
Abstract
Biosynthesis approaches for the production of higher alcohols as a source of alternative fossil fuels have garnered increasing interest recently. However, there is little information available in the literature about using undirected whole-cell mutagenesis (UWCM) in vivo to improve higher alcohols production. In this study, for the first time, we approached this question from two aspects: first preferentially improving the capacity of expression host, and subsequently optimizing metabolic pathways using multiple genetic mutations to shift metabolic flux toward the biosynthetic pathway of target products to convert intermediate 2-keto acid compounds into diversified C4~C5 higher alcohols using UWCM in vivo, with the aim of improving the production. The results demonstrated the production of higher alcohols including isobutanol, 2-methyl-1-butanol, 3-methyl-1-butanol from glucose and duckweed under simultaneous saccharification and fermentation (SSF) scheme were higher based on the two aspects compared with only the use of wild-type stain as expression host. These findings showed that the improvement via UWCM in vivo in the two aspects for expression host and metabolic flux can facilitate the increase of higher alcohols production before using gene editing technology. Our work demonstrates that a multi-faceted approach for the engineering of novel synthetic pathways in microorganisms for improving biofuel production is feasible.
Similar content being viewed by others

Introduction
Currently, one of the most promising approaches for replacing fossil fuels is the biological production of biofuels via genetically-modified microorganisms. Some examples, such as n-butanol and isobutanol, have been synthesized using genetically-engineered bacteria1,2,3,4,5. Of particular interest are longer chain hydrocarbons, such as C4~C5 higher alcohols including 3-methyl-butanol, 2-methyl-butanol, and isopentanol, which have been successfully synthesized through the use of non-synthetic E. coli and Corynebacterium based on their endogenous α-keto acid pathway6,7,8,9. These alcohols have a higher energy density close to that of gasoline, exhibit lower hygroscopicity compared to ethanol, and are currently regarded as very promising potential substitutes for fossil fuels.
One of the first challenges in the replacement of fossil fuels with biofuels derived via a biosynthesis pathway is the inefficient conversion of lignocellulosic substrate into biofuels by bacteria. To biosynthesize these C4-C5 higher alcohols by engineering bacteria such as E. coli, Bacillus subtilis, and Corynebacterium glutamicum, the fermentation substrates traditionally were pure sugar or starch without heavy metals and other impurities9,10,11,12. However, limited research has been reported using bioengineered strains to produce C4~C5 higher alcohols via fermenting hydrolysates of lignocellulose. Therefore, it is essential to investigate the capacity of bioengineered strains to directly ferment hydrolysates of lignocellulose as a more effective method of biofuel generation.
There is a growing trend to develop new, renewable, non-food plant sources as feedstock for biofuel production. Duckweed has received increasing attention as a potentially inexpensive and sustainable lignocellulose source of non-food plant biomass for producing biofuels such as ethanol and biobutanol, and has demonstrated great potential as a candidate for bioenergy feedstock6,13,14. However, there are no studies to date regarding the efficacy of bioengineered strains of Corynebacterium in producing other C4~C5 higher alcohols, especially 3-methyl-butanol and 2-methyl-butanol using hydrolysates of lignocellulose as a fermentation substrate under SSF scheme. Therefore, we selected duckweed as a fermentation substrate to investigate the ability of bioengineered strains of a Corynebacterium, C. crenatium, to produce C4~C5 higher alcohols via SSF.
Secondly, in addition to the need to overcome the inefficiency of utilizing lignocellulosic substrate to convert into biofuels, it is also essential that improvement of the expression host’s catalytic activity via introducing novel metabolic pathways with high-activity enzymes in order to maximize higher alcohols production as much as possible. One major challenge in the construction of metabolic pathways is the identification and selection of enzymes with high activity. Protein-directed engineering in vitro such as error-prone PCR and DNA shuffling provides a foundation for screening interesting mutant genes that are favorable for improving higher alcohols. However, there may be undesirable effects that may lead to a metabolic flux imbalance when overexpressing these mutant heterologous genes from other species via in vitro mutation experiment, and thus ultimately decrease the yield of higher alcohols. Unfortunately, using these methods to obtain mutant genes with high enzymatic activity while maintaining metabolic flux balance in a expression host is not always achievable.
Mutant genes with high enzymatic activity can also be obtained using UWCM in vivo. For example, some high yield auxotrophes, such as amino acid-producing Corynebacterium, and Saccharomyces cerevisiae which produces 3-methyl-1-butanol, have been obtained via perturbing whole genome using the UWCM method15,16,17,18,19,20. Using this method, multiple mutant genes are identified that have metabolic balance favorable for optimization of metabolic pathways in these auxotrophic strains. There is a dynamic equilibrium of metabolic pathways between these mutant genes and auxotrophic strains, and thus improved production. However, until now, improving higher alcohols production via overexpressing exogenous genes from the UWCM approach in vivo into a expression host has not been shown. Therefore, we proposed to use the method to obtain multiple gene mutations in an auxotrophic strain, and utilize those mutant genes to construct novel metabolic pathways, and then introduce those novel metabolic pathways into another expression host reach to improve production of C4~C5 higher alcohols.
Thirdly, the capability of host itself is also an important consideration for improving higher alcohols production in addition to the introduction of metabolic pathways with highly activated genes. In general, the expression hosts used to produce higher alcohols were typically wild-type strains prior to the use of gene editing techniques. Using improved auxotrophic strains from UWCM instead of wild-type strain as expression host to increase yield of specific products prior to using gene editing techniques, followed by the use of gene editing techniques to further reform the host may maximize product yield. However, this has not yet been reported in the literature.
Therefore, in this study, we aimed to improve higher alcohols production using two aspects. First, improved metabolic capability of C. crenatium and S. cerevisiae via UWCM, whereby the improved mutant C. crenatium was used as expression host and the mutant S. cerevisiae was used to extract multiple mutant genes. Addionally, these newly identified mutant genes involving in the biosynthesis of C4~C5 higher alcohols from mutant S. cerevisiae were used to alter the metabolic flux of the improved mutant C. crenatium by constructing novel metabolic pathways to improve C4~C5 higher alcohol production. We assessed the fermentation efficiency of bioengineered strains for producing higher alcohols using glucose and duckweed substrate under SSF.
Results and Discussion
Source of mutant genes
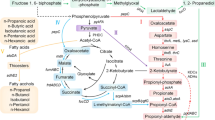
It is well-known that the formation of higher alcohols by yeast is closely related to the metabolism of amino acids including the Ehrlich metabolic pathway21 and the biological synthesis pathway22,23. The metabolic flux distribution and relevant genes involved in the formation of higher alcohols are shown in Fig. 1. Previous studies have shown that metabolic perturbation derived from mutation of genes associated with the biosynthesis and degradation pathways of leucine, isoleucine, and valine in yeast can improve C4~C5 higher alcohol yield based on these two pathways24,25,26. Mutating acetohydroxyacid synthase (AHAS) gene responsible for transforming pyruvic acid into α-acetolactic acid can reduce the generation of the diacetyl, thereby improving the production of higher alcohols. Some reports have demonstrated the mutant strains with increased higher alcohols production can be obtained using some resistance marker such as streptomycin (SM)27,28,29,30, the arginine analogue canavanine31,32,33, and 2-thiazolyl-DL-alanine34. These studies shows that acetohydroxy acid synthase (AHAS) and α-isopropylmalate synthase (IPMS) are key genes in the generation of higher alcohols, and that feedback inhibition takes place between the two genes and their respective metabolite production35,36,37 (Fig. 2). Therefore, through controlling the enzymatic processes between them to block feedback inhibition, it is possible to screen out mutant leucine-deficient strains that generate high 3-methyl-1-butanol production. Auxotrophic strain can improve the production of higher alcohols as a result of the mutation of genes related to branched chain amino acid biosynthesis. For example, mutation of ILV2 gene in valine biosynthesis can lead to improved yields of higher alcohols35,36,37.

The metabolic flux distribution and relevant enzymes involved in the formation of higher alcohols, including amino acid biosynthetic pathways, decarboxylation reduction reaction, and the Ehrlich metabolic pathway (http://www.genome.jp/dbget-bin/www_bget?pathway:sce01210).
CHA1, ILV1: L-serine/L-threonine ammonia-lyase, IRC15: dihydrolipoamide dehydrogenase, LPD1: dihydrolipoyl dehydrogenase, LEU2: 3-isopropylmalate dehydrogenase, ILV1: L-serine/L-threonine ammonia-lyase, ILV2: acetolactate synthase, ILV5: ketol-acid reductoisomerase. ILV3: dihydroxy-acid dehydratase, BAT1: branched-chain-amino-acid transaminase, BAT2: branched-chain amino acid aminotransferase, LEU4: 2-isopropylmalate synthase, LEU1: 3-isopropylmalate dehydratase, IRC15: dihydrolipoamide dehydrogenase, LPD1: dihydrolipoyl dehydrogenase, CAN1: arginine permease, ARO10: phenylpyruvate PDC1:decarboxylase pyruvate decarboxylase 1, PDC2: pyruvate decarboxylase 2, PDC5: pyruvate decarboxylase 5, PDC6: pyruvate decarboxylase 6, ADH1: alcohol dehydrogenase 1, ADH2: alcohol dehydrogenase 2, ADH6: alcohol dehydrogenase 6, SFA1: bifunctional alcohol dehydrogenase/S-(hydroxymethyl)glutathione dehydrogenase, AAD3, AAD 4: Oxidoreductases. AAD6: putative aryl-alcohol dehydrogenase 6, AAD10: putative aryl-alcohol dehydrogenase 10.

Metabolic mechanism of screening auxotrophic mutants of yeast using amino acid analogue via controlling the key enzymes based on the relationship between the formation of higher alcohols and the metabolism of amino acids.
AHAS: acetohydroxyacid synthase, IPMS: α-isopropylmalate synthase.
Learning from these previous studies, here, we used the leucine analogue 4-Aza-dl-leucine dihydrochloride (AZL) as a resistance screening compound to obtain feedback inhibition of AHAS or IPMS mutants with high 3-methyl-1-butanol production (Fig. 2). AZL has been used previously in Bacillus subtilis, Escherichia coli, and Salmonella typhimurium to identify resistant strains in which three enzymes of the leucine, isoleucine, valine metabolic pathway (isopropylmalate isomerase, isopropylmalate dehydrogenase (IPMDH), and isopropylmalate synthase (IPMS)) were inhibited38,39. These studies showed that perturbing the metabolic pathway involving AHAS regulation to obtain auxotrophic strains with some mutant genes is an effective method of generating a high yield of amino acids. And further investigated the ability of mutant strains via fermenting the hydrolysate of duckweed to identity the desirable strain with the highest 3-methyl-1-butanol. A total of 35 colonies grew after three days of incubation. The fermentative results were shown for the investigation of the capability of each colony to produce higher alcohols (Fig. 3). First, to verify the fermentative stability, these auxotrophic strains were cultured consecutively for 10 generations, and then given glucose as substrate to determine the capacity of mutant strains with stability for producing higher alcohols (Fig. 3b). Secondly, these positive auxotrophic strains with stability were used as reinspected strains to assess their capacity for fermenting duckweed hydrolysate (Fig. 3a). Compared with the original strain SC (S. cerevisiae AH109), there were 12 mutant strains possessed fermentative stability, and had significant higher titers of 3-methyl-1-butanol. Therein, the mutant strain NC-11 produced the highest production, approximately 25-fold as compared to the original strain. Additionally, using both glucose as well as hydrosylate demonstrated impressive capacity of the auxotrophic strain NC-11. Therefore, we selected the auxotrophic strain NC-11 as the candidate strain for the next step to survey which possible mutant genes are responsible for the increased biosynthesis of 3-methyl-1-butanol production. We hypothesized that these mutant genes were highly involved in Ehrlich metabolic pathway and/or the biological synthesis pathway, and would contribute to the improvement of higher alcohols in subsequent experiments. Thus supporting the extraction of these mutant genes in order to construct novel metabolic pathways.

Fermentation experiments to determine the ability of each colony to produce higher alcohols.
(Figure 5B) In order to verify the genetic stability of the mutant strains, glucose was used as a fermentation substrate. (Figure 5A) These positive mutant strains were then used as reinspected strains to assay the capacity for fermentation of duckweed hydrolysate. SC: Saccharomyces cerevisiae.
Improvement of expression host
The production of higher alcohols in synthetic microbes relies on exogenous metabolic pathways such as the decarboxylation reduction pathway that converts intermediate metabolites of the amino acid synthesis pathway or Ehrlich metabolic pathway into alcohols. Various α-keto acid intermediate compounds are converted into corresponding fatty alcohols. Therefore, increasing amino acid production can result in the accumulation of precursors, thereby providing more substrate pool for subsequent conversion, and thus improving the final total production of higher alcohols.
Here, the representative auxotrophic strain with the highest yield of amino acid was selected from a total of 35 mutant strains after UWCM via batch fermentation using glucose. The fermentation results of each strain are shown in Fig. 4. The auxotrophic strain C. crenatium MA11C generated much higher levels of the amino acids isoleucine, leucine, and valine (approximate 2-fold) compared to the original strain C. crenatium CICC 20135 (Fig. 4a,b). In particular, the representative auxotrophic strain C. crenatium MA11C produced the highest isoleucine (9.54 g/L), leucine (4.96 g/L), and valine (4.51 g/L) in the least amount of time (96 h). The substrate glucose was not completely consumed by C. crenatium MA11C, but its consumption levels were the lowest for all investigated mutant strains. For C. crenatium MA11C, the residual amount of glucose was up to 5.72 g/L, compared to the C. crenatium CICC 20135, for which the residual glucose reached 8.52 g/L (Fig. 4a,b). The changes in pH during fermentation are shown in Fig. 4(a,b). The largest pH change observed for all investigated mutant strains during fermentation was from C. crenatium MA11C (from 4.42 to 7.0; Fig. 4b). At the end of fermentation, the pH values were above 4.42. The pH during fermentation by the original strain C. crenatium CICC 20135 stayed above 5.3 (Fig. 4a).

These results demonstrate that the residual amount of glucose for the mutant strain was lower than that of the original strain, which suggests that the mutant strains have a higher uptake of glucose. This also helps to explain the improved amino acid production from the auxotrophic strain compared to the original strain. The changes in pH values also reflect the total amino acid titer; when the final pH was lower, the amino acid titer should be higher. These results indicate that high amino acid titer is likely the result of increased precursors (α-ketoacids), and that the efficiency with which the precursor was converted into higher alcohols by C. crenatium MA11C may be higher than that of the original strain C. crenatium CICC 20135. Furthermore, the increased amino acid production of C. crenatium MA11C could be due to mutation of some genes in the amino acid synthesis pathway. The results via batch fermentation repeatedly demonstrated that fermentation of the auxotrophic strain is stable after being sequentially cultured for 10 generations, thus C. crenatium MA11C is suitable for expressing heterologous genes as expression host.
Extraction of mutant genes
As above description, the improvement of higher alcohol production by mutant yeast strains is likely the result of mutation of genes associated with the metabolic pathways of higher alcohol intermediates, resulting in perturbation of metabolic flux via UWCM. The auxotrophic strain NC-11 was found to produce the highest yield of 3-methyl-1-butanol and has fermentation stability. Therefore, the auxotrophic strain was used for subsequent identification of potential genetic mutations. Genes related to the Ehrlich pathway and amino acid biosynthetic and degradation pathways have been implicated in the production of higher alcohols (Fig. 1), and our results showed that genes ILV2, ILV5, BAT2, LEU1, AR010, and ADH6 acquired mutations (Fig. 5).

Regarding mutant genes involved in the formation of higher alcohols.
(Figure 7A): the genes with red show that genes are mutated determined by our results. (Figure 7B): Sequence alignment of amino acids of mutant genes involved in biosynthesis process of higher alcohols. The gene with an asterisk indicates the gene produced mutation. The gene without asterisk indicates that the gene is native gene.
According to this results, mutant genes ILV2*, ILV5*, and LEU1* are responsible for isoleucine, leucine, and valine biosynthetic metabolic pathways. This indicates that the metabolic flux generated a change in the synthesis of 2,3-Dihydroxy-3-methylbutanoate and 2,3-Dihydroxy-3-Methylpentanoate, which are the precursors of 2-oxoisovalerate and 3-Methyl-2-oxopentanoate, respectively (Figs 5 and 6). The LEU1* also gave rise to the metabolic flux from 2-Isopropylmalate to 3-Isopropylmalate. Mutant gene BAT2* is involved in the deamination reaction pathway, indicating that metabolic flux from valine, isobutanol, and leucine to 2-oxoisovalerate, 3-Methyl-2-oxopentanoate and 4-Methyl-2-oxopentanoate, respectively, had changed (Figs 5 and 6). These mutant genes are likely to increase accumulation of intermediate metabolites and precursors of higher alcohols such as the intermediates including isopropylmalate, 2-oxobutanoate, 2-oxoisocaproate, 2-isopropyl-2-oxosuccinate, and precursors including 3-methyl-2-oxopentanoate of 2-methyl-1-butanol, 4-methyl-2-oxopentanoate of 3-methyl-1-butanol and 2-oxoisovalerate of isobutanol. These results indicate that this genes mutation led to the abnormal metabolic flux of amino acid biosynthesis and Ehrlich pathway. Therefore, this increase in amino acid production also account for increase of 3-methyl-1-butanol production in auxotrophic strain NC-11. In addition, the gene dihydrolipoyl dehydrogenase, which is responsible for the degradation of valine, isoleucine, and leucine, and the genes ARO10 and ADH6* involved in the decarboxylation reduction pathway, were also investigated (Figs 1 and 5). It was concluded that the reason that the strain NC-11 can produce high yields of 3-methyl-1-butanol may be due to the change of metabolic flux from these mutant genes working synergistically. These mutant genes were extracted and used to construct novel metabolic pathways.

The flowsheet of experiments following the methodology illustrated for screening exogenous mutant enzymes and expression host via undirected whole-cell mutagenesis (UWCM) in vivo and fermentation processes.
Higher alcohols from bioengineered strains
Engineering C. crenatium to express native genes from wild-type S. cerevisiae to produce higher alcohols have been investigated6. Although the yield was low, they have demonstrated the potential capability of this host to be useful in improving production of higher alcohols. In order to further improve the fermentation efficiency of C. crenatium, compatibility of two expression vectors in a host was first demonstrated. The gene pBL1 from expression vector pXMJ19 was inserted into the expression vector pSVT29 to construct a new expression plasmid pTVpBL. The vectors pTVpBL and PEC-XK99E were compatible and stable in the expression host. Secondly, novel metabolic pathways were constructed using mutant genes. Thirdly, the improved mutant C. crenatium was used as expression host harboring novel metabolic pathways. A schematic representation of the novel metabolic pathways is shown in Fig. 7.

The schematic plot of designing novel metabolic pathways responsible for the improvement of higher alcohol production.
Different colors rectangular frames represent different metabolic pathways. The gene with an asterisk indicats the gene produced mutation. The gene without asterisk indicats the gene is native gene. The novel metabolic pathways for accumulating precursor products is illustrated with the following pathways, labeled MEp I (LEU2-ILV2*-ILV5*), MepII (ILV2*-ILV5*-ILV3), MepIII (BAT2*), MepIV (LEU4-LEU1*), MepV (LEU1*-LEU2) and MepVI(BAT2*-LEU4-LEU1*), Metabolic pathway I, Ia, Ib including LEU2, ILV2*, ILV5*; Metabolic pathway II, IIa, IIb including ILV2*, ILV5*, ILV3; Metabolic pathway III including BAT2*; Metabolic pathway IV, IVa including LEU4, LEU1*; Metabolic pathway V, Va including LEU1*, LEU2; Metabolic pathway VI, VIa, VIb including BAT2*, LEU4, LEU1*. Another metabolic pathway responsible for the decarboxylation reduction reaction, and MepDRVII (kivd-ADH6*) Metabolic pathway VIIincluding kivd, ADH6*. LEU2: 3-isopropylmalate dehydrogenase, ILV1: L-serine/L-threonine ammonia-lyase, ILV2*: mutant acetolactate synthase, ILV5*: mutant ketol-acid reductoisomerase, ILV3: dihydroxy-acid dehydratase, BAT2*: branched-chain amino acid aminotransferase, LEU4: 2-isopropylmalate synthase, LEU1*: mutant 3-isopropylmalate dehydratase, kivd: alpha-ketoisovalerate decarboxylase, ADH6*: mutant alcohol dehydrogenase 6.
In order to guarantee the metabolic capacity of these engineered strains for producing higher alcohols, we verified whether these mutant genes involved branched-chain amino acid synthesis were overexpressed in C. crenatium MA11C. Expression levels of all mutant genes were confirmed by RT-PCR, and the results were further analyzed with a semi-quantitative method (Fig. 8). Our results demonstrated that the induced genes corresponding to respective metabolic pathways have been successfully expressed and that the expression level of these recombinant genes remained relatively different under similar cultivation conditions.

Gene expression of all recombinant genes for each metabolic pathway in the production host was analyzed using semi-quantitative RT-PCR.
Semi-quantitative RT-PCR analysis: expression profiles of all recombinant genes were evaluated using Gel-Pro analyzer 4.0 software. Labeled MEp I (LEU2-ILV2*-ILV5*), MepII (ILV2*-ILV5*-ILV3), MepIII (BAT2*), MepIV (LEU4-LEU1*), MepV (LEU1*-LEU2) and MepVI(BAT2*-LEU4-LEU1*).
Fermentation was achieved for all novel metabolic pathways using pure glucose (Fig. 9) and duckweed via the SSF procedure (Fig. 10). To construct and overexpress two metabolic pathways Mep I and Mep VII, the highest yield of 2-methyl-1-butanol at 3476.52 mg/L was obtained using glucose as the fermentation substrate. In addition, the by-products isobutanol at 735.18 mg/L and 3-methey-1-butanol at 403.53 mg/L were obtained in 96 h (Fig. 9, Table 1). Similarly, overexpressing the two metabolic pathways simultaneously led to the highest yield of 2-methyl-1-butanol (3071.5 mg/L) and byproducts of isobutanol (605.17 mg/L) and 3-methey-1-butanol (313.73 mg/L) using the duckweed via SSF. Our results showed that a higher titer can be obtained using pure glucose compared to using duckweed. The parameters of total alcohols, productivity, and total yield were higher using glucose than those using duckweed (Table 1). However, to overexpress other metabolic pathways Mep Ia + Mep VII and Mep Ib + Mep VII, the yield was clearly lower than that of Mep Iand Mep VII when using either glucose or duckweed (Figs 9 and 10). Only 288.65 mg/L 2-methyl-1-butanol, 248.31 mg/L isobutanol, and 54.208 mg/L 3-methey-1-butanol were obtained from Mep Ia + Mep VII using glucose and approximately 1588.07 mg/L 2-methyl-1-butanol, 397.46 mg/L isobutanol, and 144.4 mg/L 3-methey-1-butanol were produced from Mep Ib + Mep VII using duckweed via SSF.

Fermentation experiment to produce higher alcohols for all constructed metabolic pathways using pure glucose.
labeled MEpIa (LEU2), MEpIb (LEU2-ILV2*), MEpI (LEU2-ILV2*-ILV5*); MepIIa(ILV2*), MepIIb(ILV2*-ILV5*), MepII (ILV2*-ILV5*-ILV3); MepIII (BAT2*); MepIVa(LEU4), MepIV (LEU4-LEU1*); MepVa (LEU1*), MepV (LEU1*-LEU2); MepVIa ((BAT2*), MepVIb (BAT2*-LEU4); MepVI (BAT2*-LEU4-LEU1*); MepVII (kivd-ADH6*).

Fermentation experiment to produce higher alcohols using duckweed via SSF procedure for the most efficient mutant strain.
labeled MEp I (LEU2-ILV2*-ILV5*), MepII (ILV2*-ILV5*-ILV3), MepIII (BAT2*), MepIV (LEU4-LEU1*), MepV (LEU1*-LEU2) and MepVI(BAT2*-LEU4-LEU1*), MepVII(kivd-ADH6*).
Maximum isobutanol titer 6207.15 mg/L, and productivity 76.06 mg/L/h were achieved with glucose. Using duckweed, the resulting isobutanol level was 5607.15 mg/L and productivity was 76.06 mg/L/h, which resulted from synergistically overexpressing two metabolic pathways Mep II + Mep VII. In addition, other byproducts such as 2-methyl-1-butanol (735.46 and 705.4 mg/L), and 3-methey-1-butanol (359.6 and 289.16 mg/L), can also be obtained from glucose and duckweed, respectively, and thus lead to the highest total alcohols solution 7302.21 and 6601.7 mg/L (Figs 9 and 10, Table 1), with a productivity of 76.06, 68.77 mg/L/h and total yield of 121.7 and 66.01 mg/g for glucose and duckweed, respectively (Table 1). In addition, the other metabolic pathways Mep IIa + Mep VII and Mep IIb + Mep VII can produce 773.68 and 2881.92 mg/L isobutanol, 240.8 and 393.88 mg/L 2-methyl-1-butanol, and 139.77 and 316.45 mg/L 3-methyl-1-butanol using glucose.
To summarize the results of above two novel metabolic pathways, owing to the genes ilv2* and ilv5* are closely related to the biosynthesis of isoleucine (Figs 1 and 5), it showed that the two genes catalyze not only the conversion of ketobutyrate and pyruvate, but also that of pyruvate and pyruvate to make ketomethyl valerate and ketoiso valerate, respectively (Fig. 1). The results showed the improvement of higher alcohols can be confirmed using the mutant genes ilv2* and ilv5* to construct novel metabolic pathways to constitute Mep I and Mep II. We demonstrated that increasing the pathway flux of valine allows for the accumulation of leucine, and thus improves 2-methyl-1-butanol reached to the highest yield 3476.52 mg/L. The results also showed that reinforcing the flux of pyruvate by redirecting the carbon flux via expressing ilv2*, ilv5*, and ilv3 (Mep II) in the L-valine biosynthesis pathway can lead to significant improvement of isobutanol, reaching an increased yield 6207.15 mg/L compared to previous studies which did not utilize gene editing techniques.
Isobutanol yields were also improved by introduction of the mutant gene BAT2* via constructing metabolic pathway Mep III + Mep VII, reaching 462.76 and 412.26 mg/L isobutanol, 236.08 and 212.18 mg/L2-methyl-1-butanol and 135.02 and 99.12 mg/L from glucose and duckweed via SSF, respectively (Figs 9 and 10, Table 1). Furthermore, overexpressing the mutant gene BAT2* resulted in a higher relative proportion of C4 isobutanol than the other two C5 higher alcohols analyzed (Fig. 5). We conclude that the mutant BAT2* obtained by UWCM elicits an effect on the three branched-chain amino acids, and ultimately improved higher alcohols production. Our results revealed that mutant BAT2* supplied increased levels of precursors to valine, leucine, and isoleucine. Although BAT2 is the only transaminase of relevance in the transamination step within the biosynthesis of the three branched-chain amino acids valine, leucine and isoleucine, the results also demonstrated a greater impact on isobutanol yield than on yields of two other alcohols. Therefore we infer that the mutant gene BAT2* specifically plays an important role in the production of isobutanol.
In a similar manner, co-expression of other metabolic pathways Mep IV + Mep VII and Mep V + Mep VII, yielded different results as presented in Table 5. Other information can be seen in Fig. 9. For example, 385.39 and 491.57 mg/L isobutanol, 176.96 and 326.4 mg/L 2-methyl-1-butanol, and 727.52 and 817.97 mg/L 3-methey-1-butanol can be generated from Mep IVa + Mep VII and Mep Va + Mep VII respectively using glucose (Fig. 9). The two metabolic pathways Mep IV and Mep V have the same mutant gene LEU1*. We noted changes in the production of higher alcohols resulting from the two metabolic pathways. The yields of all higher alcohols assayed were significantly improved by Mep V as compared to those of Mep IV. The results revealed that overexpression of the LEU2 gene can produce much higher alcohols than overexpression of LEU4. This suggests that LEU2 has stronger catalytic activity for regulating metabolic flux to enhance 3-methey-1-butanol production when simultaneously overexpressed with related genes via metabolic engineering.
To construct the metabolic pathways Mep VI + Mep VII, the highest yields of 582.02 and 502.92 mg/L isobutanol, 478.47 and 503.37 mg/L 2-methyl-1-butanol, and 1115.34 and 1415.73 mg/L 3-methyl-1-butanol were obtained from pure glucose and duckweed, respectively (Fig. 10, Table 1). Similarly, 432.58 and 660.67 mg/L isobutanol, 216.29 and 452.24 mg/L 2-methyl-1-butanol, and 530.89 and 1014.6 mg/L 3-methyl-1-butanol were generated from two metabolic pathways Mep VIa + Mep VII and Mep VIb + Mep VII using pure glucose. The increased outputs from glucose metabolism led to increases in other outcomes, including, higher total alcohol solution 2501.12 vs 2096.7 mg/L, productivity 26.05 vs 21.84 (mg/L/h), total yield 41.68 vs 20.97 (mg/g) from glucose and duckweed, respectively (Table 1). Out results showed that the yield can be improved by adding two genes LEU4 and LEU1*.
The genes LEU4 and LEU2 and mutant LEU1* are involved in leucine synthesis. Specifically, LEU4 and LEU1* are responsible for chain elongation from 2-Oxoisovalerate of the final step of L-valine synthesis to the initial step of leucine formation (Fig. 1). Therefore the catalytic activity of the LEU1* gene was confirmed by constructing MepIVand Mep, resulting in increasing leucine accumulation. The results revealed an increased enzyme level for the mutant LEU1*, which also increased metabolic flux to improve 3-methyl-1-butanol production.
These results for surveying novel metabolic pathways demonstrated that the production of higher alcohols can be greatly improved when multiple mutant genes from other species are introduced via UWCM in vivo, compared to the use of only native genes. The highest higher alcohols production from our experiment is higher than previous reports. For example, the highest isobutanol 151 mg/L can be obtained using S. cerevisiae as expression host viaby overexpression of 2-ketoisovalerate decarboxylase and valine biosynthetic enzymes40,41,42. The highest isobutanol, 2-methyl-1-butanol, 3-methyl-1-butanol from C. crenatum without improvement in cell properties and harbouring mutant genes only reach to 1264.63 mg/L, 1026.61 mg/L, 748.35 mg/L43. Furthermore, we showed that production of different higher alcohols can be successfully achieved via the construction of novel metabolic pathways using mutant genes from two metabolic pathways: the Ehrlich metabolic pathway and the biological synthesis pathway. In addition, these bioengineered strains of C. crenatum can successfully ferment duckweed via the SSF scheme to produce higher alcohols. Taking a comprehensive view of biofuel generation, such a holistic integration across multiple metabolic pathways to generate bioengineered strains that increased yields of higher alcohols is a useful approach for enhancing biofuel production using lignocellulose.
The above fermentative results indicate that the improvement of higher alcohols production is because, on the one hand, the metabolic capability of expression host itself has been improved via UWCM when used improved auxotrophic strain as expression host instead of wild-type strain. Regulating metabolic mechanisms by introducing heterogenous pathways produce generally limited production in the native expression host. The synthesizing capacity of the expression host is important but native hosts are usually not fully developed before using gene knockout methods. The improved mutant strain C. crenatium MA11C demonstrated a relatively high yield of precursor molecules, leading to increased higher alcohols production. The results can be indirectly demonstrated using amino acid yield because the higher alcohol intermediates and amino acids share the same precursor (Fig. 1). The C. crenatium MA11C produced the highest yields of isoleucine, leucine and valine, providing the first confirmation of improving higher alcohols yield. The results showed that metabolic flux can be redirected to facilitate higher alcohol production via UWCM followed by genes editing biotechnology resulting in the inactivation of competing pathways for reducing or eliminating unwanted byproducts. The amount of metabolic precursors entering competing metabolic pathways can be reduced via controlling the appropriate target products by UWCM, and thus can improve the desired production. We are continuing to work on the bioengineered strain and optimizing the process conditions to enhance higher alcohols yields using follow-up gene editing technology, to remove any bottlenecks from the pathway and further reduce by-products.
On the other hand, improvement in the production of higher alcohols may be achieved by novel metabolic pathways including multiple gene mutations. In addition to allowing the accumulation of higher alcohol precursors in host, the catalytic ability of genes is also a very important contributing factor in maximizing higher alcohol yield. Many previous studies of introduced genes generally have analyzed the effects of native genes without mutations. Limited research has been conducted in which mutant genes are generated through directed evolution technology in vitro to improve enzyme catalytic activity, and the target is usually a single gene. For example, the mutated leuA was expressed in an E. coli strain previously developed for increased threonine production. The yield of longer-chain alcohol 1-hexanol was improved to 146 mg/L44. To increase efficiency, repressive regulation of the β-oxidation cycle in the presence of glucose was avoided by using a cAMP-independent mutant (crp*) to replace the native crp gene45,46. However, the use of a single gene with high catalytic activity from evolution in vitro does not always result in a high final yield of higher alcohols as this may lead to an imbalance of metabolic flux. Furthermore, overexpression of a gene alone is generally insufficient to produce the desired final product when introduced a novel metabolic pathway. Additionally, overexpressing native genes may not always be maximally efficient for improving higher alcohol production because multiple metabolic pathways often compete for a common intermediate, leading to metabolic imbalance, which is a common problem in metabolic engineering. Therefore, UWCM in vivo may be an effective strategy to obtain multiple mutant genes with a synergistic effect to avoid metabolic flux imbalance.
Here, we confirmed that improvement of higher alcohols production is possible by obtaining multiple synergistic mutations associated with biosynthesis pathway by UWCM. The resulting increase in C4~C5 alcohol production by overexpressing combined multiple mutant genes might be explained by the fact that the catalytic efficiency of mutant genes is enhanced compared to native genes. Synergistic overexpression of multiple mutant genes led to a higher production of a major alcohol, and meanwhile improved the overall production of other by-products more than native genes, suggesting that the conditions in which abundant intermediate metabolites are available allow for the increase in desired end product. The results showed that a proper integration of multiple mutant genes is an attractive proposition for improving higher alcohols produce.
In conclusion, the above two aspects should be taken fully into account together to produce a high-efficiency strategy capable of improving production of higher alcohols. To overcome the obstacles to improve higher alcohols production, a synthetic multifactorial strategy is necessary that incorporates the exploitation of the synthetic capacity of the expression host with a high rate of accumulation for precursors of higher alcohols, as well as balancing the changes in metabolic flux which result from harboring multiple exogenous mutant genes. UWCM in vivo may be an alternative solution. We conclude that this combinatorial synthetic approach for improving upon existing pathways is feasible and will facilitate further development of processes to produce high-yield compounds from renewable resources through engineering strategies of genes editing biotechnology.
Materials and Methods
Experimental methodology
In this study, we sought to demonstrate the feasibility for improving C4~C5 higher biofuels production via engineering C. crenatum overproduction of amino acid. The methodology is illustrated in the flowchart of the experimental process (Fig. 6) including the processes of mutating wild-type C. crenatium and S. cerevisiae, identifying mutant genes, constructing novel metabolic pathways47,48, and fermentation experiment. First, we conducted the UWCM in vivo using the wild-type strains of S. cerevisiae and C. crenatum to generate auxotrophic strains with high metabolic capacity. Next, we identified mutant genes from auxotrophic strain of S. cerevisiae, and optimize metabolic flux of bioengineered strains of auxotrophic C. crenatium via importing a series of metabolic pathways using mutant genes. Finally, fermentation experiment were conducted using duckweed substrate under SSF. Each experiment was conducted in triplicate in a 150-mL triangular flask, unless otherwise indicated.
Rejuvenation and propagation of microorganisms
Culture conditions for the growth, rescue, rejuvenation and seeding of the original strains are as follows. The original bacterium C. crenatum CICC 20135 was purchased from the China Center of Industrial Culture Collection (CICC, Beijing, China), and rejuvenated and propagated in a liquid nutrition gravy medium (LMGM) containing 5.0 g peptone, 3.0 g beef extract, 5.0 g NaCl, 20 g glucose, 1 L ddH2O, pH 7.0 at 30 °C in a rotary shaker for 72 h. Cultures of Lactococcus lactis cremoris CICC 1605 were obtained from the CICC and cultivated in liquid MRS medium containing 10 g peptone, 3.0 g beef extract, 3.0 g yeast extract, 2 g K2HPO4, 2 g citric acid diamine, 2 g sodium acetate, 20 g glucose, 0.6 g MgSO4·7H2O, 0.25 g MnSO4·4H2O, 1 L ddH2O, pH 6.2 at 32 °C in a rotary shaker for 72 h. Cultures of original S. cerevisiae AH109 were obtained from Clontech Laboratories, Inc. (Beijing, China) and propagated at 30 °C in liquid TGY medium in a rotary shaker for 72 h.
Analysis of duckweed on basic composition
The wild duckweed (Landoltia punctata) was randomly gathered from the surface of wild ponds that were left uncultivated for many years in Huilong Town located in Chengdu, China. It is necessary to determine the composition of duckweed in order to provide a reference value to compare with other studies. First, fresh plants were dried at 60 °C in a drying oven (DHP9050A, Shanghai, China), then pulverized into dried powder with a pulverizer (FS100S-3, Guangzhou, China). Duckweed powder was further hydrolyzed using H2SO4 at 1% (v/v) concentration. The starch content was assessed roughly according to the total sugar content (starch content = glucose content × 0.91)49,50. The total crude protein content of duckweed was measured using the Kjeldahl Method (CP = Kj N × 6.25)51,52. Cellulose content was assessed roughly using spectrophotometry with the absorbance at 620 nm53,54,55. Processing and testing of resulting materials were performed as previously described. The content of lignin was determined using acetyl bromide according to standard methods56,57. Trace elements in duckweed were determined as follows: samples were washed with deionized water, dried at 80 °C, milled to powder, and finally digested using a wet digestion method58,59. The elemental composition in the digested solution was analyzed using atomic absorption spectrometry (Z-2300, Hitachi, Japan). The resulting components of duckweed are presented in Table 2.
Acquisition and analysis of mutant genes
Mutagenesis yeast using UWCM
The original strain S. cerevisiae AH109 was propagated in YPDA medium after inoculation with seeding liquid in a constant temperature oscillation incubator at 30 °C, 200 rpm, until the growth solution concentration reached OD600 = 1.5. Growth solution was separated into 4 portions with equal distribution (each sample was 15 mL), and then centrifuged independently (3000 rpm for 1 minute). After centrifugation the supernatant was discarded and the cultured yeast cells were collect. Each sample was then pretreated as follows: Treatment 1 (NJ): 1.5% (v/v) methanol solution was added and kept for 10 min to shock cells; Treatment 2 (NS): cells were soaked in a solution containing 5% methanol and 0.2% polysorbate 80 for 15 min; Treatment 3 (NC): 10% sorbitol was added and incubated for 20 mins; Treatment 4 (PS), yeast cells were washed with 5 mL distilled water, then centrifuged at 3000 rpm for 5 mins, and collected. Yeast cells were resuspended in 5 mL solution containing 5% (v/v) glycerin and 10% (v/v) dimethyl sulfoxide (DMSO), and preserved in a −70 °C freezer overnight for subsequent use for further mutagenicity experiment.
Yeast cells were thawed and the pH was adjusted to 6.0 with phosphate buffer solution. The chemical mutagen 1-methyl-3-nitro-1- nitroso-guanidin (NTG) was dissolved in 1 mL acetone and added to each sample (5 mL). The final NTG concentration was adjusted to 500 μg/mL, and then cultured in a constant temperature incubator shaker at 30 °C for 15 min. Mutated cells were then collected by centrifugation and washed three times with distilled. 200 μL of resuspended mutated cells was plated on Yeast Nitrogen Base (YNB) solid medium containing 20 mg/mL 4-Aza-dl-leucine dihydrochloride (AZL) (Sigma Aldrich, USA), and then placed in 30 °C constant temperature incubator to culture approximately three days until the appearance of colonies. Finally, a fermentation experiment was conducted using fermented glucose and hydrolysate of duckweed to identify auxotrophic strains with the highest resulting C4~C5 alcohol concentration.
To obtain mutant yeast with high higher alcohol production
In order to identity the mutant strain with the highest efficiency for producing C4~C5 higher alcohols, we evaluated the production capacity of mutant yeast strains by conducting fermentation experiments using different fermentation substrates: pure glucose and diluted acid hydrolysates of duckweed.
For fermentation using glucose, 60 g/L glucose solution containing 5 g/L yeast extract was prepared, and a Trace Metals Mix A5 (0.222 g ZnSO4·7H2O g/L, 0.39 g/L Na2MoO4 · 2H2O, 0.079 g/L CuSO4 · 5H2O, 0.0494 g/L Co(NO3)2 · 6H2O, 5 g/L NaCl, 20 g/L (NH4)2SO4, 1.5 g/L KH2PO4, 0.3 g/L MgSO4 · 7H2O, 0.05 g/L FeSO4 · 7H2O, 0.01 g/L MnSO4 · H2O, 1.0 g/L CH3COONH) was added.
For fermentation using diluted acid duckweed hydrolysate, duckweed substrates were first pretreated using established methods of acid hydrolysis13. The products of hydrolysis were then fermented by mutant strains of S. cerevisiae AH109. The initial total glucose content of diluted acid duckweed hydrolysate was adjusted to 60 ± 3.01 g/L (before fermentation), and the pH was adjusted to 6.8 using 1% dilute phosphoric acid and 0.1% Ca(OH)2. The acid liquefied hydrolysates of duckweed were then used as the substrate for further fermentation verification experiments. The resulting carbohydrate composition after pretreatment at 0 h of fermentation is presented in Table 2.
The diluted acid hydrolysate of duckweed consisted of 20 mL modified M9 medium (2 g (NH4)2PO4, 2 g KH2PO4, 1 g K2HPO4, 1 g NH4Cl, 0.5 g NaCl, 0.5 mM MgSO4, 1 mM CaCl2, 20 mg vitamin B1, and 2 mg biotin per L of water) containing 5 g/L yeast extract, and Trace Metals Mix solution (2 g H3BO3, 2.1 g MnCl2·4H2O, 0.3 g ZnSO4·7H2O, 0.002 g MnSO4 2.5 g Na2MoO4·2H2O, 0.05 g CuSO4·5H2O, 21.2 mg Co(NO3)2·6H2O, and 0.05 g FeSO4 per L of water) in a 150-mL triangular flask.
The substrates were fermented under aerobic conditions. Briefly, 2 mL rejuvenated seeding solution from liquid medium was inoculated into 50 mL of the substrates in a 150 mL triangular flask. The antibiotics ampicillin (100 μg/mL), chloramphenicol (35 μg/mL), and kanamycin (50 μg/mL) were added into all fermentation substrates, and kept at 30 °C to ferment with shaking at 200 rpm for 96 h in a constant-temperature oscillation incubator.
Identifying and extracting mutant genes
All genes were cloned into the vector pEASY-T3 cloning Vector, which contains a LacZ gene suitable for TA-cloning (TransGen Biotech Company, CT301-01, Beijing, China). The vector was propagated in Trans1-T1 Phage Resistant Chemically Competent Cells with genotype F-φ80(lacZ)ΔM15ΔlacX74hsdR(rk-, mk+ )ΔrecA1398endA1tonA (TransGen Biotech CD501-01).
All oligonucleotides were obtained from BGI (Beijing, China) with standard purification. Primer sequences were designed with vector NTI software (Informax Vector NTI Suite 11.5) and are listed in Table 3. DNA sequencing to confirm cloning products was performed by Quintara Biosciences.
Genes were amplified using their respective primers (Table 3) from the template of the mutant strain of S. cerevisiae AH109 that demonstrated the highest yielding alcohol production using the pEASY-Blunt Simple Cloning Kit including high fidelity polymerase (CB111-01) and TransStart FastPfu DNA Polymerase (AP221-11). The PCR products were linked to a pEASY-T3 Cloning Vector (TransGen Biotech Company) with T4 DNA ligase (TransGen Biotech Company), and the sequences were detected by BGI Tech. Company (Beijing, China).
Acquisition of the expression host
Mutagenesis C. crenatum using UWCM
C. crenatum was grown up to exponential phase, the cells were centrifuged and collected at 4 °C, placed on an ice bath for 30 min, cleaned with sterile water, supplemented with 20% glycerin, and then frozen overnight. The preparation of competent cells for freezing C. crenatum was conducted according to the protocols mentioned in the Handbook of Corynebacterium glutamicum60. Wild-type C. crenatum competenct cells were collected by centrifugation at 6000 × g for 10 min at 4 °C. The supernatant was discarded, and the competent cell pellet was resuspended in 20 mL fresh LMGM medium by gentle pipetting. NTG was then added at a final concentration of 200 μg/mL. The mixture was incubated for 20 min after moderate mixing, followed by centrifugation to collect the cells. The NTG-treated cells were washed three times with PB buffer to remove residual NTG, then resuspended in 10 mL fresh LMGM medium, and incubated at 28 °C for 24 h. The NTG-treated cell suspension was diluted and plated onto LMGM solid medium. After colonies had appeared on the plate, mutants were selected to identify which auxotrophic strain had the highest isoleucine yield via fermentation experiments.
Identifing and obtaining the expression host
The expression host was determined to be the auxotrophic strain with the highest isoleucine titer identified via batch fermentation experiments. To prepare the fermentation inoculum for batch cultivation of mutant strains, we prepared a 100 mL TGYM medium containing 20 g/L glucose, 5 g/L yeast extract, 3 g/L ammonium acetate, 1 g/L sodium chloride, 1 g/L KH2PO4, 1 g/L K2HPO4, 0.2 g/L MgSO4, 0.02 g/L MnSO4·7H2O, and 0.02 g/L FeSO4·7H2O and autoclaved it at 115 °C for 20 min and cooled to 30 °C. Mutant monoclonal colonies that appeared on the plate were selected out using sterile toothpicks, transferred to sterile bottles containing TGYM medium, and cultured in a constant temperature oscillation incubator at 30 °C until the optical density (OD 600) reached 1.6.
Batch cultures were carried out for isoleucine production tests. The pH of the fermentation substrates was adjusted to 6.8 with 1% NaOH following batch culturing, which was conducted without pH control. The substrate concentration for each batch culture example was 6 0 g/L glucose, 5 g/L yeast extract, and 10 g/L peptone in a 250-mL glass anaerobic bottle (Haimen Huakai, Haimen, China) sealed with parafilm. The substrate solutions were sterilized at 115 °C for 20 min and cooled to room temperature. Subsequently, 5 mL mixture solution containing a combination of P2 trace elements (50 g/L KH2PO4, 50 g/L K2HPO4, and 220 g/L CH3COONH4), vitamins (0.1 g/L thiamin, and 0.001 g/L biotin), and minerals (20 g/L MgSO4·7H2O, 1 g/L MnSO4·H2O, 1 g/L FeSO4·7H2O, and 1 g/L NaCl) was filtered for sterilization (0.22-μm Millipore filter) and added to each bottle. The bottles were then inoculated with 0.5 mL fermentation inoculum (OD600 = 1.6), and fermented in a constant temperature oscillation incubator at 30 °C for 3 days.
Construction of metabolic pathways and engineering C. crenatum
The auxotrophic strain of C. crenatum CICC 20135 with the highest isoleucine titers was selected as expression host strain. All strains and plasmids used in the study are listed in Table 4. The plasmids are physically available from Addgene (http://www.addgene.org). The genes were amplified from the template of mutant strain of S. cerevisiae AH109 for constructing metabolic pathways using their respective primers (Table 5).
All restriction endonucleases were purchased from NEB (Shanghai, China) and T4 DNA ligase (EL0334) was supplied by MBI Fermentas (Chengdu, Beijing). All plasmids were propagated using Trans1-T1 Phage Resistant Chemically Competent Cells (TransGen Biotech Company, CD501-01, Beijing, China). The Trans1-T1 bacterial strains were cultivated in LB medium, and grown at 37 °C in a rotary shaker for 4 h. All cultures of the bioengineered strains of E. coli and mutant strains of C. crenatum CICC 20135 were then induced with 2 mM isopropyl-β-D-thiogalactoside (IPTG) and grown at 30 °C for 18 h. Antibiotics (ampicillin, 100 μg/mL; chloramphenicol, 35 μg/mL; kanamycin, 50 μg/mL) were also added when needed. The bioengineered strains were reproduced in a rotary shaker under the following conditions: E. coli in LB medium at 37 °C for 12 h, and C. crenatum in a nutrition gravy medium at 30 °C for 48 h, and moved into 4 °C to terminate the reaction.
We constructed new metabolic pathways responsible for the accumulation of higher alcohol intermediates (Fig. 7). They are designated metabolic pathway I, Ia, Ib including gene LEU2, mutant ILV2(ILV2*), and mutant ILV5(ILV5*); metabolic pathway II, IIa, IIb including gene mutant ILV2*, mutant ILV5*, and ILV3; metabolic pathway III including mutant gene BAT2(BAT2*); metabolic pathway IV, IVa including genes LEU4, and mutant LEU1(LEU1*); and metabolic pathway V, Va including gene mutant LEU1* and LEU2; metabolic pathway VI, VIa and VIb including gene mutant BAT2*, LEU4, and LEU1*, which were constructed using genes from auxotrophic strain S. cerevisiae AH109. The metabolic pathway VII responsible for oxidation-reduction reactions including genes Kivd and mutant ADH6(ADH6*) was constructed using the Kivd gene from L. lactis cremoris CICC1605 and the ADH6* gene from auxotrophic strain S. cerevisiae AH109.
The genes LEU2, ILV2*, and ILV5* of metabolic pathway I, Ia, and Ib were amplified with the primer pairs A1L2, A2I2, and A3I5, respectively. The genes ILV2*, ILV5* and ILV3 of metabolic pathwayII, IIa, and IIb were amplified with the primer pairs B1I2, B2I5, and B3I3, respectively. The gene BAT2* of metabolic pathway III was amplified with the primer pair CBA. The genes LEU4 and LEU1* of metabolic pathway IV and IVa were amplified with the primer pairs D1L4 and D2L1. The genes LEU1* and LEU2 of metabolic pathway V and Va were amplified with the primer pair E1L1 and E2L2. The genes BAT2*, LEU4, and LEU1* of metabolic pathway VI, VIa, and VIb were amplified with the primer pairs F1BA, F2LE4, and F3LE1 and finally the gene pBL1was amplified with the primer pair pBLG1. The genes Kivd and ADH6* of metabolic pathway VII were amplified with the primer pairs KiK1 and ADH1, respectively.
The plasmids PEC-XK99E and pSTV29 were prepared as the standard expression vectors for constructing metabolic pathways. To construct the following expression vectors pTVpBL, PEC-KA6, PTVpBLAL, pTVpBLALI, pTVpBLALII, pTVpBLBI, pTVpBLBII, pTVpBLBIII, pTVpBLCB, pTVpBLDL, pTVpBLDLL, pTVpBLEL, pTVpBLELL, pTVpBLFB, pTVpBLFBL, and pTVpBLFBLL, the construction process was manipulated using the following instructions. First, the gene pBL1 from the standard expression vector pXMJ19 was inserted into the standard expression vector pSVT29 using restriction enzymes SacII and ClaI to construct a new expression plasmid pTVpBL. The in-series genes Kivd and ADH6* of metabolic pathway VII were inserted into standard expression vector PEC-XK99E using restriction enzymes PstI, XbaI, and KpnI to construct expression plasmid PEC-KA6. The in-series genes LEU2-ILV2*-ILV5* of metabolic pathway I, Ia, and Ib were inserted into the new vector pTVpBL using restriction enzymes SphI, SalI, BamHI, SacI, and T4 ligase to construct plasmids pTVpBLAL, pTVpBLALI, and pTVpBLALII including different expression genes. The in-series genes ILV2*-ILV5*-ILV3 of metabolic pathway II, IIa, and IIb were inserted into vector pTVpBL using restriction enzymes PstI, SalI, BamHI, SacI and T4 ligase to construct expression plasmids pTVpBLBI, pTVpBLBII, and pTVpBLBIII responsible for expressing different genes, respectively. The gene BAT2* of metabolic pathway III was inserted into the vector pTVpBL using restriction enzymes SbfI, BamHI, and T4 ligase to construct plasmid pTVpBLCB. The in-series genes LEU4-LEU1* of metabolic pathway IV and IVa were inserted into the vector pTVpBL using restriction enzymes SbfI, XmaI, SacI, and T4 ligase to construct plasmids pTVpBLDL and pTVpBLDLL in charge of different catalytic reactions. The in-series genes LEU1*-LEU2 of metabolic pathway V and Va were inserted into the vector pTVpBL using restriction enzymes SbfI, BamHI, SphI, and T4 ligase to construct plasmids pTVpBLEL and pTVpBLELL. The in-series genes BAT2*, LEU4, and LEU1* of metabolic pathway VI, VIa, and VIb were inserted into the vector pTVpBL using restriction enzymes SbfI, BamHI, XmaI, SacI, SphI, and T4 ligase to construct plasmids pTVpBLFB, pTVpBLFBL, and pTVpBLFBLL, which are responsible for different metabolic reactions. The ribosome binding site (RBS) sequence was inserted into 6−8 nucleotides upstream of each structural gene to facilitate mRNA translation.
Competent cells of mutant strain C. crenatum were prepared as previously described in the Handbook of C. glutamicum60. All construction plasmids were introduced into mutant strains of C. crenatum using electroporation61,62. Electroporation was conducted according to the following manipulative conditions 100 Ω, 50 μF, 2.2 kV, and 8 ms using the Gene Pulser Xcell Microbial System165-2662 (BIO-RAD, Chengdu)..
Gene expression analysis
Gene expression of all recombinant genes for each metabolic pathway in the production host was analyzed using semi-quantitative RT-PCR. RNA extraction and cDNA synthesis were performed according to the operation manual of TransScript First-Strand cDNA Synthesis SuperMix kit of the manufacturer (TransGen Biotech, Beijing, China). For semi-quantitative RT-PCR analysis, expression profiles of all recombinant genes were evaluated based on semi-quantified analysis using Gel-Pro analyzer 4.0 software (Media Cybernetics, Silver Spring, MD, USA), and the calculated relative expression values for all exogenous genes were calculated using integral optical density (IOD).
Batch fermentation using bioengineered strains
Fermentation using glucose
Glucose (60 g/L) was used as a fermentation substrate. Glucose was sterilized at 115 °C for 20 h, then cooled to room temperature for use in fermentation assays. Batch fermentation cultures were carried out under aerobic conditions, and the pH of glucose before fermentation was automatically maintained at 6.5 by a pH controller for 24 h (PHC-2201; Able, Tokyo, Japan). The fermentation processes were conducted in 250 mL glass anaerobic bottles (Haimen Huakai experiment glass instrument Co., Ltd, Haimen, China) sealed with sealing film. After addition of 1 g yeast extract and 2 g peptone to each bottle, 5 mL of a combination of P2 trace elements mixture solution (50 g/L KH2PO4, 50 g/L K2HPO4, and 220 g/L CH3COONH4), vitamins (0.1 g/L para-aminobenzoic acid, 0.1 g/L thiamin, and 0.001 g/L biotin), and minerals (20 g/L (NH4)2SO4, 1.5 g/L KH2PO4, 0.3 g/L MgSO4 ∙ 7H2O, 0.05 g/L FeSO4 ∙ 7H2O, 0.01 g/L MnSO4 ∙ H2O, 1.0 g/L CH3COONH and 1 g/L NaCl) were filter sterilized (Millipore filter; 0.22 μm) and added to fermentation solutions in bottle. In addition, 2 mM IPTG and antibiotics (ampicillin, 100 μg/mL; chloramphenicol, 35 μg/mL; kanamycin, 50 μg/mL) were also added. The bottles were then inoculated with 0.5 mL fermentation inoculum (OD600 = 1.6), and fermented in constant temperature oscillation incubator at 30 °C for 3 days.
Fermentation using duckweed under SSF
For the enzymatic hydrolysis method, the fermentation substrate in duckweed trials was hydrolysate in 20 mL modified M9 medium (2 g (NH4)2PO4, 2 g KH2PO4, 1 g K2HPO4, 1 g NH4Cl, 0.5 g NaCl, 0.5 mM MgSO4, 1 mM CaCl2, 20 mg vitamin B1, and 2 mg biotin per L of water) containing 5 g/L yeast extract. The pH of the slurry of duckweed was adjusted to 6.0 with 1% H3PO4, and 0.2 mg/g α-amylase (120 KUN/g) was added, then hydrolyzed at 50 °C for 6 h. Subsequently, 0.2 mg/g β-amylase was added, and saccharified at 45 °C for 10 h. After that, 0.2 mg/g cellulase (500000 U/g, where the enzyme activity (U/g) is defined as follows: CMCA = 1 g enzyme powder decomposes the substrate CMC-Na to produce 1 mg glucose with treatment at 50 °C and pH 4.8 for 1 h; Thinkly, China) and 0.2 mg/g Optimash BG (containing 5.4 U β-glucosidase activity and 1.9 U β-xylosidase activity; Genencor, USA) were added to the solution. The reaction mixture was then buffered with 50 mM phosphate buffer at pH 5.0 and incubated on a rotary shaker (HZQ-X500; Yiheng, Shanghai, China) at 220 rpm for 12 h at 50 °C.
For the SSF procedure, the fermentation procedure was simultaneously conducted alongside enzymatic hydrolysis in a 150-mL triangular flask. Supplementary Trace Metals Mix solution (2 g H3BO3, 2.1 g MnCl2 ∙ 4H2O, 0.3 g ZnSO4 ∙ 7H2O, 0.002 g MnSO4, 2.5 g Na2MoO4 ∙ 2H2O, 0.05 g CuSO4 ∙ 5H2O, 21.2 mg Co(NO3)2 ∙ 6H2O, and 0.05 g FeSO4 per L of water) and the substrates were fermented under aerobic conditions. 2 mL rejuvenated seeding solution of bioengineered strains from liquid medium was inoculated into 50 mL of the fermentation substrate. All cultures were induced with 2 mM IPTG, kanamycin, and chloramphenicol. The pH of the fermentation substrate after enzymatic hydrolysis was automatically maintained at 6.5, with continued fermentation at 30 °C with shaking at 200 rpm for 96 h in a constant-temperature oscillation incubator.
Analysis production and data statistics
Alcohol compounds were measured with a model 6890 gas chromatograph (GC) equipped with a flame ionization detector (Agilent Technologies, Santa Clara, CA, USA) with a model 7673 A automatic injector, sampler, and controller (Hewlett-Packard). Alcohol compounds were separated out using a ZB-WAX capillary column (30 m, 0.25 mm inside diameter, 0.25 μm film thickness; Phenomenex Inc., PA, USA). The GC oven temperature was held initially at 40 °C for 5 min, then raised stepwise, by 15 °C/min, until it reached 150 °C. It was then raised by 50 °C/min up to 250 °C, and held for 4 min. Helium was used as carrier gas, with an inlet pressure of 9.3 lb/in ref. 2. The injector and detector were maintained at 220 °C. A 1-μL volume of supernatant from the culture broth was injected in split-injection mode at a 1:30 split ratio. For other secreted metabolites, the constituent compounds (20 μL) were detected with an Agilent 1100 high-performance liquid chromatography system equipped with an auto-sampler and a Bio-Rad (Hercules, CA: carbohydrate analysis column Aminex HPX-87P Column 300 × 7.8 mm catalog 125-0098 serial 426070) (5 mM H2SO4, 0.6 mL/min; column temperature at 65 °C). Glucose was detected with an ELSD 2000 CSC detector, while organic acids were detected using a photodiode array detector at 210 nm. Concentrations were determined using extrapolation from standard curves. Amino acid and other organic acid production were determined with a DIONEX UltiMate 3000 liquid chromatograph in a column packed with Aminex HPX-87H and 0.05 mM H2SO4 on Chromosorb WAW. Chromatography was conducted at an injector temperature of 175 °C, detector temperature of 180 °C, and oven temperature of 125 °C.
For each experiment, all results were repeated for three times, we calculated the mean response variables and their standard deviations (SD), unless otherwise indicated. Comparisons of variable(s) were made with Student’s t-test; values of P < 0.05 were considered to indicate statistically significant differences. Tukey’s honest significant difference test was used when the null hypothesis was rejected (P < 0.05). Statistical analyses were conducted using the software program SPSS 21.0 (IBM, USA).
Additional Information
How to cite this article: Su, H. et al. Metabolic engineering of Corynebacterium crenatium for enhancing production of higher alcohols. Sci. Rep. 6, 39543; doi: 10.1038/srep39543 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Xue, C., Zhao, X.-Q., Liu, C.-G., Chen, L.-J. & Bai, F.-W. Prospective and development of butanol as an advanced biofuel. Biotechnol Adv 31, 1575–1584, doi: 10.1016/j.biotechadv08004 (2013).
Higashide, W., Li, Y., Yang, Y. & Liao, J. C. Metabolic Engineering of Clostridium cellulolyticum for Production of Isobutanol from Cellulose. Appl Environ Microb 77, 2727–2733, doi: 10.1128/aem02454 (2011).
Xue, C. et al. A novel in situ gas stripping-pervaporation process integrated with acetone-butanol-ethanol fermentation for hyper n-butanol production. Biotechnol Bioeng 113, 120–129, doi: 10.1002/bit25666 (2016).
Dürre, P. Biobutanol: An attractive biofuel. Biotechnol J 2, 1525–1534, doi: 10.1002/biot00168 (2007).
Xue, C. et al. High-titer n-butanol production by Clostridium acetobutylicum JB200 in fed-batch fermentation with intermittent gas stripping. Biotechnol Bioeng 109, 2746–2756, doi: 10.1002/bit24563 (2012).
Su, H. et al. Engineering Corynebacterium crenatum to produce higher alcohols for biofuel using hydrolysates of duckweed (Landoltia punctata) as feedstock. Microb Cell Fact 14, 1 (2015).
Su, H. et al. Preliminary investigation of metabolic engineering in a novel host bacterium Corynebacterium crenatum for alcohol biofuel production. RSC Adv 4, 65021–65030 (2014).
Atsumi, S. & Liao, J. C. Metabolic engineering for advanced biofuels production from Escherichia coli. Curr Opin Biotechnol 19, 414–419, doi: 10.1016/j.copbio08008 (2008).
Mainguet, S. E. & Liao, J. C. Bioengineering of microorganisms for C(3) to C(5) alcohols production. Biotechnol J 5, 1297–1308, doi: 10.1002/biot00276 (2010).
Li, S., Wen, J. & Jia, X. Engineering Bacillus subtilis for isobutanol production by heterologous Ehrlich pathway construction and the biosynthetic 2-ketoisovalerate precursor pathway overexpression. Appl Microbiol Biotechnol 91, 577–589 (2011).
Blombach, B. et al. Corynebacterium glutamicum tailored for efficient isobutanol production. Appl Environ Microb 77, 3300–3310 (2011).
Smith, K. M., Cho, K.-M. & Liao, J. C. Engineering Corynebacterium glutamicum for isobutanol production. Appl Microbiol Biotechnol 87, 1045–1055 (2010).
Su, H. et al. Use of Duckweed (Landoltia punctata) as a Fermentation Substrate for the Production of Higher Alcohols as Biofuels. Energ Fuel 28, 3206–3216, doi: 10.1021/ef500335h (2014).
Su, H., Xu, G., Chen, H. & Xu, Y. Enriching duckweed as an energy crop for producing biobutanol using enzyme hydrolysis pretreatments and strengthening fermentation process using pH-stat. ACS Sustain Chem Eng 3, 2002–2011 (2015).
Xu, M. et al. Site-directed mutagenesis and feedback-resistant N-acetyl-L-glutamate kinase (NAGK) increase Corynebacterium crenatum L-arginine production. Amino Acids 43, 255–266 (2012).
Ohnishi, J. et al. A novel methodology employing Corynebacterium glutamicum genome information to generate a new L-lysine-producing mutant. Appl Microbiol Biotechnol 58, 217–223 (2002).
Hayashi, M. et al. Transcriptome analysis of acetate metabolism in Corynebacterium glutamicum using a newly developed metabolic array. Biosci Biotechnol Biochem 66, 1337–1344 (2002).
Ohnishi, J., Mizoguchi, H., Takeno, S. & Ikeda, M. Characterization of mutations induced by N-methyl-N′-nitro-N-nitrosoguanidine in an industrial Corynebacterium glutamicum strain. Mutat Res-Gen Tox En 649, 239–244 (2008).
Fujita, K., Matsuyama, A., Kobayashi, Y. & Iwahashi, H. The genome-wide screening of yeast deletion mutants to identify the genes required for tolerance to ethanol and other alcohols. FEMS Yeast Res 6, 744–750, doi: 10.1111/j1567 (2006).
Watanabe, M., Fukuda, K., Asano, K. & Ohta, S. Mutants of bakers’ yeasts producing a large amount of isobutyl alcohol or isoamyl alcohol, flavour components of bread. Appl Microbiol Biotechnol 34, 154–159 (1990).
Derrick, S. & Large, P. J. Activities of the enzymes of the Ehrlich pathway and formation of branched-chain alcohols in Saccharomyces cerevisiae and Candida utilis grown in continuous culture on valine or ammonium as sole nitrogen source. Microbiology 139, 2783–2792 (1993).
Eden, A., Van Nedervelde, L., Drukker, M., Benvenisty, N. & Debourg, A. Involvement of branched-chain amino acid aminotransferases in the production of fusel alcohols during fermentation in yeast. Appl Microbiol Biotechnol 55, 296–300, doi: 10.1007/s002530000506 (2001).
ÄYrápáá, T. Biosynthetic formation of higher alcohols by yeast. Dependence on the nitrogenous nutrient level of the medium. J Inst Brew 77, 266–276, (1971).
Styger, G., Jacobson, D. & Bauer, F. F. Identifying genes that impact on aroma profiles produced by Saccharomyces cerevisiae and the production of higher alcohols. Appl Microbiol Biotechnol 91, 713–730, doi: 10.1007/s00253-011-3237-z (2011).
Lilly, M., Bauer, F. F., Styger, G., Lambrechts, M. G. & Pretorius, I. S. The effect of increased branched-chain amino acid transaminase activity in yeast on the production of higher alcohols and on the flavour profiles of wine and distillates. FEMS Yeast Res 6, 726–743, doi: 10.1111/j.1567-1364.00057.x (2006).
Larg, S. D. a. P. J. Activities of the enzymes of the Ehrlich pathway and formation of branched-chain alcohols in Saccharomyces cerevisiae and Candida utilis grown in continuous culture on valine or ammonium as sole nitrogen source. J Gen Microbiol 139, 2783–2792 (1993).
Richards, M. & Elliott, F. R. Inhibition of Yeast Growth by Streptomycin. Nature 29, 536 (1966).
Palmer, E., Wilhelm, J. M. & Sherman, F. Phenotypic suppression of nonsense mutants in yeast by aminoglycoside antibiotics. Nature 277, 148–150 (1977).
Babczinski, P. & Zelinski, T. Mode of action of herbicidal ALS-inhibitors on acetolactate synthase from green plant cell cultures, yeast, and Escherichia coli. Pestic Sci 31, 305–323 (1991).
Oard, J. H., Zhang, N. & Sanders, D. E. Resistance to Acetolactate Synthase-Inhibiting Herbicides. US20110053777 A1 US 12/303, 888 (2011).
Walker, J. Canavanine and homoarginine as antimetabolites of arginine and lysine in yeast and algae. J Biol Chem 212, 207–215 (1955).
Fantes, P. A. & Creanor, J. Canavanine Resistance and the Mechanism of Arginine Uptake in the Fission Yeast Schizosaccharomyces pombe. Microbiology 130, 3265–3273 (1984).
Chen, Y. & Piper, P. W. Consequences of the overexpression of ubiquitin in yeast: elevated tolerances of osmostress, ethanol and canavanine, yet reduced tolerances of cadmium, arsenite and paromomycin. BBA-Mol Cell Res 1268, 59–64 (1995).
Fukuda et al. Breeding of brewing yeast producing a large amount of-phenylethyl alcohol andphenylethyl acetate. Agric Chem Soci Japan 54 (1990).
Stam, H., Hoogland, M. & Laane, C. Microbiology of Fermented Foods 16, 505–542 (Springer, US, 1997).
Singh, R., Vadlani, P. V., Harrison, M. L., Bennett, G. N. & San, K. Y. Aerobic production of isoamyl acetate by overexpression of the yeast alcohol acetyl-transferases AFT1 and AFT2 in Escherichia coli and using low-cost fermentation ingredients. Bioprocess Biosyst Eng 31, 299–306, doi: 10.1007/s00449-007-0159-3 (2008).
Watanabe, M., Tanaka, N., Mishima, H. & Takemura, S. Isolation of sake yeast mutants resistant to isoamyl monofluoroacetate to improve isoamyl acetate productivity. J Ferment Bioeng 76, 229–231 (1993).
Williams, A. L. & Williams, L. S. Control of isoleucine-valine biosynthesis in a valine-resistant mutant of Escherichia coli K-12 that simultaneously acquired azaleucine-resistance. Biochem Biophys Res Commun 131, 994–1002 (1985).
Stieglitz2, B. & Calvo, J. M. Effect of 4-Azaleucine upon Leucine Metabolism in Salmonella typhimurium. J Bacteriol 108, 95–104 (1971).
Chen, X., Nielsen, K. F., Borodina, I., Kielland-Brandt, M. C. & Karhumaa, K. Increased isobutanol production in Saccharomyces cerevisiae by overexpression of genes in valine metabolism. Biotechnol Biofuels 4, 21, doi: 10.1186/1754-6834-4-21 (2011).
Kondo, T. et al. Genetic engineering to enhance the Ehrlich pathway and alter carbon flux for increased isobutanol production from glucose by Saccharomyces cerevisiae. J Biotechnol 159, 32–37, doi: j.jbiotec01022 (2012).
Lee, W.-H. et al. Isobutanol production in engineered Saccharomyces cerevisiae by overexpression of 2-ketoisovalerate decarboxylase and valine biosynthetic enzymes. Bioprocess Biosyst Eng 35, 1467–1475, doi: 10.1007/s00449-012-0736-y (2012).
Su, H. et al. Engineering Corynebacterium crenatum to produce higher alcohols for biofuel using hydrolysates of duckweed (Landoltia punctata) as feedstock. Microb Cell Fact 14, 16, doi: 10.1186/s12934-015-0199-3 (2015).
Zhang, K., Sawaya, M. R., Eisenberg, D. S. & Liao, J. C. Expanding metabolism for biosynthesis of nonnatural alcohols. P Natl Acad Sci USA 105, 20653–20658 (2008).
Cho, B.-K., Knight, E. M. & Palsson, B. Ø. Transcriptional regulation of the fad regulon genes of Escherichia coli by ArcA. Microbiology 152, 2207–2219 (2006).
Dellomonaco, C., Rivera, C., Campbell, P. & Gonzalez, R. Engineered respiro-fermentative metabolism for the production of biofuels and biochemicals from fatty acid-rich feedstocks. Appl Environl Microb 76, 5067–5078 (2010).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M., KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462 (2016).
Kanehisa, M. & Goto, S., KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30 (2000).
Rose, R. et al. Starch determination by perchloric acid vs enzymes: evaluating the accuracy and precision of six colorimetric methods. J Agric Food Chem 39, 2–11, doi: 10.1021/jf00001a001 (1991).
Pirt, S. J. & Whelan, W. J. The determination of starch by acid hydrolysis. J Sci Food Agric 2, 224–228, doi: 10.1002/jsfa2740020507 (1951).
Thiex, N. J., Manson, H., Anderson, S. & Persson, J.-Å. Determination of Crude Protein in Animal Feed, Forage, Grain, and Oilseeds by Using Block Digestion with a Copper Catalyst and Steam Distillation into Boric Acid: Collaborative Study. J AOAC Int 85, 309–317 (2002).
Xiao, Y., Fang, Y., Jin, Y., Zhang, G. & Zhao, H. Culturing duckweed in the field for starch accumulation. Ind Crops Prod 48, 183–190, doi: 10.1016/j.indcrop04017 (2013).
Viles, F. J. & Silverman, L. Determination of Starch and Cellulose with Anthrone. Anal Chem 21, 950–953, doi: 10.1021/ac60032a019 (1949).
Hansen, J. & Møller, I. Percolation of starch and soluble carbohydrates from plant tissue for quantitative determination with anthrone. Anal Biochem 68, 87–94 (1975).
Black, H. Determination of Sodium Carboxymethylcellulose in Detergent Mixtures by Anthrone Method. Anal Chem 23, 1792–1795 (1951).
Iiyama, K. & Wallis, A. F. Determination of lignin in herbaceous plants by an improved acetyl bromide procedure. J Sci Food Agric 51, 145–161 (1990).
Iiyama, K. & Wallis, A. F. A. An improved acetyl bromide procedure for determining lignin in woods and wood pulps. Wood Sci.Technol. 22, 271–280, doi: 10.1007/BF00386022 (1988).
Tinggi, U., Reilly, C. & Patterson, C. Determination of manganese and chromium in foods by atomic absorption spectrometry after wet digestion. Food Chem 60, 123–128 (1997).
Voegborlo, R. & Adimado, A. A simple classical wet digestion technique for the determination of total mercury in fish tissue by cold-vapour atomic absorption spectrometry in a low technology environment. Food Chem 123, 936–940 (2010).
Eggeling, L. & Bott, M. Handbook of Corynebacterium glutamicum. (CRC press, 2010).
Haynes, J. A. & Britz, M. L. The effect of growth conditions of Corynebacterium glutamicum on the transformation frequency obtained by electroporation. J Gen Microbiol 136, 255–263 (1990).
Van der Rest, M., Lange, C. & Molenaar, D. A heat shock following electroporation induces highly efficient transformation of Corynebacterium glutamicum with xenogeneic plasmid DNA. Appl Microbiol Biotechnol 52, 541–545 (1999).
Acknowledgements
This work was supported by the earmarked fund for National Natural Science Foundation of China (Grant Nos. 21502186), Science and technology project of Chongqing city land, resources and Housing Administration Bureau (CQGT-KJ-2015022).
Author information
Authors and Affiliations
Contributions
H.F.S. participated in the conception, design, data collection and analysis, and drafted the manuscript, provided fund support. G.W.W. assisted the laboratory work and provided fund support. J.F.L. participated in discussion of the results and revision of the manuscript. All authors read and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Su, H., Lin, J. & Wang, G. Metabolic engineering of Corynebacterium crenatium for enhancing production of higher alcohols. Sci Rep 6, 39543 (2016). https://doi.org/10.1038/srep39543
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep39543
- Springer Nature Limited
This article is cited by
-
Biosynthesis pathways of expanding carbon chains for producing advanced biofuels
Biotechnology for Biofuels and Bioproducts (2023)
-
Toward bioproduction of oxo chemicals from C1 feedstocks using isobutyraldehyde as an example
Biotechnology for Biofuels and Bioproducts (2022)
-
Enriching the Production of 2-Methyl-1-Butanol in Fermentation Process Using Corynebacterium crenatum
Current Microbiology (2020)
-
Engineering Corynebacterium glutamicum Mutants for 3-Methyl-1-butanol Production
Biochemical Genetics (2019)
-
Synthetic biology approaches to access renewable carbon source utilization in Corynebacterium glutamicum
Applied Microbiology and Biotechnology (2018)