum ammonia levels in patients with extrahepatic hyperammonemia [45]. Plasma and brain tissue ammonia levels were significantly elevated in dog and rat models of diabetic coma [126]. Endotoxin increased the sensitivity of piglets to acute cerebral hypoxia. Escherichia coli endotoxin administration to piglets increased neuronal loss and metabolic disturbances, as well as increased the probability of brain death followed by carotid artery occlusion [152]. Endotoxin administration to ferrets exacerbated brain damage due to carotid artery occlusion [153]. Endotoxin potentiated brain edema during altitude hypoxia [154].
Thus, the available data indicate that endotoxin and ammonia produced by the normal gut microbiota are involved in the pathogenesis of abnormal conditions that can complicate acute poisoning, such as shock, sepsis, secondary acute lung injuries, and acute cerebral failure.
SECONDARY DYSFUNCTION OF THE INTESTINAL BARRIER AND STOCHASTICITY OF COMPLICATIONS OF ACUTE POISONINGS
The phenomenon of an altered tolerance of acute damaging exposures against the background of preformed chronic pathology has long been used in functional diagnostics of the latter. This phenomenon also occurs in acute poisoning. Xenobiotic-induced damage to the IB may be superimposed on chronic pathological conditions, in which it is damaged, such as intestinal dysbiosis, irritable bowel syndrome, Crohn’s disease, nonspecific ulcerative colitis, etc. This can exacerbate endotoxemia in acute exogenous poisoning. Such an overlap, shown schematically in Fig. 4, may contribute to the individual variability in the severity of their complications.

Effect of chronic gut intestinal barrier dysfunction on the outcome of acute exogenous poisoning. a—absence, and b—presence of preformed chronic gut barrier dysfunction.
INTESTINAL BARRIER PROTECTION IN ACUTE EXOGENOUS POISONINGS
The intestinal chyme is an aggressive environment for the damaged intestinal mucosa, and its local damaging effect increases with intestinal dysbiosis [95]. Therefore, the planned measures on IB protection should be aimed at optimizing intestinal microbiota composition and increasing the local resistance of the mucous membrane to toxicants of bacterial origin. The main approaches to optimizing intestinal microbiota composition are prebiotic, metabiotic, probiotic and autoprobiotic therapies.
Prebiotics are nutrients that selectively promote the vegetation of host-beneficial members of the gut microbiota whose therapeutic effects can be exemplified by gut microbiota recovery in mice and guinea pigs with antibiotic-associated dysbiosis [155]. Metabiotic therapy, a variant of prebiotic therapy, implies the use of metabolites of functional microorganisms. Metabolites of Lactobacillus plantarum accelerated gut microbiota recovery in mice with antibiotic-associated dysbiosis by 5965 times and in guinea pigs by 308 times. The prebiotic drug Stimbifid (a complex of fructooligo- and fructopolysaccharides), when applied on the same experimental models, accelerated gut microbiota recovery by 7118 and 1047 times, respectively [155].
Probiotics are drugs derived from functional microorganisms referring to collection strains. A classic example of probiotic therapy is the use of Lactobacillus bulgaricus proposed by I.I. Mechnikov [29]. Preparations of live Lactobacilli, Enterococci, Bifidobacteria, and E. coli are widely used for this purpose [156]. Anaerobic Gram-positive bacteria—Lactobacilli, Bifidobacteria, Propionobacteria—suppress conditionally pathogenic intestinal microorganisms, as well as the production of ammonia, amines, substrates for the synthesis of trimethylamine-N-oxide, indoxyl sulfate, p-cresyl sulfate, which have both systemic and local toxicity [45]. Officinal microbiological preparations containing probiotic microorganisms are applicable as a source thereof. At the same time, such microorganisms are unable to be involved in the formation of a “microbial–tissue complex”, as well as in long-term colonization of the human intestine [155]. In this regard, autoprobiotic therapy, based on the application of certain indigenous bacteria isolated from the organism and returned to it after cultivation and accumulation in vitro, seems to be promising. The therapeutic efficacy of Enterococci [157] and Bifidobacteria [158], used in this way, has been shown in experimental dysbiosis.
Low-toxicity drugs, suitable for long-term application, are also promising for boosting the local intestinal resistance to toxic substances of bacterial origin. It is reasonable to search for them among the drugs having adaptogenic properties. The beneficial properties of plantain juice as a remedy to correct acute hyperammonemic and neurotoxic effects of cyclophosphamide have been experimentally proven. Against the background of just a single intragastric administration of an officinal preparation of plantain juice to rats at a dose equivalent to 47 mL of that for humans, the hyperammonemic effect of cyclophosphamide administered at a supralethal dose (2.1 LD50) was 1.5 times lower, the average hang time during which the rats could hold out on a wire grid in the four limb hanging test increased by 20%, while the average life expectancy of these animals increased by 30% [159].
Promising means of emergency prevention of IB lesions are the preparations able to form a protective film on the surface of the mucous membrane. Such substances can be pectins whose adhesive properties find use in hydrocolloid wound plasters [160]. When pectin is administered by ingestion, a protective film of calcium pectates forms on the mucous membranes [75], preventing substances produced by gut microflora from getting into blood. Gelatin tannate has a similar ability [161]. The protective effect of these substances is mainly expected during the drug transit in the small intestine: from 10 min to 5 h post ingestion.
The condition necessary for maintaining the intestinal barrier function in severe acute poisoning is the elimination of intestinal hypoxia and inflammation, as confirmed by the anti-inflammatory effect of hyperbaric oxygenation in sepsis [162]. Clinical trials of oxygen-enriched gas mixtures containing inert gases, nitrogen oxide or hydrogen at low concentrations also show a lot of promise; the properties of these additives can modulate the effects of oxygen therapy. Xenon, nitric oxide and hydrogen have neuroprotective and cardiotonic properties, while argon and hydrogen sulfide have neuroprotective properties only [163]. Anti-inflammatory drugs can be supplementary to oxygen therapy: in experimental sepsis modeling, both IB dysfunction and endotoxemia were prevented by hydrocortisone administration [164].
CONCLUSION
1. In a healthy person, the intestinal chyme contains products of bacterial origin, whose amount and systemic toxicity profile, when losing the gradients of their concentration between the chyme and blood, ensure the formation of critical states of the organism, such as shock, sepsis, secondary acute lung injuries, and acute cerebral insufficiency.
2. Severe acute exogenous poisoning creates prerequisites for secondary intestinal barrier dysfunction and endotoxemia of intestinal origin.
3. Endotoxemia, including endotoxinemia and hyperammonemia, is involved in the formation of complications of acute poisoning, as manifested in critical states of an organism listed in paragraph 1.
4. Planned measures to protect the intestinal barrier from damage in the following acute poisoning should be aimed at optimizing the composition of gut microbiota and suppressing its production of cytotoxic substances, as well as boosting local intestinal resistance, by prescribing low-toxicity drugs intended for a long-term administration.
5. The main approaches to optimizing gut microbiota composition are prebiotic, metabiotic, probiotic and autoprobiotic therapies. Low-toxicity preparations having adaptogenic properties and suitable for long-term application are promising for increasing local intestinal resistance to toxic substances of bacterial origin.
6. Emergency prevention of secondary intestinal barrier dysfunction involves the use of drugs shielding the mucous membrane from the chyme and eliminating intestinal hypoxia and inflammation.
Abbreviations
- Bacterial exotoxins:
-
toxic proteins and peptides released by living bacteria into the external environment
- Brain–blood barrier (BBB):
-
a set of structural elements separating the central nervous system (CNS) and blood
- Endogenous substance:
-
a compound produced by the cells of the host or its symbiotic microflora
- Endotoxemia:
-
accumulation of toxic endogenous substances in blood, including toxic low molecular weight metabolites of bacterial and/or tissue origin, endotoxin, and bacterial exotoxins
- Endotoxicosis:
-
a set of clinical manifestations of endotoxaemia
- Endotoxin:
-
the sum of outer cell wall lipopolysaccharides of gram-negative bacteria
- Endotoxinemia:
-
endotoxin accumulation in blood
- Intestinal barrier (IB):
-
a set of structural elements separating the intestinal chyme and blood
- Low molecular weight bacterial metabolites:
-
substances of bacterial origin with a molecular weight of less than 1500 Da
- Primary intestinal barrier dysfunction:
-
preformed (inborn or acquired) chronic intestinal barrier disorder
- Secondary intestinal barrier dysfunction:
-
damage to the intestinal barrier caused by external exposure
- Toxicant:
-
any substance that causes damage to the organism in a non-mechanical way when exposed at a given dose
- Xenobiotic:
-
a foreign substance
REFERENCES
Grishin SM (2018) Crimes committed by medical workers as a result of improper performance of their professional duties (based on the case-law of the European part of Russia 2015–2017). Medicina (Mex) 6: 1–14. https://doi.org/10.29234/2308-9113-2018-6-1-1-14
Ponti G, Maccaferri M, Ruini C, Tomasi A, Ozben T (2020) Biomarkers associated with COVID-19 disease progression. Crit Rev Clin Lab Sci 57: 389–399. https://doi.org/10.1080/10408363.2020.1770685
Ivnitsky JuJu, Schäfer TV, Rejniuk VL, Golovko AI (2022) Endogenous humoral determinants of vascular endothelial dysfunction as triggers of acute poisoning complications. J Appl Toxicol Online ahead of print. https://doi.org/10.1002/jat.4312
Vutukuri R, Brunkhorst R, Kestner R-I, Hansen L, Bouzas NF, Pfeilschifter J, Devraj K, Pfeilschifter W (2018) Alteration of sphingolipid metabolism as a putative mechanism underlying LPS-induced BBB disruption. J Neurochem 144: 172–185. https://doi.org/10.1111/jnc.14236
Hill MJ (1995) Role of gut bacteria in human toxicology and pharmacology. Taylor & Francis, London; Bristol, PA.
Tang WHW, Kitai T, Hazen SL (2017) Gut Microbiota in Cardiovascular Health and Disease. Circ Res 120: 1183–1196. https://doi.org/10.1161/CIRCRESAHA.117.309715
Choi T-Y, Choi YP, Koo JW (2020) Mental Disorders Linked to Crosstalk between The Gut Microbiome and The Brain. Exp Neurobiol 29: 403–416. https://doi.org/10.5607/en20047
Lopetuso LR, Scaldaferri F, Bruno G, Petito V, Franceschi F, Gasbarrini A (2015) The therapeutic management of gut barrier leaking: the emerging role for mucosal barrier protectors. Eur Rev Med Pharmacol Sci 19: 1068–1076.
Gurwara S, Dai A, Ajami NJ, Graham DY, White DL, Chen L, Jang A, Chen E, El-Serag HB, Petrosino JF, Jiao L (2020) Alcohol use alters the colonic mucosa-associated gut microbiota in humans. Nutr Res 83: 119–128. https://doi.org/10.1016/j.nutres.2020.09.004
Liu Z, Yang D, Gao J, Xiang X, Hu X, Li S, Wu W, Cai J, Tang C, Zhang D, Dong Z (2020) Discovery and validation of miR-452 as an effective biomarker for acute kidney injury in sepsis. Theranostics 10: 11963–11975. https://doi.org/10.7150/thno.50093
Gad SC (2018) Toxicology of the Gastrointestinal Tract. CRC Press, Boca Raton, Florida
Lechuga S, Naydenov NG, Feygin A, Cruise M, Ervasti JM, Ivanov AI (2020) Loss of β-Cytoplasmic Actin in the Intestinal Epithelium Increases Gut Barrier Permeability in vivo and Exaggerates the Severity of Experimental Colitis. Front Cell Dev Biol 8: 588836. https://doi.org/10.3389/fcell.2020.588836
Magnotti LJ, Upperman JS, Xu DZ, Lu Q, Deitch EA (1998) Gut-derived mesenteric lymph but not portal blood increases endothelial cell permeability and promotes lung injury after hemorrhagic shock. Ann Surg 228: 518–527. https://doi.org/10.1097/00000658-199810000-00008
Schäfer TV, Ivnitsky JuJu, Rejniuk VL (2011) Cyclophosphamide-induced leakage of gastrointestinal ammonia into the common bloodstream in rats. Drug Chem Toxicol 34: 25–31. https://doi.org/10.3109/01480545.2010.483518
Milan Manani S, Virzì GM, Giuliani A, Baretta M, Corradi V, De Cal M, Biasi C, Crepaldi C, Ronco C (2020) Lipopolysaccharide Evaluation in Peritoneal Dialysis Patients with Peritonitis. Blood Purif 49: 434–439. https://doi.org/10.1159/000505388
Møller S, Kimer N, Barløse M, Bendtsen F (2020) Pathophysiological-based treatments of complications of cirrhosis. Scand J Gastroenterol 55: 383–394. https://doi.org/10.1080/00365521.2020.1744709
Komarov BD, Luzhnikov EA, Shimanko II (1981) Surgical treatments for acute poisoning. Meditsina, Moscow. (In Russ).
Shi K, Wang F, Jiang H, Liu H, Wei M, Wang Z, Xie L (2014) Gut bacterial translocation may aggravate microinflammation in hemodialysis patients. Dig Dis Sci 59: 2109–2117. https://doi.org/10.1007/s10620-014-3202-7
Travis S, Menzies I (1992) Intestinal permeability: functional assessment and significance. Clin Sci 82: 471–488. https://doi.org/10.1042/cs0820471
Samoilov VO (2013) Medical biophysics. SpezLit, St-Petersburg (In Russ).
Lin HC, Pimentel M (2005) Bacterial concepts in irritable bowel syndrome. Rev Gastroenterol Disord 5(3): S3–S9.
Sender R, Fuchs S, Milo R (2016) Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLOS Biol 14: e1002533. https://doi.org/10.1371/journal.pbio.1002533
Hunt RH, Camilleri M, Crowe SE, El-Omar EM, Fox JG, Kuipers EJ, Malfertheiner P, McColl KEL, Pritchard DM, Rugge M, Sonnenberg A, Sugano K, Tack J (2015) The stomach in health and disease. Gut 64: 1650–1668. https://doi.org/10.1136/gutjnl-2014-307595
Walker V (2012) Severe hyperammonaemia in adults not explained by liver disease. Ann Clin Biochem Int J Lab Med 49: 214–228. https://doi.org/10.1258/acb.2011.011206
Adamberg K, Jaagura M, Aaspõllu A, Nurk E, Adamberg S (2020) The composition of faecal microbiota is related to the amount and variety of dietary fibres. Int J Food Sci Nutr 71: 845–855. https://doi.org/10.1080/09637486.2020.1727864
Rai R, Saraswat VA, Dhiman RK (2015) Gut Microbiota: Its Role in Hepatic Encephalopathy. J Clin Exp Hepatol 5: S29–S36. https://doi.org/10.1016/j.jceh.2014.12.003
O’Grady J, Murphy CL, Barry L, Shanahan F, Buckley M (2020) Defining gastrointestinal transit time using video capsule endoscopy: a study of healthy subjects. Endosc Int Open 8: E396–E400. https://doi.org/10.1055/a-1073-7653
Rodríguez-Hernández P, Cardador MJ, Arce L, Rodríguez-Estévez V (2020) Analytical Tools for Disease Diagnosis in Animals via Fecal Volatilome. Crit Rev Anal Chem 12: 1–16. https://doi.org/10.1080/10408347.2020.1843130
Metchnikoff E (1907) Essais optimistes. A. Maloine, Paris
Ivnitsky YuYu, Schaefer TV, Reinyuk VL (2012) Endogenous ammonia in a toxic process. Palmarium Acad Publish. (In Russ).
Summerskill WHJ, Wolpert E (1970) Ammonia Metabolism in the Gut. Am J Clin Nutr 23: 633–639. https://doi.org/10.1093/ajcn/23.5.633
Gips CH, Qué GS, Wibbens-Alberts M (1973) The arterial ammonia curve after oral and intraduodenal loading with ammonium acetate. Absorption in the stomach. Neth J Med 16: 14–17.
Dobson GP, Veech RL, Passonneau JV, Huang MT (1990) In vivo portal-hepatic venous gradients of glycogenic precursors and incorporation of D-[3-3H]glucose into liver glycogen in the awake rat. J Biol Chem 265: 16350–16357.
Ali R, Nagalli S (2021) Hyperammonemia. StatPearls Publ.
Hahn M, Massen O, Nencki M, Pawlow J (1893) Die Eck’sche Fistel zwischen der unteren Hohlvene und der Pfortader und ihre Folgen für den Organismus. Arch Für Exp Pathol Pharmakol 32: 161–210. https://doi.org/10.1007/BF01995065
Pagana K, Pagana T, Pagana T (2021) Mosby’s manual of diagnostic and laboratory tests, 7th ed. Elsevier Inc, Philadelphia.
Agostini L, Down PF, Murison J, Wrong OM (1972) Faecal ammonia and pH during lactulose administration in man: comparison with other cathartics. Gut 13: 859–866. https://doi.org/10.1136/gut.13.11.859
Scott TR (2013) Pathophysiology of cerebral oedema in acute liver failure. World J Gastroenterol 19: 9240. https://doi.org/10.3748/wjg.v19.i48.9240
Ott P, Vilstrup H (2014) Cerebral effects of ammonia in liver disease: current hypotheses. Metab Brain Dis 29: 901–911. https://doi.org/10.1007/s11011-014-9494-7
Skowrońska M, Albrecht J (2013) Oxidative and nitrosative stress in ammonia neurotoxicity. Neurochem Int 62: 731–737. https://doi.org/10.1016/j.neuint.2012.10.013
Tranah TH, Vijay GKM, Ryan JM, Shawcross DL (2013) Systemic inflammation and ammonia in hepatic encephalopathy. Metab Brain Dis 28: 1–5. https://doi.org/10.1007/s11011-012-9370-2
Jayakumar AR, Norenberg MD (2018) Hyperammonemia in Hepatic Encephalopathy. J Clin Exp Hepatol 8: 272–280. https://doi.org/10.1016/j.jceh.2018.06.007
McClung HJ, Sloan HR, Powers P, Merola AJ, Murray R, Kerzner B, Pollack JD (1990) Early changes in the permeability of the blood-brain barrier produced by toxins associated with liver failure. Pediatr Res 28: 227–231. https://doi.org/10.1203/00006450-199009000-00014
Ochoa-Sanchez R, Rose CF (2018) Pathogenesis of Hepatic Encephalopathy in Chronic Liver Disease. J Clin Exp Hepatol 8: 262–271. https://doi.org/10.1016/j.jceh.2018.08.001
Bested AC, Logan AC, Selhub EM (2013) Intestinal microbiota, probiotics and mental health: from Metchnikoff to modern advances: Part II—Contemporary contextual research. Gut Pathog 5: 3. https://doi.org/10.1186/1757-4749-5-3
Brown GC (2019) The endotoxin hypothesis of neurodegeneration. J Neuroinflammation 16: 180. https://doi.org/10.1186/s12974-019-1564-7
Iba T, Levy JH, Hirota T, Hiki M, Sato K, Murakami T, Nagaoka I (2018) Protection of the endothelial glycocalyx by antithrombin in an endotoxin-induced rat model of sepsis. Thromb Res 171: 1–6. https://doi.org/10.1016/j.thromres.2018.09.042
Minami T, Oda K, Gima N, Yamazaki H (2007) Effects of lipopolysaccharide and chelator on mercury content in the cerebrum of thimerosal-administered mice. Environ Toxicol Pharmacol 24: 316–320. https://doi.org/10.1016/j.etap.2007.08.004
Pfalzgraff A, Weindl G (2019) Intracellular Lipopolysaccharide Sensing as a Potential Therapeutic Target for Sepsis. Trends Pharmacol Sci 40: 187–197. https://doi.org/10.1016/j.tips.2019.01.001
Yoshida N, Yamashita T, Kishino S, Watanabe H, Sasaki K, Sasaki D, Tabata T, Sugiyama Y, Kitamura N, Saito Y, Emoto T, Hayashi T, Takahashi T, Shinohara M, Osawa R, Kondo A, Yamada T, Ogawa J, Hirata K (2020) A possible beneficial effect of Bacteroides on faecal lipopolysaccharide activity and cardiovascular diseases. Sci Rep 10: 13009. https://doi.org/10.1038/s41598-020-69983-z
Wideman RF, Erf GF, Chapman ME (2001) Intravenous Endotoxin Triggers Pulmonary Vasoconstriction and Pulmonary Hypertension in Broiler Chickens. Poult Sci 80: 647–655. https://doi.org/10.1093/ps/80.5.647
Bermejo-Martin J, Martín-Fernandez M, López-Mestanza C, Duque P, Almansa R (2018) Shared Features of Endothelial Dysfunction between Sepsis and Its Preceding Risk Factors (Aging and Chronic Disease). J Clin Med 7: 400. https://doi.org/10.3390/jcm7110400
Corrêa TD, Pereira AJ, Takala J, Jakob SM (2020) Regional venous-arterial CO2 to arterial-venous O2 content difference ratio in experimental circulatory shock and hypoxia. Intensive Care Med Exp 8: 64. https://doi.org/10.1186/s40635-020-00353-9
Yeo IJ, Yun J, Son DJ, Han S-B, Hong JT (2020) Antifungal drug miconazole ameliorated memory deficits in a mouse model of LPS-induced memory loss through targeting iNOS. Cell Death Dis 11: 623. https://doi.org/10.1038/s41419-020-2619-5
Wang L, Cao Y, Gorshkov B, Zhou Y, Yang Q, Xu J, Ma Q, Zhang X, Wang J, Mao X, Zeng X, Su Y, Verin AD, Hong M, Liu Z, Huo Y (2019) Ablation of endothelial Pfkfb3 protects mice from acute lung injury in LPS-induced endotoxemia. Pharmacol Res 146: 104292. https://doi.org/10.1016/j.phrs.2019.104292
Jadhav K, Cohen TS (2020) Can You Trust Your Gut? Implicating a Disrupted Intestinal Microbiome in the Progression of NAFLD/NASH. Front Endocrinol 11: 592157. https://doi.org/10.3389/fendo.2020.592157
Solé C, Guilly S, Da Silva K, Llopis M, Le-Chatelier E, Huelin P, Carol M, Moreira R, Fabrellas N, De Prada G, Napoleone L, Graupera I, Pose E, Juanola A, Borruel N, Berland M, Toapanta D, Casellas F, Guarner F, Doré J, Solà E, Ehrlich SD, Ginès P (2021) Alterations in Gut Microbiome in Cirrhosis as Assessed by Quantitative Metagenomics: Relationship With Acute-on-Chronic Liver Failure and Prognosis. Gastroenterology 160: 206–218. e13. https://doi.org/10.1053/j.gastro.2020.08.054
Nežić L, Škrbić R, Amidžić L, Gajanin R, Milovanović Z, Nepovimova E, Kuča K, Jaćević V (2020) Protective Effects of Simvastatin on Endotoxin-Induced Acute Kidney Injury through Activation of Tubular Epithelial Cells’ Survival and Hindering Cytochrome C-Mediated Apoptosis. Int J Mol Sci 21: E7236. https://doi.org/10.3390/ijms21197236
Soria LR, Marrone J, Molinas SM, Lehmann GL, Calamita G, Marinelli RA (2014) Lipopolysaccharide impairs hepatocyte ureagenesis from ammonia: Involvement of mitochondrial aquaporin-8. FEBS Lett 588: 1686–1691. https://doi.org/10.1016/j.febslet.2014.03.012
Skowrońska M, Zielińska M, Wójcik-Stanaszek L, Ruszkiewicz J, Milatovic D, Aschner M, Albrecht J (2012) Ammonia increases paracellular permeability of rat brain endothelial cells by a mechanism encompassing oxidative/nitrosative stress and activation of matrix metalloproteinases: Ammonia-induced brain endothelium damage. J Neurochem 121: 125–134. https://doi.org/10.1111/j.1471-4159.2012.07669.x
Zijlstra FJ, van Meeteren ME, Garrelds IM, Meijssen MAC (2003) Effect of pharmacologically induced smooth muscle activation on permeability in murine colitis. Mediators Inflamm 12: 21–27. https://doi.org/10.1080/0962935031000096944
Wilms E, Jonkers DMAE, Savelkoul HFJ, Elizalde M, Tischmann L, de Vos P, Masclee AAM, Troost FJ (2019) The Impact of Pectin Supplementation on Intestinal Barrier Function in Healthy Young Adults and Healthy Elderly. Nutrients 11: 1554. https://doi.org/10.3390/nu11071554
Schäfer TV, Rejniuk VL, Krasnov KA, Ivnitsky JuJu (2011) Increased permeability of a small intestine to luminal hydrophylic medium-sized molecules in cyclophosphamide-treated rats. Medline.ru. Russ Biomed J 12: 1437–1449. (In Russ).
Jiang Y, Bian Y, Lian N, Wang Y, Xie K, Qin C, Yu Y (2020) iTRAQ-Based Quantitative Proteomic Analysis of Intestines in Murine Polymicrobial Sepsis with Hydrogen Gas Treatment. Drug Des Devel Ther 14: 4885–4900. https://doi.org/10.2147/DDDT.S271191
Di Palo DM, Garruti G, Di Ciaula A, Molina-Molina E, Shanmugam H, De Angelis M, Portincasa P (2020) Increased Colonic Permeability and Lifestyles as Contributing Factors to Obesity and Liver Steatosis. Nutrients 12: E564. https://doi.org/10.3390/nu12020564
Wick MJ, Harral JW, Loomis ZL, Dempsey EC (2018) An Optimized Evans Blue Protocol to Assess Vascular Leak in the Mouse. J Vis Exp 139: 57037. https://doi.org/10.3791/57037
Alves NG, Motawe ZY, Yuan SY, Breslin JW (2018) Endothelial Protrusions in Junctional Integrity and Barrier Function. Curr Top Membr 82: 93–140. https://doi.org/10.1016/bs.ctm.2018.08.006
Ijiri K, Potten CS (1983) Response of intestinal cells of differing topographical and hierarchical status to ten cytotoxic drugs and five sources of radiation. Br J Cancer 47: 175–185. https://doi.org/10.1038/bjc.1983.25
Kutsenko SA (2004) Fundamentals of Toxicology. Foliant Publishing House, St-Petersburg. (In Russ).
Bishehsari F, Magno E, Swanson G, Desai V, Voigt RM, Forsyth CB, Keshavarzian A (2017) Alcohol and Gut-Derived Inflammation. Alcohol Res Curr Rev 38: 163–171.
Zhao Y, Zhou C, Wu C, Guo X, Hu G, Wu Q, Xu Z, Li G, Cao H, Li L, Latigo V, Liu P, Cheng S, Liu P (2020) Subchronic oral mercury caused intestinal injury and changed gut microbiota in mice. Sci Total Environ 721: 137639. https://doi.org/10.1016/j.scitotenv.2020.137639
Akbarali HI, Dewey WL (2017) The gut-brain interaction in opioid tolerance. Curr Opin Pharmacol 37: 126–130. https://doi.org/10.1016/j.coph.2017.10.012
Puel O, Galtier P, Oswald I (2010) Biosynthesis and Toxicological Effects of Patulin. Toxins 2: 613–631. https://doi.org/10.3390/toxins2040613
Karádi O, Nagy Z, Bódis B, Mózsik G (2001) Atropine-induced gastrointestinal cytoprotection dependences to the intact of vagal nerve against indomethacin-induced gastrointestinal mucosal and microvascular damage in rats. J Physiol (Paris) 95: 29–33. https://doi.org/10.1016/s0928-4257(01)00006-7
Ishisono K, Mano T, Yabe T, Kitaguchi K (2019) Dietary Fiber Pectin Ameliorates Experimental Colitis in a Neutral Sugar Side Chain-Dependent Manner. Front Immunol 10: 2979. https://doi.org/10.3389/fimmu.2019.02979
Ge L, Lin Z, Le G, Hou L, Mao X, Liu S, Liu D, Gan F, Huang K (2020) Nontoxic-dose deoxynivalenol aggravates lipopolysaccharides-induced inflammation and tight junction disorder in IPEC-J2 cells through activation of NF-κB and LC3B. Food Chem Toxicol Int J Publ Br Ind Biol Res Assoc 145: 111712. https://doi.org/10.1016/j.fct.2020.111712
Semenov EI, Mishina NN, Papunidi KKh (2019) Unaccounted anaphylactic reaction to effect of mycotoxins. Medline.ru. Russ Biomed J 20: 36–43. (In Russ).
McMillen KK, Coghlin-Dickson T, Adintori PA (2021) Optimization of nutrition support practices early after hematopoietic cell transplantation. Bone Marrow Transplant 56: 314–326. https://doi.org/10.1038/s41409-020-01078-9
Ivnitsky YuYu, Schäfer TV, Tyaptin AA, Rejniuk VL (2019) Changes in the chemical composition of blood and brain of rats under the conditions of modeling of the myeloablation regimen of cyclophosphamide administration. Toxicol Rev 3: 13–18. https://doi.org/10.36946/0869-7922-2019-3-13-18
Ivnitsky JuJu, Schäfer TV, Rejniuk VL (2011) Promotion of the Toxic Action of Cyclophosphamide by Digestive Tract Luminal Ammonia in Rats. ISRN Toxicol 2011: 1–4. https://doi.org/10.5402/2011/450875
Rejniuk VL, Schäfer TV, Ivnitsky JuJu (2010) Increase of Ammonia Pool in the Gastrointestinal Tract of Rats Potentiates Acute Toxicity of Cyclophosphamide. Bull Exp Biol Med 149: 718–720. https://doi.org/10.1007/s10517-010-1034-9
Khуzhnyak SV, Bezdrobna LK, Stepanova LI, Morozova VS, Voitsitskіy VM (2014) Oxidative phosphorylation in mitochondria of small-intestinal enterocytes at chronic and single exposure to low power ionizing radiation. Probl Rad Med Radiobiol 19: 482–489.
Khanna K, Mishra KP, Chanda S, Eslavath MR, Ganju L, Kumar B, Singh SB (2019) Effects of Acute Exposure to Hypobaric Hypoxia on Mucosal Barrier Injury and the Gastrointestinal Immune Axis in Rats. High Alt Med Biol 20: 35–44. https://doi.org/10.1089/ham.2018.0031
Hill GW, Gillum TL, Lee BJ, Romano PA, Schall ZJ, Hamilton AM, Kuennen MR (2020) Prolonged treadmill running in normobaric hypoxia causes gastrointestinal barrier permeability and elevates circulating levels of pro- and anti-inflammatory cytokines. Appl Physiol Nutr Metab Physiol Appl Nutr Metab 45: 376–386. https://doi.org/10.1139/apnm-2019-0378
Abuga KM, Muriuki JM, Williams TN, Atkinson SH (2020) How Severe Anaemia Might Influence the Risk of Invasive Bacterial Infections in African Children. Int J Mol Sci 21: E6976. https://doi.org/10.3390/ijms21186976
Khazoom F, L’Écuyer S, Gilbert K, Gagné M-A, Bouchard C, Rose CF, Rousseau G, Charbonney E (2020) Impact of uric acid on liver injury and intestinal permeability following resuscitated hemorrhagic shock in rats. J Trauma Acute Care Surg 89: 1076–1084. https://doi.org/10.1097/TA.0000000000002868
Saxena A, Lopes F, McKay DM (2018) Reduced intestinal epithelial mitochondrial function enhances in vitro interleukin-8 production in response to commensal Escherichia coli. Inflamm Res 67: 829–837. https://doi.org/10.1007/s00011-018-1172-5
Bär F, Bochmann W, Widok A, von Medem K, Pagel R, Hirose M, Yu X, Kalies K, König P, Böhm R, Herdegen T, Reinicke AT, Büning J, Lehnert H, Fellermann K, Ibrahim S, Sina C (2013) Mitochondrial gene polymorphisms that protect mice from colitis. Gastroenterology 145: 1055–1063. e3. https://doi.org/10.1053/j.gastro.2013.07.015
Zvenigorodskaya LA, Samsonova NG, Toporkov AS (2010) Chronic ischemic disease of the digestive system: an algorithm for diagnosis and treatment. Russ Med Zhurn 9: 544–548. (In Russ).
Uno Y (2019) Hypothesis: Mechanism of irritable bowel syndrome in inflammatory bowel disease. Med Hypotheses 132: 109324. https://doi.org/10.1016/j.mehy.2019.109324
Lychkova AE (2005) Gradients of serotoninergic innervation of the large intestine. Bull Exp Biol Med 139: 550–553. https://doi.org/10.1007/s10517-005-0342-y
Deane AM, Chapman MJ, Reintam Blaser A, McClave SA, Emmanuel A (2019) Pathophysiology and Treatment of Gastrointestinal Motility Disorders in the Acutely Ill. Nutr Clin Pract Off Publ Am Soc Parenter Enter Nutr 34: 23–36. https://doi.org/10.1002/ncp.10199
Ivnitsky JuJu, Rejniuk VL, Schäfer TV, Malakhovsky VN (2006) Fulminant hyperammonaemia induced by thiopental coma in rats. Toxicology 224: 184–190. https://doi.org/10.1016/j.tox.2006.04.002
Schäfer TV, Ivnitsky JuJu, Rejniuk VL (2022) Modelling myeloablative cytostatic therapy with cyclophosphamide is accompanied by gastrointestinal stasis in rats. Med Extrem Situat 1: 51–55. https://doi.org/10.47183/mes.2022.001
Visek WJ (1972) Effects of urea hydrolysis on cell life-span and metabolism. Fed Proc 31: 1178–1193.
Zhao J, He Y, Xu P, Liu J, Ye S, Cao Y (2020) Serum ammonia levels on admission for predicting sepsis patient mortality at D28 in the emergency department: A 2-center retrospective study. Medicine (Baltimore) 99: e19477. https://doi.org/10.1097/MD.0000000000019477
Horioka K, Tanaka H, Isozaki S, Konishi H, Fujiya M, Okuda K, Asari M, Shiono H, Ogawa K, Shimizu K (2020) Acute Colchicine Poisoning Causes Endotoxemia via the Destruction of Intestinal Barrier Function: The Curative Effect of Endotoxin Prevention in a Murine Model. Dig Dis Sci 65: 132–140. https://doi.org/10.1007/s10620-019-05729-w
Lee SI, Kang KS (2017) Function of capric acid in cyclophosphamide-induced intestinal inflammation, oxidative stress, and barrier function in pigs. Sci Rep 7: 16530. https://doi.org/10.1038/s41598-017-16561-5
Herbers AHE, de Haan AFJ, van der Velden WJFM, Donnelly JP, Blijlevens NMA (2014) Mucositis not neutropenia determines bacteremia among hematopoietic stem cell transplant recipients. Transpl Infect Dis Off J Transplant Soc 16: 279–285. https://doi.org/10.1111/tid.12195
Mathieson PW, Thiru S, Oliveira DB (1992) Mercuric chloride-treated brown Norway rats develop widespread tissue injury including necrotizing vasculitis. Lab Investig J Tech Methods Pathol 67: 121–129.
Winnard KP, Dmitrieva N, Berkley KJ (2006) Cross-organ interactions between reproductive, gastrointestinal, and urinary tracts: modulation by estrous stage and involvement of the hypogastric nerve. Am J Physiol-Regul Integr Comp Physiol 291: R1592–R1601. https://doi.org/10.1152/ajpregu.00455.2006
Matkevich VA, Luzhnikov EA, Belova MV, Yevdokimova NV, Syromyatnikova ED, Kurilkin YA (2015) The role of intestinal translocation in the origin of endotoxemia in acute poisoning and detoxification effect of intestinal lavage. Russ Sklifosovsky J Emerg Med Care 4: 16–21.
Matkevich VA, Potskhveriya MM, Goldfarb YuS, Simonova AYu (2018) Violations of homeostasis parameters in acute poisonings and ways of their correction. Toxicol Rev 3: 18–26. https://doi.org/10.36946/0869-7922-2018-3-18-26
Aburto-Fernández MDC, Araujo-López A, García-Godoy IU, Alvarado-González A, Gutiérrez-Samperio JL, de Orendáin AÁ-M, Ocio MAR, Arteaga-Villalba LR, Herrera-Díaz A, Jiménez-Ríos CO, Lerma-Alvarado RM (2019) Secondary colonic necrosis. Case report. Cir Cir 87: 33–37. https://doi.org/10.24875/CIRU.18000656
Chaung WW, Brenner M, Yen H-T, Ochani ML, Jacob A, Wang P (2019) Recombinant human milk fat globule-EGF factor VIII (rhMFG-E8) as a therapy for sepsis after acute exposure to alcohol. Mol Med Camb Mass 25: 52. https://doi.org/10.1186/s10020-019-0118-x
Johansson J-E, Hasséus B, Johansson P, Eklöf C, Ohman D, Stockelberg D (2009) Gut protection by palifermin during autologous haematopoietic SCT. Bone Marrow Transplant 43: 807–811. https://doi.org/10.1038/bmt.2008.388
Guinan EC, Palmer CD, Mancuso CJ, Brennan L, Stoler-Barak L, Kalish LA, Suter EE, Gallington LC, Huhtelin DP, Mansilla M, Schumann RR, Murray JC, Weiss J, Levy O (2014) Identification of single nucleotide polymorphisms in hematopoietic cell transplant patients affecting early recognition of, and response to, endotoxin. Innate Immun 20: 697–711. https://doi.org/10.1177/1753425913505122
Laemmle A, Hahn D, Hu L, Rüfenacht V, Gautschi M, Leibundgut K, Nuoffer J-M, Häberle J (2015) Fatal hyperammonemia and carbamoyl phosphate synthetase 1 (CPS1) deficiency following high-dose chemotherapy and autologous hematopoietic stem cell transplantation. Mol Genet Metab 114: 438–444. https://doi.org/10.1016/j.ymgme.2015.01.002
Uygun V, Karasu G, Daloğlu H, Hazar V, Yeşilipek A (2015) Idiopathic hyperammonemia after hematopoietic stem cell transplantation: A case report. Pediatr Transplant 19: E104–E105. https://doi.org/10.1111/petr.12467
Webster LT (1965) Hepatic coma—a biochemical disorder of the brain. Gastroenterology 49: 698–702.
Kimura A, Yoshida I, Ono E, Matsuishi T, Yoshino M, Yamashita F, Yamamoto M, Hashimoto T, Shinka T, Kuhara T (1986) Acute encephalopathy with hyperammonemia and dicarboxylic aciduria during calcium hopantenate therapy: a patient report. Brain Dev 8: 601–605. https://doi.org/10.1016/s0387-7604(86)80006-7
Slyundin DG, Alekhnovich AV, Ivanov VB, Livanov AS, Anuchin VV (2010) The pharmacological correction of hyperammoniemisis conditions at the criminal poisoning clozapine. Medline.ru. Russ Biomed J 11: 518–525.
Kramer L, Bauer E, Schenk P, Steininger R, Vigl M, Mallek R (2003) Successful treatment of refractory cerebral oedema in ecstasy/cocaine-induced fulminant hepatic failure using a new high-efficacy liver detoxification device (FPSA-Prometheus). Wien Klin Wochenschr 115: 599–603. https://doi.org/10.1007/BF03040456
Goldberg JE, Hamilton WK (1959) Blood ammonia levels during ether and cyclopropane anesthesia. Anesthesiology 20: 836–841. https://doi.org/10.1097/00000542-195911000-00015
Yamamoto H (1993) Relationship among cyanide-induced encephalopathy, blood ammonia levels, and brain aromatic amino acid levels in rats. Bull Environ Contam Toxicol 50: 274–281. https://doi.org/10.1007/BF00191733
Ivnitsky JuJu, Schäfer TV, Rejniuk VL (2010) Redistribution of gastrointestinal ammonia into blood in alcohol coma rat: the role in lethal outcome. Patol fiziol eksperim terapiya 3: 34–37. (In Russ).
Watanabe A, Kuwabara Y (1994) Hyperammonemia induced in rats by inhalation anesthesia with ether. Res Exp Med (Berl) 194: 157–164. https://doi.org/10.1007/BF02576376
Proudfoot AT, Bradberry SM, Vale JA (2006) Sodium fluoroacetate poisoning. Toxicol Rev 25: 213–219. https://doi.org/10.2165/00139709-200625040-00002
Mitani S, Kadowaki S, Komori A, Sugiyama K, Narita Y, Taniguchi H, Ura T, Ando M, Sato Y, Yamaura H, Inaba Y, Ishihara M, Tanaka T, Tajika M, Muro K (2017) Acute hyperammonemic encephalopathy after fluoropyrimidine-based chemotherapy: A case series and review of the literature. Medicine (Baltimore) 96: e6874. https://doi.org/10.1097/MD.0000000000006874
Blennow G, Folbergrova J, Nilsson B, Siesjö BK (1979) Cerebral metabolic and circulatory changes in the rat during sustained seizures induced by DL-homocysteine. Brain Res 179: 129–146. https://doi.org/10.1016/0006-8993(79)90497-9
Maslinski PG, Loeb JA (2004) Pica-associated cerebral edema in an adult. J Neurol Sci 225: 149–151. https://doi.org/10.1016/j.jns.2004.07.016
Foster WA, Schoenhals JA (1995) Hyperammonemia with severe methanol intoxication. West J Med 163: 377–379.
Singh AK, Banister EW (1983) Tissue ammonia and amino acids in rats at various oxygen pressures. J Appl Physiol 54: 438–444. https://doi.org/10.1152/jappl.1983.54.2.438
Simma B, Meister B, Deutsch J, Sperl W, Fend F, Öfner D, Margreiter R, Vogel W (1995) Fulminant hepatic failure in a child as a potential adverse effect of trimethoprim-sulphamethoxazole. Eur J Pediatr 154: 530–533. https://doi.org/10.1007/BF02074828
Woo PYM, Woo AWY, Lam SW, Ko NMW, Ho JWK, Chu ACH, Kwan MCL, Chan Y, Wong H-T, Chan K-Y (2020) Incidence, Presentation, and Risk Factors for Sodium Valproate–Associated Hyperammonemia in Neurosurgical Patients: A Prospective, Observational Study. World Neurosurg 144: e597–e604. https://doi.org/10.1016/j.wneu.2020.09.027
Kozlov NB (1971) Ammonia, its exchange and role in pathology. Medicina, Moscow. (In Russ).
Gao C, Wang D, Li W, Guo X, Ma J, Xie Y (2020) [Value of blood ammonia on predicting the severity and prognosis of patients with sepsis: a prospective observation study]. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue 32: 1315–1319. https://doi.org/10.3760/cma.j.cn121430-20200305-00210
Singla A, Kaur S, Kaur N, Gill C (2016) Arterial ammonia levels: Prognostic marker in traumatic hemorrhage. Int J Appl Basic Med Res 6: 255. https://doi.org/10.4103/2229-516X.192601
Hagiwara A, Sakamoto T (2009) Clinical Significance of Plasma Ammonia in Patients With Traumatic Hemorrhage. J Trauma Inj Infect Crit Care 67: 115–120. https://doi.org/10.1097/TA.0b013e3181a5e63e
Lin C-H, Chi C-H, Wu S-Y, Hsu H-C, Chang Y-H, Huang Y-Y, Chang C-J, Hong M-Y, Chan T-Y, Shih H-I (2013) Prognostic values of blood ammonia and partial pressure of ammonia on hospital arrival in out-of-hospital cardiac arrests. Am J Emerg Med 31: 8–15. https://doi.org/10.1016/j.ajem.2012.04.037
Fujiwara Y, Ohnishi K, Horlad H, Saito Y, Shiraishi D, Takeya H, Yoshii D, Kaieda S, Hoshino T, Komohara Y (2020) CD163 deficiency facilitates lipopolysaccharide-induced inflammatory responses and endotoxin shock in mice. Clin Transl Immunol 9: e1162. https://doi.org/10.1002/cti2.1162
Luna M, Kamariski M, Principi I, Bocanegra V, Vallés PG (2021) Severely ill pediatric patients with Shiga toxin-associated hemolytic uremic syndrome (STEC-HUS) who suffered from multiple organ involvement in the early stage. Pediatr Nephrol 36: 1499–1509. https://doi.org/10.1007/s00467-020-04829-4
Hu C, Sun J, Du J, Wen D, Lu H, Zhang H, Xue Y, Zhang A, Yang C, Zeng L, Jiang J (2019) The Hippo-YAP pathway regulates the proliferation of alveolar epithelial progenitors after acute lung injury. Cell Biol Int 43: 1174–1183. https://doi.org/10.1002/cbin.11098
Thuijls G, Derikx JPM, Haan J-J de, Grootjans J, Bruïne A de, Masclee AAM, Heineman E, Buurman WA (2010) Urine-based Detection of Intestinal Tight Junction Loss. J Clin Gastroenterol 44: e14–e19. https://doi.org/10.1097/MCG.0b013e31819f5652
Khramykh TP, Dolgikh VT (2009) Endotoxemia in hemorrhagic hypotension. Patol fiziol eksperim terapiya 1: 28–30. (In Russ).
Kumar M, Kaeley N, Nagasubramanyam V, Bhardwaj BB, Kumar S, Kabi A, Arora P, Dhar M (2019) Single center experience of managing methanol poisoning in the hilly state of Uttarakhand: A cross sectional study. Int J Crit Illn Inj Sci 9: 172–176. https://doi.org/10.4103/IJCIIS.IJCIIS_49_19
Chew CH, Cheng L-W, Huang W-T, Wu YM, Lee C-W, Wu M-S, Chen C-C (2020) Ultrahigh packing density next generation microtube array membrane: A novel solution for absorption-based extracorporeal endotoxin removal device. J Biomed Mater Res B Appl Biomater 108: 2903–2911. https://doi.org/10.1002/jbm.b.34621
Novosad SA, Sapiano MRP, Grigg C, Lake J, Robyn M, Dumyati G, Felsen C, Blog D, Dufort E, Zansky S, Wiedeman K, Avery L, Dantes RB, Jernigan JA, Magill SS, Fiore A, Epstein L (2016) Vital Signs: Epidemiology of Sepsis: Prevalence of Health Care Factors and Opportunities for Prevention. MMWR Morb Mortal Wkly Rep 65: 864–869. https://doi.org/10.15585/mmwr.mm6533e1
Śmiechowicz J (2022) The Rationale and Current Status of Endotoxin Adsorption in the Treatment of Septic Shock. J Clin Med 11: 619. https://doi.org/10.3390/jcm11030619
Chen S, Xiu G, Zhou J, Liu P, Chen X, Sun J, Ling B (2020) [Role of high mobility group box 1 in intestinal mucosal barrier injury in rat with sepsis induced by endotoxin]. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue 32: 803–807. https://doi.org/10.3760/cma.j.cn121430-20200109-00126
Colbert JF, Schmidt EP (2016) Endothelial and Microcirculatory Function and Dysfunction in Sepsis. Clin Chest Med 37: 263–275. https://doi.org/10.1016/j.ccm.2016.01.009
Soltanian A, Mosallanejad B, Razi Jalali M, Najafzadeh Varzi H, Ghorbanpoor M (2020) Comparative evaluation of the therapeutic effects of silymarin and hydrocortisone on clinical and hematological alterations, and organ injury (liver and heart) in low-dose canine lipopolysaccharide-induced sepsis model. Vet Res Forum 11: 235–241. https://doi.org/10.30466/vrf.2018.83961.2105
Pool R, Gomez H, Kellum JA (2018) Mechanisms of Organ Dysfunction in Sepsis. Crit Care Clin 34: 63–80. https://doi.org/10.1016/j.ccc.2017.08.003
Maniatis NA, Kotanidou A, Catravas JD, Orfanos SE (2008) Endothelial pathomechanisms in acute lung injury. Vascul Pharmacol 49: 119–133. https://doi.org/10.1016/j.vph.2008.06.009
Wang W, Weng J, Yu L, Huang Q, Jiang Y, Guo X (2018) Role of TLR4-p38 MAPK-Hsp27 signal pathway in LPS-induced pulmonary epithelial hyperpermeability. BMC Pulm Med 18: 178. https://doi.org/10.1186/s12890-018-0735-0
Forrester SJ, Xu Q, Kikuchi DS, Okwan-Duodu D, Campos AC, Faidley EA, Zhang G, Lassègue B, Sadikot RT, Griendling KK, Hernandes MS (2019) Poldip2 deficiency protects against lung edema and vascular inflammation in a model of acute respiratory distress syndrome. Clin Sci Lond Engl 133: 321–334. https://doi.org/10.1042/CS20180944
Al‐Ani B, ShamsEldeen AM, Kamar SS, Haidara MA, Al‐Hashem F, Alshahrani MY, Al‐Hakami AM, Kader DHA, Maarouf A (2022) Lipopolysaccharide induces acute lung injury and alveolar haemorrhage in association with the cytokine storm, coagulopathy and AT1R/JAK/STAT augmentation in a rat model that mimics moderate and severe Covid‐19 pathology. Clin Exp Pharmacol Physiol 49: 483–491. https://doi.org/10.1111/1440-1681.13620
Bloom PP, Rodriguez-Lopez J, Witkin AS, Al-Samkari H, Kuter DJ, Mojtahed A, Luther J (2020) Ammonia Predicts Hepatic Involvement and Pulmonary Hypertension in Patients With Hereditary Hemorrhagic Telangiectasia. Clin Transl Gastroenterol 11: e00118. https://doi.org/10.14309/ctg.0000000000000118
Liang X-Y, Jia T-X, Zhang M (2021) Intestinal bacterial overgrowth in the early stage of severe acute pancreatitis is associated with acute respiratory distress syndrome. World J Gastroenterol 27: 1643–1654. https://doi.org/10.3748/wjg.v27.i15.1643
Shilov VV, Aleksandrov MV, Vasilev SA, Aleksandrova TV, Chernyi VS (2010) Acute cerebral failure at the serious poisoning. Medline.ru. Russ Biomed J 11(25):315–321. (In Russ).
Sheikh MF, Unni N, Agarwal B (2018) Neurological Monitoring in Acute Liver Failure. J Clin Exp Hepatol 8: 441–447. https://doi.org/10.1016/j.jceh.2018.04.013
Pang R, Martinello KA, Meehan C, Avdic-Belltheus A, Lingam I, Sokolska M, Mutshiya T, Bainbridge A, Golay X, Robertson NJ (2020) Proton Magnetic Resonance Spectroscopy Lactate/N-Acetylaspartate Within 48 h Predicts Cell Death Following Varied Neuroprotective Interventions in a Piglet Model of Hypoxia-Ischemia With and Without Inflammation-Sensitization. Front Neurol 11: 883. https://doi.org/10.3389/fneur.2020.00883
Wood T, Moralejo D, Corry K, Fisher C, Snyder JM, Acuna V, Holden-Hunt A, Virk S, White O, Law J, Parikh P, Juul SE (2019) A Ferret Model of Inflammation-sensitized Late Preterm Hypoxic-ischemic Brain Injury. J Vis Exp (153): e60131. https://doi.org/10.3791/60131
Zhou Y, Huang X, Zhao T, Qiao M, Zhao X, Zhao M, Xu L, Zhao Y, Wu L, Wu K, Chen R, Fan M, Zhu L (2017) Hypoxia augments LPS-induced inflammation and triggers high altitude cerebral edema in mice. Brain Behav Immun 64: 266–275. https://doi.org/10.1016/j.bbi.2017.04.013
Chicherin IYu, Pogorelsky IP, Lundovskikh IA, Gavrilov KE, Shabalina MR, Darmov IV (2013) Autoprobiotic therapy. J Infectology 5: 43—54. https://doi.org/10.22625/2072-6732-2013-5-4-43-54
Shenderov BA (2011) Probiotics and Functional Foods. In: Food Engineering. EOLSS Publishers, Oxford; UK.
Suvorov A (2013) Gut Microbiota, Probiotics, and Human Health. Biosci Microbiota Food Health 32: 81–91. https://doi.org/10.12938/bmfh.32.81
Gromova LV, Ermolenko EI, Sepp AL, Dmitrieva YV, Alekseeva AS, Lavrenova NS, Kotyleva MP, Kramskaya TA, Karaseva AB, Suvorov AN, Gruzdkov AA (2021) Gut Digestive Function and Microbiome after Correction of Experimental Dysbiosis in Rats by Indigenous Bifidobacteria. Microorganisms 9: 522. https://doi.org/10.3390/microorganisms9030522
Schäfer TV, Ivnitsky JuJu, Rejniuk VL (2015) The influence of the plantain juice on the manifestations and outcome of acute cyclophosphamide intoxication in rats. Biopreparaty 55(3): 61–64. (In Russ)].
Sabando C, Ide W, Rodríguez-Díaz M, Cabrera-Barjas G, Castaño J, Bouza R, Müller N, Gutiérrez C, Barral L, Rojas J, Martínez F, Rodríguez-Llamazares S (2020) A Novel Hydrocolloid Film Based on Pectin, Starch and Gunnera tinctoria and Ugni molinae Plant Extracts for Wound Dressing Applications. Curr Top Med Chem 20: 280–292. https://doi.org/10.2174/1568026620666200124100631
Çağan E, Ceylan S, Mengi Ş, Çağan HH (2017) Evaluation of Gelatin Tannate Against Symptoms of Acute Diarrhea in Pediatric Patients. Med Sci Monit Int Med J Exp Clin Res 23: 2029–2034. https://doi.org/10.12659/msm.903158
Rinaldi B, Cuzzocrea S, Donniacuo M, Capuano A, Di Palma D, Imperatore F, Mazzon E, Di Paola R, Sodano L, Rossi F (2011) Hyper baric oxygen therapy reduces the toll-like receptor signaling pathway in multiple organ failures. Intensive Care Med 37: 1110–1119. https://doi.org/10.1007/s00134-011-2241-1
Alshami A, Einav S, Skrifvars MB, Varon J (2020) Administration of inhaled noble and other gases after cardiopulmonary resuscitation: A systematic review. Am J Emerg Med 38: 2179–2184. https://doi.org/10.1016/j.ajem.2020.06.066
Assimakopoulos SF, Papadopoulou I, Bantouna D, de Lastic A-L, Rodi M, Mouzaki A, Gogos CA, Zolota V, Maroulis I (2021) Fecal Microbiota Transplantation and Hydrocortisone Ameliorate Intestinal Barrier Dysfunction and Improve Survival in a Rat Model of Cecal Ligation and Puncture-Induced Sepsis. Shock Augusta Ga 55: 666–675. https://doi.org/10.1097/SHK.0000000000001566
ACKNOWLEDGMENTS
The authors are grateful to Prof. S.A. Kutsenko, former head of the Department of Military Toxicology and Medical Defense, for providing the author’s copy of his monograph “Fundamentals of Toxicology” [69] whose materials were extremely useful for the discussion of selective toxicity of xenobiotics. [69], as well as to Prof. K.V. Yaremenko, consultant of the N.N. Petrov Scientific and Research Center for Oncology, for kind sharing her invaluable clinical experience in the prevention of enterotoxicity of cytostatic agents. The authors are also thankful to the anonymous reviewers of their previous article [3] for a detailed discussion of the physiological activity of substances produced by the normal gut microflora, and to the anonymous reviewer of the present article for the help in highlighting those aspects of the problem that relate to the composition of the gut microbiota.
Funding
This review article had no sponsor support.
Author information
Authors and Affiliations
Contributions
Ju.Ju.I.—conceptualization, structuring the article, formulating its generalizations and conclusions; T.V.S.—participation in writing all sections of the article, searching and analyzing the scientific literature; V.L.R.—participation in writing the section “Intestinal barrier damage in acute poisoning”, searching and analyzing the scientific literature; O.A.V.—participation in searching and analyzing the scientific literature, preparing the text and list of references.
Corresponding author
Ethics declarations
CONFLICT OF INTEREST
The authors declare that they have neither evident nor potential conflicts of interest or interpersonal relationships that might affect the publication of this article.
Additional information
Translated by A. Polyanovsky
Russian Text © The Author(s), 2022, published in Rossiiskii Fiziologicheskii Zhurnal imeni I.M. Sechenova, 2022, Vol. 108, No. 7, pp. 807–835https://doi.org/10.31857/S0869813922070020.
Rights and permissions
About this article

Cite this article
Ivnitsky, J.J., Schäfer, T.V., Rejniuk, V.L. et al. Secondary Dysfunction of the Intestinal Barrier in the Pathogenesis of Complications of Acute Poisoning. J Evol Biochem Phys 58, 1075–1098 (2022). https://doi.org/10.1134/S0022093022040123
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0022093022040123