
Abstract
Background
The pathogenic mechanisms of venous thromboembolism (VT) remain to be defined. This study aimed to identify differentially expressed genes (DEGs) that could serve as potential therapeutic targets for VT.
Methods
Two human datasets (GSE19151 and GSE48000) were analyzed by the robust rank aggregation method. Gene ontology and Kyoto encyclopedia of genes and genomes pathway enrichment analyses were conducted for the DEGs. To explore potential correlations between gene sets and clinical features and to identify hub genes, we utilized weighted gene coexpression network analysis (WGCNA) to build gene coexpression networks incorporating the DEGs. Then, the levels of the hub genes were analyzed in the GSE datasets. Based on the expression of the hub genes, the possible pathways were explored by gene set enrichment analysis and gene set variation analysis. Finally, the diagnostic value of the hub genes was assessed by receiver operating characteristic (ROC) analysis in the GEO database.
Results
In this study, we identified 54 upregulated and 10 downregulated genes that overlapped between normal and VT samples. After performing WGCNA, the magenta module was the module with the strongest negative correlation with the clinical characteristics. From the key module, FECH, GYPA, RPIA and XK were chosen for further validation. We found that these genes were upregulated in VT samples, and high expression levels were related to recurrent VT. Additionally, the four hub genes might be highly correlated with ribosomal and metabolic pathways. The ROC curves suggested a diagnostic value of the four genes for VT.
Conclusions
These results indicated that FECH, GYPA, RPIA and XK could be used as promising biomarkers for the prognosis and prediction of VT.
Similar content being viewed by others

Introduction
Venous thromboembolism (VT), which includes pulmonary embolism (PE) and deep vein thrombosis (DVT), has been reported to be the third most commonly occurring cardiovascular disease (CVD) worldwide, following coronary heart disease and hypertension [1]. Annually, approximately 600,000 incidents are diagnosed in the United States. Furthermore, VT leads to complications, such as reappearance, chronic thromboembolic pulmonary hypertension, postthrombotic syndrome, and death [2,3,4,5,6]. Early diagnosis and treatment for VT patients are crucial to effectively reduce mortality and improve prognosis.
In recent decades, considerable attention has been given to exploring new biomarkers and potential molecular mechanisms for VT diagnosis and therapy [7,8,9,10]. Based on the GEO database, RPL9, RPL5, RPS20, TP53, and RPL23 were enriched in the ribosome pathway, validating them as potential targets for VT therapy [9]. This is a noteworthy finding, as it suggests that the PAI-1 4G/5G polymorphism might be a prospective VT risk biomarker, especially in the Asian population, according to a meta-analysis [11]. Moreover, it has been reported that COX7C and UQCRQ may play vital roles in a single VT, while ADRBK1, NDUFA5, and ATP5O may be possible targets for recurrent VT [12]. Therefore, it is essential to identify new novel biomarkers significantly correlated with VT diagnosis to improve the effectiveness of therapeutic approaches.
In this research, we examined 2 GEO datasets and found 64 significant differentially expressed genes (DEGs) between normal and VT samples. A weighted gene coexpression network analysis (WGCNA) was performed to evaluate the key module correlated with VT. Moreover, the preservation of gene modules was evaluated as preserved. Four hub genes, FECH, GYPA, RPIA, and XK, were found to be highly associated with VT, including low risk, moderate risk, high risk, single, and recurrent VT. Additionally, four genes appeared to be highly correlated with ribosome and metabolism pathways based on the GSEA and GSVA data. The diagnostic values of the four genes were validated by ROC curves. Thus, we identified the four hub genes FECH, GYPA, RPIA, and XK as new biomarkers and verified the prognostic and predicted values for VT patients.
Methods
Collection of data
The RNA expression profiles were obtained from two eligible microarray datasets (GSE19151 [13] and GSE48000 [14]) that contained 107 healthy samples and 160 VT samples. The patients with VT were separated into 3 groups: (1) ‘low-risk’ patients had one or more provoked VTs; (2) ‘moderate-risk’ patients had a single unprovoked VT; and (3) ‘high-risk’ patients had ≥ 2 unprovoked VTs.
Identification of robust DEGs
The R program “limma” was used to standardize the data and evaluate DEGs based on dataset series matrix files [15]. The DEGs that met the criteria of adjusted p value < 0.05 and log 2-fold change (FC) > 0.5 were filtered by robust rank aggregation (RRA) [16].
Gene ontology (GO) and kyoto encyclopedia of genes and genomes (KEGG) pathway analyses
Using the R package “clusterProfiler”, we conducted GO enrichment analysis for the DEGs; the analysis included molecular function (MF), cellular components (CC), and biological process (BP). KEGG pathway analysis was performed to investigate the high-level functions and utilities of the biological system.
WGCNA and identification of the key module
We chose the top 25% of genes with the most variance from GEO to build a coexpression network in R using the WGCNA program to identify VT-associated modules. Once outliers were removed at a cutoff point of 10,000, the data were grouped using Pearson’s correlation [17]. The optimum power value was determined once the independence level was 0.9 and a slope of approximately 1 was chosen. Subsequently, for network creation and module identification, we adjusted the soft-threshold power to 4, the cutoff height to 0.25, and the least module size to 10. Gene significance was used to quantify the associations between individual genes and traits.
Module preservation analysis
The module preservation function (nPermutations = 200) was used to generate module preservation and quality metrics to assess the stability of the obtained module utilizing the WGCNA package [18]. GSE19151 was a validation dataset that comprised mRNA expression data from 133 specimens. The modules showing elevated Zsummary and reduced medianRank scores were considered highly conservative and stable modules, respectively [19].
Gene set enrichment analysis (GSEA) and gene set variation analysis (GSVA)
We used the R package “clusterprofiler” to conduct GSEA on hub genes using sequencing data [20]. Furthermore, the “GSVA” R program was utilized to identify the pathways most closely associated with hub genes [21]. The samples were categorized into two cohorts based on the median expression per hub gene (low expression vs. high expression). p < 0.01 was considered statistically significant. The chosen reference gene set was “c2.cp.kegg.v6.2.symbols.gmt”, which was acquired from the Molecular Signature Database (MSigDB).
Validation of the prediction of hub genes
We used the “pROC” R package to construct receiver operating characteristic (ROC) curves and compute the area under the ROC curve (AUC) to assess the projected values of hub genes [22].
Results
Differentially expressed mRNAs related to VT
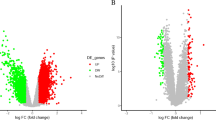
We compared the gene expression between normal and VT samples by analyzing GSE19151 and GSE48000 (Fig. 1). There were 375 upregulated and 187 downregulated genes in GSE19151 and 1285 upregulated and 230 downregulated genes in GSE48000 (Fig. 2a). The DEGs are presented in the Heatmaps (Fig. 2b). The Venn diagrams show the 54 upregulated and 10 downregulated genes that overlap the two different GEO datasets (Fig. 2c, Additional file 1).

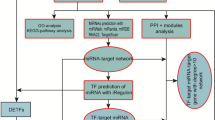
The workflow of this study

Analysis of DEGs in VT. a Volcano plot visualizing DEGs between normal and VT samples in GSE19151 and GSE48000; b the expression of DEGs in the heatmap of normal and VT samples; Venn diagrams showing the identification of c overlapped upregulated and d downregulated DEGs in two different datasets
Pathway enrichment analysis of DEGs
The top five enriched pathways for DEGs were ribosomal, structural constituent of ribosome, small ribosomal subunit, ribosomal subunit, and mitochondrial ribosome pathways (Fig. 3a, Additional file 2). In addition, the metabolic pathways were visualized in schemes depicting the ribosomal pathway (Fig. 3b). In the ribosomal pathway, RPS15, RPL15, RPL13, and RPS21 were upregulated. These data reveal that these genes might be crucial for VT.

Functional enrichment of DEGs. a GO terms identified in the GO analysis for DEGs; b Visualization of the ribosome pathway. Red nodes represent upregulated DEGs
WGCNA
A WGCNA was performed on GSE19151, integrating the DEGs generated from the RRA analysis to identify the main modules that highly correlate with the clinical characteristics of VTs (Fig. 4a). As indicated in Fig. 4b and c, beta (β) = 4 (scale-free R2 = 0.8) was additionally adjusted as the soft threshold for computation of adjacencies. Moreover, an aggregate of 14 modules was discovered after merging similar modules (Fig. 4c). Based on a heatmap of module–trait associations, the yellow, green–yellow, pink, and magenta modules were the four with the strongest association with VT (Fig. 4d). Additionally, we found that the significance of the yellow, green–yellow, pink, and magenta modules was higher than that of the others module, implying that these modules might have a significant relationship with VT (Fig. 4e). Furthermore, in the magenta module, the association and p value between module affiliation and gene significance values were 0.39 and 0.0013, respectively (Fig. 4f).

Identification of key modules associated with clinical traits by WGCNA. a Clustering dendrograms of samples; b Analysis of the scale-free fit index and the mean connectivity for various soft-thresholding powers; c Dendrogram of all DEGs clustered with dissimilarity measure based on topological overlap; d Heatmap of the correlation between module eigengenes and clinical traits. Each row denoted a module eigengene, each column represented a clinical trait and each cell contained the correlation coefficient and p value; e Gene significance in different modules (bottom); f Scatter plot of genes in yellow module
Network preservation analysis
We utilized GSE19151 as a validation set to execute preservation analysis to assess the preservation of gene modules. The preservation was evaluated using an integrated modulePreservation algorithm in the WGCNA package. A Z score ranging between 2 and 10 was considered to represent mild to moderate preservation, while scores above 10 represent excellent preservation [23,24,25,26]. Meanwhile, a module with a substantially lower rank appears to have better observable preservation metrics than a module with a greater median rank. Combining the median rank, Z score, and module–trait correlations, the magenta module, comprising 65 DEGs, was found to be the module with the strongest negative correlation with clinical characteristics (Fig. 5a, Additional file 3). The most enriched pathways of the DEGs in the magenta module were 2 iron, 2 sulfur cluster binding, efflux transmembrane transporter activity, and carbonate dehydratase activity (Fig. 5b, Additional file 4).

The medianRank and Zsummary values of the module preservation using the GSE19151 dataset. a The medianRank graph and the Zsummary graph. The dashed blue and green lines indicate the thresholds Zsummary = 2 and Zsummary = 10, respectively; b The plot for the top 9 GO enrichment annotations of all genes in magenta module
Validation of hub genes
We chose four hub genes (FECH, GYPA, RPIA, and XK) to explore their correlations with clinical values. They were shown to be more highly expressed in the VT group than in the normal group (Fig. 6a). There was a significant difference in the low-, moderate- and high-risk groups compared to the normal group (Fig. 6b). Based on the data from GSE19151, we found that the levels of hub genes were much higher in the single VT (Fig. 6c) and recurrent VT (Fig. 6d) groups than in the normal groups. The data suggested that four genes were strongly correlated with VT and might be associated with VT recurrence.

Correlation analysis between the expression of EFCH, GYPA, RPIA and XK and clinicopathological parameters in GSE19151 and GSE48000. a the expression of hub genes and VT; b low risk and high risk; c single VT; d recurrent VT
GSEA and GSVA of four hub genes
To further identify the possible functions of FECH, GYPA, RPIA, and XK in VT, we conducted GSEA and GSVA with GSE19151. Genes in the high-expression cohorts of FECH, GYPA, RPIA, and XK were highly enriched in ribosome (Fig. 7a), graft versus host disease (Fig. 7b), primary immunodeficiency (Fig. 7c), and B cell receptor signaling pathways, respectively (Fig. 7d). Based on the analysis of GSVA, FECH was associated with vascular smooth muscle contraction (Fig. 7e). GYPA was enriched in aminoacyl tRNA biosynthesis (Fig. 7f). PRIA was related to nitrogen metabolism (Fig. 7g), and XK was associated with porphyrin and chlorophyll metabolism (Fig. 7h). After comprehensively considering the results of GSEA and GSVA, we concluded that these four genes might be highly correlated with ribosomal and metabolic pathways.

GSEA and GSVA of hub genes in datasets. a–d Top 3 gene sets (according to GSEA enrichment score) enriched in the high-expression group of a EFCH; b GYPA; c RPIA; d XK. GSVA-derived clustering heatmaps of differentially expressed pathways for e EFCH; f GYPA; g RPIA; h XK
The predicted values of hub genes in VT
ROC curves showed the predicted value of these genes as biomarkers for the incidence of VT (FECH AUC: 0.765, GYPA AUG: 0.837, RPIA AUG: 0.668, and XK AUG: 0.819) and the possibility of single and recurrent VT (FECH AUC: 0.655, GYPA AUG: 0.642, RPIA AUG: 0.641, and XK AUG: 0.634) (Fig. 8a, b). The ROC curves suggest that these four genes have predictive values for low risk VT (FECH AUC: 0.688, GYPA AUG: 0.696, RPIA AUG: 0.701, and XK AUG: 0.719) (Fig. 8c) and moderate risk VT (FECH AUC: 0.642, GYPA AUG: 0.663, RPIA AUG: 0.682, and XK AUG: 0.658) (Fig. 8d).

The diagnostic values of hub genes in VT. ROC curves and AUC statistics to evaluate the diagnostic efficiency of hub genes on the incidence of VT (a); the possibility of single and recurrent VT (b); low risk (c) and moderate risk (d)
Discussion
VT is reported to be the third most common CVD worldwide following coronary heart disease and hypertension. Although numerous investigations have been conducted to investigate treatment targets for VT, discrepancies between the DEGs discovered have been observed in various studies [27,28,29,30,31]. This study is the first to integrate RRA and WGCNA to identify new biomarkers associated with VT. Two GEO datasets were included in our study, and 54 upregulated and 10 downregulated genes were determined to overlap. After investigating their enrichment in GO and KEGG pathways, we discovered that these DEGs may be involved in ribosomal and metabolic pathways. We were able to identify the coexpression module linked to clinical characteristics using WGCNA. Genes in this module were enriched in 2 iron, 2 sulfur cluster binding, efflux transmembrane transporter activity, and carbonate dehydratase activity.
We identified four hub genes, FECH, GYPA, RPIA, and XK, from the magenta module. High expression of these four genes was significantly associated with VT, low-, moderate- and high-risk VT, and recurrence of VT, indicating the prognostic value of these hub genes. Ferrochelatase (FECH) is the terminal enzyme in heme biosynthesis and plays vital roles in choroidal neovascularization, retinal neovascularization, and erythropoietic protoporphyria [32,33,34]. It was reported that FECH was involved in the metabolic pathway [35], and our work concurs with this. Glycophorins (GYPA) are one of the primary sialoglycoproteins of the human erythrocyte membrane and serve as receptors for pathogens, including Plasmodium falciparum erythrocyte-binding antigen 175 (EBA-175), influenza virus, and hepatitis A virus (HAV). GYPA plays a vital role in the high activity of solute carrier family 4 member 1 (SLC4A1) and translocation of SLC4A1 to the plasma membrane [36]. Our study showed that GYPA was strongly associated with the occurrence of VT, acting as a possible and not well-studied biomarker for VT. Furthermore, the enzyme ribose 5-phosphate isomerase A (RPIA) plays an essential role in carbohydrate metabolism [37, 38] in Enterococcus faecalis and is a prognostic biomarker for human hepatocellular carcinoma [39]. Our data indicated that RPIA was involved in the incidence of VT. The X-linked Kx blood group (XK) protein has eukaryotic and prokaryotic membrane transport protein structural features. Mutations in XK are highly correlated with McLeod syndrome with defects in the hematopoietic and neuromuscular systems. Thus, FECH, GYPA, RPIA, and XK were identified in our study as potential biomarkers for VT.
To explore their potential biological functions in VT, we conducted GSEA and GSVA, which revealed that these four novel genes might be positively involved in ribosomal and metabolic pathways. It has been reported that RPIA is a vital mediator in the process of carbohydrate metabolism and nucleotide metabolism in cancer [37, 38, 40]. Additionally, many related pathways, such as the B cell receptor signaling pathway, antigen processing and presentation, and allograft rejection, were found to be enriched in the high-expression cohorts of hub genes, revealing their potential roles in VT.
Conclusions
By combining RRA and WGCNA, we found some robust DEGs and significant gene modules in VT. Four hub genes (FECH, GYPA, RPIA, and XK) were significantly upregulated in the VT cohort, and GSEA and GSVA indicated that these genes might contribute to the incidence of VT. Moreover, these four hub genes were found to be diagnostic biomarkers for the prediction of VT. Further experiments are required to determine the possible mechanisms of these biomarkers in VT.
Availability of data and materials
The gene expression profiles of GSE19151 and GSE48000 were downloaded from Gene Expression Omnibus (GEO).
Abbreviations
- VT:
-
Venous thromboembolism
- RRA:
-
Robust rank aggregation
- DEGs:
-
Differentially expressed genes
- WGCNA:
-
Weighted gene co-expression network analysis
- GSVA:
-
Gene set enrichment analysis
- GSEA:
-
Gene set variation analysis
- PE:
-
Pulmonary embolism
- DVT:
-
Deep vein thrombosis
- CVD:
-
Cardiovascular disease
- GO:
-
Gene ontology (GO)
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- MF:
-
Molecular function
- CC:
-
Cellular components
- BP:
-
Biological process
- ROC:
-
Construct receiver operating characteristic
- AUC:
-
Area under the ROC curve
- FECH:
-
Ferrochelatase
- GYPA:
-
Glycophorins
- RPIA:
-
Ribose 5-phosphate isomerase A
- XK:
-
X-linked Kx blood group
- EBA-175:
-
Erythrocyte-binding antigen 175
- HAV:
-
Influenza virus, and hepatitis A virus
- SLC4A1:
-
Solute carrier family 4 member 1
References
Goldhaber SZ, Bounameaux H. Pulmonary embolism and deep vein thrombosis. Lancet. 2012;379(9828):1835–46.
Naess IA, Christiansen SC, Romundstad P, Cannegieter SC, Rosendaal FR, Hammerstrom J. Incidence and mortality of venous thrombosis: a population-based study. J Thromb Haemost. 2007;5(4):692–9.
Day ISCfWT. Thrombosis: a major contributor to the global disease burden. J Thromb Haemost. 2014;12(10):1580–90.
Schulman S, Lindmarker P, Holmstrom M, Larfars G, Carlsson A, Nicol P, Svensson E, Ljungberg B, Viering S, Nordlander S, et al. Post-thrombotic syndrome, recurrence, and death 10 years after the first episode of venous thromboembolism treated with warfarin for 6 weeks or 6 months. J Thromb Haemost. 2006;4(4):734–42.
Arshad N, Bjori E, Hindberg K, Isaksen T, Hansen JB, Braekkan SK. Recurrence and mortality after first venous thromboembolism in a large population-based cohort. J Thromb Haemost. 2017;15(2):295–303.
Ende-Verhaar YM, Cannegieter SC, Vonk Noordegraaf A, Delcroix M, Pruszczyk P, Mairuhu AT, Huisman MV, Klok FA. Incidence of chronic thromboembolic pulmonary hypertension after acute pulmonary embolism: a contemporary view of the published literature. Eur Respir J. 2017;49(2):1601792.
Morelli VM, Braekkan SK, Hansen JB. Role of microRNAs in venous thromboembolism. Int J Mol Sci. 2020;21(7):2602.
Heit JA, Armasu SM, McCauley BM, Kullo IJ, Sicotte H, Pathak J, Chute CG, Gottesman O, Bottinger EP, Denny JC, et al. Identification of unique venous thromboembolism-susceptibility variants in African–Americans. Thromb Haemost. 2017;117(4):758–68.
Wang G, Zhao W, Yang Y, Yang G, Wei Z, Guo J. Identification of biomarkers of venous thromboembolism by bioinformatics analyses. Medicine (Baltimore). 2018;97(14):e0152.
Zhou ZH, Chen Y, Zhao BH, Jiang Y, Luo Q. Early postpartum venous thromboembolism: risk factors and predictive index. Clin Appl Thromb Hemost. 2019. https://doi.org/10.1177/1076029618818777.
Zhang Q, Jin Y, Li X, Peng X, Peng N, Song J, Xu M. Plasminogen activator inhibitor-1 (PAI-1) 4G/5G promoter polymorphisms and risk of venous thromboembolism—a meta-analysis and systematic review. Vasa. 2020;49(2):141–6.
Zhou W, Zhang K, Chen D, Gao P, Wang Q. Gene microarray analyses for potential biomarkers of single and recurrent venous thromboembolism. Mol Med Rep. 2015;12(5):7358–66.
Lewis DA, Stashenko GJ, Akay OM, Price LI, Owzar K, Ginsburg GS, Chi JT, Ortel TL. Whole blood gene expression analyses in patients with single versus recurrent venous thromboembolism. Thromb Res. 2011;128(6):536–40.
Lewis DA, Suchindran S, Beckman MG, Hooper WC, Grant AM, Heit JA, Manco-Johnson M, Moll S, Philipp CS, Kenney K, et al. Whole blood gene expression profiles distinguish clinical phenotypes of venous thromboembolism. Thromb Res. 2015;135(4):659–65.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47.
Kolde R, Laur S, Adler P, Vilo J. Robust rank aggregation for gene list integration and meta-analysis. Bioinformatics. 2012;28(4):573–80.
Zhong H, Wang J, Zhu Y, Shen Y. Comprehensive analysis of a nine-gene signature related to tumor microenvironment in lung adenocarcinoma. Front Cell Dev Biol. 2021;9:700607.
Zhu Z, Jin Z, Deng Y, Wei L, Yuan X, Zhang M, Sun D. Co-expression network analysis identifies four hub genes associated with prognosis in soft tissue sarcoma. Front Genet. 2019;10:37.
Langfelder P, Luo R, Oldham MC, Horvath S. Is my network module preserved and reproducible? PLoS Comput Biol. 2011;7(1):e1001057.
Wei Y, Chen X, Ren X, Wang B, Zhang Q, Bu H, Qian J, Shao P. Identification of MX2 as a novel prognostic biomarker for sunitinib resistance in clear cell renal cell carcinoma. Front Genet. 2021;12:680369.
Shou Y, Yang L, Yang Y, Zhu X, Li F, Xu J. Identification of signatures of prognosis prediction for melanoma using a hypoxia score. Front Genet. 2020;11:570530.
Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, Muller M. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011;12:77.
Yang B, Wei S, Ma YB, Chu SH. Integrated transcriptomic analysis reveals the molecular mechanism of meningiomas by weighted gene coexpression network analysis. Biomed Res Int. 2020;2020:4927547.
Bakhtiarizadeh MR, Mirzaei S, Norouzi M, Sheybani N, Vafaei Sadi MS. Identification of gene modules and hub genes involved in mastitis development using a systems biology approach. Front Genet. 2020;11:722.
Panahi B, Hejazi MA. Weighted gene co-expression network analysis of the salt-responsive transcriptomes reveals novel hub genes in green halophytic microalgae Dunaliella salina. Sci Rep. 2021;11(1):1607.
Tang X, Huang X, Wang D, Yan R, Lu F, Cheng C, Li Y, Xu J. Identifying gene modules of thyroid cancer associated with pathological stage by weighted gene co-expression network analysis. Gene. 2019;704:142–8.
Wang X, Sundquist K, Svensson PJ, Rastkhani H, Palmer K, Memon AA, Sundquist J, Zoller B. Association of recurrent venous thromboembolism and circulating microRNAs. Clin Epigenet. 2019;11(1):28.
Riva N, Vella K, Hickey K, Bertu L, Zammit D, Spiteri S, Kitchen S, Makris M, Ageno W, Gatt A. Biomarkers for the diagnosis of venous thromboembolism: D-dimer, thrombin generation, procoagulant phospholipid and soluble P-selectin. J Clin Pathol. 2018;71(11):1015–22.
Mauracher LM, Posch F, Martinod K, Grilz E, Daullary T, Hell L, Brostjan C, Zielinski C, Ay C, Wagner DD, et al. Citrullinated histone H3, a biomarker of neutrophil extracellular trap formation, predicts the risk of venous thromboembolism in cancer patients. J Thromb Haemost. 2018;16(3):508–18.
Chen X, Cao J, Ge Z, Xia Z. Correlation and integration of circulating miRNA and peripheral whole blood gene expression profiles in patients with venous thromboembolism. Bioengineered. 2021;12(1):2352–63.
Dzikowska-Diduch O, Domienik-Karlowicz J, Gorska E, Demkow U, Pruszczyk P, Kostrubiec M. E-selectin and sICAM-1, biomarkers of endothelial function, predict recurrence of venous thromboembolism. Thromb Res. 2017;157:173–80.
Basavarajappa HD, Sulaiman RS, Qi X, Shetty T, Sheik Pran Babu S, Sishtla KL, Lee B, Quigley J, Alkhairy S, Briggs CM, et al. Ferrochelatase is a therapeutic target for ocular neovascularization. EMBO Mol Med. 2017;9(6):786–801.
Pran Babu SPS, White D, Corson TW. Ferrochelatase regulates retinal neovascularization. FASEB J. 2020;34(9):12419–35.
Long ZB, Wang YW, Yang C, Liu G, Du YL, Nie GJ, Chang YZ, Han B. Identification of FECH gene multiple variations in two Chinese patients with erythropoietic protoporphyria and a review. J Zhejiang Univ Sci B. 2016;17(10):813–20.
Wang P, Sachar M, Guo GL, Shehu AI, Lu J, Zhong XB, Ma X. Liver metabolomics in a mouse model of erythropoietic protoporphyria. Biochem Pharmacol. 2018;154:474–81.
Steiper ME, Walsh F, Zichello JM. The SLC4A1 gene is under differential selective pressure in primates infected by Plasmodium falciparum and related parasites. Infect Genet Evol. 2012;12(5):1037–45.
Ramos Y, Rocha J, Hael AL, van Gestel J, Vlamakis H, Cywes-Bentley C, Cubillos-Ruiz JR, Pier GB, Gilmore MS, Kolter R, et al. PolyGlcNAc-containing exopolymers enable surface penetration by non-motile Enterococcus faecalis. PLoS Pathog. 2019;15(2):e1007571.
Ramos Y, Morales DK. Exopolysaccharide-mediated surface penetration as new virulence trait in Enterococcus faecalis. Commun Integr Biol. 2019;12(1):144–7.
Ciou SC, Chou YT, Liu YL, Nieh YC, Lu JW, Huang SF, Chou YT, Cheng LH, Lo JF, Chen MJ, et al. Ribose-5-phosphate isomerase A regulates hepatocarcinogenesis via PP2A and ERK signaling. Int J Cancer. 2015;137(1):104–15.
Buj R, Chen CW, Dahl ES, Leon KE, Kuskovsky R, Maglakelidze N, Navaratnarajah M, Zhang G, Doan MT, Jiang H, et al. Suppression of p16 Induces mTORC1-mediated nucleotide metabolic reprogramming. Cell Rep. 2019;28(8):1971–80.
Acknowledgements
We thank all participants for their supports.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: ZHW, FGJ, JZH, WKQ and YHT, Analyzed the data: WJ, LYK and CB, Wrote the paper: FGJ and WKQ. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declared no potential competing interests with respect to the research, authorship, and/or publication of this article.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. DEGs of GSE19151 and GSE48000.
Additional file 2
. GO terms of DEGs.
Additional file 3
. 65 DEGs in the magenta module.
Additional file 4
. The enriched pathways of the DEGs in the magenta module.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Fan, G., Jin, Z., Wang, K. et al. Identification of four hub genes in venous thromboembolism via weighted gene coexpression network analysis. BMC Cardiovasc Disord 21, 577 (2021). https://doi.org/10.1186/s12872-021-02409-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12872-021-02409-4