
Abstract
Venous thromboembolism (VTE) is a complex, multifactorial life-threatening disease that involves vascular endothelial cell (VEC) dysfunction. However, the exact pathogenesis and underlying mechanisms of VTE are not completely clear. The aim of this study was to identify the core genes and pathways in VECs that are involved in the development and progression of unprovoked VTE (uVTE). The microarray dataset GSE118259 was downloaded from the Gene Expression Omnibus database, and 341 up-regulated and 8 down-regulated genes were identified in the VTE patients relative to the healthy controls, including CREB1, HIF1α, CBL, ILK, ESM1 and the ribosomal protein family genes. The protein–protein interaction (PPI) network and the transcription factor (TF)-miRNA-target gene network were constructed with these differentially expressed genes (DEGs), and visualized using Cytoscape software 3.6.1. Eighty-nine miRNAs were predicted as the targeting miRNAs of the DEGs, and 197 TFs were predicted as regulators of these miRNAs. In addition, 237 node genes and 4 modules were identified in the PPI network. The significantly enriched pathways included metabolic, cell adhesion, cell proliferation and cellular response to growth factor stimulus pathways. CREB1 was a differentially expressed TF in the TF-miRNA-target gene network, which regulated six miRNA-target gene pairs. The up-regulation of ESM1, HIF1α and CREB1 was confirmed at the mRNA and protein level in the plasma of uVTE patients. Taken together, ESM1, HIF1α and the CREB1-miRNA-target genes axis play potential mechanistic roles in uVTE development.
Similar content being viewed by others

Introduction
Venous thromboembolism (VTE) refers to a condition wherein blood clots are formed in the vasculature that block the blood reflux, leading to deep venous thrombosis (DVT) and pulmonary embolism (PE)1. The annual incidence of VTE ranges from 75/100,000 to 136/100,000 in the Western countries, and 10.6% and 23% of these patients within 30 days or 1 year respectively2,3. In Hong Kong, the annual incidence rate is 41.7/100,000 and the 30-day mortality rate is 39.7%4. Approximately 0.1–9% of the PE patients develop chronic thromboembolic pulmonary hypertension and almost 50% of the DVT patients progress to post-thrombotic syndrome within 3 months5. Studies increasingly show that vascular injury and vascular endothelial damage are the risk factors of VTE in both adult and pediatric populations6,7,8. Endothelial cells (ECs) form the vascular intima that lines all blood vessels. Healthy ECs release anti-thrombotic factors such as thrombomodulin and tissue plasminogen activator, whereas injured ECs release coagulants like the von Willebrand factor9. In addition, adhesion between leukocytes and the activated endothelium in injured vessels may activate the coagulation cascade and lead to platelet accumulation10. Although there is evidence of a link between endothelial dysfunction and the initiation and progression of VTE11, the underlying molecular mechanisms and signaling pathways are poorly understood.
Micro RNAs (miRNAs) are a class of small, non-coding RNAs consisting of 18 to 23 nucleotides that regulate target gene expression at the post-transcriptional level, either by blocking target mRNA translation or by promoting its degradation. The miRNA-encoding genes are first transcribed into primary miRNAs by RNA polymerase II, which are subsequently processed into pre-miRNAs by a nuclear protein complex called microprocessor. The pre-miRNAs are cleaved by dicer and loaded into argonaute protein, resulting in the formation of mature miRNAs and the RNA-induced silencing complex that regulates the expression of target genes. However, the regulation of miRNAs biogenesis and function is highly complex, and some miRNAs are generated by drosha-independent or dicer-independent mechanisms. In addition, multiple factors affect miRNAs processing, stability and function, such as post-translational modification of argonaute proteins and changes in the miRNA sequence12,13. Studies increasingly show that miRNAs are involved in the pathogenesis of cardiovascular diseases, and are potential diagnostic biomarkers and therapeutic targets14,15. A recent study reported a substantial number of differentially expressed plasma miRNAs between VTE patients and healthy controls16. For example, miR-424-5p and miRNA-320a/b were up-regulated in VTE patients, and their respective area under the curve (AUC) in the receiver-operating characteristic (ROC) curve analysis for diagnosing VTE were 0.62, 0.70 and 0.7917,18. In addition, miR-145 is known to reduce thrombogenesis by targeting the coagulation factor XI in vivo19, and some miRNAs influence the viability of endothelial progenitor cells in DVT patients and enhance thrombus recanalization and resolution20. However, the potential role of miRNAs in the vascular ECs (VECs) of patients with VTE needs further investigation.
Transcription factors (TFs) regulate gene expression by binding to the target gene promotor sequences adjacent to transcription initiation sites or distal enhancer elements. Recent developments in whole-genome sequencing and functional genomics have identified TFs associated with various physiological and pathological processes, including cardiovascular diseases21 and VTE. For example, hypoxia-inducible factor (HIF)1α, a TF targeting vascular endothelial growth factor (VEGF)22, promoted angiogenic factor production and accelerated thrombus resolution and vein recanalization in a murine inferior vena cava model23. In addition, HIF1α can promote the initiation or development of VTE by enhancing pro-inflammatory and innate immune responses via VEGF and serpin family E member 1 (PAI-1)24. These studies indicate that HIF1α plays an important role in maintaining the balance between thrombolysis and thrombosis. The cAMP response element binding protein (CREB) also drives thrombospondin-1 transcription, and is involved in the formation and maintenance of arterial thrombosis25,26. Wang et al. showed that the TF Yin Yang (YY)1 up-regulated vitronectin expression by binding to the rs2227721-G (rs is an id of dbSNP database and G is a base located at rs222721) variant of its promoter. The G to T mutation in rs2227721 decreased the binding efficiency between YYI and vitronectin promoter and increased risk of DVT, indicating that YY1 is a repressor of thrombosis27. Recent studies also show that TFs can regulate gene expression by binding to the promoter of cognate miRNAs28. A potential role of TFs and the TF-miRNAs axis in the VECs of VTE patients has not been reported so far.
We conducted bioinformatics analysis to identify the differentially expressed genes (DEGs) and pathways in the VECs of patients with unprovoked VTE (uVTE) relative to healthy controls. Furthermore, we also predicted potential miRNAs and TFs regulating the VTE-associated genes and pathways. In addition, the expression levels of some potentially crucial genes were confirmed at the mRNA and protein levels in plasma of uVTE patients with routine molecular biology assays. Our findings provide novel insights into targeted VTE therapy.
Methods
Ethics statement
Ethics approval was waived for microarray expression profiling since publicly available datasets were used for the analysis. Data acquisition and application were as per the guidelines of the gene expression omnibus (GEO) database. All human studies were approved by the Ethics Committee of Liuzhou Worker’s Hospital, Fourth Affiliated Hospital of Guangxi Medical University (Approval Number: 20180518), and conducted according to the relevant guidelines and regulations. Written informed consent was obtained from all subjects.
Study population
A total of 12 patients with uVTE and 12 age- and sex-matched healthy volunteers were enrolled (Supplementary Information-Table S1). All patients had VTE diagnosed as acute, symptomatic DVT of the lower extremities. All uVTE had been objectively confirmed by D-dimer and color doppler examination. Fasting blood samples were collected from all participants and then centrifuged at 3000×g for 10 min at 4 °C. Serum samples were then collected and stored at − 80 °C for qRT-PCR analysis and ELISA.
Identification of DEGs and hierarchical clustering analysis
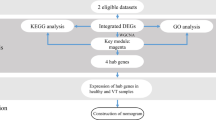
The workflow of this study is shown in Fig. 1. The microarray dataset GSE118259 based on the GPL10558 Platform (Illumina HumanHT-12 V4.0 expression beadchip) was downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo/, accessed November, 2019). It includes data from a total of 13 endothelial colony-forming cell samples isolated from uVTE patients (n = 8) and healthy controls (n = 5). The DEGs between the datasets were screened using the GEO2R online tool with |logFC|> 1 and adj.p value < 0.05 as the cut-off criteria. A volcano plot was constructed and hierarchical clustering analysis was performed using the ggplot2 package and the pheatmap package in R respectively.

Flow diagram of the analysis procedure.
Functional and pathway enrichment analyses
The Database for Annotation, Visualization and Integrated Discovery version 6.8 (DAVID; https://david.ncifcrf.gov/, accessed November, 2019) was used for functional classification and annotation of the DEGs29,30. Gene Ontology (GO), and the Kyoto Encyclopedia of Genes and Genomes (KEGG) and REACTOME pathway enrichment analyses were performed. Homo Sapiens was selected as species and background and p values < 0.05 were considered statistically significant. Gene set enrichment analysis (GSEA) was performed to explore potential mechanisms based on the Molecular Signatures Database (MSigDB) including the c2 (c2. cp. kegg. V7.4. symbol. gmt and c2. cp. wikipathway. V7.4. symbol. gmt) and c5 (c5. go. bp or cc or mf V7.4. symbol. gmt) (http://software.broadinstitute.org). The background of GSEA was Illumina HumanHT-12 V4.0 expression beadchip. The number of permutations was set at 1,000 and nominal P-value < 0.05 and false discovery rate (FDR) < 0.25 were considered statistically significant.
PPI network of DEGs and module analysis
The Search Tool for the Retrieval Interacting Genes (STRING) version 11.0 (https://www.string-db.org/, accessed December, 2019) was used to analyze the interaction of protein-encoding genes31,32,33. All DEGs were uploaded to the STRING online tool to construct the PPI network, which was then visualized using Cytoscape software 3.6.1. “Homo sapiens” and an interaction score > 0.4 (medium confidence) were the parameters. Module analysis of the PPI network was performed with the MOCDE app of Cytoscape software 3.6.1. The parameters for MCODE analysis were MCODE score > 3.5 and the number of nodes > 10.
Construction of the miRNA-target DEG regulatory network
The miRNAs targeting the DEGs were predicted using the miRWalk1.0 (http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk/, accessed December, 2019)34, miRanda (http://www.microrna.org/microrna/home.do, accessed December, 2019)35, miRDB (http://www.mirdb.org/, accessed December, 2019)36, RNA22 (https://cm.jefferson.edu/rna22/, accessed December, 2019)37 and TargetScan (http://www.targetscan.org/vert_72/, accessed December, 2019) databases38. The miRNA- argeting DEG pairs predicted by all five databases were imported into the Cytoscape software 3.6.1 to construct the miRNA-target DEG regulatory network.
Construction of the TF‑miRNA‑target gene regulatory network
The TFs regulating miRNAs were predicted using the iRegulon plug-in (http://apps.cytoscape.org/ apps/iRegulon) of the Cytoscape software 3.6.1, and the Transfac (http://www.gene-regulation.com/pub/databases.html, accessed January, 2020) and Encode (https://www.encodeproject.org/, accessed January, 2020) databases39,40. An enrichment score threshold > 3, a minimum identity of 0.0 between orthologous genes, and a maximum false discovery rate (FDR) of 0.001 on motif similarity were the thresholds. The predicted TFs that were also identified as DEGs were designated differentially expressed transcription factors (DETFs), and used to construct the DETF‑miRNA‑target gene network. In addition, the protein-encoding DEGs with a degree of more than 10 in the PPI network were selected to build the TF-miRNA-target gene network in accordance with the miRNA-target DEG and the TF-miRNA pairs. All networks were built and visualized using Cytoscape software 3.6.1.
RNA extraction and qRT-PCR analysis
Total RNA was isolated from serum samples using TRIzol according to the manufacturer’s instructions (Invitrogen, Carlsbad, California, USA). For each sample, the total RNA content was determined by measuring the absorbance at 260 nm and the purity was ascertained in terms of A260/A280. First strand cDNAs of the miRNAs and mRNAs were synthesized using the Mir-X miRNA first strand synthesis kit and PrimeScript RT reagent kit with gDNA eraser (Takara Biotechnology, Dalian, China) respectively. RT-PCR was performed using 2 µl of each cDNA template and the FastStart SYBR Green Kit (Roche, East Sussex, UK) on the ABI 7500 Fast cycler (Applied Biosystems, Darmstadt, Germany)41. The primer sequences are shown in Supplementary Information-Table S2.
ELISA
The serum levels of gene expression were measured using the appropriate ELISA kits (Solarbio Life Sciences, Beijing, China). Optical density (OD) was measured at 450 nm according to the manufacturer’s instruction.
Statistical analysis
SPSS statistical software for Windows, version 22.0 (SPSS, Chicago, IL, USA), was used for statistical analysis. Groups were compared using t-test and P < 0.05 was considered statistically significant. Data were shown as mean ± standard deviation (SD).
Results
Screening of DEGs and hierarchical clustering analysis
Based on the cutoff criteria of |logFold change| (logFC) > 1 and adj.p value < 0.05, a total of 341 up-regulated and 8 down-regulated endothelial colony-forming cell-specific genes were identified between the patient and control datasets. HIF1α, CBL, ILK and ESM1 had a higher fold change, as shown in the volcano plot (Fig. 2A) and in Supplementary Information-Table S3. The hierarchy cluster analysis clearly showed that the DEGs can distinguish between the VTE patients and control group (Fig. 2B). These results suggest that HIF1α, CBL, ILK and ESM1 play possibly an important role in VTE.

Identification of DEGs and hierarchical clustering analysis. (A) The volcano plot of GSE118259, with red nodes indicating up-regulated genes with logFC > 1 and p value < 0.05, green nodes down-regulated genes with logFC < -1 and p value < 0.05, and blue nodes similarly expressed genes. (B) Heat map of DEGs. The bottom horizontal axis shows sample names and the top horizontal axis shows the sample clusters. Each row represents a single gene and the right vertical axis represents DEG clusters. Colors closer to red indicate high gene expression levels and those closer to green indicate low gene expression levels. Differentially expressed genes, DEGs, Fold change, FC. Created with R soft 3.5.3 (https://mirrors.tuna.tsinghua.edu.cn/CRAN/).
Functional and pathway enrichment analysis
To further elucidate the biological changes in uVTE, the DEGs were annotated for the GO biological process (BP), GO cellular component (CC) and GO molecular function (MF) terms. The significant GO BP terms were cell–cell adhesion (GO:0098609), cell proliferation (GO:0008283), cellular response to DNA damage stimulus (GO:0006974), SRP-dependent cotranslational protein targeting to membrane (GO:0006614), oxidative phosphorylation (GO:0006119), positive regulation of vascular endothelial growth factor production ( GO:0010575) and cellular response to platelet-derived growth factor stimulus and epidermal growth factor stimulus (GO:0036120 and GO:0071364). For the GO CC terms, the DEGs were mainly enriched in focal adhesion (GO:0005925), cell–cell junction (GO:0005911), cell–cell adherens junction (GO:0005913), cytoplasm (GO:0005737), nucleus (GO:0005634) and extracellular exosome (GO:0070062). The main GO MF terms were cadherin binding involved in cell–cell adhesion (GO:0098641), protein binding (GO:0005515), poly(A) RNA binding (GO:0044822), structural constituent of ribosome (GO:0003735) and nucleotide binding (GO:0000166). In addition, metabolic pathways (hsa01100) including metabolism of protein (R-HSA-156827, R-HSA-72689 and R-HSA-72706), metabolism of RNA (R-HSA-975957), Ribosome (hsa03010) and oxidative phosphorylation (hsa00190) were highly enriched among the DEGs (Fig. 3 and Supplementary Information-Table S4). As shown in the GSEA results, the DEGs were significantly enriched in oxidative phosphorylation, peroxisome, positive regulation of behavior, positive regulation of tissue remodeling, negative regulation of interleukin 8 production, cell proliferation, regulation of toll like receptor signaling pathway (Fig. 4 and Table Supplementary Information-Table S5). Cell adhesion was significantly enriched in multiple GO analyses. Given that CBL, ILK, ESM1 were also enriched in cell adhesion, they may regulate VTE progression by influencing the biological behavior of endothelial cells.

GO and pathway enrichment analyses of DEGs. (A–D) represent significant GO Biological Process (BP), GO Molecular Function (MF) and GO Cellular Component (CC) terms, and enriched pathways of the DEGs respectively. Differentially expressed genes, DEGs, Gene ontology, GO. Created with DAVID version 6.8 (https://david.ncifcrf.gov/, accessed November, 2019) and R soft 3.5.3 (https://mirrors.tuna.tsinghua.edu.cn/CRAN/).

GSEA results of uVTE and control group samples. (A–E) GSEA results of reference gene sets regarding GO terms in c5 GO V7.4. symbol. gmt. (F–M) GSEA results of reference gene sets regarding pathways in c2 KEGG V7.4. symbol. gmt and c2 wikipathway V7.4. symbol. gmt. GSEA gene set enrichment analysis, NES normalized enrichment score, uVTE unprovoked Venous Thromboembolism, KEGG Kyoto Encyclopedia of Genes and Genomes, Go Ontology, GO. Created with GSEA soft_4.1.0 (http://www.gsea-msigdb.org/gsea/downloads.jsp).
PPI network and Modular Analysis
The PPI network was constructed to evaluate the relationship between the protein-encoding DEGs, and consisted of 1105 edges corresponding to 237 protein-encoding DEGs. Only ZNF395 was down-regulated, while the remaining 236 DEGs (Fig. 5A) including HIF1α, ILK, CBL and CREB1 were upregulated, of which only CREB1 had a degree ≥ 10. In addition, module analysis using Cytoscape MCODE enriched four modules from the PPI network based on the degree of importance. Module 1 mainly consisted of the ribosomal protein family and 113 edges (Fig. 5B) that were related to the metabolism of proteins, metabolism of RNAs, translational initiation, focal adhesion and binding GO BP terms (Table 1 and Supplementary Information-Table S6). Module 2 contained 29 nodes and 90 edges (Fig. 5C), which were involved in the GO BP terms of poly (A) RNA binding, protein binding and ATP binding (Table 1 and Supplementary Information-Table S6). Module 3 comprised of 17 nodes and 36 edges (Fig. 5D), which were mainly enriched in the GO BP terms of mRNA splicing and RNA binding (Table 1 and Supplementary Information-Table S6), while module 4 was made up of 13 nodes and 22 edges (Fig. 5E) that participated in protein binding and cell proliferation (Table 1 and Supplementary Information-Table S6). These results further indicated that HIF1α, ILK, CBL and CREB1 may play a key role in VTE and and endothelial cell metabolism.

PPI network of the DEGs and module analysis. (A) PPI network constructed using 237 protein-encoding DEGs and 1105 edges. Node size indicates node degree, and the red, green, pink and yellow nodes represent module 1, module 2, module 3 and module 4 genes respectively. (B) Module 1 consists of 16 nodes and 113 edges. (C) Module 2 consists of 29 nodes and 90 edges. (D) Module 3 consists of 17 nodes and 36 edges. (E) Module 4 consists of 13 nodes and 22 edges. In module analysis, node size indicates node degree, edge size and edge color based on combined score. Protein–protein interaction, PPI, Differentially expressed genes, DEGs. Created with STRING soft version 11.0 (https://string-db.org) and cytoscape software 3.6.1 (www.cytoscape.org).
The microRNA–target gene regulatory network
To identify the potential miRNAs regulating initiation and progression of VTE, we screened 89 miRNAs from the miRWalk 1.0 database and 121 miRNA-DEG pairs were identified. Each miRNA-target DEG pair was simultaneously predicted by the miRanda, miRDB, miRWalk, RNA22 and TargetScan databases. The miRNA target DEG network consisted of 89 miRNAs, 52 DEGs and 121 edges, and included the has-miR-18b-HIF1α, has-miR-489-CREB1, has-miR-433-CREB1, has-miR-222-NXN, has-miR-222-SANP23, has-miR-222-YWHAG, has-miR-125b-PRRC1, has-miR-206-HIGD1A, has-miR-29b-ZFP36L1, has-miR-34a-DNM1L and has-miR-135a-ZRANB2 pairs. In addition, HIGD1A, SHE and SLC38A2 were putative hub targeting genes with seven different miRNAs, and hsa-miR-181b was the putative targeting miRNA for 4 DEGs (Fig. 6).

MiRNA-target DEGs interaction networks. Red nodes represent miRNAs and blue nodes represent target DEGs. Arrows indicate miRNA-target relationship. Differentially expressed genes, DEGs. Created with cytoscape software 3.6.1 (www.cytoscape.org).
TF‑miRNA‑target DEG regulatory network analysis
Based on the iRegulon app of Cytoscape software, a total of 197 TFs were predicted as regulators of miRNAs in the miRNA-target gene regulatory network, of which CREB1, ZNF281, NR2F2, ETS1, FLI1 and YY1 were the DETFs. The TF-miRNA regulatory network consisted of 2839 TF-miRNA pairs, 197 TFs and 58 miRNAs (Fig. 7A). CREB1 regulated 6 miRNAs–hsa-miR-34a, hsa-miR-29b, hsa-miR-222, hsa-miR-206, hsa-miR-135a and hsa-miR-125b – in the TF-miRNA network. Among the miRNA- DEG pairs, the protein-encoding DEGs with a degree ≥ 10 in the PPI network (CREB1, HIF1A, ILK, YY1, MYC etc.) were screened to construct the TF‑miRNA‑target DEG regulatory network. There were 12 miRNA-DEG pairs, 528 TF-miRNA pairs, 197 nodes and 540 edges in this network (Fig. 7B). CREB1 and other five DETFs were also used to construct a DETF‑miRNA‑target network, which consisted of 60 nodes, 111 edges, and the 6 CREB1-miRNA pairs and their corresponding 8 miRNA-target gene pairs (Fig. 7C). Finally, based on all the protein-coding DEGs and the miRNAs targeting and targeted by CREB1, a comprehensive CREB1 regulatory network was constructed (Fig. 8). The results indicated that some DEGs, such as CREB1, HIF1A etc., are associated with multiple regulatory networks in the progression of VTE.

Transcription factor (TF) regulatory network. (A) TF-miRNA regulatory network. the red and green nodes indicate miRNAs and TFs, respectively. (B) TF-miRNA-target DEGs regulatory network. Blue nodes represent DEGs with a degree > 10 in the PPI network. (C) DETFs-miRNA-target regulatory network. Blue nodes represent DEGs, and the red and green nodes indicate miRNAs and DETFs respectively. The arrow indicates the TF-miRNA target relationship and the T shape represents the miRNA-target DEG relationship. TF, Transcription factor, DEGs, Differentially expressed genes, PPI, Protein–protein interaction, DETFs, Differentially expressed Transcription Factors. Created with cytoscape software 3.6.1 (www.cytoscape.org).

CREB1 regulatory network. Green node indicates CREB1. Red nodes indicate predicted target miRNAs of CREB1. Blue nodes represent target DEGs of miRNAs. Purple nodes represent predicted miRNAs regulating CREB1. Yellow nodes represent DEGs interacting with CREB1 in the PPI network. The arrow indicates the CREB1-miRNA target relationship and the T shape represents the miRNA-target relationship. The circle represents the miRNA-CREB1 target relationship and the diamond represents CREB1-DEGs relationship. Differentially expressed genes, DEGs, Protein–protein interaction, PPI. Created with cytoscape software 3.6.1 (www.cytoscape.org).
Potential crucial genes levels in plasma of uVTE patients
To further confirm the biological role of these identified DEGs in VTE, we measured the levels of ESM1, HIF1α, CBL, ILk, CREB1, hsa-miR-18b, hsa-miR-34a, hsa-miR-135a, hsa-miR-29b, has-miR-489 and has-miR-433 levels in the plasma samples of uVTE patients. As shown in Fig. 9, ESM1, HIF1α, CREB1, hsa-miR-34a and hsa-miR-135a were upregulated, whereas hsa-miR-18b was downregulated in the patient samples. Furthermore, has-miR-18b was negatively correlated with HIFa (Fig. 9). Consistent with this, the ESM1, HIF1α and CREB1 protein levels were also up-regulated in uVTE patients (Fig. 10 and Supplementary Information-Table S7). In contrast, CBL, ILk and other miRNAs were not statistically significant and may need further verification on a larger cohort. Taken together, ESM1, HIF1α and CREB1 are crucial genes involved in VTE progression.

Expression levels of some crucial genes in the plasma of uVTE patients. (A–E) ESM1, HIF1α and CREB1 mRNA levels in the plasma of uVTE patients and healthy controls. (D–F) hsa-miR-18b, hsa-miR-34a, hsa-miR-135a levels in the plasma of uVTE patients and healthy controls. G, Correlation analysis between HIFa and has-miR-18b. Unprovoked Venous Thromboembolism, uVTE, quantitative Real-time- Polymerase Chain Reaction, qRT-PCR.

The proteins levels of some crucial genes in the plasma of uVTE patients. (A–C) ESM1, HIF1α and CREB1 protein levels in the plasma of uVTE patients and healthy controls. Unprovoked Venous Thromboembolism, uVTE, Enzyme Linked Immune Sorbent Assay, ELISA.
Discussion
We identified several putative uVTE-associated genes and pathways, along with one TF-miRNA-target gene axis, in human ECs. CREB1, ESM1, HIF1α, CBL, ILK and ribosomal protein family mRNAs were differentially expressed between the uVTE and control groups. In addition, ESM1, CBL, ILK, CREB1 and HIF1α were significantly enriched in biological processes of cell adhesion and cellular response to growth factor stimulus respectively. CREB1 was identified as a DETF involved in the TF-miRNA-target gene axis. The altered expression levels of ESM1, HIF1α, CREB1, hsa-miR-34a, hsa-miR-135a and hsa-miR-18b in uVTE was confirmed in patient plasma samples. Taken together, the aforementioned factors likely regulate the miRNA/target gene axis which activates EC adhesion and initiates VEC injury in uVTE.
Bella et al42 also found that the TNFSF15–TNFRSF25 axis is upregulated in uVTE and involved in endothelial dysfunction. Although the same microarray data was used in both studies, the results were different, which can be attributed to the distinct methodologies and aims. While Bella et al. focused on the genes related to the regulation of endothelial function, we applied systems biology to identify multiple crucial genes and pathways and construct a regulatory network.
Vascular endothelial damage is a major risk factor of thrombosis, and may disrupt the balance between coagulation and anticoagulant systems following decreased fibrinolysis43. In this study, ECs derived from VTE patients and healthy controls were analyzed, and a large number of DEGs were enriched in metabolism of protein, metabolism of RNA, mRNA transcription and translation, and cellular response to DNA damage stimulus pathways, as well as the GO term of cell proliferation. We hypothesize therefore that ECs increase the proliferative capacity of ECs after endothelial damage as a self-healing mechanism. In a stasis-induced rat DVT study, the majority of DEGs between the thrombosis and control groups were mainly expressed by ECs, and therefore enriched in EC-related functions. In addition, the top three enriched pathways or biological processes of DEGs were also related to metabolism and cell growth44. We also constructed a PPI network of the DEGs, and found that some genes in module 1 and module 3, for e.g. ribosomal proteins and thioredoxin like 1 (TXNL1), are associated with apoptosis45,46. However, some genes in module 2 and module 4 are associated with cell proliferation and cell migration, such as proteasome 26S subunit, non-ATPase 14 (PSMD14), MYC proto-oncogene and YY147,48,49. These results indicate that altered EC viability may lead to vascular injury and even VTE.
Since neutrophils are the first cells to adhere to injured vessels via the activated endothelium10,50, changes in EC adhesion may be associated with thrombosis. Consistent with this, the DEGs in our study were enriched in GO terms like cell–cell adhesion, cell adhesion molecule binding and protein binding involved in cell adhesion, further underscoring their role in VTE. ESM1, CBL and ILK were enriched in the biological process of cell adhesion, and ESM1 was the top-ranked DEG in this study. Since it is an established predictor of sepsis severity51, ESM1 is also likely associated with the coagulation cascade. Mosevoll et al. reported that plasma ESM1 levels, in combination with plasma E-selectin, C-reactive protein and D-dimer, can be used to distinguish DVT52. Consistent with this, ESM1 mRNA and protein levels were significantly up-regulated in the plasma of uVTE patients. Therefore, we hypothesize that ESM1 participates in VTE by enhancing EC adhesion. CBL and ILK promote thrombosis by regulating platelet activation, and CBL increases platelet aggregation and thrombosis via αIIbβ3 and GPVI signaling53,54. ILK levels are increased in arterial thrombosis patients, and ILK-/- mice showed reduced platelet activation and subsequent aggregation, leading to unstable thrombosis55,56. Both CBL and ILK were upregulated in the uVTE patients in our study, albeit not significantly. A larger cohort may be required to further verify their expression levels in uVTE patients. Finally, factors involved in cell adhesion are significantly up-regulated in the plasma of VTE patients as well as mouse models57,58,59,60,61. Taken together, excessive EC adhesion is the major underlying mechanism of VTE, and is mediated by ESM1, CBL and ILK.
GO analysis also showed the enrichment of biological processes such as cellular response to platelet-derived growth factor (PDGF) stimulus, epidermal growth factor (EGF) stimulus and VEGF. Consistent with our findings, a previous study showed that the target genes of circulating miRNAs in DVT patients were enriched in the VEGF signaling pathway62. In addition, high-throughput affinity plasma proteomic profiling of VTE patients showed that PDGF-β was an independent risk factor of thrombosis-related disorders63, and HIF1α and CREB1 were enriched in growth factor related-GO BP terms with VEGF as the common target in murine macrophages22,64. VEGF is a risk factor of VTE, and regulates proliferation and migration of ECs. High levels of VEGF have been reported in cancer-related VTE65,66. It also induces tissue factor, the initiator of the coagulation cascade, and activates the fibrinolytic system by inducing the tissue-type plasminogen activator (tPA), urokinase-type plasminogen activator (uPA) and plasminogen activator inhibitor 167. Interestingly, CREB and its coactivator protein p300 can maintain functional integrity of the HIF-1a/VEGF transcription machinery and subsequent angiogenesis68. In this study, we found that CREB1 and HIFa were also up-regulated in the plasma of uVTE patients. Taken together, enrichment of growth factor-related GO terms in the uVTE-associated DEGs are closely associated with endothelial dysfunction, and the CREB-HIF-1a/VEGF axis likely plays a crucial role in VTE.
In the PPI network, some genes in module 2 were associated with HIF1α, which promotes SOCS box containing 1 (WSB1) expression in hypoxic cells, indicating that module 2 mediates HIF-1 signaling pathway to initiate VTE69. In addition, HIF1α was also a predicted target of hsa-miR-18b and was negatively correlated with has-miR-18b in this study. Similar to our study, Dolt et al. found that the HIF1α mRNA 3'UTR is highly conserved across humans, mice and rats, which is indicative of its biological significance70. Wang et al. and Chen et al. found that overexpression or inhibition of miR-18b by mimics or inhibitors respectively can suppress or promote HIFα expression in normal human trophoblast cell lines and malignant melanoma cell lines. Furthermore, Chen et al. confirmed that HIFα is a direct target gene of miR-18b based on luciferase reporter assay. In addition, CREB1 was predicted as the targeting gene of has-miR-489 and has-miR-433, and 6 other miRNAs were predicted as CREB1 targets. Hsa-miR-34a and hsa-miR-135a were also up-regulated in the plasma of uVTE patients, whereas hsa-miR-18b was down-regulated. Several DEGs encoding ribosomal proteins were also identified and enriched in module 1. The ribosomal family proteins are upregulated in the plasma of VTE patients71,72, and are known to trigger venous thrombus formation in aged mice via p53 activation73. Furthermore, ribosomal proteins are also detected in circulating platelets and are associated with VTE74,75. Therefore, based on the above results, we hypothesize that the hsa-miR-18b/HIF1α/VEGF axis, the CREB1-miRNA-target genes axis and the ribosomal protein family genes are associated with VTE and need to be investigated further.
Our findings provide new insights into the pathogenesis of uVTE and identify potential diagnostic and therapeutic targets/pathways. However, further experimental studies and independent cohort studies are needed to validate these findings.
Data availability
The data used to support the findings of this study are available in the supplementary information.
References
Day, I. S. C. F. W. T. Thrombosis: A major contributor to global disease burden. Thromb. Res. 134, 931–938. https://doi.org/10.1016/j.thromres.2014.08.014 (2014).
Wang, K. L. et al. The diagnosis and treatment of venous thromboembolism in Asian patients. Thromb. J. 16, 4. https://doi.org/10.1186/s12959-017-0155-z (2018).
Cohen, A. et al. Managing venous thromboembolism in Asia: Winds of change in the era of new oral anticoagulants. Thromb. Res. 130, 291–301. https://doi.org/10.1016/j.thromres.2012.05.025 (2012).
Law, Y., Chan, Y. C. & Cheng, S. W. K. Epidemiological updates of venous thromboembolism in a Chinese population. Asian J. Surg. 41, 176–182. https://doi.org/10.1016/j.asjsur.2016.11.005 (2018).
Tagalakis, V., Patenaude, V., Kahn, S. R. & Suissa, S. Incidence of and mortality from venous thromboembolism in a real-world population: The Q-VTE Study Cohort. Am. J. Med. 126(832), e813-821. https://doi.org/10.1016/j.amjmed.2013.02.024 (2013).
Bagot, C. N. & Arya, R. Virchow and his triad: A question of attribution. Br. J. Haematol. 143, 180–190. https://doi.org/10.1111/j.1365-2141.2008.07323.x (2008).
Allen, C. J. et al. Risk factors for venous thromboembolism after pediatric trauma. J. Pediatr. Surg. 51, 168–171. https://doi.org/10.1016/j.jpedsurg.2015.10.033 (2016).
Frank, B. et al. Venous thromboembolism after major venous injuries: Competing priorities. J. Trauma Acute Care Surg. 83, 1095–1101. https://doi.org/10.1097/TA.0000000000001655 (2017).
Blann, A. D. How a damaged blood vessel wall contibutes to thrombosis and hypertension. Pathophysiol. Haemost. Thromb. 33, 445–448 (2003).
Darbousset, R. et al. Tissue factor-positive neutrophils bind to injured endothelial wall and initiate thrombus formation. Blood 120, 2133–2143. https://doi.org/10.1182/blood-2012-06-437772 (2012).
Poredos, P. & Jezovnik, M. K. Endothelial dysfunction and venous thrombosis. Angiology 69, 564–567. https://doi.org/10.1177/0003319717732238 (2018).
Gebert, L. F. R. & MacRae, I. J. Regulation of microRNA function in animals. Nat. Rev. Mol. Cell. Biol. 20, 21–37. https://doi.org/10.1038/s41580-018-0045-7 (2019).
Treiber, T., Treiber, N. & Meister, G. Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat. Rev. Mol. Cell Biol. 20, 5–20. https://doi.org/10.1038/s41580-018-0059-1 (2019).
Lucas, T., Bonauer, A. & Dimmeler, S. RNA therapeutics in cardiovascular disease. Circ. Res. 123, 205–220. https://doi.org/10.1161/CIRCRESAHA.117.311311 (2018).
Zhou, S. S. et al. miRNAS in cardiovascular diseases: Potential biomarkers, therapeutic targets and challenges. Acta Pharmacol. Sin. 39, 1073–1084. https://doi.org/10.1038/aps.2018.30 (2018).
Starikova, I. et al. Differential expression of plasma miRNAs in patients with unprovoked venous thromboembolism and healthy control individuals. Thromb. Res. 136, 566–572. https://doi.org/10.1016/j.thromres.2015.07.005 (2015).
Jiang, Z. et al. Combination of circulating miRNA-320a/b and D-dimer improves diagnostic accuracy in deep vein thrombosis patients. Med. Sci. Monit. 24, 2031–2037. https://doi.org/10.12659/msm.906596 (2018).
Wang, X. et al. Diagnostic potential of plasma microRNA signatures in patients with deep-vein thrombosis. Thromb. Haemost. 116, 328–336. https://doi.org/10.1160/TH16-01-0071 (2016).
Sahu, A. et al. MicroRNA-145 impedes thrombus formation via targeting tissue factor in venous thrombosis. EBioMedicine 26, 175–186. https://doi.org/10.1016/j.ebiom.2017.11.022 (2017).
Kong, L. et al. Upregulation of miR-483–3p contributes to endothelial progenitor cells dysfunction in deep vein thrombosis patients via SRF. J. Transl. Med. 14, 15. https://doi.org/10.1186/s12967-016-0775-2 (2016).
Zhou, H. et al. Activating transcription factor 3 in cardiovascular diseases: A potential therapeutic target. Basic Res. Cardiol. 113, 37. https://doi.org/10.1007/s00395-018-0698-6 (2018).
Zhang, Z., Yin, D. & Wang, Z. Contribution of hypoxia-inducible factor-1alpha to transcriptional regulation of vascular endothelial growth factor in bovine developing luteal cells. Anim. Sci. J. 82, 244–250. https://doi.org/10.1111/j.1740-0929.2010.00832.x (2011).
Evans, C. E. et al. Upregulation of hypoxia-inducible factor 1 alpha in local vein wall is associated with enhanced venous thrombus resolution. Thromb. Res. 128, 346–351. https://doi.org/10.1016/j.thromres.2011.05.006 (2011).
Bovill, E. G. & van der Vliet, A. Venous valvular stasis-associated hypoxia and thrombosis: What is the link?. Annu. Rev. Physiol. 73, 527–545. https://doi.org/10.1146/annurev-physiol-012110-142305 (2011).
Kuijpers, M. J. et al. Supporting roles of platelet thrombospondin-1 and CD36 in thrombus formation on collagen. Arterioscler. Thromb. Vasc. Biol. 34, 1187–1192. https://doi.org/10.1161/ATVBAHA.113.302917 (2014).
Prakash, P., Kulkarni, P. P. & Chauhan, A. K. Thrombospondin 1 requires von Willebrand factor to modulate arterial thrombosis in mice. Blood 125, 399–406. https://doi.org/10.1182/blood-2014-06-581942 (2015).
Wang, Y. et al. Promoter variants of VTN are associated with vascular disease. Int. J. Cardiol. 168, 163–168. https://doi.org/10.1016/j.ijcard.2012.09.100 (2013).
Zhang, F. et al. miR-25–3p, positively regulated by transcription factor AP-2alpha, regulates the metabolism of C2C12 cells by targeting Akt1. Int. J. Mol. Sci. 19, 773. https://doi.org/10.3390/ijms19030773 (2018).
da Huang, W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57. https://doi.org/10.1038/nprot.2008.211 (2009).
da Huang, W., Sherman, B. T. & Lempicki, R. A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13. https://doi.org/10.1093/nar/gkn923 (2009).
Yi, Y. et al. RAID v20: An updated resource of RNA-associated interactions across organisms. Nucleic Acids Res. 45, D115–D118. https://doi.org/10.1093/nar/gkw1052 (2017).
Zhang, T. et al. RNALocate: A resource for RNA subcellular localizations. Nucleic Acids Res. 45, D135–D138. https://doi.org/10.1093/nar/gkw728 (2017).
Cui, T. et al. MNDR v2.0: An updated resource of ncRNA-disease associations in mammals. Nucleic Acids Res. 46, D371–D374. https://doi.org/10.1093/nar/gkx1025 (2018).
Dweep, H., Gretz, N. & Sticht, C. miRWalk database for miRNA-target interactions. Methods Mol. Biol. 1182, 289–305. https://doi.org/10.1007/978-1-4939-1062-5_25 (2014).
John, B. et al. Human MicroRNA targets. PLoS Biol. 2, e363. https://doi.org/10.1371/journal.pbio.0020363 (2004).
Wong, N. & Wang, X. miRDB: An online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 43, D146-152. https://doi.org/10.1093/nar/gku1104 (2015).
Loher, P. & Rigoutsos, I. Interactive exploration of RNA22 microRNA target predictions. Bioinformatics 28, 3322–3323. https://doi.org/10.1093/bioinformatics/bts615 (2012).
Agarwal, V., Bell, G. W., Nam, J. W. & Bartel, D. P. Predicting effective microRNA target sites in mammalian mRNAs. Elife https://doi.org/10.7554/eLife.05005 (2015).
Kaplun, A. et al. Establishing and validating regulatory regions for variant annotation and expression analysis. BMC Genom. 17, 393. https://doi.org/10.1186/s12864-016-2724-0 (2016).
Consortium, E. P. A user’s guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol. 9, e1001046. https://doi.org/10.1371/journal.pbio.1001046 (2011).
Jiang, W. et al. The involvement of miR-29b-3p in arterial calcification by targeting matrix metalloproteinase-2. Biomed. Res. Int. 2017, 6713606. https://doi.org/10.1155/2017/6713606 (2017).
Della Bella, S. et al. Pathologic up-regulation of TNFSF15-TNFRSF25 axis sustains endothelial dysfunction in unprovoked venous thromboembolism. Cardiovasc. Res. 116, 698–707. https://doi.org/10.1093/cvr/cvz131 (2020).
Verhamme, P. & Hoylaerts, M. F. The pivotal role of the endothelium in haemostasis and thrombosis. Acta Clin. Belg. 61, 213–219. https://doi.org/10.1179/acb.2006.036 (2006).
Jin, Q.-Q. et al. Integrating microRNA and messenger RNA expression profiles in a rat model of deep vein thrombosis. Int. J. Mol. Med. 40, 1019–1028. https://doi.org/10.3892/ijmm.2017.3105 (2017).
Qi, Y. et al. Ribosomal protein L23 negatively regulates cellular apoptosis via the RPL23/Miz-1/c-Myc circuit in higher-risk myelodysplastic syndrome. Sci. Rep. 7, 2323. https://doi.org/10.1038/s41598-017-02403-x (2017).
Wang, C. et al. Newcastle disease virus V protein inhibits apoptosis in DF-1 cells by downregulating TXNL1. Vet. Res. 49, 102. https://doi.org/10.1186/s13567-018-0599-6 (2018).
Byrne, A. et al. Knockdown of human deubiquitinase PSMD14 induces cell cycle arrest and senescence. Exp. Cell Res. 316, 258–271. https://doi.org/10.1016/j.yexcr.2009.08.018 (2010).
Duan, X. et al. FOXP3 inhibits MYC expression via regulating miR-198 and influences cell viability, proliferation and cell apoptosis in HepG2. Cancer Med. https://doi.org/10.1002/cam4.1780 (2018).
Wu, S. et al. Transcription factor YY1 promotes cell proliferation by directly activating the pentose phosphate pathway. Can. Res. 78, 4549–4562. https://doi.org/10.1158/0008-5472.can-17-4047 (2018).
Zarbock, A., Ley, K., McEver, R. P. & Hidalgo, A. Leukocyte ligands for endothelial selectins: Specialized glycoconjugates that mediate rolling and signaling under flow. Blood 118, 6743–6751. https://doi.org/10.1182/blood-2011-07-343566 (2011).
Mihajlovic, D. M. et al. Endocan is useful biomarker of survival and severity in sepsis. Microvasc. Res. 93, 92–97. https://doi.org/10.1016/j.mvr.2014.04.004 (2014).
Mosevoll, K. A., Lindas, R., Wendelbo, O., Bruserud, O. & Reikvam, H. Systemic levels of the endothelium-derived soluble adhesion molecules endocan and E-selectin in patients with suspected deep vein thrombosis. Springerplus 3, 571. https://doi.org/10.1186/2193-1801-3-571 (2014).
Buitrago, L., Tsygankov, A., Sanjay, A. & Kunapuli, S. P. Cbl proteins in platelet activation. Platelets 24, 419–427. https://doi.org/10.3109/09537104.2012.715216 (2013).
Daniel, J. L. et al. Cbl-b is a novel physiologic regulator of glycoprotein VI-dependent platelet activation. J. Biol. Chem. 285, 17282–17291. https://doi.org/10.1074/jbc.M109.080200 (2010).
Jones, C. I. et al. Integrin-linked kinase regulates the rate of platelet activation and is essential for the formation of stable thrombi. J. Thromb. Haemost. 12, 1342–1352. https://doi.org/10.1111/jth.12620 (2014).
Tucker, K. L. et al. A dual role for integrin-linked kinase in platelets: Regulating integrin function and -granule secretion. Blood 112, 4523–4531. https://doi.org/10.1182/blood-2008-03-148502 (2008).
Zapponi, K. C. S. et al. Increased adhesive properties of neutrophils and inflammatory markers in venous thromboembolism patients with residual vein occlusion and high D-dimer levels. Thromb. Res. 133, 736–742. https://doi.org/10.1016/j.thromres.2014.01.035 (2014).
Obi, A. T. et al. Gram-negative pneumonia alters large-vein cell-adhesion molecule profile and potentiates experimental stasis venous thrombosis. J. Vasc. Res. 53, 186–195. https://doi.org/10.1159/000447299 (2016).
Mosevoll, K. A., Lindas, R., Tvedt, T. H., Bruserud, O. & Reikvam, H. Altered plasma levels of cytokines, soluble adhesion molecules and matrix metalloproteases in venous thrombosis. Thromb. Res. 136, 30–39. https://doi.org/10.1016/j.thromres.2015.04.002 (2015).
de Haan, H. G. et al. Genetic variants in Cell Adhesion Molecule 1 (CADM1): A validation study of a novel endothelial cell venous thrombosis risk factor. Thromb. Res. 134, 1186–1192. https://doi.org/10.1016/j.thromres.2014.09.018 (2014).
Antonopoulos, C. N., Sfyroeras, G. S., Kakisis, J. D., Moulakakis, K. G. & Liapis, C. D. The role of soluble P selectin in the diagnosis of venous thromboembolism. Thromb. Res. 133, 17–24. https://doi.org/10.1016/j.thromres.2013.08.014 (2014).
Jiang, Z. et al. Circulating microRNA expression and their target genes in deep vein thrombosis: A systematic review and bioinformatics analysis. Medicine 96, e9330. https://doi.org/10.1097/MD.0000000000009330 (2017).
Bruzelius, M. et al. PDGFB, a new candidate plasma biomarker for venous thromboembolism: Results from the VEREMA affinity proteomics study. Blood 128, e59–e66. https://doi.org/10.1182/blood-2016-05-711846 (2016).
Jeon, S. H. et al. The PKA/CREB pathway is closely involved in VEGF expression in mouse macrophages. Mol. Cells 23, 23–29 (2007).
Posch, F. et al. Soluble vascular endothelial growth Factor (sVEGF) and the risk of venous thromboembolism in patients with cancer: Results from the Vienna Cancer and Thrombosis Study (CATS). Clin. Cancer Res. 22, 200–206. https://doi.org/10.1158/1078-0432.ccr-14-3358 (2015).
Malaponte, G. et al. Increased levels of NF-kB-dependent markers in cancer-associated deep venous thrombosis. PLoS ONE 10, e0132496. https://doi.org/10.1371/journal.pone.0132496 (2015).
Dogan, M. & Demirkazik, A. Venous thromboembolism in patients with cancer and its relationship to the coagulation cascade and vascular endothelial growth factor. Support. Cancer Ther. 3, 28–34. https://doi.org/10.3816/SCT.2005.n.022 (2005).
Ding, L., Yang, M., Zhao, T. & Lv, G. Roles of p300 and cyclic adenosine monophosphate response element binding protein in high glucose-induced hypoxia-inducible factor 1alpha inactivation under hypoxic conditions. J. Diabetes Investig. 8, 277–285. https://doi.org/10.1111/jdi.12592 (2017).
Tong, Y. et al. HIF1 regulates WSB-1 expression to promote hypoxia-induced chemoresistance in hepatocellular carcinoma cells. FEBS Lett. 587, 2530–2535. https://doi.org/10.1016/j.febslet.2013.06.017 (2013).
Dolt, K. S. et al. cDNA cloning, gene organization and variant specific expression of HIF-1 alpha in high altitude yak (Bos grunniens). Gene 386, 73–80. https://doi.org/10.1016/j.gene.2006.08.004 (2007).
Wang, G. et al. Identification of biomarkers of venous thromboembolism by bioinformatics analyses. Medicine 97, e0152. https://doi.org/10.1097/MD.0000000000010152 (2018).
Zhou, T., Zhang, Y., Wu, P., Sun, Q. & Guo, Y. Screening feature genes of venous thromboembolism with DNA microarray. Chem. Biol. Drug Des. 86, 821–828. https://doi.org/10.1111/cbdd.12557 (2015).
Bochenek, M. L. et al. The endothelial tumor suppressor p53 is essential for venous thrombus formation in aged mice. Blood Adv. 2, 1300–1314 (2018).
Tsao, C. H. Rough endoplasmic reticulum and ribosomes in blood platelets. Scand. J. Haematol. 8, 134–140 (1971).
von Bruhl, M. L. et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 209, 819–835. https://doi.org/10.1084/jem.20112322 (2012).
Acknowledgements
This work was supported in part by the self-financing research project of the Health and Family Planning Commission of Guangxi Zhuang Autonomous Region (no. Z20180518).
Author information
Authors and Affiliations
Contributions
P.H. and Y.S. conceived and instructed the work. Y.S., Q.L., Z.Z. and X.W. checked the associated database and analyze raw data. Y.S. wrote and revised the manuscript. All of the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Su, Y., Li, Q., Zheng, Z. et al. Identification of genes, pathways and transcription factor-miRNA-target gene networks and experimental verification in venous thromboembolism. Sci Rep 11, 16352 (2021). https://doi.org/10.1038/s41598-021-95909-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-95909-4
- Springer Nature Limited
This article is cited by
-
Pulmonary artery embolism: comprehensive transcriptomic analysis in understanding the pathogenic mechanisms of the disease
BMC Genomics (2023)
-
CREB1 Silencing Protects Against Inflammatory Response in Rats with Deep Vein Thrombosis Through Reducing RPL9 Expression and Blocking NF-κB Signaling
Journal of Cardiovascular Translational Research (2023)