
Abstract
Brazil is the world's largest producer of common bean. Knowledge of the genetic diversity and relatedness of accessions adapted to Brazilian conditions is of great importance for the conservation of germplasm and for directing breeding programs aimed at the development of new cultivars. In this context, the objective of this study was to analyze the genetic diversity, population structure, and linkage disequilibrium (LD) of a diversity panel consisting of 219 common bean accessions, most of which belonging to the Mesoamerican gene pool. Genotyping by sequencing (GBS) of these accessions allowed the identification of 49,817 SNPs with minor allele frequency > 0.05. Of these, 17,149 and 12,876 were exclusive to the Mesoamerican and Andean pools, respectively, and 11,805 SNPs could differentiate the two gene pools. Further the separation according to the gene pool, bayesian analysis of the population structure showed a subdivision of the Mesoamerican accessions based on the origin and color of the seed tegument. LD analysis revealed the occurrence of long linkage blocks and low LD decay with physical distance between SNPs (LD half decay in 249 kb, corrected for population structure and relatedness). The GBS technique could effectively characterize the Brazilian common bean germplasms, and the diversity panel used in this study may be of great use in future genome-wide association studies.
Similar content being viewed by others

Introduction
The common bean (Phaseolus vulgaris L.) is one of the five cultivated species of the Phaseolus genus and is one of the most consumed legumes worldwide. It is the most important legume grain for direct human consumption and the main source of protein and micronutrients in several countries1. Globally, around 31 million tons of bean grains are produced per year, with the Americas accounting for 32.4% of the total production. Brazil is the world's largest producer of common bean, and other countries that are among the largest producers are India, Myanmar, China, United States, and Mexico2.
The common bean is known to have originated in Mexico and the Southern Andes, where it was domesticated independently to give rise to two gene pools, i.e., the Andean and Mesoamerican groups, which are morphologically and genetically different3,4,5,6. Different parts of the world prefer either the Andean or Mesoamerican grains. The Mesoamerican common beans are more common in North America, Central America, and the lowland part of South America, whereas the Andean common beans are preferred in parts of Africa, Europe, and Andean part of South America7,8. In Brazil, Mesoamerican common beans are preferred, of which the carioca and black beans represent the most consumed commercial groups9,10. Carioca beans are the most widely produced in Brazil, accounting for approximately 70% of the national common bean production, whereas black beans represent about 15% of the total production11.
Genetic diversity studies are of great importance for breeding programs, as they provide valuable information for effective conservation and application of available germplasm12. Such studies facilitate the understanding of genetic relationships between accessions, identification of redundancies and admixtures in the germplasm, and determination of genitor pairs with adequate genetic distance.
Molecular markers have been widely used in plant breeding programs. Several different types of markers are available; however, their applications have been restricted in the past due to limitations such as low density, labor intensity, technical requirements, and high cost of large-scale analysis13,14. The advent of next-generation sequencing (NGS) technologies has resulted in an exponential increase in the number of genetic variants that can be discovered in a single experiment15. The publication of the complete genome sequence of the common bean by Schmutz et al.5 facilitated the discovery of single nucleotide polymorphisms (SNPs) and genetic mapping, further allowing the construction of maps from short reads of different genotypes using the genome sequence as a reference16.
Among the NGS methods, the genotyping by sequencing (GBS) technique has emerged as a new approach to mitigate the constraints of previously employed markers17. GBS is a robust, high-performance, cost-effective, and simple technique for obtaining thousands of markers from a large number of individuals, and allows the identification of SNPs using a reduced representation library18,19,20. SNPs are the most abundant and universal sequence variations in all genomes, which makes them very useful markers for genetic analyses in plants21.
The GBS technique is often employed in plant breeding, and is frequently used in genetic diversity studies, mapping (linkage and association) studies, and genomic selection (GS)13. Genome-wide association studies (GWAS) are a powerful tool for identifying candidate genomic regions associated with traits of interest. Some of the most important parameters for successful GWAS are the representativity of the diversity panel, the size of the panel, the levels and genomic distribution of linkage disequilibrium (LD), and the population structure or genetic relationships among individuals22,23,24. The diversity panel should represent most of the available genetic and phenotypic diversity, and LD should be analyzed to determine the density of markers required for GWAS25.
Studies on genetic diversity and population structure have already been conducted for several crops, including wheat26,27,28, flaxseed29, pepper30 and rice31. Several diversity panels have also been developed for the common bean crop, including accessions from different regions of the world32,33,34,35,36,37. Based on these initial studies, several GWAS have further been conducted for different traits of interest, such as yield, plant architecture, nutritional content of grains, cooking time, resistance to diseases, and tolerance to abiotic factors7,32,37,38,39,40,41,42,43,44,45,46,47. Some GWAS have been conducted in Brazil37,40,48, however, panels consisting of different genotypes, can contribute to a better understanding about the genetic diversity and relationships of the germplasm available for genetic breeding.
In view of the above, the objective of the present study was to analyze the genetic diversity, population structure, and LD of the Brazilian Diversity Panel (BDP), which is a common bean diversity panel representing a large proportion of the genetic diversity of Brazilian common bean populations. It is composed mainly of materials from the carioca and black bean commercial groups, which are the most consumed cultivars in the country, and is expected to be used for GWAS in the future.
Results
Genotyping by sequencing
Using the GBS method optimized for common beans by Ariani et al.18, a total of 392,585,199 good barcoded reads were obtained from the sequenced accessions, of which 364,454,550 could be aligned with the Andean reference genome (G198335), resulting in an average mapping rate of 93%. Initially, 461,199 SNPs were obtained, of which 49,817 SNPs were retained after filtering. Eleven accessions had a low rate of genotyping (less than 10% of genotyped positions) and were excluded from the BDP, for this reason 219 accessions were used in the subsequent analyses.
SNPs were unevenly distributed throughout the genome, and fewer SNPs were observed in regions near the centromere than in regions near the telomeres on the chromosome (Fig. 1a). The mean number of SNPs per chromosome was 4528, ranging from 3361 to 5910 SNPs on the Pv06 and Pv02 chromosomes, respectively (Table 1). Physical chromosome length was positively correlated with the number of SNPs (r = 0.74, p < 0.01).

Identification and annotation of 49,817 single nucleotide polymorphisms (SNPs) obtained from the genotyping of 219 common bean accessions. (a) Distribution of SNP density along the common bean genome in a 200 kb sliding window. (b) Annotation of SNPs and proportion of genomic traits. (c) Transversion/transition ratio. Figure produced in R v.4.0.
Of the total SNPs obtained, 33.8% were located in intragenic regions (17.5% in intron and 16.3% in exons), 12.5% in coding DNA sequences, 1.2% in 5′ UTR regions, and 2.6% in 3′ UTR regions (Fig. 1b). Thirty percent of the annotated genes in the reference genome of Phaseolus vulgaris v2.0 were tagged by at least one SNP (tagged genes) (Table 1). A positive correlation between the number of genes and tagged genes per chromosome was observed (r = 0.96, p < 0.01).
Of the different types of polymorphism, transitions (63.1%) were more frequent than transversions (36.9%), resulting in a transition/transversion rate of 1.71 (Fig. 1c). The percentages of A/G and C/T transitions were very similar (32% and 31%, respectively), as were those of polymorphism due to A/T, A/C, G/T, and G/C transversions (11%, 9%, 9%, and 8%, respectively). Considering only the SNPs inside genes the transition/transversion rate was 1.27, smaller compared to the overall rate, for the reason that the percentage of transversions (44.1%) was greater than the overall.
Genetic diversity and population structure
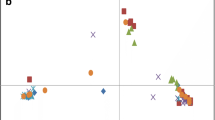
The population structure of all accessions included in the BDP were analyzed using 819 SNPs that were retained after LD filtering (r2 < 0.2). The results of the principal component analysis (PCA) showed that the accessions could be segregated into two distinct groups, based on the gene pools (Andean and Mesoamerican) (Fig. 2a).

Genetic differentiation between Andean and Mesoamerican gene pools. (a) Principal component analysis of 219 accessions of Andean and Mesoamerican origin including different commercial groups (black, carioca, cream, red, etc.). (b) Venn diagram of the total set of SNPs and SNPs belonging to the Andean and Mesoamerican groups. (c) Distribution of the Fst values of each SNP (colored according to the population in which they occur). (d) Total number of differentiating SNPs on each chromosome and number of differentiating SNPs located within genes. (e) Distribution of the 11,805 differentiating SNPs of the Andean and Mesoamerican groups along the common bean genome in a 200 kb sliding window. Figures (a), (c), (d) and (e) produced in R v.4.0 and Figure b produced in JVENN (http://jvenn.toulouse.inra.fr).
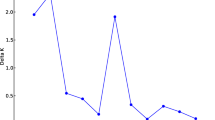
The two gene pools were also segregated in the Bayesian population structure analysis. However, based on the ΔK49 criterion, the number of groups (K) with the highest value of ΔK was three (K = 3), which demonstrated a subdivision of the Mesoamerican group (Fig. 3). Based on the membership coefficient (≥ 0.6), 90.9% of the accessions could be assigned to a specific group, and only 20 accessions were categorized as admixtures. The accessions of Andean origin formed a group, and the Mesoamerican accessions were divided into two distinct groups and the admixture group. In the two Mesoamerican groups formed solely by individuals with a membership coefficient ≥ 0.6, the accessions were distinguished by the color of the seed tegument; one group was composed primarily of carioca-type grain accessions, whereas the other group included accessions with black, purple, red, cream, and other tegument colors. The admixture group comprised accessions that had resulted from hybridization between the previous two groups. The accessions of commercial groups other than black and carioca (i.e., purple, red, cream, and others) were predominantly grouped with the black commercial group; however, there was a tendency to cluster according to the color of the flower, which is purple in the black group, white in the carioca group, and variable (white, pink, and purple) in other accessions. Three accessions initially identified as Mesoamerican were assigned to the Andean group in these analyses and were therefore treated as Andean in subsequent analyses.

Analysis of the population structure using 219 accessions belonging to the Brazilian common bean diversity panel with K = 3: (1) corresponds to the group of common beans of Andean origin; (2) mostly formed by Mesoamerican accessions of black, cream, red, and other seed tegument colors; (3) mostly formed by Mesoamerican accessions from the carioca commercial group; and (4) mostly formed by Mesoamerican accessions with membership coefficient < 0.6 for the previous groups. Figure produced in R v.4.0.
Removal of accessions of Andean origin from the panel left 207 accessions of the Mesoamerican origin. Among these 207 accessions, 25,136 SNPs with MAF > 0.05 could be identified, i.e., the number of SNPs per chromosome was reduced on average by 50% relative to the number of SNPs identified when the Andean accessions were included in the panel. The chromosomes exhibiting the greatest reduction in the number of SNPs were Pv05 and Pv11, whereas Pv09 and Pv01 presented the smallest reduction.
Genetic differentiation between Andean and Mesoamerican gene pools
The two gene pools shared 7987 SNPs, whereas 17,149 and 12,876 SNPs were unique to the Mesoamerican and Andean groups (Fig. 2b), respectively. The mean pairwise fixation index (Fst) for each of these SNP groups was 0.39, 0.34, and 0.94, respectively (Fig. 2c). A total of 11,805 SNPs differentiating the Andean and Mesoamerican groups were detected, with a mean Fst of 0.97. The mean Fst between the Andean and Mesoamerican pools was 0.77 when all the SNPs were included. The Mesoamerican group showed greater mean nucleotide diversity (π = 0.31) than the Andean group (π = 0.22). Regarding Tajima’s D, the Mesoamerican gene pool showed a positive value (D = 1.50), while the Andean gene pool showed a negative value (D = − 0.50) (Table 2).
Most SNPs that differentiate the Andean and Mesoamerican pools were located on chromosomes Pv01 (2492 SNPs), Pv08 (1781 SNPs), Pv09 (1554 SNPs), Pv02 (1506 SNPs), and Pv10 (1387 SNPs) (Fig. 2d,e), and 30.8% were located within genes, with 2187 genes including at least one differentiating SNP. Most of these SNPs inside genes were located on chromosomes Pv02 (648 SNPs), Pv01 (595 SNPs), Pv09 (504 SNPs), Pv06 (476 SNPs), and Pv08 (435 SNPs) chromosomes (Fig. 2d). Among the SNPs located in coding regions, 26% were synonymous SNPs and 74% were non-synonymous (being 68% missense variants). Of the genes containing the differentiating SNPs, 279 were putative candidates for domestication, of which 179 are known to be involved in the domestication of the Mesoamerican group, 91 in that of the Andean group, and 9 in the domestication of both these groups5.
Genetic differentiation among the Mesoamerican accessions
As seen in the PCA and Bayesian analysis of population structure, the accessions of Mesoamerican origin were also segregated into two main groups in the phylogenetic tree, based on the tegument color, with one group consisting of the carioca commercial group and the other including the accessions with black, cream, red, white and purple tegument (Figs. 4a, 5).

Principal component analyses and Venn diagrams. (a) Principal component analysis of 207 accessions of common beans of Mesoamerican origin with different seed tegument colors. (b) Venn diagram for the different sets of SNPs related to seed tegument color. (c) Principal component analysis of 207 accessions of common beans from different research institutions. (d) Venn diagram for the different sets of SNPs related to the institutions of origin. Figure (a) and (c) produced in R v.4.0 and Figures b and d produced in JVENN (http://jvenn.toulouse.inra.fr).

Dendrogram showing the genetic relatedness among 207 common bean accessions belonging to the Brazilian Diversity Panel. The different colors identify the accessions according to the color of the seed tegument. Purple = black tegument, Orange = carioca-type tegument, Green = cream tegument, Pink = red tegument, and Blue = others. Figure produced in FigTree v1.4.4.
The separation of Mesoamerican individuals by seed color showed that each group had a variable number of SNPs, and only 12,795 SNPs were common to all these color groups (Fig. 4b). The cream-colored accessions exhibited the highest number of SNPs (27,579) and the lowest π (0.29) value. The red-colored group had the highest π (0.35), whereas π values of the carioca and black groups were similar (0.32 and 0.31, respectively) (Table 2). According to Fst, the carioca and red groups were the most different, with an Fst value of 0.22, whereas comparisons between the other colors yielded low Fst values. The Tajima’s D values were all positive in relation to the seed tegument color as well as for the institution of origin (Table 2).
Regarding the institution of origin, a clustering trend was observed for the accessions of the International Center for Tropical Agriculture (CIAT), Agronomic Institute of Pernambuco (IPA), and the more recent inbred lines of the Rural Development Institute of Paraná—IAPAR–EMATER (IAPAR) (Fig. 4c). The number of SNPs was variable for each institution; however, the π was similar. A total of 16,196 SNPs was shared in the accessions of all institutions, whereas 8210 were exclusive, i.e., belonged to only one institution (Fig. 4d). The Agronomic Institute of Campinas (IAC) accessions presented the highest number of exclusive markers (6112), whereas the Brazilian Agricultural Research Corporation (EMBRAPA) accessions included only 313 unique markers. Comparison of the accessions from different institutions did not yield high differentiation indexes (Fst), with the highest value being observed between CIAT and IAC (0.12) and the lowest value between IAPAR and EMBRAPA (0.01) (Table 2).
Linkage disequilibrium
LD decay and half-decay distances were calculated for individual chromosomes and for the whole genome. In both cases, the differences between conventional r2 and population structure-corrected r2 (r2s) were small. Considering the whole genome (all chromosomes), the half-decay distance was 1361 kb and 1180 kb for r2 and r2s, respectively. The r2 was remarkably different when compared with r2v (r2 corrected for relatedness) and r2vs (r2 corrected for population structure and relatedness) (Fig. 6). The latter two measures exhibited very similar decay values, with half-decay occurring at 249 kb.

Analysis of linkage disequilibrium (LD) decay as a function of physical distance without correction (r2), and after correcting for population structure (r2s), relatedness (r2v), and for both population structure and relatedness (r2vs). Figure produced in R v.4.0.
In the analysis of r2v and r2vs of individual chromosomes, the half-decay distance ranged from 183 to 397. The highest decay values were noted for chromosomes Pv10, Pv08, Pv01, and Pv06 chromosomes (183, 187, 193 and 198 kb, respectively), whereas the lowest decay values were presented by chromosomes Pv05, Pv04, Pv07, and Pv09 (397, 322, 317 and 310, respectively) (Fig. 6).
Discussion
The common bean is a very important crop in Brazil and is cultivated in all states of the country, mainly by family farmers. Considering the history of common bean cultivation in the country, domesticated common beans are highly diverse, although Brazil is not a primary center of diversity50,51. In this context, the present study was developed to understand the genetics and population structure of a newly created common bean diversity panel that includes a large part of the diversity of the most consumed common bean types in Brazil. These results will assist future GWAS for determining genomic regions or genes associated with several economically important traits.
The GBS methodology proposed by Ariani et al.18 was used in this study, which could effectively detect numerous SNPs in the analyzed accessions. These authors found that the CviAII enzyme was more effective than the commonly used ApeKI enzyme. As a methylation-insensitive enzyme, CviAII exhibited a higher number of restriction sites and acted preferentially on non-repetitive parts of the genome, allowing the identification of thousands of markers spaced unevenly throughout the common bean genome, with a density distribution resembling that of the distribution of genes.
Initially, 461,199 SNPs were identified. However, 89% of the markers did not satisfy the filtering criteria (Non-biallelic, indels, MAF < 0.05, coefficient of inbreeding < 0.9 and less than 10% of genotyped positions) and were not used in subsequent analyses. Polymorphisms were widely distributed across the 11 chromosomes and were highly correlated with the length and number of genes on each chromosome. The transition/transversion rate was consistent with that observed in other studies on common bean and other species28,35,52,53. Transitions are usually more frequent than transversions in several species, which indicates that the former are better tolerated during natural selection, which may be due to the fact that they are synonymous mutations in protein-coding sequences29,54.
Because LD may affect the inference of the population structure, an LD filter was further applied, which resulted in a decrease of the number of SNPs. This is due to the fact that the common bean is an autogamous plant with very long blocks of markers in LD35,55,56.
Genetic differentiation between common bean accessions based on the gene pool has been well documented in several previous studies3,34,35,57,58,59,60. The relationship between the genetic similarity of the Mesoamerican accessions and the color of the seed tegument was also observed by Valdisser et al.61 and Gioia et al.62. In Brazil, breeding programs for the carioca and black commercial groups have different objectives63. Moreover, genetic breeding of the carioca group is much more advanced than that of the black group, because of its greater importance in the country due to consumers and market preferences. Efforts to improve the carioca bean are directed towards the grain size traits, to satisfy the consumers’ preference for larger grains. However, the grain size is negatively correlated with yield, in case of the black group, selection is based mainly on yield, resulting in cultivars with smaller grains64.
Several SNPs exclusive to either of the gene pools were observed, in addition to the differentiating SNPs between the two pools. Other authors have also reported that the proportion of polymorphic loci tends to be higher in populations composed of accessions from the two centers of origin, and it tends to decrease when they are studied separately61,65,66. The two gene pools differ in both phenotypic and molecular characteristics, which is supported by the high rates of genetic differentiation obtained in the present analysis and in other studies50,51,61,67. In addition, the Mesoamerican gene pool exhibits higher nucleotide diversity than the Andean, possibly because a strong bottleneck occurred during the dispersal of Southern Andean common beans from Mesoamerica, which drastically reduced its nucleotide diversity4,5,33,34,60,67.
To identify genomic signatures of selection between the Andean and Mesoamerican pools, the Fst was estimated for each SNP. The Fst of nucleotide positions that were polymorphic only when the two gene pools were studied together was close to 1, and these SNPs were therefore highly discriminating between the gene pools. The Fst of SNPs present only in the Andean group was also high, similar to that of the discriminating markers. This may be due to the small number of Andean accessions included in this study.
There is significant evidence supporting the independent domestication of the Andean and Mesoamerican gene pools. Schmutz et al.5 identified 1835 candidate genes for domestication in the Mesoamerican group and 748 candidate genes in the Andean group. Of these, only 59 genes were common to both groups. These genes are mainly located on chromosomes Pv01, Pv02, Pv07, Pv09, and Pv10. In the present study, 11% of all the candidate genes for domestication harbored differentiating SNPs. These genes have also been identified in other studies aimed at finding selection signatures between the Andean and Mesoamerican34,35 accessions. The candidate genes for domestication are directly or indirectly associated with the main characteristics that distinguish the two gene pools, such as flowering time, plant size, and seed size.
The low rate of differentiation of the accessions based on the institution of origin may be related to the protocols of the breeding programs. Breeding programs tend to be conservative and almost always employ the Mesoamerican germplasm, with little exploration of exotic germplasms; in addition, they use a selected group of elite parents, which further narrows the genetic base57,61,62,63. Because of significant exchange of germplasm between institutions, there was no formation of well-defined groups among the accessions from the different institutions of origin57, only a trend for clustering was observed for accessions belonging to CIAT, IPA, and the more recent inbred lines of IAPAR.
LD measurement is very important in association mapping studies for identifying loci associated with quantitative traits. Importantly, the population structure and relatedness between the analyzed accessions may cause a bias in LD estimation. Frequent selection, admixture of populations, and crossing of a small number of cultivars in breeding programs reduces genetic diversity and affects LD patterns68. These factors can affect different genomic regions in several ways, which can introduce heterogeneity of LD through the genome. This makes the resolution and power achieved in GWAS dependent on the species and the population under study. LD decay is slower in autogamous species, such as common bean and soybean, in which recombination is less effective than in allogamous species68,69.
In this study, the LD corrected for population structure (r2s) was not significantly different from the conventional r2. However, r2v and r2vs exhibited a faster LD decay when compared with the conventional r2. The fact that r2v was considerably lower than conventional r2 demonstrates the need to remove the effect of relatedness to reduce the overestimation of LD. The similarity between the estimated r2 and r2s (LD half-decay with 296 kb) shows that the BDP is not highly structured, which is consistent with the results of other studies on common bean diversity panels40,51,57. As observed by Diniz et al.57 in panels composed mainly of improved genotypes, the degree of relatedness between individuals was very high.
The present study demonstrated that GBS is a powerful approach for analyzing the population structure and genetic diversity in common bean. The newly developed diversity panel, which represents a large proportion of the Brazilian common bean diversity, exhibited high genetic diversity, and was shown to be adequate for future studies to identify genomic regions related to traits of interest (GWAS).
Methods
Plant material
The BDP, including 230 common bean accessions that represent a large component of the common bean genetic diversity in Brazil, was used in this study (Table S1). The diversity panel is composed of modern and old cultivars developed between 1968 and 2019 by different research institutions (Table 2), in addition to inbred lines and landraces, all of which belong to the germplasm bank of the Rural Development Institute of Paraná –IAPAR–EMATER (IAPAR). Among the CIAT accessions present in this panel, most of them are inbred lines from breeding programs directed to the needs of Brazil and/or are accessions that compose the genealogy of cultivars developed by Brazilian institutions. Most accessions in this panel are of Mesoamerican origin, which exhibit significant diversity in the color of the seed tegument and include different commercial classes, with the carioca and black bean groups being the most representative. In addition, 10 accessions of Andean origin were included in this study for comparison.
Genotyping by sequencing (GBS)
DNA extraction and the preparation of GBS libraries for sequencing was performed following the protocol developed for common bean by Ariani et al.18. DNA was extracted from lyophilized leaves collected from a single plant of each accession grown in a green house. The extracted DNA was purified using the Genomic DNA Clean and Concentrator kit (Zymo Research, CA, USA), according to the manufacturer's instructions. The DNA quality was checked using NanoDrop Lite (Thermo Fisher Scientific), and only samples with an absorbance ratio (A260/A280) greater than 1.7 were used for preparing the libraries. Genomic DNA was quantified using Quant-iT PicoGreen dsDNA Assay Kit (Thermo Fisher Scientific), and 100 ng of the DNA from each genotype was used for preparing the libraries.
The genomic DNA was digested using the restriction enzyme CviAII (recognition site C'ATG); after the preparation process, the samples were multiplexed into two libraries with up to 144 accessions each, including as control a blank sample and the genotype of P. vulgaris used to construct the reference genome (G19833) in each of the two libraries67. The presence of adapter dimers in the sequencing libraries was checked using DNA High Sensitivity Kit (Agilent 2100 Bionalyzer, Agilent Technologies).
The genomic libraries were sequenced using the Illumina HiSeq 4000 sequencer (Illumina, San Diego, CA, USA) with the 100-bp-single-end protocol, at the DNA Technologies and Expression Analysis Core Laboratory, located in the Genome Center, University of California, Davis, CA.
Analysis of sequencing data
SNPs were called using the Tassel-5-GBS pipeline version 270, with the standard software settings, except for the minimum quality score (-mnQs 20) and minimum count (-c 10) parameters. The obtained sequences were aligned with the reference genome of Phaseolus vulgaris v2.0 obtained from the Phytozome website (https://phytozome.jgi.doe.gov, accessed on March 10, 2019), using the Burrows-Wheeler Alignment (BWA) (-aln option) tool version 0.7.1071. Non-biallelic SNPs, and SNPs with indels, minor allele frequency (MAF) < 0.05, coefficient of inbreeding < 0.9, and those SNPs and accessions containing < 10% of genotyped positions were removed using VCFtools version 0.1.1572. Because common bean is an autogamous species, after the initial filtering, the occurrence of heterozygotes was insignificant, but heterozygous SNPs were treated as missing data, as they may indicate sequencing errors. After filtering, the SNPs were imputed using Beagle software version 573, and only SNPs anchored to chromosomes in the common bean reference genome were used.
The SNPs were annotated according to the common bean genomic annotation (GFF3 file, version 2.1) available on the Phytozome website (https://phytozome.jgi.doe.gov, accessed on January 10, 2019), using a custom R74 script developed by Hu et al.75 (https://github.com/zhenbinHU/Sorghum_SNP_dataset, accessed on June 17, 2019).
Genetic diversity and population structure
The 219 accessions belonging to the BDP that passed by the quality control mentioned above were included in the initial analyses. The population structure was inferred using the Bayesian clustering algorithm in Structure v2.3.476 software from the command line python program StrAuto77. The admixture model with 50,000 burn-ins, 200,000 MCMC, and 10 replications for hypothetical numbers of subpopulations (K) between 1 and 10 was used. The statistical parameter ΔK49 was used to determine the number of groups. Only the accessions with a membership coefficient equal or higher than 0.6 were assigned to a genetic group, and those with membership coefficient lower than 0.6 were clustered in the admixture group. The admixture model assumes that the markers are not strongly linked; hence, the SNPs were filtered based on LD, using the indep-pairwise option of the PLINK78 software, and only SNPs with LD ≤ 0.2 were retained for population structure analysis. These data filtered for LD were also used for PCA, using the snpgdsPCA function of the SNPRelate79 package in R.
After verifying the center of origin, only individuals of Mesoamerican origin were retained, and the SNPs were again filtered to exclude monomorphics, SNPs with MAF < 0.05 and LD ≥ 0.2, using VCFtools version 0.1.1572 and PLINK78. These data were then used for PCA and population structure analysis, as previously described. In addition, phylogenetic inference was estimated using TASSEL v580, based on identity-by-state (IBS) distance and using Neighbor-Joining as the clustering method. The generated tree was customized using FigTree v1.4.481.
To detect molecular differences in relation to the center of origin, color of the seed tegument, and institution of origin, new files were created from the initial file (including all SNPs) containing the different groups, and only polymorphic SNPs and those with MAF > 0.05 were retained. Subsequently, a Venn diagram was constructed to detect the differentiating SNPs for each of the three parameters using the JVENN tool82. Fst index83, nucleotide diversity (π) and Tajima’s D84, were also calculated using VCFtools version 0.1.1572 and averaged on 100-kb genomic bins.
Linkage disequilibrium
LD between SNPs was estimated using the LDcorSV85 package in R. This package corrects for the bias due to population structure and relatedness while estimating LD. In addition to the conventional r2, r2 corrected for population structure (r2s), r2 considering kinship (r2v), and r2 including both population structure and kinship (r2vs) were calculated. Only individuals belonging to the Mesoamerican group were used for these calculations. The STRUCTURE result at K = 2 for common beans of Mesoamerican origin was used as the population structure, and for relatedness the kinship matrix was calculated using the rrBLUP86 package in R. LD decay was calculated using the nonlinear method proposed by Hill and Weir87, and adjusted with the nls function in R.
References
Broughton, W. J. et al. Beans (Phaseolus spp.)—model food legumes. Plant Soil 252, 55–128 (2003).
FAO, F. and A. O. FAOSTAT: FAO Statistical Databases. http://www.fao.org/faostat/ (2020).
Kwak, M. & Gepts, P. Structure of genetic diversity in the two major gene pools of common bean (Phaseolus vulgaris L., Fabaceae). Theor. Appl. Genet. 118, 979–992 (2009).
Bitocchi, E. et al. Molecular analysis of the parallel domestication of the common bean (Phaseolus vulgaris) in Mesoamerica and the Andes. New Phytol. 197, 300–313 (2013).
Schmutz, J. et al. A reference genome for common bean and genome-wide analysis of dual domestications. Nat. Genet. 46, 707–713 (2014).
Vlasova, A. et al. Genome and transcriptome analysis of the Mesoamerican common bean and the role of gene duplications in establishing tissue and temporal specialization of genes. Genome Biol. 17, 1–18 (2016).
Oladzad, A. et al. Single and multi-trait GWAS identify genetic factors associated with production traits in common bean under abiotic stress environments. G3 Genes Genomes Genet. 9, 1881–1892 (2019).
Cichy, K. A., Caldas, G. V., Snapp, S. S. & Blair, M. W. QTL analysis of seed iron, zinc, and phosphorus levels in an andean bean population. Crop Sci. 49, 1742–1750 (2009).
Ribeiro, N. D. et al. Evaluation of special grains bean lines for grain yield, cooking time and mineral concentrations. Crop Breed. Appl. Biotechnol. 14, 15–22 (2014).
Perseguini, J. M. K. C. et al. Developing a common bean core collection suitable for association mapping studies. Genet. Mol. Biol. 38, 67–78 (2015).
MAPA, Ministerio da Agricultura Pecuária e Abastecimento. Plano nacional de desenvolvimento da cadeia do feijão e pulses (2018).
Cheng, J. et al. Development of a SNP array and its application to genetic mapping and diversity assessment in pepper (Capsicum spp.). Sci. Rep. 6, 1–11 (2016).
He, J. et al. Genotyping-by-sequencing (GBS), an ultimate marker-assisted selection (MAS) tool to accelerate plant breeding. Front. Plant Sci. 5, 484 (2014).
Bhattarai, U. & Subudhi, P. K. Identification of drought responsive QTLs during vegetative growth stage of rice using a saturated GBS-based SNP linkage map. Euphytica 214, 1–17 (2018).
Stapley, J. et al. Adaptation genomics: The next generation. Trends Ecol. Evol. 25, 705–712 (2010).
Gujaria-Verma, N. et al. Gene-based SNP discovery in tepary bean (Phaseolus acutifolius) and common bean (P. vulgaris) for diversity analysis and comparative mapping. BMC Genomics 17, 239 (2016).
Siadjeu, C., Mayland-Quellhorst, E. & Albach, D. C. Genetic diversity and population structure of trifoliate yam (Dioscorea dumetorum Kunth) in Cameroon revealed by genotyping-by-sequencing (GBS). BMC Plant Biol. 18, 1–14 (2018).
Ariani, A., Bern y Miery Teran, J. C. & Gepts, P. Genome-wide identification of SNPs and copy number variation in common bean (Phaseolus vulgaris L.) using genotyping-by-sequencing (GBS). Mol. Breed. 36, 87 (2016).
Elshire, R. J. et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6, 1–10 (2011).
Schröder, S. et al. Optimization of genotyping by sequencing (GBS) data in common bean (Phaseolus vulgaris L.). Mol. Breed. 36, 1–9 (2016).
Wang, S. et al. Sequence-based ultra-dense genetic and physical maps reveal structural variations of allopolyploid cotton genomes. Genome Biol. 16, 108 (2015).
Burghardt, L. T., Young, N. D. & Tiffin, P. A guide to genome-wide association mapping in plants. Curr. Protoc. Plant Biol. 2, 22–38 (2017).
Korte, A. & Ashley, F. The advantages and limitations of trait analysis with GWAS: A review Self-fertilisation makes Arabidopsis particularly well suited to GWAS. Plant Methods 9, 29 (2013).
Zhang, D., Bai, G., Zhu, C., Yu, J. & Carver, B. F. Genetic diversity, population structure, and linkage disequilibrium in U.S. elite winter wheat. Plant Genome J. 3, 117 (2010).
Nicolas, S. D. et al. Genetic diversity, linkage disequilibrium and power of a large grapevine (Vitis vinifera L) diversity panel newly designed for association studies. BMC Plant Biol. 16, 1–19 (2016).
Bhatta, M., Morgounov, A., Belamkar, V., Poland, J. & Baenziger, P. S. Unlocking the novel genetic diversity and population structure of synthetic Hexaploid wheat. BMC Genomics 19, 1–12 (2018).
Eltaher, S. et al. Genetic diversity and population structure of F3:6 Nebraska Winter wheat genotypes using genotyping-by-sequencing. Front. Genet. 9, 1–9 (2018).
Alipour, H. et al. Genotyping-by-sequencing (GBS) revealed molecular genetic diversity of Iranian wheat landraces and cultivars. Front. Plant Sci. 8, 1–14 (2017).
Luo, Z. et al. Genetic diversity and population structure of a Camelina sativa spring panel. Front. Plant Sci. 10, 1–12 (2019).
Pereira-Dias, L., Vilanova, S., Fita, A., Prohens, J. & Rodríguez-Burruezo, A. Genetic diversity, population structure, and relationships in a collection of pepper (Capsicum spp.) landraces from the Spanish centre of diversity revealed by genotyping-by-sequencing (GBS). Hortic. Res. 6, 54 (2019).
Xu, Q. et al. The genetic diversity and structure of indica rice in China as detected by single nucleotide polymorphism analysis. BMC Genet. 17, 1–8 (2016).
Moghaddam, S. M. et al. Genome-wide association study identifies candidate loci underlying agronomic traits in a middle American diversity panel of common bean. Plant Genome 9, 1–21 (2016).
Cichy, K. A. et al. A Phaseolus vulgaris diversity panel for Andean bean Improvement. Crop Sci. 55, 2149–2160 (2015).
Campa, A., Murube, E. & Ferreira, J. J. Genetic diversity, population structure, and linkage disequilibrium in a Spanish common bean diversity panel revealed through genotyping-by-sequencing. Genes (Basel) 9, 518 (2018).
Lioi, L., Zuluaga, D. L., Pavan, S. & Sonnante, G. Genotyping-by-sequencing reveals molecular genetic diversity in Italian common bean landraces. Diversity 11, 1–14 (2019).
Raggi, L., Caproni, L., Carboni, A. & Negri, V. Genome-wide association study reveals candidate genes for flowering time variation in common bean (Phaseolus vulgaris L.). Front. Plant Sci. 10, 1–14 (2019).
Perseguini, J. M. K. C. et al. Genome-wide association studies of anthracnose and angular leaf spot resistance in common bean (Phaseolus vulgaris L.). PLoS ONE 11, 1–19 (2016).
Tock, A. J. et al. Genome-wide linkage and association mapping of halo blight resistance in common bean to race 6 of the globally important bacterial pathogen. Front. Plant Sci. 8, 1–17 (2017).
Katuuramu, D. N. et al. Genome-wide association analysis of nutritional composition-related traits and iron bioavailability in cooked dry beans (Phaseolus vulgaris L.). Mol. Breed. 38, 44 (2018).
Resende, R. T. et al. Genome-wide association and Regional Heritability Mapping of plant architecture, lodging and productivity in Phaseolus vulgaris. G3 Genes Genomes Genet. 8, 2841–2854 (2018).
Moghaddam, S. M. et al. Genetic architecture of dietary fiber and oligosaccharide content in a middle american panel of edible dry bean. Plant Genome 11, 1–11 (2018).
Kamfwa, K., Cichy, K. A. & Kelly, J. D. Genome-wide association study of agronomic traits in common bean. Plant Genome 8, 1–12 (2015).
Cichy, K. A., Wiesinger, J. A. & Mendoza, F. A. Genetic diversity and genome-wide association analysis of cooking time in dry bean (Phaseolus vulgaris L.). Theor. Appl. Genet. 128, 1555–1567 (2015).
Kamfwa, K., Cichy, K. A. & Kelly, J. D. Genome-wide association analysis of symbiotic nitrogen fixation in common bean. Theor. Appl. Genet. 128, 1999–2017 (2015).
Zuiderveen, G. H., Padder, B. A., Kamfwa, K. & Song, Q. Genome-wide association study of anthracnose resistance in Andean beans (Phaseolus vulgaris). PLoS ONE https://doi.org/10.1371/journal.pone.0156391 (2016).
Soltani, A. et al. Genetic architecture of flooding tolerance in the dry bean middle-American diversity panel. Front. Plant Sci. 8, 1–15 (2017).
Soltani, A. et al. Genetic analysis of flooding tolerance in an Andean diversity panel of dry bean (Phaseolus vulgaris L.). Front. Plant Sci. 9, 767 (2018).
Valdisser, P. A. et al. Genome-wide association studies detect multiple QTLs for productivity in Mesoamerican diversity panel of common bean under drought stress. Front. Plant Sci. https://doi.org/10.3389/fpls.2020.574674 (2020).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 14, 2611–2620 (2005).
Burle, M. L., Fonseca, J. R., Kami, J. A. & Gepts, P. Microsatellite diversity and genetic structure among common bean (Phaseolus vulgaris L.) landraces in Brazil, a secondary center of diversity. Theor. Appl. Genet. 121, 801–813 (2010).
Valdisser, P. A. M. R. et al. In-depth genome characterization of a Brazilian common bean core collection using DArTseq high-density SNP genotyping. BMC Genomics 18, 1–19 (2017).
Gaur, R. et al. High density linkage mapping of genomic and transcriptomic SNPs for synteny analysis and anchoring the genome sequence of chickpea. Sci. Rep. 5, 1–11 (2015).
Pavan, S. et al. A distinct genetic cluster in cultivated chickpea as revealed by genome-wide marker discovery and genotyping. Plant Genome 10, 1–9 (2017).
Guo, C. et al. Transversions have larger regulatory effects than transitions. BMC Genomics 18, 1 (2017).
Berny Mier y Teran, J. C. et al. Root and shoot variation in relation to potential intermittent drought adaptation of Mesoamerican wild common bean (Phaseolus vulgaris L.). Ann. Bot. https://doi.org/10.1093/aob/mcy221 (2018).
Blair, M. W. et al. Uneven recombination rate and linkage disequilibrium across a reference SNP map for common bean (Phaseolus vulgaris L.). PLoS ONE 13, 1–21 (2018).
Diniz, A. L. et al. Evidence for strong kinship influence on the extent of linkage disequilibrium in cultivated common beans. Genes (Basel) 10, 1–16 (2019).
Pipan, B. & Meglič, V. Diversification and genetic structure of the western-to-eastern progression of European Phaseolus vulgaris L. germplasm. BMC Plant Biol. 19, 1–16 (2019).
Raatz, B. et al. Analyses of African common bean (Phaseolus vulgaris L.) germplasm using a SNP fingerprinting platform: Diversity, quality control and molecular breeding. Genet. Resour. Crop Evol. 66, 707–722 (2019).
Valentini, G., Gonçalves-Vidigal, M. C., Elias, J. C. F., Moiana, L. D. & Mindo, N. N. A. Population structure and genetic diversity of common bean accessions from Brazil. Plant Mol. Biol. Rep. 36, 897–906 (2018).
Valdisser, P. A. M. R. et al. SNP discovery in common bean by restriction-associated DNA (RAD) sequencing for genetic diversity and population structure analysis. Mol. Genet. Genomics 291, 1277–1291 (2016).
Gioia, T., Logozzo, G., Marzario, S., Zeuli, P. S. & Gepts, P. Evolution of SSR diversity from wild types to U.S. Advanced cultivars in the Andean and Mesoamerican domestications of common bean (Phaseolus vulgaris). PLoS ONE 14, 1–21 (2019).
Delfini, J. et al. Distinctness of Brazilian common bean cultivars with carioca and black grain by means of morphoagronomic and molecular descriptors. PLoS ONE 12, e0188798 (2017).
Delfini, J. et al. Estimation of genetic parameters and prediction of genotypic values in common beans using mixed models. Emirates J. Food Agric. 30, 1026–1035 (2018).
Cortés, A. J., Chavarro, M. C. & Blair, M. W. SNP marker diversity in common bean (Phaseolus vulgaris L.). Theor. Appl. Genet. 123, 827–845 (2011).
Blair, M. W. et al. A high-throughput SNP marker system for parental polymorphism screening, and diversity analysis in common bean (Phaseolus vulgaris L.). Theor. Appl. Genet. 126, 535–548 (2013).
Ariani, A., Mier Teran, J. C. B. & Gepts, P. Spatial and temporal scales of range expansion in wild Phaseolus vulgaris. Mol. Biol. Evol. 35, 119–131 (2017).
Contreras-Soto, R. I., de Oliveira, M. B., Costenaro-da-Silva, D., Scapim, C. A. & Schuster, I. Population structure, genetic relatedness and linkage disequilibrium blocks in cultivars of tropical soybean (Glycine max). Euphytica 213, 173 (2017).
Flint-Garcia, S. A., Thornsberry, J. M. & Buckler, E. S. Structure of linkage disequilibrium in plants. Annu. Rev. Plant Biol. 54, 357–374 (2003).
Glaubitz, J. C. et al. TASSEL-GBS: A high capacity genotyping by sequencing analysis pipeline. PLoS ONE 9, e90346 (2014).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595 (2010).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Browning, B. L., Zhou, Y. & Browning, S. R. A one-penny imputed genome from next-generation reference panels. Am. J. Hum. Genet. 103, 338–348 (2018).
R Core Team. R: A language and environment for statistical computing (2020).
Hu, Z., Olatoye, M. O., Marla, S. & Morris, G. P. An integrated genotyping-by-sequencing polymorphism map for over 10,000 sorghum genotypes. Plant Genome 12, 180044 (2019).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Chhatre, V. E. & Emerson, K. J. StrAuto: Automation and parallelization of STRUCTURE analysis. BMC Bioinform. 18, 1–5 (2017).
Purcell, S. et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Zheng, X. et al. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 28, 3326–3328 (2012).
Bradbury, P. J. et al. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635 (2007).
Rambaut, A. FigTree v1.4.4 (2018).
Bardou, P., Mariette, J., Escudié, F., Djemiel, C. & Klopp, C. SOFTWARE Open Access jvenn: An interactive Venn diagram viewer. BMC Bioinform. 15, 1–7 (2014).
Weir, B. S. & Cockerham, C. C. Estimating F-statistics for the analysis of population structure. Evolution (N.Y.) 38, 1358–1370 (1984).
Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–595 (1989).
Desrousseaux, D., Sandron, F., Siberchicot, A., Cierco-Ayrolles, C. & Mangin, B. LDcorSV: Linkage disequilibrium corrected by the structure and the relatedness. R package version 1.3.2 (2017).
Endelman, J. B. Ridge regression and other kernels for genomic selection with R package rrBLUP. Plant Genome J. 4, 250 (2011).
Hill, W. G. & Weir, B. S. Variances and covariances of squared linkage disequilibria in finite populations. Theor. Popul. Biol. 33, 54–78 (1988).
Acknowledgements
The authors would like to thank the Instituto de Desenvolvimento Rural do Paraná (IDR-Paraná) and the University of California, Davis (through the Gepts’ Lab) for support this research and the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) for the scholarship in Brazil and abroad (Finance Code 001).
Author information
Authors and Affiliations
Contributions
J.D., V.M.C. and L.S.A.G. conceived and designed the study; J.D. and J.S.N. collected plant material, extracted DNA and performed the genotyping; J.D., G.C.S. and L.S.A.G. performed bioinformatics and statistical analyses; J.D. drafted the manuscript; J.D., V.M.C., J.S.N., P.M.R, G.C.S., P.G., and L.S.A.G. edited and revised the final manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Delfini, J., Moda-Cirino, V., dos Santos Neto, J. et al. Population structure, genetic diversity and genomic selection signatures among a Brazilian common bean germplasm. Sci Rep 11, 2964 (2021). https://doi.org/10.1038/s41598-021-82437-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-82437-4
- Springer Nature Limited
This article is cited by
-
A genomic toolkit for winged bean Psophocarpus tetragonolobus
Nature Communications (2024)
-
Analysis of population structure and genetic diversity of Camellia tachangensis in Guizhou based on SNP markers
Molecular Biology Reports (2024)
-
Genome-Wide Association Studies for Identification of Novel QTLs Related to Agronomic Traits in Horsegram (Macrotyloma uniflorum)
Plant Molecular Biology Reporter (2024)
-
Whole-genome resequencing of common bean elite breeding lines
Scientific Reports (2023)
-
Molecular markers for assessing the inter- and intra-racial genetic diversity and structure of common bean
Genetic Resources and Crop Evolution (2023)