
Abstract
Localized dynamics of RAS, including regions distal to the nucleotide-binding site, is of high interest for elucidating the mechanisms by which RAS proteins interact with effectors and regulators and for designing inhibitors. Among several oncogenic mutants, methyl relaxation dispersion experiments reveal highly synchronized conformational dynamics in the active (GMPPNP-bound) KRASG13D, which suggests an exchange between two conformational states in solution. Methyl and 31P NMR spectra of active KRASG13D in solution confirm a two-state ensemble interconverting on the millisecond timescale, with a major Pγ atom peak corresponding to the dominant State 1 conformation and a secondary peak indicating an intermediate state different from the known State 2 conformation recognized by RAS effectors. High-resolution crystal structures of active KRASG13D and KRASG13D-RAF1 RBD complex provide snapshots of the State 1 and 2 conformations, respectively. We use residual dipolar couplings to solve and cross-validate the structure of the intermediate state of active KRASG13D, showing a conformation distinct from those of States 1 and 2 outside the known flexible switch regions. The dynamic coupling between the conformational exchange in the effector lobe and the breathing motion in the allosteric lobe is further validated by a secondary mutation in the allosteric lobe, which affects the conformational population equilibrium.
Similar content being viewed by others

Introduction
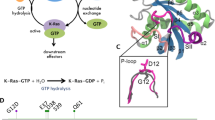
RAS proteins are small GTPases, cycling between the GTP-bound form (active) and the GDP-bound form (inactive). Regulatory proteins facilitate the cycling: guanine nucleotide exchange factors (GEFs) promote conversion from the GDP-bound form to the GTP-bound form, and GTPase activating proteins (GAPs) return RAS to the GDP-bound form. RAS proteins are the central node in signaling pathways controlling cell proliferation, survival, and differentiation through interactions with various effectors, such as RAF, PI3K, and others. Oncogenic RAS mutants are the main driving factors in many lethal cancers1. KRAS, one of the three RAS isoforms (KRAS, HRAS, NRAS), is the most frequently mutated RAS protein in cancer patients, and G12, G13, and Q61 are three hot spots among the oncogenic mutations. These mutants sample the RAS conformational space differently. For example, the KRAS G13D mutant (KRASG13D), which has a reduced oncogenic phenotype compared to G12D, was found to favor State 1 conformation (with the switch I region open). It was recently shown that KRASG13D has a stronger binding affinity to the guanine nucleotide exchange factor, Son of Sevenless (SOS), than wild-type KRAS (KRASWT)2, and can allosterically increase the nucleotide exchange rate of KRAS at the active site more than twice of that in KRASWT. On the other hand, the rate of SOS-independent nucleotide exchange for GDP-bound KRASG13D is an order of magnitude faster than that of GDP-bound KRASWT 3,4, which could result in auto-activation by spontaneous GDP/GTP exchange. A similarly elevated SOS-independent nucleotide exchange rate was also observed for HRASG13D (15-fold compared to HRASWT)4,5. Although KRASG13D is partially sensitive to NF1-mediated GTP hydrolysis6, it will be clinically important to develop a selective inhibitor targeting the oncogenic mutant KRASG13D. In recent years G-quadruplex structure (G4) formation in the KRAS promoter7,8 was identified in cancer cells with effects on active oncogene expression, increased genomic instability, and telomere maintenance. This has opened yet another therapeutic avenue of finding ligands that would stabilize G4s to downregulate KRAS9,10,11.
The G-domain of RAS proteins comprises two lobes (Fig. 1a), the effector lobe (residues 1–86) and the allosteric lobe (residues 87–166). In all RAS proteins, the G-domain is followed by a C-terminal hypervariable region (residues 167–188/189) involved in plasma membrane association following isoform-dependent palmitoylation and farnesylation. The effector lobe is fully conserved across all RAS isoforms, while the allosteric lobe has 90% sequence identity among the RAS isoforms. Two conformational states in RAS proteins, detected initially by crystal structures and solution-state NMR12,13, correspond to an ‘open’ versus a ‘closed’ position of the dynamic Switch I relative to the bound nucleotide in the active state. They are deemed States 1 and 2 conformations, with the latter adopted in complexes with effector proteins. Various literature reviews provide a wealth of information on the available KRAS structural data giving insights into conformational dynamics from experimental data and molecular dynamics (MD) simulations1,14,15,16,17. In the known conformational space of KRAS, the differences between State 1 and 2 conformations in most crystal structures are highly localized to the Switch I region (residues 30–38, showing two states corresponding to the open/closed configurations) and the Switch II region (residues 59–76), which exhibit a sizeable conformational variability among various crystal structures, while the rest of the backbone is highly similar (RMSD of ~0.3–0.5 Å when both switch regions are excluded). The high similarity of the crystal structures is evident not only for the WT form but also across the deposited structures of various KRAS mutants (including G13D) and even across their GDP vs. GTP-analogs structures, which suggests this represents the protein’s ground state18. Although several studies on RAS proteins by solution-state NMR have shown the presence of allostery connecting the effector and allosteric lobes using chemical shift perturbations5,19 and relaxation dispersion experiments20,21,22, it is not clear how those observations can be translated to structural changes outside of the switch regions in KRAS in solution, which is the focus of our present NMR study. It had been shown that the dynamics of the HRAS isoform can be affected by using non-hydrolyzable GTP analogs23, and recent dynamic studies using GTP with HRAS22 and with KRAS24 showed that two different minor states of GTP-bound RAS can be produced under different experimental conditions. Interestingly, although HRAS and KRAS proteins have an identical primary sequence in the effector lobe, they display different structural features in the reported minor states, highlighting the complex dynamics of RAS proteins in solution. The observed functional differences among various oncogenic mutants are likely rooted in subtle conformational and dynamic differences14. To provide insights and inspire therapeutic strategies, it would be valuable to elucidate the solution structure of any RAS high-energy state (excited, intermediate, or transition state) relative to the well-known ground state structures (lowest energy, corresponding to existing crystal structures).

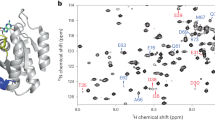
a Methyl probes are distributed in the effector and allosteric lobes of KRAS (PDB: 6VC8). Results for KRASWT, KRASG12D, KRASG13D, and KRASQ61H proteins with δ1-labeled methyl groups in isoleucine residues are plotted in panels b, c, d, and e, respectively. Each methyl group data set contains three measurements at three different temperatures (30, 25, and 20 °C) that are fitted separately, assuming a simple two-site exchange model with reaction rates (k1 and k−1) following the Arrhenius equation and a fixed chemical shift difference (Δω) at different temperatures. The errors of the fitting are reported as the standard deviations among the 10 best-fitting results out of 100 that start from randomly selected initial points. The methyl data are reported only when the amplitudes of their relaxation dispersion profiles at 25 °C are well above the experimental errors (Supplementary Fig. 1). The calculated exchange rates (kex = k1 + k−1) are plotted against temperatures. Residues I-21, I-24, I-36, I-55, I-100, I-139, and I-142 are shown in blue, green, purple, black, red, orange, and cyan, respectively.
While RAS proteins are known to sample two major conformational states, States 1 and 2, previous NMR studies19,21 have shown that RAS proteins also exhibit many sub-states that interconvert on millisecond and microsecond (ms-μs) timescales in solution. When several excited states are present in solution, it is difficult to acquire sufficient NMR data to characterize each of them. Often, crystallography provides a single snapshot representing the ground state. To examine the complex dynamics, we explored several oncogenic mutants and detected uniquely synchronized conformational dynamics in KRASG13D bound to GMPPNP (a non-hydrolyzable GTP-analog) by methyl Carr-Purcell Meiboom-Gill (CPMG)25 relaxation dispersion experiments, which can reveal conformational exchanges when the exchange rates between distinct conformational states are within the detectable timescale. By recording methyl NMR spectra at low temperatures to reduce the exchange rates, we could confirm the exchange between two conformational states on the ms timescale present in both effector and allosteric lobes of GMPPNP-bound KRASG13D. Furthermore, the 31P spectrum collected at low temperature showed a major Pγ atom peak corresponding to the State 1 conformation and a secondary peak indicating an intermediate state, or a transition state, different from State 2.
The solution state residual dipolar couplings (RDCs) contain information on population-weighted bond orientations of fast exchanging conformers with non-split NMR peaks, assuming Gaussian fluctuations are integrated over the entire NMR timescale of the measurement, i.e., tens of milliseconds26,27. Thus, the structural restraints for the intermediate state can be determined from RDCs, acquired in different alignment media, when the structure of the ground state is known. To provide ground state conformations for States 1 and 2, we solved crystal structures of GMPPNP-bound KRASG13D and KRASG13D-RAF1 RBD complex at 1.2 and 2.0 Å resolutions, respectively. We then performed a population-weighted, two-state (N = 2) ensemble structural refinement using experimental RDC restraints and obtained a structural ensemble representing the minor GMPPNP-bound KRASG13D intermediate conformational state. The structural ensemble of the intermediate state in GMPPNP-bound KRASG13D was cross-validated by the improvement in the fit of each RDC set when iteratively excluded from the structural calculation restraints. A similarly calculated two-state structural ensemble of KRASWT could not be cross-validated by RDCs, due to the lack of synchronized dynamics. Finally, we introduced secondary mutations to further validate the structure of the intermediate state and the dynamic coupling between the effector and allosteric lobes. Since wild-type and mutant KRAS in complex with effectors adopts the conformational State 2 in all crystal structures determined to date, presumably by both conformational selection and induced fit, the structure of an intermediate state could likely shed light into these interactions, with potential relevance for drug design.
In summary, we detected an intermediate state of active KRASG13D by NMR relaxation dispersion and chemical shifts. We used RDCs to solve and cross-validate this intermediate state structure with a conformation distinct from those of States 1 and 2 outside the known flexible switch regions. We obtained high quality representations of States 1 and 2 by crystallography. The dynamic coupling between the effector and allosteric lobes is further validated by a secondary mutation in the allosteric lobe, which affects the conformational population equilibrium.
Results
Synchronized conformational dynamics in GMPPNP-bound KRASG13D revealed by relaxation dispersion experiments
Although the dynamics of RAS proteins had been extensively studied in solution by 31P NMR, which reports two major conformational states in the effector lobe (Fig. 1a), recent studies19,21 demonstrated the presence of global dynamics as well as local dynamics throughout the whole protein (both effector and allosteric lobes) on the ms-μs timescale. We also showed that altered conformational dynamics on the ms-μs timescale subtly tune the biological functions of mutant KRAS using sensitivity-enhanced methyl relaxation dispersion experiments21. While investigating the conformational dynamics of oncogenic mutants using this method (Fig. 1b–e and Supplementary Fig. 1), we found that GMPPNP-bound KRASG13D displays a rare synchronized conformational dynamics behavior connecting the effector and allosteric lobes. The conformational exchange rates are reduced relative to GDP-bound KRASG13D, which shows fast and diverse conformational dynamics (Supplementary Fig. 2). This overall trend of reduced conformational exchange rates of KRASG13D in the presence of γ phosphate is similar to that of KRASWT (Supplementary Fig. 2). Still, the conformational dynamics of GMPPNP-bound KRASG13D is surprisingly more synchronized than that of other KRAS oncogenic mutants (Fig. 1b–e). In our analysis, although we could globally fit the relaxation dispersion data of GMPPNP-bound- KRASG13D with a simple two-site exchange model at 25 °C, the data were insufficient to disentangle the population distribution and chemical shift differences between the two dominant conformational states (Supplementary Fig. 3a). However, by combining all relaxation dispersion data from different temperatures, we could determine that the minor state represents ~15% of the population but with a significantly elevated χ2 when assuming the simple two-site exchange and Arrhenius equation (Supplementary Fig. 3b). The elevated χ2 suggests that this assumption is incompatible with our experimental data; therefore, our relaxation dispersion data suggest fast exchanges among substates within the major conformational states on the ms-μs timescale may exist. At the same time, by investigating the amide relaxation dispersion data, we also observed the ms-μs backbone conformational dynamics throughout the G-domain of GMPPNP-bound KRASG13D (Supplementary Fig. 4). Thus, the combined relaxation dispersion data confirm that the ms-μs conformational dynamics are not restricted to the effector lobe of KRAS proteins, which contains the switch regions but are distributed throughout the G-domain.
The two-state conformational exchange of GMPPNP-bound KRASG13D on the millisecond timescale revealed by spectral analysis
Thermodynamically, temperature change has little effect on the enthalpic term (structures of the conformational states). In contrast, it substantially impacts the entropic term (exchanges among different conformational states). Because the global analysis of our CPMG data on GMPPNP-bound KRASG13D could not independently validate the two-state exchange model, we further investigated the lineshape changes of methyl resonances when slowing the exchange rates by lowering the temperature (Fig. 2a). Similar lineshape analysis was used to investigate ms conformational dynamics in previous 31P NMR studies28,29. Indeed, we could observe methyl resonance splitting starting at 5 °C for KRASG13D but not for KRAST35S (a KRAS mutant known to stabilize State 1) (Fig. 2a). After adding 10% glycerol to increase viscosity and lowering the temperature to 2 °C to further decrease the conformational exchange rate, complete separation between the major peak (representing the major conformational state) and the minor peak (representing the minor conformational state) could be observed for the methyl resonance of I-55 (Fig. 2a). The dominant population is in the State 1 conformation (with peaks matching those of GMPPNP-bound KRAST35S), and the peak heights suggest that the minor state represents ~30% of the population (Fig. 2a). At low temperatures, all methyl groups with relaxation dispersion profiles in GMPPNP-bound KRASG13D (Supplementary Fig. 1c) show complete or partial resonance splitting into two peaks or resonance broadening, as expected for a range of chemical shift differences corresponding to the two interconverting conformational states (Fig. 2a). Combined with our relaxation dispersion data, this spectral analysis confirmed the existence of a synchronized two-state conformational exchange on the ms timescale spanning both lobes of GMPPNP-bound KRASG13D. In contrast, all methyl spectra of GMPPNP-bound KRASWT, KRASG12D, and KRASQ61H, at 5 °C, showed more complex dynamic behaviors on the ms timescale (Fig. 2b), as expected from our data analysis of CPMG experiments (Fig. 1b–e). In the case of KRASWT, the methyl resonances from I-21, I-24, and I-55 in the effector lobe split into two major peaks, while the methyl resonance from I-100 in the allosteric lobe split into one major peak and two minor peaks. In the case of KRASG12D, the methyl resonances from I-21, I-24, and I-55 in the effector lobe split into two major peaks and one minor peak, while the methyl resonance from I-139 in the allosteric lobe split into two major peaks. The resonance from I-100 in the same lobe splits into two major peaks and two minor peaks. In the case of KRASQ61H, the methyl resonances from I-21, I24, and I-55 in the effector lobe split into two major peaks. In comparison, the methyl resonance from I-100 in the allosteric lobe splits into two broad peaks, indicating more complex ms dynamics in the allosteric lobe. At 25 °C, we also examined the amide chemical shifts of GMPPNP-bound KRASWT, KRASG13D, and KRAST35S. We observed that some resonances (but not all) show a linear path following the increasing population of State 1 conformation in mutant KRAS (Fig. 2c). The conformational state populations in mutant KRAS were determined by the 31P spectra (Fig. 3). As reported previously28,29, similar resonance splitting was also observed in the 31P spectra of nucleotide-bound RAS proteins at low temperatures (Fig. 3). Based on the 31P spectra, the population of State 1 conformation is ~35% in KRASWT, ~70% in KRASG13D, and ~100% in KRAST35S. However, in our amide HSQC spectra (Fig. 2c), we also observed that many other resonances do not show a linear path following an increasing population of the State 1 conformation. Since the amide chemical shifts of KRAST35S represent the State 1 conformation and those of KRASWT represent the structural averaging between the State 1 and 2 conformations, this suggests that the conformation of the minor state in GMPPNP-bound KRASG13D is not simply an average conformation between the State 1 and 2 conformations. While analyzing 31P NMR spectra of the nucleotide γ phosphate in GMPPNP-bound KRAS proteins at 5 °C, we again observed two conformational states of GMPPNP-bound KRASG13D in solution on the millisecond timescale. The major population is in the State 1 conformation, and the minor one is in an intermediate state (Fig. 3), likely with a semi-open state of the Switch I region. The peak heights suggested that the minor state in GMPPNP-bound KRASG13D represents ~30% of the population, in agreement with the intensity ratio of the split methyl resonances at low temperatures (Fig. 2a). The existence of an intermediate state in GMPPNP-bound KRASG13D supports the proposed mechanism of the interaction between RAS proteins and RAF1-RBD in a previous transient kinetic analysis of RAS proteins30, where an initial rapid equilibrium step (conformational selection) is followed by an isomerization reaction (induced fit).

a The two-state exchange on the ms timescale revealed by methyl-TROSY spectra of KRASG13D and KRAST35S by lowering the temperature. In the right panel, methyl signals of KRASG13D and KRAST35S are overlaid, and the methyl resonances in KRASG13D with detectable relaxation dispersion profiles (Supplementary Fig. 1) are labeled. The assignments of all methyl resonances are shown in the KRAST35S 15 °C spectrum in panel a. GMPPNP-bound KRASG13D and KRAST35S are shown in red and blue, respectively. b The complex local dynamics on the ms timescale revealed by methyl-TROSY spectra of KRASWT, KRASG12D, and KRASQ61H at 5 °C. The methyl resonances with detectable relaxation dispersion profiles (Supplementary Fig. 1) are labeled. c Overlaid amide spectral regions of GMPPNP-bound KRASG13D, KRAST35S, and KRASWT, at 25 °C, are shown in red, blue, and green, respectively. All assigned resonances are labeled, an unassigned arginine NηHη side-chain resonance is marked by the asterisk.

An intermediate state distinct from States 1 and 2 is revealed in the 31P NMR spectra of the GMPPNP-bound KRASG13D at 5 °C.
Crystal structures of GMPPNP-bound KRASG13D and KRASG13D-RAF1 RBD represent the State 1 and 2 conformations, respectively
To gain detailed insights into the State 1 and 2 conformations of active KRASG13D, including the Switch regions, we solved high-resolution crystal structures of GMPPNP-bound KRASG13D and KRASG13D in complex with RAF1 RBD at 1.2 Å and 2.0 Å, respectively (Table 1). The electron density associated with GMPPNP, magnesium, and switch regions was unambiguous in the high-resolution maps. This contrasts with the recently reported 3.4 Å resolution structure of GMPPNP-bound KRASG13D (PDB: 6E6F; RMSD: 0.61 Å), in which the flexible Switch II could not be modeled4. In the GMPPNP-bound KRASG13D structure, Switch I is in State 1, since it moves away from the GMPPNP, and T35 does not contact the Mg2+ ion (Fig. 4a). This Switch I conformation in this structure is different from the lower resolution structure (PDB: 6E6F) despite both being in the State 1 conformation (the dynamic Switch I is captured in two different snapshots as observed in two different crystal forms) and is clearly different from the State 2 conformation of KRASG13D bound to RAF1-RBD (Fig. 4b). The overall structure of the KRASG13D-RAF1-RBD complex is similar to the GMPPNP-bound KRASWT (PDB: 6GOD) and KRASWT-RAF1-RBD (PDB: 6VJJ) structures previously reported in State 2 conformation, except that Y32 in the Switch I region occupies a different rotameric conformation due to steric clash with D13 (Fig. 4c). The difference in Switch II is likely due to different crystal packing interactions and is not believed to be associated with the G13D mutation, different states of Switch I, or the presence of RAF1-RBD. The sidechain of D13 in GMPPNP-bound KRASG13D points toward Switch II (χ1: −51.3°), which is similar to that in the lower resolution structure (PDB: 6E6F; χ1: −62.9°) but opposite to that in the GDP-bound structure (PDB: 4TQA; χ1: −148.7°) or the RAF1-RBD-bound structure (χ1: −149.6°)3. This suggests that the D13 side chain is flexible and samples two rotameric conformations. In all cases, the D13 side chain interacts with a positively charged moiety, such as K117, or with a Mg2+ ion in the solvent introduced by the crystallization condition. A structural comparison of GMPPNP-bound KRASWT in the State 2 conformation with KRASG13D in the GDP-bound conformation and GMPPNP-bound State 1 and 2 conformations shows four different snapshots of the Switch I region (Fig. 4d–f). Sidechain atoms of T35 and Y32 transition in opposite directions in these snapshots, illustrating how the conformation ensembles of the Switch I region change from inactive to active (State 1) and then from State 1 to State 2 conformations. The conformation of the T35 side chain is maintained in the State 2 conformation of WT and G13D mutant of KRAS; however, the Y32 side chain occupies a different rotameric conformation due to a steric clash with D13 in the KRASG13D structure (Fig. 4f).

a The overall structure of GMPPNP-bound KRASG13D in State 1 conformation. KRASG13D (PDB 8EBZ) is colored light blue, with the Switch I and II regions highlighted in blue and purple, respectively. GMPPNP is shown as sticks, and Mg2+ (green) and water (red) molecules are shown as spheres. b The overall structure of GMPPNP-bound KRASG13D (green; State 2 conformation) in complex with RAF1-RBD (cyan) (PDB 8EPW). c Structural superposition of WT (orange; PDB 6GOD) and G13D (green; PDB 8EPW) mutant of KRAS in State 2 conformation showing differences in switch regions. Side-chain atoms of Y32 and T35 are shown to highlight conformational differences in these structures. d Structural superposition of KRASWT (State 2; PDB 6GOD) with KRASG13D in State 1 (PDB 8EBZ) and State 2 (PDB 8EPW) conformations showing differences in switch regions. e Structural comparison of KRASWT (State 2; PDB 6GOD) with KRASG13D in GDP (light magenta; PDB 4TQA) and GMPPNP-bound (PDB 8EBZ, 8EPW) forms showing differences in the switch regions. f Enlarged view of the Switch I region from panel E shows the transition of the Switch I region from GDP to State 1, State 1 to State 2, and State 2 conformational similarities (T35) and differences (Y32) between WT and G13D mutant of KRAS.
Structural analysis of GMPPNP-bound KRASG13D by RDCs
All RAS crystal structures show consistent conformational variability exclusively in the flexible Switch I and II regions due to variations in crystal packing. Our relaxation dispersion data and NMR chemical shift analysis demonstrated the presence of conformational exchange between two dominant states outside the flexible switch regions because our methyl probes and measured RDCs span the entire G-domain (i.e., throughout both effector and allosteric lobes). We measured residual dipolar couplings (RDCs) to elucidate the structural features of the excited state in solution distal from the Switch I and II regions. Since experimental RDCs report time-averaged conformational states31, we examined this two-state exchange model of GMPPNP-bound KRASG13D in solution by measuring RDCs of various bond vectors in two alignment media (Pf1 and bicelles). Although the 1DNH and 1DCαHα RDCs of the GMPPNP-bound KRASG13D, measured in both Pf1 and bicelles, aligned well with high-resolution crystal structures, the best fit RMSD was 2-3 times larger than the corresponding RDC measurement precision (Supplementary Figs. 5 and 6). This suggested that the crystal structures do not completely represent the solution state of KRASG13D and led us to generate an N = 2 ensemble of solution structure models that would fit the RDC restraints within (but not tighter than) the experimental precision, as shown in Supplementary Fig. 5. Therefore, the resulting structural ensemble refined by RDCs is a more accurate representation of the conformational states sampled by GMPPNP-bound KRASG13D in solution.
The structure of the intermediate state of GMPPNP-bound KRASG13D and its validation
The experimental RDCs of GMPPNP-bound KRASG13D could not be measured for most residues in the switch regions due to the complete exchange broadening of amide resonances with large chemical shift differences. Therefore, we focused on refining the backbone of the GMPPNP-bound KRASG13D using the observable residues to obtain the intermediate state conformation. The refinement protocol fixed the major conformer to the State 1 conformation (represented by the GMPPNP-bound KRASG13D crystal structure), as indicated by the 31P spectra. We calculated RDC-refined structures of the GMPPNP-bound KRASG13D starting from the corresponding crystal structures (State 1 conformation) and allowing a maximum 3 Å displacement of the heavy atom coordinates under simulated annealing using all experimental RDC restraints. We carefully adjusted the relative force constants of the RDC energy terms so that the calculated structures would fit the RDCs within the experimental errors (less than 1 Hz for NH and less than 2 Hz for CαHα, Supplementary Fig. 5), but without overfitting. Additionally, backbone dihedral angle restraints obtained from NMR chemical shifts using the Talos-N32 program were included in the structure calculations. We also performed a grid search by repeating the N = 2 ensemble refinement and RDC cross-validation with various relative population ratios and found an optimal ratio of 30:70, in agreement with the 31P and methyl spectra at 5 °C (Figs. 2 and 3). This population ratio resulted in the lowest dihedral angle potential energies, while the other energy terms (except the RDC term) remained essentially unchanged. Separate structural calculations to cross-validate the obtained intermediate state of GMPPNP-bound KRASG13D were performed by excluding each RDC set to calculate the Rfree quality factor33. This factor quantitates the agreement between each experimental RDC set and the corresponding RDC values back-calculated from the structural ensemble obtained when that set was not used as a structural restraint, as shown in Supplementary Fig. 6. Notably, the calculated conformations representing the intermediate state satisfy the RDC cross-validation tests, as shown in Supplementary Figs. 6 and 7. These Rfree values for the N = 2 ensemble (weighted RDC fit over both major and minor conformations) exhibit small but consistent improvements over the R values of the major conformation only (i.e., ground state, represented by the crystal structure of State 1). The N = 2 cross-validation improvements are more pronounced for the RDCs with smaller relative experimental errors, i.e., NH and CαHα in Pf1 and NH in bicelles, but less pronounced for CαHα in bicelles due to a slight instability in the bicelles sample, which began to slowly hydrolyze during the experiment (the buffer pH was above neutral), and for the much smaller 1DNC’ and 2DNC’ couplings. When selecting only the residues showing the most significant changes in the spatial orientation of the RDC vectors (i.e., N-H, Cα-Hα, N-C’i, N-C’i-1 of residues 7, 13–15, 23–29, 43–54, 60–63, 73–81, 86, 92–95, 105–113, 120–124, 137–140, 144–150), the cross-validation improvements in the Rfree quality factor (or in the RMSD of the fit) are easier to visualize (Supplementary Fig. 7). Thus, the statistical improvement in the N = 2 ensemble refinement of GMPPNP-bound KRASG13D over the starting crystal structure validates the existence of an excited state in solution. The intermediate state (Fig. 5) is represented by the ten lowest energy structures (out of 100 calculated) and has an RMSD of 1.0 Å from the mean structure. The pairwise RMSDs between these and the ground state (i.e., the starting crystal structure) are between 1.0 and 1.3 Å (with no significant displacements of the switch regions, as discussed above). The average backbone RMSD values between the ground state (crystal structure) and the excited state (refined structural ensemble) are shown in Supplementary Fig. 8.

The intermediate state of the GMPPNP-bound KRASG13D (minor 30% conformer, colored wheat) overlaid to the State 1 conformation (major 70% conformer, crystal structure, colored blue).
Structural analysis of GMPPNP-bound KRASWT by RDCs
To further validate the above results, we applied the same approach to GMPPNP-bound KRASWT, which has more complex conformational dynamics shown by relaxation dispersion data (Fig. 1a). We similarly examined the N = 2 ensemble refinement of the GMPPNP-bound KRASWT with RDCs (using NH and CαHα measured in Pf1 and bicelles) and found an optimal ratio of approximately 60:40. Although the result agreed with the 31P spectrum (Fig. 3), the RDC cross-validation showed no significant improvement over the fit to the starting high-resolution crystal structure in the State 2 conformation (Supplementary Table 1). The calculated minor state of GMPPNP-bound KRASWT showed similar structural displacements from the starting crystal structure as did the excited state of KRASG13D. Still, only the GMPPNP-bound KRASG13D structures exhibited statistically significant Rfree improvements of the RDCs. When comparing directly their corresponding experimental RDCs (NH and CαHα measured in Pf1 and bicelles), the most prominent outliers, having consistent RDC differences 1.5 times larger than the pairwise RMSD (mapped on the structure in Supplementary Fig. 9), correlated well with the regions of the GMPPNP-bound KRASG13D intermediate state showing displacements from the ground state (Supplementary Figs. 8 and 9). This suggests that a simple population-weighted average of just two conformational states represents more accurately the average solution structure of KRASG13D than that of KRASWT. For GMPPNP-bound KRASWT, a similar N = 2 ensemble refined by RDCs likely represents an oversimplification of a more complex distribution of intermediate states. Therefore, our data suggest that the KRASWT crystal structures might already accurately represent the average of its complex dynamic fluctuations in solution outside the switch regions, while this was not the case for KRASG13D.
Validation of the synchronized motion and the structure of the intermediate (excited) state in GMPPNP-bound KRASG13D
We introduced secondary mutations in the allosteric lobe to confirm the existence of the synchronized motion and the intermediate state in GMPPNP-bound KRASG13D. These mutations could cause structural perturbations in the effector lobe and shift the population equilibrium, which could be monitored by the changes in the nucleotide 31P NMR spectra. Close inspection of the RDC-refined structure of the intermediate state in the allosteric lobe revealed a hydrophobic cavity near residue A130, which is present in all conformers of the calculated ensemble (Fig. 6a, b). Four mutations (A130V, A130L, A130I, and A130F) with varying sizes of the hydrophobic sidechains were selected and introduced in KRASG13D to favor the formation of the cavity. All the double mutants were found to be less stable than KRASG13D. Due to the low solubility of KRASG13D,A130F, only three out of four double mutants could be further analyzed. In the most stable double mutant KRASG13D,A130I, we found that the slight population shift (~10%) induced by a second mutation A130I (Fig. 6c) resulted in a two-fold increase in the binding affinity to RAF1-RBD compared with the single mutant KRASG13D (Fig. 6d). To confirm the relatively small differences in the high-affinity binding of KRAS mutants to RAF1-RBD (Fig. 6d), we repeated the ITC experiments on KRASG13D and KRASG13D,A130I with a higher salt concentration buffer to observe the change in titration curves caused by weakening the interactions between KRAS mutants and RAF1-RBD. Indeed, the higher salt concentration did reduce the binding strength of KRAS to RAF1-RBD but maintained the two-fold increase in the binding affinity to RAF1-RBD for the KRASG13D,A130I double mutant over that of the KRASG13D mutant (Supplementary Fig. 10). The other double mutants, KRASG13D,A130V and KRASG13D,A130L, with almost identical population ratios to KRASG13D,A130I (Supplementary Fig. 11), show roughly similar binding affinities to RAF1-RBD (Supplementary Fig. 10). Consistent with our previous study21, the population distribution per se cannot determine the binding affinity of KRAS mutants to RAF1-RBD, and the contribution of the entropic term likely accounts for the variations in KD. In the background of the G13D mutation, the binding between KRAS mutants and RAF1-RBD is enthalpically driven under our experimental conditions, unlike that of KRASWT (Supplementary Table 2). These results confirm the dynamic coupling between the effector and allosteric lobes and validate the RDC-refined structure of the intermediate state of GMPPNP-bound KRASG13D.

a The displaced conformation of the α4 helix in the allosteric lobe of the intermediate state is shown on the crystal structure (PDB: 8EBZ). b In the allosteric lobe, a small hydrophobic pocket near A130 appears in the RDC-refined structure of the excited state. c The introduction of the secondary mutation A130I, with a larger hydrophobic sidechain, increases the population of the intermediate state as monitored by nucleotide 31P NMR spectra. d Binding affinities between GMPPNP-bound KRAS mutants and RAF1-RBD obtained by ITC measurements. The error in the KD values corresponds to the standard error of fitting of a singlicate dataset with 19 injections.
Discussion
Proteins function often involves an exchange between multiple conformations that are sparsely populated and occur transiently. If the timescale of the conformational exchange is on the order of ms-μs, determining these relevant conformational states is a complex endeavor, especially when it is difficult to trap them by mutations or interactions with ligands, or when the exchange rates are not sufficiently slow to result in distinct chemical shifts for each conformation. Here we describe a novel approach to identify and elucidate an excited conformation in the presence of complex ms-μs conformational dynamics of RAS proteins. Since the dominant State 1 of GMPPNP-bound KRASG13D is not a single conformational state on the ms-μs timescale21, accurate chemical shift difference (Δω) values cannot be extracted from relaxation dispersion experiments. Consequently, previous NMR approaches34,35,36 to determine the structure of a sparsely populated excited state in solution using accurate determination of Δω values do not apply to this case. Even if the CPMG data cannot be globally fitted to provide detailed dynamic information, our data analysis identified a synchronized motion in the GMPPNP-bound oncogenic mutant KRASG13D. Although two minor states and the conformational exchanges involved in intrinsic nucleotide exchange have been revealed by GTP-bound RAS proteins22,24, the results of this work and our previous study21 demonstrate the plasticity of KRAS proteins when interacting with their effectors instead. Using a high-resolution crystal structure and multiple sets of residual dipolar couplings, we determined a synchronized ‘breathing’ motion spanning subtle structural changes distributed through this oncogenic mutant’s G-domain (Fig. 5 and Supplementary Fig. 9), which couples with the conformational exchange in Switch I region (Figs. 1, 3, and 5). Finally, we introduced a series of additional mutations at A130 to validate the presence of a small hydrophobic pocket in the allosteric lobe by monitoring the population shifts in the effector lobe using 31P NMR spectra (Fig. 6c and Supplementary Fig. 11). Even though all these double mutants of KRAS display similar conformational population distribution, they have different binding affinities to the RAF1-RBD effector, as shown by ITC data (Supplementary Fig. 10). We observed a similar situation in our previous study21, where three different mutations stabilizing the State I conformation in KRAS displayed different dynamics coupled with different binding affinities to RAF1-RBD. The result agrees with an earlier study of the same mutations in HRAS37. Our ITC data and previous results21,37 suggest that the population distribution of RAS conformations in solution is not the sole factor affecting the binding of RAS proteins to the effectors. On the other hand, our 31P spectra on KRASG13D (Fig. 3) and a previous transient kinetic study30 support that the binding of RAS proteins to their effectors involves both conformational selection and induced fit. The induced fit from the intermediate state to the bound state and additional unpredicted entropic contributions could explain the variation in the binding affinities of different KRAS double mutants to RAF1-RBD (Supplementary Fig. 10).
The statistical improvement in the RDC cross-validation tests observed only for the GMPPNP-bound KRASG13D shows that complex dynamics do not average out the structural information of its excited state on the ms-μs timescale. In the GMPPNP-bound KRASG13D, a consistent deviation between the ground state and the excited state can be observed in the allosteric lobe (Fig. 5), which has not been previously reported due to the lower resolution of the solved structures. In the RDC restrained model, the slight departures of β5 and β6 strands of the excited state relative to the ground state result in a displaced α4 helix away from the ground state (crystal structure) position.
In our study, the I46-D47-G48 (β2-β3 hairpin) amides are broadened in the NMR spectra of GMPPNP-bound KRASG13D and KRASWT. This motif exhibits a displaced conformation in the minor intermediate state (Fig. 5). Conformational variation of this β2-β3 hairpin has also been observed in various crystal structures, including some determined at high pressure38. Moreover, MD simulations have detected correlated fluctuations of these β2-β3 strands, located between the Switch I and Switch II regions in active KRAS, with the hairpin driving Y157 (α5 helix) motions39. Lastly, the β2–β3 and α5 were deemed to form a conformational switch in HRAS, with mutations in D47-E49 causing hyperactive RAS40 and, indeed, the G48A mutation was detected in a patient with lung adenocarcinoma40. Another region showing a different minor conformation relative to the ground state is α3, which exhibits a reduced bend at H95. This straighter α3 helix and the displaced Loop L7 (Fig. 5) correspond to the α3-L7 motif, which, together with both switch regions, is involved in the HRAS binding of GEFs41. Conversely, a slightly more bent α3 helix is observed in a KRASG12D,K104Q mutant, which causes an allosteric effect on the stability of the α2 helix in Switch II when compared to KRASWT 42. Furthermore, fluctuation in this α3-L7 motif was found to drive Switch I and II fluctuations in KRAS-GTP in MD simulations39. The structural differences observed in the minor state in the α4 helix, the straighter α3 helix and displaced loop 7, the β2-β3 hairpin that wraps around α5, the minor changes in β strand positions and β-sheet twist, together with the less compact packing of the helices in the allosteric lobe (when compared to the crystal structures in State 1/State 2 conformations), likely contribute to the observed functional differences of GMPPNP-bound KRASG13D vs. GMPPNP-bound KRASWT.
In summary, our methyl relaxation dispersion experiments revealed highly synchronized conformational dynamics in the active KRASG13D. Our high-resolution crystal structures provide snapshots of this mutant in State 1 and 2 conformations. Using RDCs and a population-weighted two-state ensemble refinement method, we solved the structure of the intermediate state, showing regions of structural fluctuations with functional relevance. This excited (intermediate) state, distinct from the known dominant States 1 and 2 in KRASWT and its G12D and Q61H mutants, is validated by three pieces of evidence. Firstly, the consistent improvement in the RDC cross-validation of the KRASG13D two-state ensemble comprising State 1 and this distinct excited state (Supplementary Figs. 6 and 7) in contrast with the KRASWT test case that uses States 1 and 2, which shows no similar improvement (Supplementary Table 1). This is because only KRASG13D is a simple two-conformation state system, while KRASWT is a more complex multi-state system with less synchronized conformational motion. Secondly, the distinct chemical shift of the 31Pγ signal of KRASG13D in Fig. 3 confirms that the nucleotide-binding pocket samples an excited conformation different from the known States 1 and 2 in KRASWT and most of its mutants. Thirdly, the non-linearity of the trajectory of some KRASG13D amide chemical shifts when superimposed on the spectra of KRAST35S and KRASWT (representing respectively the State 1 and a conformational average of States 1 and 2, Fig. 2c) confirms that these differences occur throughout the rigid backbone of the protein (distal from the Switch regions). The structural analysis of the intermediate (excited) state in GMPPNP-bound KRASG13D suggests the formation of a new pocket in the allosteric lobe, which could be exploited for developing new therapeutics against KRASG13D-driven cancers. Our approach can be applied to other proteins with similar dynamic behavior.
Materials and methods
DNA for protein production
Genes for protein expression were generated from DNA constructs initially synthesized as Gateway Entry clones (ATUM, Newark, CA). Constructs consisted of E. coli gene-optimized fragments containing an upstream tobacco etch virus (TEV) protease site (ENLYFQ/G) followed by the coding sequence of human KRAS4b(1-169) or human RAF1(52-131). Entry clones were transferred to an E. coli destination vector containing an amino-terminal His6-MBP (pDest-566, Addgene #11517) tag by Gateway LR recombination (Thermo Scientific, Waltham, MA). Constructs generated were:
R989-X02-566: His6-MBP-tev-G-Hs.KRAS4b(1-169) [Addgene:159539]
R989-X09-566: His6-MBP-tev-G-Hs.KRAS4b(1-169) G12D [Addgene:159541]
R989-X62-566: His6-MBP-tev-G-Hs.KRAS4b(1-169) Q61H
R988-X60-566: His6-MBP-tev-G-Hs.KRAS4b(1-169) T35S
R989-X11-566: His6-MBP-tev-G-Hs.KRAS4b(1-169) G13D [Addgene:159543]
R929-X18-566: His6-MBP-tev-G-Hs.KRAS4b(1-169) G13D/A130I
R929-X19-566: His6-MBP-tev-G-Hs.KRAS4b(1-169) G13D/A130L
R929-X20-566: His6-MBP-tev-G-Hs.KRAS4b(1-169) G13D/A130V
R929-X21-566: His6-MBP-tev-G-Hs.KRAS4b(1-169) G13D/A130F
R702-X66-566: His6-MBP-tev-Hs.RAF1(52-131).
Protein expression and purification
RAF1(52-131) was expressed using the auto-induction media protocol, and KRAS proteins were expressed using the Dynamite media protocol43. Gly-Hs.KRAS4b(1-169) G13D was expressed following published protocols for 15N incorporation44. Gly-Hs.KRAS4b(1-169) G13D was expressed following published protocols for 13C/15N incorporation with modifications45. Specifically, ZnCl2 was omitted, and IPTG induction was done at either 16 °C or 20 °C to provide a higher yield. Highly deuterated, 13C-methyl/15N-labeled KRAS proteins were expressed as previously described21,46. All proteins were purified as previously outlined for Gly-Hs.KRAS (1-169)47; MgCl2 was omitted when purifying RAF1(52-131).
NMR relaxation dispersion experiments
All relaxation dispersion experiments were carried out on a 700 MHz (16.4 T) Bruker Avance NEO spectrometer equipped with a helium-cooled TCI cryoprobe. All methyl relaxation dispersion experiments25,48,49 were performed using U-[15N, 2H], 13CH3-δ1-Ile-labeled KRASG13D samples (1 − 169) with a concentration of about 250 μM in a 20 mM HEPES (pH 7.4), 150 mM NaCl, 5 mM MgCl2, and 2 mM TCEP buffer. A γB1 frequency of ∼15 kHz was used for the 13C π pulse in a 20 ms constant-time spin-echo period of 13C single-quantum CPMG experiments, with νCPMG frequencies of 100, 200, 400, 500, 800, and 1000 Hz, with repetition of the 100, 400, and 1000 Hz data points. On the other hand, in methyl adiabatic relaxation dispersion experiments (HARD), only four different composite hyperbolic secant (HS) pulses were used to measure different relaxation rates in adiabatic R1ρ (HS1, HS2, HS4, and HS8) and R2ρ (HS1, HS4, HS8, and HS4_1ms) experiments. All the data were processed by NMRPipe and NMRFAM-Sparky50 before further analysis. During the final analysis, CPMG data and HARD data were fit with 2-site exact analytic solution and 2-site solution surface repectively. All the fits started with an extensive grid search and ended with an accelerated gradient descent by assuming the Arrhenius equation and a fixed Δω value at different temperatures.
Amide relaxation dispersion experiments were performed using U-[15N] labeled GMPPNP-bound- KRASG13D(1 − 169) samples with a concentration of about 400 μM in the same buffer conditions. TROSY 15N single-quantum CPMG experiments51 were carried out with a 20 ms constant time, 2 s recycle delay, 32 scans, 128 complex points in the 15N dimension (35 ppm), and 2048 complex points in the 1H dimension (14 ppm) at 25 °C. A γB1 frequency of ∼7 kHz was used for the 15N π pulse in the constant-time spin-echo period, with νCPMG values of 100, 200, 400, 500, 800, and 1000 Hz, with repetition of the 100, 400, and 1000 Hz data points. All the data were processed with a linear prediction of an additional 128 complex points in the 15N dimension using NMRPipe52 and subsequently analyzed in NMRFAM-Sparky50. Experimental values of the relaxation dispersion intensities and analysis data are provided as a Supplementary Data 2.
31P NMR
1D-31P NMR spectra were acquired at 5 °C on a Bruker 500 MHz NMR spectrometer (202 MHz 31P frequency) with a 5 mm Prodigy broadband cryogenic probe using 70° flip angle pulses, 1200 scans, an interscan delay of 7 s, an acquisition time of 84 ms, and a WALTZ-16 proton decoupling sequence. The 1D-31P NMR experiments were repeated with similar conditions (except those of double-mutants), and the resulting spectra showed no visible difference from the original ones.
Assignments
Resonance assignments of KRASG13D, in both GDP and GMPPNP-bound- states, were obtained from the analysis of a standard set of two-dimensional (2D) and three-dimensional (3D) NMR spectra: 2D HN-HSQC, 3D HNCACB, 3D CBCA(CO)NH, and 3D C(CO)NH and deposited in BMRB (Entry: 51642). All 3D experiments were recorded using non-uniform sampling (NUS) at a sampling rate of 40%, and the spectra were reconstructed using the SMILE53 and NMRPipe52 software packages.
Residual dipolar couplings
KRASG13D and KRASWT NMR samples contained 0.25–1.2 mM of uniformly [15N]- or [13C/15N]-labeled protein in GDP and GMPPNP-bound states in 20 mM HEPES pH 7.4, 150 mM NaCl, 2 mM MgCl2, 1 mM TCEP and 5% D2O in susceptibility matched 5 mm Shigemi microtubes. Anisotropic samples additionally contained 20 mg/ml Pf1 phage particles (Asla Biotech, Riga, Latvia), resulting in an HDO deuterium signal splitting of 13.1 Hz in lines with 1 Hz linewidths, or 6% w/v of q = 3.0 DMPC/DHPC bicelles (Avanti Polar Lipids, Alabaster, AL), resulting in an HDO deuterium signal splitting of 10.2 Hz in lines with 1 Hz linewidths. All RDC NMR spectra were acquired on a Bruker 700 MHz NMR spectrometer at 25 °C (except the bicelles RDC spectra, measured at 32 °C). 1DNH and 1DCaHa RDCs were measured using a 3D-HNCO variant of the ARTSY54 experiment and a 3D HCA(CO)N antiphase 1H-coupled in the 13Cα dimension experiment, respectively. The 1DNC’ and 2DHNC’ were measured from a 3D IPAP-J-HNCO pulse sequence included in the Bruker spectrometer pulse sequence library. All RDC experiments were acquired with sufficient S/N to ensure low experimental errors, as represented by the error bars in Supplementary Figs. 5, 6, and 7 (some become smaller than the dot size for experimental RDCs with large ranges of magnitude). Experimental values of all RDCs are provided as a Supplementary Data 1.
Since each RDC set measured in a particular alignment medium does not sample all possible spatial bond orientations in the alignment tensor frame (or, equivalently, in the molecular frame), the RDC-fitted magnitude of the corresponding alignment tensor using singular value decomposition (SVD) is often underestimated by about 10%. In contrast, the rhombicity of the tensor is unaffected. We performed a grid search that imposed slightly larger alignment tensor magnitudes to compensate for this ‘sparsity underestimate’ and determined the optimal magnitude of the alignment tensors for each alignment medium using all experimental RDCs measured in that medium and the crystal structure (State 1). These corrected alignment tensor magnitudes provided the best RDC fit to the State I crystal structure (lowest R factors33) for all types of RDCs measured in each alignment medium. The result was indeed between 7% and 12% larger than those obtained in a simple SVD fit of any individual set of RDCs. We subsequently fixed these optimal values of each alignment tensor throughout the structure calculations of the two-state ensemble.
Structure calculations
We used Xplor-NIH55 to perform an N = 2 ensemble RDC refinement protocol in torsion angle coordinates, during which one ensemble member was fixed throughout to the crystal structure of State 1 (ground state). Since we were not refining the positions of the switch regions (due to the lack of NMR restraints) and the high-resolution crystal structures of State 1 and State 2 conformations are highly similar (RMSD of only 0.1–0.2 Å when the switch regions are excluded), either structural KRASG13D model could have been equally employed as a starting point of the N = 2 ensemble refinement. Consequently, the calculated GMPPNP-bound- KRASG13D minor state represents the intermediate state, while its switch regions remain close to their State 1 conformation. The refinement protocol utilized population-weighted RDC and Talos-N dihedral angle restraints from NMR experiments in addition to standard Xplor-NIH energy terms, using a script that allows up to 3 Å departures from the initial (crystal structure) atomic positions in a non-crystallographic symmetry type of potential. All 3D structural representations were made in PyMol56.
Crystallization and structure determination of active KRASG13D and KRASG13D-RAF1-RBD complex
To crystallize KRASG13D bound to GMPPNP, we first carried out nucleotide exchange to replace GDP with GMPPNP using the protocol described previously57. Crystallization screenings were carried out using the sitting-drop vapor diffusion method by mixing the protein (8 mg/ml) with an equal volume of reservoir solution. Initial crystals were obtained using a reservoir solution consisting of 100 mM Tris pH 8.5, 20% PEG 400, 20% PEG 8000, and 50 mM MgCl2. To improve crystal quality, microseeding was used to generate single 3D crystals from the initial rod clusters. Crystals were flash-frozen in liquid nitrogen without any additional cryo-protectant. A crystallization sample of KRASG13D bound to RAF1-RBD was prepared as described previously58, and crystals were obtained using the reservoir solution containing 0.15 M KBr and 30% PEG-MME2K. Diffraction data were collected on a 24-ID-E beamline at Advanced Photon Source (APS), Argonne National Laboratory (ANL). Data were integrated and scaled using XDS59. Structure solution was obtained with molecular replacement using Phaser as implemented in the Phenix programs suite, using the GMPPNP-bound KRASQ61H mutant (PDB: 3GFT, the best GMPPNP-bound KRAS model available at the time) as the search model for KRASG13D bound to GMPPNP and KRAS-RAF1(RBD) complex (PDB: 6VJJ) as a search model for the structure of KRASG13D bound to RAF1-RBD60. Iterative model building and refinement were performed with COOT61 and Phenix.Refine62. Crystal parameters, data collection statistics, and refinement statistics are summarized in Table 1. The SBGrid consortium63 provided crystallographic and structural analysis software support.
ITC measurements
A MicroCal PEAQ-ITC calorimeter (Malvern) was used to perform ITC binding studies. GMPPNP-bound KRAS and RAF1-RBD used in ITC measurements were extensively dialyzed in a buffer containing 150 mM NaCl or 300 mM NaCl with 20 mM HEPES (pH 7.3), 5 mM MgCl2, and 1 mM TCEP. The concentrations of the proteins were measured using absorbance at 280 nm in a NanoDrop 2000c spectrophotometer (Thermo Fisher Scientific). Before each ITC run, protein samples were centrifuged at 14,000 g for 5 min at 4 °C to remove debris and air bubbles. ITC titrations were performed at 25 °C by an initial injection of 0.4 μL followed by 18 injections of 2.2 μL of RAF1-RBD proteins at 150 s intervals into a cell containing KRAS mutant proteins. Data analysis was performed using a “one set of sites” model using the MicroCal PEAQ-ITC analysis software (v1.41, Malvern Panalytical) to obtain thermodynamic parameters (ΔG, ΔH, and −TΔS), curve fitting, equilibrium dissociation constant, and molar ratio calculations. All binding and thermodynamic data obtained from ITC experiments (n = 1) are tabulated in Supplementary Table 2. Errors in the KD and ΔH values correspond to the standard error of fitting. The ITC raw data are provided as Supplementary Data 3 and 4.
Statistics and reproducibility
All the NMR data, X-ray crystallography data, and ITC data were carried out as single measurements (no replicates).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
GMPPNP-bound KRASG13D assignments were performed using NMRFAM-Sparky50 and were deposited in BMRB (Entry: 51642). The atomic coordinates and structure factors of the GMPPNP-bound KRASG13D and KRASG13D in complex with RAF1-RBD were deposited in the Protein Data Bank with accession codes 8EBZ and 8EPW, respectively. Experimental values of all RDCs, relaxation dispersion intensities and analysis data, and ITC raw data are provided as Supplemental Excel spreadsheets. All other data are available from the corresponding authors on reasonable request.
References
Simanshu, D. K., Nissley, D. V. & McCormick, F. RAS proteins and their regulators in human disease. Cell 170, 17–33 (2017).
Moghadamchargari, Z. et al. Molecular assemblies of the catalytic domain of SOS with KRas and oncogenic mutants. Proc. Natl Acad. Sci. 118, e2022403118 (2021).
Hunter, J. C. et al. Biochemical and structural analysis of common cancer-associated KRAS mutations. Mol. Cancer Res. 13, 1325–1335 (2015).
Johnson, C. W. et al. Isoform-specific destabilization of the active site reveals a molecular mechanism of intrinsic activation of KRas G13D. Cell Rep. 28, 1538–1550.e1537 (2019).
Smith, M. J., Neel, B. G. & Ikura, M. NMR-based functional profiling of RASopathies and oncogenic RAS mutations. Proc. Natl Acad. Sci. USA 110, 4574–4579 (2013).
Rabara, D. et al. KRAS G13D sensitivity to neurofibromin-mediated GTP hydrolysis. Proc. Natl Acad. Sci. USA 116, 22122–22131 (2019).
Cogoi, S. & Xodo, L. E. G4 DNA in ras genes and its potential in cancer therapy. Biochim Biophys. Acta 1859, 663–674 (2016).
Kerkour, A. et al. High-resolution three-dimensional NMR structure of the KRAS proto-oncogene promoter reveals key features of a G-quadruplex involved in transcriptional regulation. J. Biol. Chem. 292, 8082–8091 (2017).
Carvalho, J. et al. Study of the interaction between indole-based compounds and biologically relevant G-quadruplexes. Biochimie 135, 186–195 (2017).
Carvalho, J. et al. Fluorescent light-up acridine orange derivatives bind and stabilize KRAS-22RT G-quadruplex. Biochimie 144, 144–152 (2018).
Caterino, M. et al. Selective binding of a bioactive porphyrin-based photosensitizer to the G-quadruplex from the KRAS oncogene promoter. Int J. Biol. Macromol. 145, 244–251 (2020).
Geyer, M. et al. Conformational transitions in p21ras and in its complexes with the effector protein Raf-RBD and the GTPase activating protein GAP. Biochemistry 35, 10308–10320 (1996).
Ito, Y. et al. Regional polysterism in the GTP-Bound Form of the Human c-Ha-Ras Protein. Biochemistry 36, 9109–9119 (1997).
Pantsar, T. The current understanding of KRAS protein structure and dynamics. Comput Struct. Biotechnol. J. 18, 189–198 (2020).
Simanshu, D. K. & Morrison, D. K. A structure is worth a thousand words: new insights for RAS and RAF regulation. Cancer Discov. 12, 899–912 (2022).
Lu, S. et al. Ras conformational ensembles, allostery, and signaling. Chem. Rev. 116, 6607–6665 (2016).
Grant, B. J. et al. Novel allosteric sites on Ras for lead generation. PLoS One 6, e25711 (2011).
Al-Hashimi, H. M. Biochemistry. Exciting structures. Science 329, 1295–1296 (2010).
Kalbitzer, H. R. et al. Intrinsic allosteric inhibition of signaling proteins by targeting rare interaction states detected by high-pressure NMR spectroscopy. Angew. Chem. Int. Ed. Engl. 52, 14242–14246 (2013).
O’Connor, C. & Kovrigin, E. L. Global conformational dynamics in ras. Biochemistry 47, 10244–10246 (2008).
Chao, F. A. et al. Insights into the Cross Talk between effector and allosteric lobes of KRAS from methyl conformational dynamics. J. Am. Chem. Soc. 144, 4196–4205 (2022).
Chen, X. et al. Extending the lifetime of native GTP-bound ras for site-resolved NMR measurements: quantifying the allosteric dynamics. Angew. Chem. Int. Ed. Engl. 58, 2730–2733 (2019).
Long, D. et al. A comparative CEST NMR study of slow conformational dynamics of small GTPases complexed with GTP and GTP analogues. Angew. Chem. Int. Ed. Engl. 52, 10771–10774 (2013).
Palfy, G. et al. The importance of Mg(2+) -free state in nucleotide exchange of oncogenic K-Ras mutants. Chemistry 28, e202201449 (2022).
Lundstrom, P., Vallurupalli, P., Religa, T. L., Dahlquist, F. W. & Kay, L. E. A single-quantum methyl 13C-relaxation dispersion experiment with improved sensitivity. J. Biomol. NMR 38, 79–88 (2007).
Clore, G. M. & Schwieters, C. D. How much backbone motion in ubiquitin is required to account for dipolar coupling data measured in multiple alignment media as assessed by independent cross-validation? J. Am. Chem. Soc. 126, 2923–2938 (2004).
Clore, G. M. & Schwieters, C. D. Amplitudes of protein backbone dynamics and correlated motions in a small alpha/beta protein: correspondence of dipolar coupling and heteronuclear relaxation measurements. Biochemistry 43, 10678–10691 (2004).
Spoerner, M. et al. Conformational states of Ras complexed with the GTP analogue GppNHp or GppCH2p: implications for the interaction with effector proteins. Biochemistry 44, 2225–2236 (2005).
Spoerner, M. et al. Conformational states of human rat sarcoma (Ras) protein complexed with its natural ligand GTP and their role for effector interaction and GTP hydrolysis. J. Biol. Chem. 285, 39768–39778 (2010).
Sydor, J. R., Engelhard, M., Wittinghofer, A., Goody, R. S. & Herrmann, C. Transient kinetic studies on the interaction of Ras and the Ras-binding domain of c-Raf-1 reveal rapid equilibration of the complex. Biochemistry 37, 14292–14299 (1998).
Bax, A. & Grishaev, A. Weak alignment NMR: a hawk-eyed view of biomolecular structure. Curr. Opin. Struct. Biol. 15, 563–570 (2005).
Shen, Y. & Bax, A. Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR 56, 227–241 (2013).
Clore, G. M. & Garrett, D. S. R-factor, free R, and complete cross-validation for dipolar coupling refinement of NMR structures. J. Am. Chem. Soc. 121, 9008–9012 (1999).
Stiller, J. B. et al. Structure determination of high-energy states in a dynamic protein ensemble. Nature 603, 528–535 (2022).
Xie, T., Saleh, T., Rossi, P. & Kalodimos, C. G. Conformational states dynamically populated by a kinase determine its function. Science 370, eabc2754 (2020).
Vallurupalli, P., Hansen, D. F. & Kay, L. E. Structures of invisible, excited protein states by relaxation dispersion NMR spectroscopy. Proc. Natl Acad. Sci. USA 105, 11766–11771 (2008).
Spoerner, M., Herrmann, C., Vetter, I. R., Kalbitzer, H. R. & Wittinghofer, A. Dynamic properties of the Ras switch I region and its importance for binding to effectors. Proc. Natl Acad. Sci. USA 98, 4944–4949 (2001).
Girard, E. et al. Equilibria between conformational states of the Ras oncogene protein revealed by high pressure crystallography. Chem. Sci. 13, 2001–2010 (2022).
Vatansever, S., Gümüş, Z. H. & Erman, B. Intrinsic K-Ras dynamics: a novel molecular dynamics data analysis method shows causality between residue pair motions. Sci. Rep. 6, 37012 (2016).
Marabese, M. et al. G48A, a new KRAS mutation found in lung adenocarcinoma. J. Thorac. Oncol. 11, 1170–1175 (2016).
Day, G. J., Mosteller, R. D. & Broek, D. Distinct subclasses of small GTPases interact with guanine nucleotide exchange factors in a similar manner. Mol. Cell Biol. 18, 7444–7454 (1998).
Yang, M. et al. Allosteric regulation of switch-II controls K-Ras oncogenicity. Preprint at https://doi.org/10.1101/2022.09.20.508702 (2022).
Taylor, T., Denson, J. P. & Esposito, D. Optimizing Expression and Solubility of Proteins in E. coli Using Modified Media and Induction Parameters. Methods Mol. Biol. 1586, 65–82 (2017).
Agamasu, C. et al. KRAS Prenylation Is Required for Bivalent Binding with Calmodulin in a Nucleotide-Independent Manner. Biophys. J. 116, 1049–1063 (2019).
Travers, T. et al. Molecular recognition of RAS/RAF complex at the membrane: Role of RAF cysteine-rich domain. Sci. Rep. 8, 8461 (2018).
Li, J. & Byrd, R. A. A simple protocol for the production of highly deuterated proteins for biophysical studies. J. Biol. Chem. 298, 102253 (2022).
Kopra, K. et al. Homogeneous dual-parametric-coupled assay for simultaneous nucleotide exchange and KRAS/RAF-RBD interaction monitoring. Anal. Chem. 92, 4971–4979 (2020).
Chao, F.-A., Li, Y., Zhang, Y. & Byrd, R. A. Probing the broad time scale and heterogeneous conformational dynamics in the catalytic core of the Arf-GAP ASAP1 via methyl adiabatic relaxation dispersion. J. Am. Chem. Soc. 141, 11881–11891 (2019).
Chao, F.-A., Khago, D. & Byrd, R. A. Achieving pure spin effects by artifact suppression in methyl adiabatic relaxation experiments. J. Biomol. NMR 74, 223–228 (2020).
Lee, W., Tonelli, M. & Markley, J. L. NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31, 1325–1327 (2015).
Loria, J. P., Rance, M. & Palmer, A. G. III A. TROSY CPMG sequence for characterizing chemical exchange in large proteins. J. Biomol. NMR 15, 151–155 (1999).
Delaglio, F. et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 (1995).
Ying, J., Delaglio, F., Torchia, D. A. & Bax, A. Sparse multidimensional iterative lineshape-enhanced (SMILE) reconstruction of both non-uniformly sampled and conventional NMR data. J. Biomol. NMR 68, 101–118 (2017).
Fitzkee, N. C. & Bax, A. Facile measurement of (1)H-(1)5N residual dipolar couplings in larger perdeuterated proteins. J. Biomol. NMR 48, 65–70 (2010).
Schwieters, C. D., Bermejo, G. A. & Clore, G. M. Xplor-NIH for molecular structure determination from NMR and other data sources. Protein Sci. 27, 26–40 (2018).
DeLano, W. L. The PyMOL Molecular Graphics System, Version 2.0 (Schrödinger, LLC., 2002).
Dharmaiah, S. et al. Structures of N-terminally processed KRAS provide insight into the role of N-acetylation. Sci. Rep. 9, 10512 (2019).
Tran, T. H. et al. KRAS interaction with RAF1 RAS-binding domain and cysteine-rich domain provides insights into RAS-mediated RAF activation. Nat. Commun. 12, 1176 (2021).
Kabsch, W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr D. Biol. Crystallogr 66, 133–144 (2010).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl Crystallogr 40, 658–674 (2007).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr D. Biol. Crystallogr 66, 486–501 (2010).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D. Biol. Crystallogr 66, 213–221 (2010).
Morin, A. et al. Collaboration gets the most out of software. Elife 2, e01456 (2013).
Acknowledgements
We acknowledge J-P Denson, Matt Drew, Peter Frank, Brianna Higgins, Jennifer Mehalko, Simon Messing, Ashley Mitchell, Jose Sanchez-Hernandez, Mukul Sherekar, Kelly Snead, and Bill Gillette for their help in cloning, protein expression, protein purification, and electrospray ionization mass spectroscopy. CDS was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases. We thank Ad Bax and Andy Byrd for stimulating discussions and Janusz Koscielniak for his continuous support of our NMR Facility. Crystallographic data were collected at the Northeastern Collaborative Access Team beamlines (24-ID-E), funded by the U.S. National Institutes of Health (NIGMS P30 GM124165). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (D.O.E.) Office of Science User Facility operated for the D.O.E. Office of Science by Argonne National Laboratory under Contract DE-AC02-06CH11357. This project was partly financed with federal funds from the National Cancer Institute, National Institutes of Health Contract HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, and the mention of trade names, commercial products, or organizations does not imply endorsement by the U.S. Government.
Funding
Open Access funding provided by the National Institutes of Health (NIH).
Author information
Authors and Affiliations
Contributions
NMR experiments were designed and carried out by F.C. and G.C. The population weighted N = 2 structural ensemble refinement protocol was designed and implemented for Xplor-NIH by C.D.S. ITC experiments and crystallographic work were carried out by S.D., A.H.C., T.H.T, and D.K.S. Sample preparation and quality control were performed by F.C., G.C., T.T., N.R., and D.E. D.V.N. and F.M. provided scientific guidance. Results were analyzed by F.C., A.H.C., D.K.S., and G.C. The manuscript was prepared by F.C., A.H.C., D.K.S., and G.C.
Corresponding authors
Ethics declarations
Competing interests
All authors, except F.M., declare no competing interests. F.M. is a consultant for Amgen, Daiichi, Frontiers Med, Exuma Biotech, Ideaya Biosciences, Kura Oncology, Leidos Biomedical Research, PellePharm, Pfizer, P.M.V. Pharma, and Quanta Therapeutics. F.M. is a consultant and co-founder for BridgeBio, Olema Pharmaceuticals, and Quartz. F.M. has received research grants from Daiichi Sankyo and Gilead Sciences and has a current grant from Boehringer-Ingelheim.
Peer review
Peer review information
Communications Biology thanks Gyula Pálfy, Nicholas Martinez and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Gene Chong. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chao, FA., Chan, A.H., Dharmaiah, S. et al. Reduced dynamic complexity allows structure elucidation of an excited state of KRASG13D. Commun Biol 6, 594 (2023). https://doi.org/10.1038/s42003-023-04960-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-023-04960-6
- Springer Nature Limited