
Abstract
Background
To illustrate the mechanism of miRNA and mRNA in coronary artery diseasen (CAD), differentially expressed microRNAs (DEmiRNAs) and genes (DEGs) were analyzed.
Methods
The mRNA transcription profiles of GSE20680 (including 87 blood samples of CAD and 52 blood samples of control), GSE20681 (including 99 blood samples of CAD and 99 blood samples of control) and GSE12288 (including 110 blood samples of CAD and 112 blood samples of control) and the miRNA transcription profiles of GSE59421 (including 33 blood samples of CAD and 37 blood samples of control), GSE49823 (including 12 blood samples of CAD and 12 blood samples of control) and GSE28858 (including 13 blood samples of CAD and 13 blood samples of control) were downloaded from Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/). Then, the homogenous expressed mRNAs and miRNAs across the three mRNA transcription profiles and three miRNA transcription profiles were screened using the Fishers exact test in MetaDE. ES package. The weighted gene co-expression network analysis (WGCNA) was used to analyze gene modules. Additionally, the integrated miRNAs–targets regulatory network using the DEmiRNA and their targets was constructed using Cytoscape.
Results
A total of 1201 homogenously statistically significant DEGs were identified including 879 up-regulated and 322 down-regulated DEGs, while a total of 47 homogenously statistically significant DEmiRNAs including 37 up-regulated and 10 down-regulated DEmiRNAs in CAD compared with the controls across the three mRNA transcription profiles and the three miRNA transcription profiles. A total of 5067 genes were clustered into 9 modules in the training dataset, among which, 8 modules were validated. In the miRNAs-targets network, there existed 267 interaction relationships among 5 miRNAs (hsa-miR-361-5p, hsa-miR-139-5p, hsa-miR-146b-5p, hsa-miR-502-5p and hsa-miR-501-5p) and 213 targets. CAV1 could be the target of hsa-miR-361-5 while HSF2 was the target of both hsa-miR-361-5p and hsa-miR-146b-5p. CAV1 was significantly enriched in the GO term of regulation of cell proliferation.
Conclusion
hsa-miR-361-5p, has-miR-146b-5p, CAV1 and HSF2 could play an important role in CAD.
Similar content being viewed by others
Background
Coronary artery disease (CAD) as the most common type of cardiovascular disease, can lead to a complete blockage of blood flowing to the heart, resulting in a heart attack. For its high morbidity and mortality rate, CAD has probably the most serious cardiovascular disorder threatening people’s health, worldwidely [1].
MicroRNAs (miRNAs) with the length from 18 to 25 nucleotides, is a class of non-protein coding RNA, which has been shown to be involved in a wide variety of biological processes through suppressing the mRNA expression or translation [2]. MiRNAs have been found to express in various tissues and cell types and play important roles in physiological processes including cell growth, proliferation, differentiation, apoptosis, metabolism, and homeostasis as biological regulators [3]. MiRNAs were mostly found in body fluids such as blood and the circulating miRNA expression profiles have been shown to differ significantly between healthy and disease including cancers/tumor, diabetes, neurodegenerative diseases, cardiovascular diseases [3, 4]. So, the blood as one of the most popular non-invasive samples was widely used for identifying potential diagnostic and prognostic biomarkers in human diseases. For CAD, numerous miRNAs have been reported to be circulating biomarkers in the diagnosis of CAD and their functional role in cardiovascular primary prevention has been suggested [5]. For example, the expression levels of miR-126, miR-17, miR-92a, and the inflammation-associated miR-155 in plasma were significantly reduced in patients with CAD compared with healthy controls. [6]. The serum miR-197 and miR-223 could be used to predict cardiovascular death in a cohort of patients with symptomatic CAD [7]. The blood miRNA-19a has been identified as a potential novel biomarker for diagnosis of acute myocardial infarction [8]. However, the miRNAs identified in a single dataset may be limited.
In our study, using three mRNA microarray data of GSE20680 [9], GSE20681 [10] and GSE12288 [10], as well as three miRNA microarray data of GSE59421 [11], GSE49823 [12] and GSE28858 [13], we aimed to further screen the potential biomarkers related to CAD with different analysis methods.
Materials and methods
RNA sequencing data
Since our goal was to screen the miRNA and mRNA related to CAD, the key words of coronary artery disease was used to search the Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/). As a result, the mRNA transcription profiles of GSE20680 (platform: GPL4133, including 87 blood samples of CAD and 52 blood samples of control), GSE20681 (platform: GPL4133, including 99 blood samples of CAD and 99 blood samples of control) and GSE12288 (platform: GPL4133, including 110 blood samples of CAD and 112 blood samples of control) were downloaded from the GEO. Additionally, the miRNA transcription profiles of GSE59421 (platform: GPL10850, including 33 blood samples of CAD and 37 blood samples of control), GSE49823 (platform: GPL15467, including 12 blood samples of CAD and 12 blood samples of control) and GSE28858 (platform: GPL8179, including 13 blood samples of CAD and 13 blood samples of control) were downloaded from GEO. All the data were downloaded on January 16th, 2020 (Table 1).
Meta-analysis for differentially expressed RNAs
In order to obtain more reliable miRNAs and mRNAs associated with CAD, a meta-analysis of the microarray datasets was performed.
The homogenous expressed mRNAs and miRNAs across the three mRNA transcription profiles and the three miRNA transcription profiles were screened using the MetaDE. ES algorithm of MetaDE package in R with the thresholds of homogeneity were set as tau2 = 0 and Qpval > 0.05 [14,15,16]. Subsequently, the homogenously statistically significant differentially expressed genes (DEGs) and differentially expressed miRNAs (DEmiRNAs) were screened with the threshold of false discovery rate (FDR) of < 0.05 using Fishers exact test in metaDE. Moreover, the Fold Change of the DEGs and DEmiRNAs must be simultaneously > 1 or < 1 in each of three mRNA transcription profiles and the three miRNA transcription profiles. The heatmap.sig.genes in MetaDE was used to conduct the bidirectional hierarchical clustering.
CAD related genes and gene modules screening by WGCNA
WGCNA (weighted gene co-expression network analysis, version 1.61, https://cran.r-project.org/web/packages/WGCNA/index.html) package in R language was used to analyze gene modules and genes that were highly correlated with CAD [15]. The GSE12288 was used as the training dataset to conduct the WGCNA while the GSE20680 and GSE20681 were used as the validation dataset. The power value when the correlation coefficient squared value of connection degree k and p(k) for the first time to reach 0.9 were selected, that was, power = 14; under this power parameter, the mean connectivity degree of the geness was 1, which conformed to the small-world property in a scale-free network. The cutHeight = 0.99 and the gene number ≥ 100 within a module were used as the threshold. The gene moludes with Z-score > 5 and p ≤ 0.05 were refered as the preserved gene modules.
miRNA-mRNA regulatory network construction
Firstly, the targets of the homogenously statistically significant DEmiRNAs were predicted using starBase database (Version 2.0, http://starbase.sysu.edu.cn/). And the targets which were the the homogenously statistically significant DEGs were retained. Secondly, the miRNA-mRNA pairs with the negative correlation were retained. Thirdly, the targets belonging to the gene modules were highlighted. Lastly, the screened miRNA-mRNA pairs were visualized using Cytoscape software (Version 3.6.1, http://www.cytoscape.org) [17]. DAVID (version 6.8, https://david.ncifcrf.gov/) was used to perform the GO and KEGG pathway enrichment for the mRNA in the network [18, 19].
Results
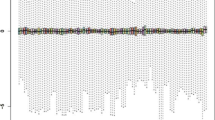
A total of 1201 homogenously statistically significant DEGs were identified including 879 up-regulated and 322 down-regulated DEGs. Additionally, a total of 47 homogenously statistically significant DEmiRNAs including 37 up-regulated and 10 down-regulated DEmiRNAs were screened. The bidirectional hierarchical clustering revealed that DEGs and DEmiRNAs could distinguish the CADs from the controls very well (Fig. 1).

The two-way clustering of differentially expressed RNAs (DErs) between the controls and the coronary artery diseas. a For the mRNA; b for the miRNAs; up horizontal axis, the samples; vertical axis, datasets; the color from red to green indicated the changes in expression level from high to low
Modules and genes screened by WGCNA
According to the method, 5067 highly correlated genes were clustered into 9 modules (black, blue, brown, grey, green, red, pink, turquoise and yellow) in the training dataset GSE12288. Among which, 8 modules including modules black, blue, brown, green, red, pink, turquoise and yellow were validated using the validation datasets (Table 2). The Gene dendrogram of the training and validation datasets was shown in Additional file 1: Fig. S1.
MiRNA-mRNA network construction
In the miRNAs-targets network, there existed 267 interaction relationships among 5 miRNAs (hsa-miR-361-5p, hsa-miR-139-5p, hsa-miR-146b-5p, hsa-miR-502-5p and hsa-miR-501-5p) and 213 targets (Fig. 2). Among the which, CAV1 could be the target of hsa-miR-361-5 while HSF2 was the target of both hsa-miR-361-5p and hsa-miR-146b-5p.

The miRNA-targets regulation network. The triangle nodes represented the up-regulated miRNAs and mRNAs; the quadrangle nodes represented the down-regulated miRNAs and mRNAs; The nodes with colors represented that the targets belonging to the gene modules
Functional enrichment analysis showed that the targets were significantly enriched in 16 GO terms, such as regulation of cell preliferation and 9 KEGG pathways (Fig. 3).

Enriched functions of the targets in the miRNA-mRNA regulation network. a For KEGG pathway; b for GO terms; Gene numbers are displayed on the x-axis. The color represents the − log (FDR)
Discussion
In this study, by using three miRNA microarray and three mRNA microarray of blood samples of CAD and control, a total of 1201 homogenously statistically significant DEGs including 879 up-regulated and 322 down-regulated DEGs and 47 homogenously statistically significant DEmiRNAs including 37 up-regulated and 10 down-regulated DEmiRNAs were identified. Additionally, the WGCNA, miRNA-mRNA regulatory network, functionally enrichment were used to identify the key miRNAs and mRNAs related to CAD. Finally, 5 miRNAs including hsa-miR-361-5p, hsa-miR-139-5p, hsa-miR-146b-5p, hsa-miR-502-5p and hsa-miR-501-5p were identified.
According to the analysis, we found that hsa-miR-146b-5p was significantly up-regulated in the CAD patients. It has been found that miR-146a/b could be involved in CAD by regulating the TLR4 downstream molecules IRAK1 (interleukin-1-receptor-associated kinase 1) and TRAF6 (tumour-necrosis-factor-receptor-associated factor 6) and its level was significantly higher in the CAD group than in the non-CAD group (all P < 0.01) [20]. Additionally, miR-146b-5p has also been reported to promote the proliferation, migration and the phenotype transition of vascular smooth muscle cells (VSMCs), which played pivotal roles in vascular remodeling in atherosclerosis [21]. Dysregulation of miR-146a-5p/RHOJ and miR-146b-5p/RHOJ axis in the plasma and ECFCs of CAD patients could be used as biomarkers or therapeutic targets for CAD and other angiogenesis-related diseases [22]. As a result, has -miR-146b-5p could be the potential factor in CAD.
For hsa-miR-361-5p, it was also significantly up-regulated in the CAD patients. Former study has demonstrated the dysregulation of miR-361-5p/VEGF Axis in the plasma and endothelial progenitor cells of patients with CAD [23]. LncRNA MEG3-derived miR-361-5p could regulate VSMCs proliferation and apoptosis by targeting ABCA1 [24]. In the miRNA-mRNA network, hsa-miR-361-5p could regulate the expression of CAV1. The functinal enrichent showed that CAV1 was significantly enriched in the GO term of regulation of cell proliferation. CAV1 which encodes caveolin-1 expressed in cell types relevant to atherosclerosis, was found to be associated with significant risk of CAD when its aberrant expression [25]. So, CAV1 targeted by hsa-miR-361-5p could involved in the mechanism of CAD through the regulation of VSMCs proliferation.
As for hsa-miR-139-5p, hsa-miR-502-5p and hsa-miR-501-5p, there was no study reporting their directed relationship to CAD. However, it has been found that aberrant expression of hsa-miR-139-5p could lead to apoptosis [26] which has been observed in coronary atherosclerosis [27]. hsa-miR-502-5p has been reported to inhibit autophage [28] and the alteration of autophagic genes has also be discovered in CAD [29]. So, these three miRNAs could play an important role in CAD.
In our study, we also found that HSF2 was significantly down-regulated in CAD and was the target of both hsa-miR-361-5p and hsa-miR-146b-5p. HSF2, as a heat shock transcription factors, is more prominently activated during mouse heart development [30]. Former study has demonstrated that HSF2 could induce cardiac hypertrophy during hypertension-induced heart failure by the activation of IGF-IIR [31]. So, we speculated that HSF2 targeted by hsa-miR-361-5p and miR-146b-5p could be related to CAD.
The study’s limitations should be noted. On the one hand, the methods for screening the biomarkers was based on the statistical method rather than the biological experiment. On the other hand, the miRNA and mRNA data were not from the same project. So the additional experiment such as real-time quantitative PCR was needed to validate the miRNA and mRNA expression levels of the same project.
Conclusions
Above all, we speculated that hsa-miR-361-5p, has-miR-146b-5p, CAV1 and HSF2 could play an important role in CAD. However, further research is required to validate the results.
Availability of data and materials
The raw data were collected and analyzed by the Authors, and are not ready to share their data because the data have not been published.
Abbreviations
- DEmiRNAs:
-
Differentially expressed microRNAs
- CAD:
-
Coronary artery disease
- miRNAs:
-
MicroRNAs
- WGCNA:
-
Weighted gene co-expression network analysis
- VSMCs:
-
Vascular smooth muscle cells
References
Piccolo R, et al. Stable coronary artery disease: revascularisation and invasive strategies. Lancet. 2015;386(9994):702–13.
van Rooij E, Olson EN. MicroRNAs: powerful new regulators of heart disease and provocative therapeutic targets. J Clin Invest. 2007;117(9):2369–76.
Huang W. MicroRNAs: biomarkers, diagnostics, and therapeutics. Methods Mol Biol. 2017;1617:57–67.
Weber JA, et al. The microRNA spectrum in 12 body fluids. Clin Chem. 2010;56(11):1733–41.
Schulte C, Zeller T. microRNA-based diagnostics and therapy in cardiovascular disease-summing up the facts. Cardiovasc Diagn Ther. 2015;5(1):17–36.
Fichtlscherer S, et al. Circulating microRNAs in patients with coronary artery disease. Circ Res. 2010;107(5):677–84.
Schulte C, et al. miRNA-197 and miRNA-223 predict cardiovascular death in a cohort of patients with symptomatic coronary artery disease. PLoS ONE. 2015;10(12):e0145930.
Zhong J, et al. Circulating microRNA-19a as a potential novel biomarker for diagnosis of acute myocardial infarction. Int J Mol Sci. 2014;15(11):20355–64.
Elashoff MR, et al. Development of a blood-based gene expression algorithm for assessment of obstructive coronary artery disease in non-diabetic patients. BMC Med Genom. 2011;4:26.
Sinnaeve PR, et al. Gene expression patterns in peripheral blood correlate with the extent of coronary artery disease. PLoS ONE. 2009;4(9):e7037.
Kok MG, et al. Normalization panels for the reliable quantification of circulating microRNAs by RT-qPCR. FASEB J. 2015;29(9):3853–62.
Ren J, et al. Signature of circulating microRNAs as potential biomarkers in vulnerable coronary artery disease. PLoS ONE. 2013;8(12):e80738.
Sondermeijer BM, et al. Platelets in patients with premature coronary artery disease exhibit upregulation of miRNA340* and miRNA624*. PLoS ONE. 2011;6(10):e25946.
Qi C, et al. Identification of metastasis-associated genes in colorectal cancer using metaDE and survival analysis. Oncol Lett. 2016;11(1):568–74.
Wang X, et al. An R package suite for microarray meta-analysis in quality control, differentially expressed gene analysis and pathway enrichment detection. Bioinformatics. 2012;28(19):2534–6.
Tuo Y, An N, Zhang M. Feature genes in metastatic breast cancer identified by MetaDE and SVM classifier methods. Mol Med Rep. 2018;17(3):4281–90.
Shannon P, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57.
da Huang W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucl Acids Res. 2009;37(1):1–13.
Takahashi Y, et al. Expression of miR-146a/b is associated with the Toll-like receptor 4 signal in coronary artery disease: effect of renin-angiotensin system blockade and statins on miRNA-146a/b and Toll-like receptor 4 levels. Clin Sci (Lond). 2010;119(9):395–405.
Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res. 2016;118(4):692–702.
Chang TY, et al. Dysregulation of endothelial colony-forming cell function by a negative feedback loop of circulating miR-146a and -146b in cardiovascular disease patients. PLoS ONE. 2017;12(7):e0181562.
Wang HW, et al. Dysregulated miR-361–5p/VEGF axis in the plasma and endothelial progenitor cells of patients with coronary artery disease. PLoS ONE. 2014;9(5):e98070.
Wang M, et al. LncRNA MEG3-derived miR-361-5p regulate vascular smooth muscle cells proliferation and apoptosis by targeting ABCA1. Am J Transl Res. 2019;11(6):3600–9.
Chen S, et al. Genomic variant in CAV1 increases susceptibility to coronary artery disease and myocardial infarction. Atherosclerosis. 2016;246:148–56.
Sun C, et al. Hsa-miR-139-5p inhibits proliferation and causes apoptosis associated with down-regulation of c-Met. Oncotarget. 2015;6(37):39756–92.
Best PJ, et al. Apoptosis. Basic concepts and implications in coronary artery disease. Arterioscler Thromb Vasc Biol. 1999;19(1):14–22.
Zhai H, et al. Inhibition of autophagy and tumor growth in colon cancer by miR-502. Oncogene. 2013;32(12):1570–9.
Zhang J, et al. Altered expression of lysosomal hydrolase, acid alpha-glucosidase, gene in coronary artery disease. Coron Artery Dis. 2016;27(2):104–8.
Sistonen L, Sarge KD, Morimoto RI. Human heat shock factors 1 and 2 are differentially activated and can synergistically induce hsp70 gene transcription. Mol Cell Biol. 1994;14(3):2087–99.
Huang CY, et al. p53-mediated miR-18 repression activates HSF2 for IGF-IIR-dependent myocyte hypertrophy in hypertension-induced heart failure. Cell Death Dis. 2017;8(8):e2990.
Acknowledgements
None.
Funding
None.
Author information
Authors and Affiliations
Contributions
LL and JZZ participated in the design of this study, and they both performed the statistical analysis. MW carried out the study and collected important background information. HMX drafted the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
The Gene dendrogram of the training and validation datasets.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, L., Zhang, J., Wu, M. et al. Identification of key miRNAs and mRNAs related to coronary artery disease by meta-analysis. BMC Cardiovasc Disord 21, 443 (2021). https://doi.org/10.1186/s12872-021-02211-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12872-021-02211-2