
Abstract
Ramie (Boehmeria nivea L. Gaud) suffers from long-term continuous cropping. Here, using Illumina high-throughput sequencing technology, we aimed to identify bacteria and fungi associated with continuous cropping in ramie fields in Yuanjiang, Xianning, Sichuan, and Jiangxi. The rarefaction results showed that Jiangxi had significantly lower bacterial α-diversity than that of the other areas. Firmicutes, Proteobacteria, and Acidobacteria were the dominant bacterial phyla, and Ascomycota, Basidiomycota, and Zygomycota were the dominant fungal phyla. In Jiangxi, Firmicutes accounted for 79.03% of all valid reads, which could have significant decreased microbial diversity and negative effects of continuous ramie cropping. We used traditional methods to examine soil nutrients. Sichuan had a relatively high pH and available P and K, but low total N; opposite findings were recorded in Jiangxi. The redundancy analysis revealed that the urease activity, PH, available K, and total N significantly correlated with bacterial community abundance, whereas only total N significantly correlated with fungal community abundance (P < 0.01). Overall, the effect of soil environmental factors on the bacterial diversity of continuous ramie cropping was greater than that on fungal diversity. In the future, we will focus on the effect of rhizosphere bacteria to solve the obstacle in continuous ramie cropping.
Similar content being viewed by others

Introduction
In continuous cropping, the same or a similar species is planted continuously in the same field1. Generally, long-term continuous cropping decreases crop yield and quality2,3,4, alters soil microbial communities5,6, affects soil biochemical properties7,8, and enriches soil-borne plant pathogens in soil9,10. Soil microorganims are not only an important part of soil, but also the main drivers of soil nutrient cycling11. Soil microorganisms play a crucial role in regulating the fertility of soil, health of plants, and cycling of C, N, and other nutrients11,12. Previous studies have shown that long-term continuous cropping can alter soil microbial communities13,14,15. In a continuous pea-cropping field, the soil microbial community was smaller, and the abundance of beneficial gram-positive bacteria and arbuscular mycorrhizal (AM) fungi was reduced16. Similarly, Sun et al. found that in continuous cropping of banana, bacterial community diversity continuously decreased and bacterial community composition and structure were affected8. Numerous studies have revealed that soil attributes are influenced by continuous cropping17,18. In addition, environmental factors, such as soil pH, affect soil microbial communities19,20. Wang et al. found that root exudate composition and soil pH affected soil microbial community in different plant growth stages21. Thus, it is crucial to analyse the relationship between soil microbial community and environmental factors. However, only a few studies have focused on differences in different areas under continuous cropping with the same plant.
Ramie, also known as “China grass,” is a perennial plant that belongs to the family Urticaceae. Ramie is a traditional fibre crop in China and an important natural fibre crop in India and other Southeast Asian and Pacific Rim countries. Owing to its high protein and amino acid content and rapid growth rate, ramie is used as a feed crop22. However, because of issues associated with continuous cropping, the cultivation areas of ramie have decreased sharply in recent years. Ramie is the main plant cultivated in the Hunan, Hubei, Sichuan, and Jiangxi Provinces of China. Because of differences in the climate and soil type, the rhizosphere microorganisms of continuous cropping ramie may change each year, thereby influencing the growth of ramie. Owing to the influence of different climatic conditions and soil types, the species and number of rhizosphere microorganisms in different planting areas of ramie deserve further study.
Crop rotation offers one way to solve continuous cropping problems; however, it is not applicable to ramie as it is a perennial crop. As soil microorganisms play an important role in plant health and crop yield, modulation of soil microbes provides an option for tackling continuous-cropping obstacles. In the early stage, we studied the changes in rhizosphere microorganisms in continuous cropping of ramie in the Hunan Province3. The main rhizosphere microorganisms negatively affecting the continuous cropping of ramie have been identified. However, microbial communities in ramie fields in different areas are poorly characterised. In this study, to obtain a comprehensive understanding of the bacterial and fungal community structures, we compared all continuous-cropping ramie fields located in four regions (Yuanjiang, Xianning, Sichuan, and Jiangxi) in China. To comparatively explore bacterial and fungal communities in different ramie planting areas, we subjected bacterial and fungal communities from continuous ramie cropping fields in Yuanjiang, Xianning, Sichuan, and Jiangxi (China) to high-throughput sequencing, and we used redundancy analysis (RDA) to analyse relationships between soil microbial communities and soil properties.
Results
Ramie-field soil basic properties
Basic properties of soils from continuous ramie cropping fields in Yuanjiang, Xianning, Sichuan, and Jiangxi are shown in Table 1. Soil pH ranged from 5.33 to 6.67. Soil pH was similar in Xianning and Jiangxi, and in Yuanjiang and Sichuan. Urease activity and TN were the highest in Yuanjiang, with values of 0.62 mg/kg/h and 1.52 g/kg respectively, and the lowest in Sichuan, with values of 0.43 mg/kg/h and 0.76 g/kg respectively. The highest available P level of 38.61 mg/kg was recorded in Sichuan and the lowest of 24.82 mg/kg in Jiangxi. Available K was the highest in Yuanjiang, with 162.60 mg/kg, and the lowest in Jiangxi, with 92.94 mg/kg (Table 1). Statistical analysis showed that the soil PH values of Xianning, Jiangxi, Yuanjiang, and Sichuan were significantly different and that there were significant differences between the total N (TN) and available K in all four regions (Table 1).
Overall diversity of microbial communities in ramie fields in different regions
The overall diversity of microbial communities is shown in Table 2. In total, 385,225 raw reads and 360,544 clean reads were obtained for bacteria, and 456,224 raw reads and 444,898 clean reads were obtained for fungi. The clean bacterial reads included 43,095 reads for Yuanjiang, 64,954 for Xianning, 110,987 for Sichuan, and 141,508 for Jiangxi. The clean fungal reads included 80,267 reads for Yuanjiang, 200,601 for Xianning, 95,671 for Sichuan, and 68,359 for Jiangxi. The α-diversities indicated that the microbial diversity was high in all soil samples. The observed bacterial species ranged from 546 to 2,606. The number of bacterial species was the highest in Sichuan and the lowest in Jiangxi. Observed fungal species ranged from 82 to 466. The number of fungal species was the highest in Xianning and the lowest in Jiangxi.
Moreover, the calculated bacterial and fungal α-diversity species richness (chao1), Simpson, and Shannon indices were all different in the four regions. For bacteria, the results of ANOVA of the Shannon, Simpson, and Chao1 diversity indices showed significant differences, with Jiangxi showing significantly lower diversity than other regions (Table 2). Similarly, for fungi, these indices showed significant differences between the four regions of continuous ramie cropping (Table 2).
Soil microbial community composition in continuous cropping ramie fields in the different areas
Sequences that could not be classified into any known group were assigned as unclassified. The bacterial OTUs were assigned to 52 different phyla, 513 families, or 862 genera (Table S1). Five different phyla (Firmicutes, Proteobacteria, Acidobacteria, Other, and Gemmatimonadetes) out of the 52 total phylotypes were common to the four libraries, accounting for more than 85% of the total reads in each library (Fig. 1A, Table S1). Firmicutes was the most dominant phylum in all soil samples. In Jiangxi, this phylum accounted for 79.03% of all valid reads, which was more than that in other samples. Firmicutes accounted for 30.89%, 37.47%, and 29.46% of valid reads for Sichuan, Xianning, and Yuanjiang, respectively (P < 0.01, Table S2). Proteobacteria was the second dominant phylum. Compared with Firmicutes, Proteobacteria had a significantly lower abundance in Jiangxi (9.08%). In Xianning and Yuanjiang, Proteobacteria abundance was approximately 26% (P < 0.01, Table S2). Other bacteria, including Actinobacteria, Gemmatimonadetes, Bacteroidetes, Chloroflexi, Verrucomicrobia, WS3, Nitrospirae, Planctomycetes, TM7, WPS-2, AD3, Elusimicrobia, Cyanobacteria, Chlamydiae, Chlorobi, FCPU426, and TM6 were all the least abundant in Jiangxi.

Relative abundance of the dominant bacterial (A) and fungal (B) taxa in four different area continuous cropping ramie soil samples at the phylum level. which were identified using the RDP classifier. Sequences not classified into any known group were designated as “other”.
We detected nine fungal phyla, with Ascomycota, Basidiomycota, and Zygomycota being the dominant (Fig. 1B). Ascomycota accounted for 37.96%–64.17% of all valid reads. Ascomycota members were the most abundant in Sichuan, with 64.17%, and the least abundant in Xianning, with 37.96%. Basidiomycota accounted for 10.25%, 15.23%, 27.44%, and 46.79% of the valid reads for Sichuan, Yuanjiang, Jiangxi, and Xianning, respectively (P < 0.05, Table S3). Zygomycota was the third dominant phylum in all samples, with an average relative abundance of 9.79% (P < 0.01, Table S3).
Differences in microbial community among continuous-cropping ramie fields in different areas
To explore differences in microbial communities among all soil samples, Venn diagrams were generated based on OTUs, using Mothur. In total, 93,326 bacterial OTUs were detected in all samples, among which 21,949, 12,260, 22,230, and 23,343 OTUs were detected specifically in samples from Sichuan, Jiangxi, Yuanjiang, and Xianning, respectively, and 829 were shared by all samples (Fig. 2A). Totally, 18,406 fungal OTUs were detected, of which 6,006, 3,587, 3,593, and 3,326 were detected only in samples from Sichuan, Jiangxi, Yuanjiang, and Xianning, respectively, and 256 were shared by all samples (Fig. 2B).

Venn diagram showing the number of unique bacterial (A) and fungal OTUs (B) detected in four different areas continuous cropping ramie soil samples.
The relative abundance of the 30 most dominant bacterial and fungal genera was visualised in heatmaps generated using custom R scripts. The bacteria Bacillus and Lactococcus presented a high relative abundance in all soil samples (Fig. 3A, P < 0.01, Table S2). Enterococcus was highly abundant in all samples, except in those from Xianning. Except for Bacillus, Lactococcus, Enterococcus, Alkaliphilus, and Camobacterium, other dominant genera had a low abundance (P < 0.01, Table S2). The fungi Mortierella, Candida, Cryptococcus, and Aureobasidium exhibited a high abundance in the four fields (Fig. 3B). Fusarium was highly abundant in Sichuan, Xianning, and Yuanjiang, but low in Jiangxi (P < 0.05, Table S3). Except for Mortierella, Candida, Cryptococcus, Aureobasidium, Guehomyces, Trichosporon, Malassezia, and Aspergillus, other genera presented a low abundance in Jiangxi. The heatmap showed that soil microbial composition was distinct in the different areas. The principal component analysis (PCA) was used to identify the community structure differences in different areas under continuous ramie cropping (Fig. 4). Two-dimensional plots of the coefficients of the first two principal components were generated to illustrate relationships among soil samples. As for bacteria, PC1 and PC2 contributed 13.974% and 12.993%, respectively (Fig. 4A). The PC1 value of Yuanjiang samples was similar to that of Sichuan samples, and Xianning samples had the highest PC1 value and Jiangxi samples had the lowest PC1 value. Although Jiangxi samples had the lowest PC1 value, the PC2 value was the highest. As for fungi, PC1 contributed 10.126% and PC2 contributed 8.25% (Fig. 4B). All soil samples had similar PC1 values. The PC2 values of the 12 samples were similar, but there were some differences; Xianning samples had the highest PC2 value and Jiangxi samples had the lowest PC2 value. In line with the heatmap results, the PCA results indicated that bacterial and fungal abundance differed among the four ramie-cropping areas.

Heatmap analysis of bacterial (A) and fungi (B) based on the relative abundances of dominant genera from different areas continuous-cropping soil samples.

PCA of the OTUs detected major variations in the bacterial (A) and fungal (B) communities in four different area continuous cropping ramie soil samples.
Correlation between community structure and environmental factors
In all samples, the bacterial OTUs were significantly positively correlated with the performance of several environmental factors; however, for fungi, only total T was significantly correlated with fungal community abundance (Table 3). To further identify the major environmental variables controlling the soil microbial community structure, the RDA was performed (Fig. 5A). The RDA based on OTU reads and all studied environmental variables was carried out for the continuous cropping ramie fields in the different regions. Relationships between bacterial communities and soil properties are shown in Fig. 5A (axis 1 = 26.6%, axis 2 = 18.6%), and relationships between soil properties and fungal communities are shown in Fig. 5B (axis 1 = 12.2%, axis 2 = 11.3%). The length of the arrow in the RDA plot indicates the degree of correlation between the environmental factor and sample distribution. The analysis revealed that the urease activity and TN exhibited the most significant correlation with bacterial community composition in all samples, whereas soil T was the least correlated with bacterial community composition in all soil samples (Fig. 5A, Table 3). The effects of urease activity, TN, and soil T were in the order Jiangxi > Xianning > Yuanjiang > Sichuan, and those of available K and P, and pH were in the order Sichuan > Yuanjiang > Jiangxi > Xianning. The effects of TN and P were in the order Sichuan > Jiangxi > Xianning > Yuanjiang (Fig. 5B). The RDA results demonstrated that environmental factors significantly affect soil microbial community composition.

Redundancy analysis (RDA) based on bacterial (A) and fungal OUT (B) data with chemical parameters in four different area continuous cropping ramie soil.
Discussion
In this study, using high-throughput sequencing, we analysed soil bacterial and fungal communities in continuous-cropping ramie fields in Yuanjiang, Xianning, Sichuan, and Jiangxi. According to the α-diversity analysis, the overall diversity of bacterial and fungal community compositions differed among the soil samples. For bacteria, the Shannon, Simpson and Chao1 α-diversity indices revealed that the diversity of Jiangxi was significantly lower than that of the other three regions (Table 2). This may be due to different soil properties and climactic conditions in the different regions. For bacteria, Firmicutes, Proteobacteria, and Acidobacteria were the dominant phyla, which was in accordance with the findings of our previous study3. These phyla were also dominant in continuous-cropping fields of tobacco6, peanut15, and soybean23. Proteobacteria members play key roles in C, S, and N cycling in soil24. In healthy soil with adequate fertilisation, the abundance of Proteobacteria increases, whereas it decreases when nutrients are exhausted21. However, in our study, the percentage of the dominant phyla was different among the four regions under continuous ramie cropping. The abundance of Firmicutes was the highest in Jiangxi, accounting for 79.03% of all valid reads, which was higher than that in other samples (Fig. 1A). Data from numerous studies show that most rhizosphere species belong to the phylum Firmicutes25,26,27. It may be that the proportion of Firmicutes is so large that the α-diversity index in the previous analysis in Jiangxi was significantly lower than that in the other three places. The increase in Firmicutes may also be one of the main barriers to the continuous cropping of ramie in Jiangxi; this point needs further investigation. Bacillus, Lactococcus, Koribacter, Enterococcus, and Alkaliphilus were the dominant bacterial genera, and Bacillus was the most dominant genus in all samples. Bacillus, which belongs to Firmicutes, can promote plant growth and control soil-borne diseases as a beneficial microbe28. For example, Bacillus restrains bacterial wilt caused by Ralstonia solanacearum29,30,31,32. Furthermore, Bacillus-inoculated fertiliser reportedly increases soil bacterial diversity33. Hence, Bacillus is a good choice for improving the soil microbial community. Interestingly, except for Bacillus, Lactococcus, Enterococcus, Alkaliphilus, Camobacterium, and Paenibacillus, the abundance of 24 other genera were lower in Jiangxi. The decrease in these species directly led to the decrease in rhizosphere microbial diversity in Jiangxi. Ascomycota, Basidiomycota, and Zygomycota were the dominant fungal phyla, which was consistent with the findings of previous studies in citrus34 Ascomycota and Basidiomycota are important fungi in most soils35,36, and the species in both phyla are involved in C cycling by degrading organic substances37,38. We observed significant differences in the relative abundance of Ascomycota and Basidiomycota in our samples; especially in Sichuan, the abundance of Ascomycota was the highest compared with that in the other three regions. The RDA results revealed that the urease activity and TN had the lowest effects in Sichuan. Thus, we speculate that under the condition of low TN in Sichuan, Ascomycota may participate in C cycling by degrading organic substances.
Soil properties, including soil pH and available N, are influenced by continuous cropping18,39. Furthermore, soil microbial communities are affected by environmental factors, including soil pH, TN, and T40,41,42,43. Hence, we used the RDA to analyse relationships between soil microbial composition and environmental factors (including soil pH, soil T, available P, available K, TN, and urease activity). The analysis revealed that different environmental factors differentially affected the bacterial and fungal communities. Furthermore, we carried out the correlation analysis between environmental and number of operational taxonomic units (OTUs) of continuous cropping ramie in the different areas (Table 3). All six environmental factors were significantly correlated with bacterial communities; however, there was no significant correlation between fungi and any of the tested parameters, except with TN (Table 3). Numerous studies have found environmental factors to differentially affect bacterial and fungal communities44,45,46. However, in this study, environmental factors were found to have a greater effect on bacterial diversity than on fungal diversity. The urease activity was the most significantly correlated with bacterial and fungal communities (Fig. 5). Urease catalyses the breakdown of urea into CO2 and NH3, and might be a good index of soil quality47,48. Bacteria as well as plants secrete urease49. Continuous cropping causes a decline in the urease activity, and significant correlations between urease activity and bacterial networks have been previously found50,51. We found that TN was significantly correlated with microbial communities in all samples (Fig. 5). Previous studies have demonstrated that N primarily regulates the bacterial community52,53. Lei et al. found that the urease activity increased when the N application rate was increased from 247 to 433 mg/kg54. Liang et al. reported that a medium N level increased the urease activity55. Thus, N fertilisation provides a good means to increase the urease activity to improve the soil microbiota. However, high quantities of ammonia reduce the urease activity56; therefore, TN is a factor that has the greatest influence on the diversity of rhizosphere bacteria and fungi. The N fertilisation dose is crucial in different regions of continuous cropping ramie.
In conclusion, our study indicated that microbial community diversity and composition in continuous-cropping ramie fields differed among Yuanjiang, Xianning, Sichuan, and Jiangxi. However, some common features also exist; Firmicutes, Proteobacteria, and Acidobacteria were the dominant bacterial phyla, accounting for more than 85% of the total reads in each area, and Ascomycota, Basidiomycota, and Zygomycota were the dominant fungal phyla. Furthermore, environmental factors, including the urease activity, TN, and soil T, affected the microbial communities. However, based on our findings, the effect of soil environmental factors on the bacterial diversity of continuous cropping ramie was greater than that on fungal diversity. According to the correlation analysis of root microorganisms and environmental factors in several different places, the TN and urease in soil were the key factors influencing microbial diversity and thus the main targets to solve the problem of continuous cropping and the growth of ramie. We suggest that regulating the microbial community by modulating the urease activity and TN content might provide a means to tackle problems caused by continuous cropping.
Methods
Site description and sample collection
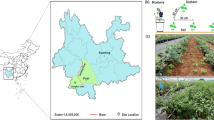
The experimental sites were located in Yuanjiang (Hunan Province, 112°38′84.35″N, 28°83′76.29″E), Xianning (Hubei Province, 114°25′21.29″N, 29°91′10.33″E), Sichuan (Sichuan Province, 107°51′84.07″N, 31°22′25.1″E), and Jiangxi (Jiangxi Province, 114°42′15.3″N, 27°78′83.96″E). All soil samples were collected in May 2016 from four fields with more than eight years of continuous ramie cropping. May is the season of vigorous ramie growth and the period when ramie rhizosphere microorganisms are highly active. Therefore, sampling at this time is representative. In each study area, the variety of ramie cultured is Zhongzhu no.1. Ramie plants with similar growth in four different planting areas were selected as sampling objects. We collected soil samples close to the root systems of five plants and pooled them into one sample. From each area, we collected three pooled samples, which were labelled as the province name followed by “R1,” “R2,” and “R3” (e.g., for the Yuanjiang area, samples were named yuanjiangR1, yuanjiangR2, and yuanjiangR3). Soil samples were sifted through a 2-mm sieve and homogeneously mixed. Twelve soil samples were stored in plastic bags and transferred on ice to the laboratory. One half of each soil sample was stored at −70 °C for biological and biochemical analyses and the other was air-dried at room temperature for one week for chemical analysis. Each sample was analysed in triplicate.
Analysis of soil basic properties
Soil basic properties, including pH, TN, available P, available K, soil temperature (Soil T), and urease activity were analysed. Basic chemical properties were analysed according to published procedures34, soil pH was measured in a 1:5 (w/w) soil:CO2-free distilled water suspension. TN was determined using the Kjeldahl method. Available P was determined by molybdenum antimony blue colorimetry after digesting the sample with a mixture of perchloric acid and sulphuric acid. The Olsen method was used to measure available K. Soil T was monitored using a portable probe attached to the Li-8100 system. Soil urease activity was determined by an improved sodium phenate and sodium hypochlorite colorimetric method57.
DNA extraction
The total genomic DNA was extracted from the soil samples using the E.N.Z.A. Soil DNA Kit (Omega Bio-tek), following the manufacturer’s protocol (http://omegabiotek.com/store/product/soil-dna-kit/). DNA quantity and quality were evaluated by spectrophotometry (NanoDrop) and agarose gel electrophoresis, respectively. The DNA was diluted to 1 ng/μL and stored at −20 °C until further analysis.
PCR amplification and Illumina high-throughput sequencing analysis
Bacterial 16S and fungal ITS rRNA gene sequences were amplified using barcoded primers and HiFi Hot Start Ready Mix (Kapa Biosystems). For bacterial diversity analysis, we used the primers 338F (5′-ACTCCTACGGGAGGCAGCAG-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′), which amplify the flexible V3-V4 regions of the 16S rRNA gene. For fungal diversity analysis, we used the primers fITS7 (5′-GTGARTCATCGAATCTTTG-3′) and ITS4 (5′-TCCTCCGCTTATTGATATGC-3′), which amplify the ITS2 region. PCR amplicon quality was evaluated by gel electrophoresis, and the amplicons were purified using AMPure XP beads (Agencourt). The amplicons were then amplified in a second round of PCR, subjected to purification using AMPure XP beads, and quantified using the Qubit dsDNA Assay Kit. Equimolar amounts of purified amplicons were pooled for sequencing. Amplicons were subjected to high-throughput sequencing on an Illumina Mi-Seq platform (Illumina, San Diego, CA, USA) at OE Biotech (Shanghai, China). All sequence data have been deposited in the NCBI Sequence Read Archive database under accession number PRJNA543166. After raw paired-end reads were quality-filtered with Trimmomatic software, FLASH software was used for paired-end read assembly58. QIIME software (version 1.8.0) and UPARSE pipeline were applied to analyse the sequences59. Then, UPARSE pipeline was used to explore OTUs at 97% similarity60 Using a representative sequence of each OTU, taxonomic composition as a.ssigned using the RDP classifier61.
Data analyses
For all parameters tested, multiple comparisons were conducted using the one-way analysis of variance followed by Tukey’s honest significant difference multiple-range tests. For α-diversity, all analyses were based on OTU clusters, with a cut-off of 3% dissimilarity. The Chao1 index was calculated to estimate the richness in each sample. Diversity within each sample was estimated using the nonparametric Shannon diversity index. Rarefaction curves based on the average number of observed OTUs were generated using Mothur software to compare the relative levels of bacterial and fungal OTU diversity across continuous-cropping ramie field soil samples. For β-diversity, hierarchical cluster dendrograms (with Bray–Curtis distance dissimilarities) were generated using Mothur, based on the OTU composition. The dendrograms were used to compare bacterial and fungal community structures among all soil samples. Heatmaps and Venn diagrams were generated using custom R scripts. Weighted and unweighted UniFrac distance metrics (based on the phylogenetic structure) were used to generate principle coordinate analysis (PCA) plots to assess similarities in community composition among the different samples. The RDA was used to study relationships between bacterial and fungal communities and soil properties. Histograms were created in SPSS and Microsoft Excel 2010.
References
Shipton, P. J. Monoculture and soilborne plant pathogens. Annual Review of Phytopathology 15(1), 387–407 (1977).
Yu, J. Q., Shou, S. Y., Qian, Y. R., Zhu, Z. J. & Hu, W. H. Autotoxic potential of cucurbit crops. Plant and Soil 223(1–2), 149–153 (2000).
Zhu, S. et al. Potential use of high-throughput sequencing of soil microbial communities for estimating the adverse effects of continuous cropping on ramie (Boehmeria nivea L. Gaud). PloS One. 13(5), e0197095 (2018).
Kreye, C., Bouman, B. A. M., Faronilo, J. E. & Llorca, L. Causes for soil sickness affecting early plant growth in aerobic rice. Field Crops Research 114(2), 182–187 (2009).
Zhou, X. & Wu, F. Dynamics of the diversity of fungal and Fusarium communities during continuous cropping of cucumber in the greenhouse. FEMS Microbiology Ecology 80(2), 469–478 (2012).
She, S. et al. Significant relationship between soil bacterial community structure and incidence of bacterial wilt disease under continuous cropping system. Archives of Microbiology 199(2), 267–275 (2017).
Tang, J., Xue, Z., Daroch, M. & Ma, J. Impact of continuous Salvia miltiorrhiza cropping on rhizosphere actinomycetes and fungi communities. Annals of Microbiology 65(3), 1267–1275 (2015).
Sun, J. et al. Soil microbial and chemical properties influenced by continuous cropping of banana. Scientia Agricola 75(5), 420–425 (2015).
Yang, J., Ruegger, P. M., McKenry, M. V., Becker, J. O. & Borneman, J. Correlations between root-associated microorganisms and peach replant disease symptoms in a California soil. PLoS One 7(10), e46420 (2012).
Liu, X. et al. Microbial community diversities and taxa abundances in soils along a seven-year gradient of potato monoculture using high-throughput pyrosequencing approach. PLoS One 9, e86610 (2014).
Berendsen, R. L., Pieterse, C. M. J. & Bakker, P. A. H. M. The rhizosphere microbiome and plant health. Trends in Plant Science 17(8), 478–486 (2012).
Miransari, M. Soil microbes and the availability of soil nutrients. Acta Physiologiae Plantarum 35(11), 3075–3084 (2013).
Berg, G. & Smalla, K. Plant species and soil type cooperatively shape the structure and function of microbial communities in the rhizosphere. FEMS Microbiology Ecology 68(1), 1–13 (2009).
Lu, L. et al. Fungal networks in yield-invigorating and-debilitating soils induced by prolonged potato monoculture. Soil Biology and Biochemistry 65, 186–194 (2013).
Li, X., Ding, C., Zhang, T. & Wang, X. Fungal pathogen accumulation at the expense of plant-beneficial fungi as a consequence of consecutive peanut monoculturing. Soil Biology and Biochemistry 72, 11–18 (2014).
Jeffries, P., Gianinazzi, S., Perotto, S., Turnau, K. & Barea, J. M. The contribution of arbuscular mycorrhizal fungi in sustainable maintenance of plant health and soil fertility. Biology and Fertility of Soils 37(1), 1–16 (2003).
Rezapour, S., Taghipour, A. & Samadi, A. Modifications in selected soil attributes as influenced by long-term continuous cropping in a calcareous semiarid environment. Natural Hazards 69(3), 1951–1966 (2013).
Yu, Y. et al. Soil pH, organic matter, and nutrient content change with the continuous cropping of Cunninghamia lanceolata plantations in South China. Journal of Soils and Sediments 17(9), 2230–2238 (2017).
Zhang, T., Wang, N. F., Liu, H. Y., Zhang, Y. Q. & Yu, L. Y. Soil pH is a key determinant of soil fungal community composition in the Ny-Ålesund Region, Svalbard (High Arctic). Frontiers in Microbiology 7, 227 (2016).
Kim, J. M. et al. Soil pH and electrical conductivity are key edaphic factors shaping bacterial communities of greenhouse soils in Korea. Journal of Microbiology 54(12), 838–845 (2016).
Wang, R. et al. Microbial community composition is related to soil biological and chemical properties and bacterial wilt outbreak. Scientific Reports 7(1), 343 (2017).
Squibb, R., Méndez, J., Guzma’nM & Scrimshaw, N. Ramie-a high protein forage crop for tropical areas. Grass Forage Science 9, 313–322 (1954).
Li, C., Li, X., Kong, W., Wu, Y. & Wang, J. Effect of monoculture soybean on soil microbial community in the Northeast China. Plant and Soil 330(1–2), 423–433 (2010).
Nosheen et al. Protein Quantity and Quality of Safflower Seed Improved by NP Fertilizer and Rhizobacteria (Azospirillum and Azotobacter spp.). Frontiers in Plant science 7, 104 (2016).
Dias, A. C. F. et al. Potato cultivar type affects the structure of ammonia oxidizer communities in field soil under potato beyond the rhizosphere. Soil Biology and Biochemistry 50, 85–95 (2012).
Garbeva, P., Van Elsas, J. D. & Van Veen, J. A. Rhizosphere microbial community and its response to plantspecies and soil history. Plant and Soil 302(12), 19–32 (2008).
Smalla, K. et al. Bulk and rhizosphere soil bacterial communities studied by denaturing gradient gel electrophoresis: plant-dependent enrichment and seasonal shifts revealed. Applied and Environmental Microbiology 67(10), 4742–4751 (2001).
Raaijmakers, J., Paulitz, T., Steinberg, C., Alabouvette, C. & Moenne-loccoz, Y. The rhizosphere: a playground and battlefield for soilborne pathogens and beneficial microorganisms. Plant and Soil 321(1–2), 341–361 (2009).
Guo, J. H. et al. Biocontrol of tomato wilt by plant growth-promoting rhizobacteria. Biological Control 29(1), 66–72 (2004).
Hu, H. Q., Li, X. S. & He, H. Characterization of an antimicrobial material from a newly isolated Bacillus amyloliquefaciens from mangrove for biocontrol of Capsicum bacterial wilt. Biological Control 54(3), 359–365 (2010).
Tan, S. et al. Two Bacillus amyloliquefaciens strains isolated using the competitive tomato root enrichment method and their effects on suppressing Ralstonia solanacearum and promoting tomato plant growth. Crop Protection 43, 134–140 (2013).
Chen, D. et al. Isolation of Bacillus amyloliquefaciens S20 and its application in control of eggplant bacterial wilt. Journal of Environmental Management 137, 120–127 (2014).
Huang, X., Zhang, N., Yong, X., Yang, X. & Shen, Q. Biocontrol of Rhizoctonia solani damping-off disease in cucumber with Bacillus pumilus SQR-N43. Microbiological Research 167(3), 135–143 (2012).
Xu, J. et al. The structure and function of the global citrus rhizosphere microbiome. Nature Communications 9(1), 4894 (2018).
Hussain, Q. et al. Variation of bacterial and fungal community structures in the rhizosphere of hybrid and standard rice cultivars and linkage to CO2 flux. FEMS Microbiology Ecology 78(1), 116–128 (2011).
Wallenstein, M. D., McMahon, S. & Schimel, J. Bacterial and fungal community structure in Arctic tundra tussock and shrub soils. FEMS Microbiology Ecology 59(2), 428–435 (2007).
Unterseher, M., Peršoh, D. & Schnittler, M. Leaf-inhabiting endophytic fungi of European Beech (Fagus sylvatica L.) co-occur in leaf litter but are rare on decaying wood of the same host. Fungal Diversity 60(1), 43–54 (2013).
Purahong, W. et al. Life in leaf litter: novel insights into community dynamics of bacteria and fungi during litter decomposition. Molecular Ecology 25(16), 4059–4074 (2016).
Zhao, M. et al. Effects of increased nitrogen deposition and rotation length on long-term productivity of Cunninghamia lanceolata plantation in southern China. PLoS One 8(2), e55376 (2013).
Timling, I. et al. Distribution and drivers of ectomycorrhizal fungal communities across the North American Arctic. Ecosphere 3(11), 1–25 (2012).
Xu, H. et al. Does urbanization shape bacterial community composition in urban park soils? A case study in 16 representative Chinese cities based on the pyrosequencing method. FEMS Microbiology Ecology 87(1), 182–192 (2014).
Bartram, A. et al. Exploring links between pH and bacterial community composition in soils from the Craibstone Experimental Farm. FEMS Microbiology Ecology 87(2), 403–415 (2014).
Yeoh, Y. K. et al. The core root microbiome of sugarcanes cultivated under varying nitrogen fertilizer application. Environmental Microbiology 18(5), 1338–1351 (2016).
Zhang, W. et al. Soil microbial responses to experimental warming and clipping in a tallgrass prairie. Global Change Biology 11(2), 266–277 (2005).
DeAngelis, K. M. et al. Long-term forest soil warming alters microbial communities in temperate forest soils. Frontiers in Microbiology 6, 104 (2015).
Zhou, W. P., Shen, W. J., Li, Y. E. & Hui, D. F. Interactive effects of temperature and moisture on composition of the soil microbial community. European Journal of Soil Science 68(6), 909–918 (2017).
Das, S. K. & Varma, A. Role of Enzymes in Maintaining Soil Health[M]//Soil Enzymology (pp. 25–42. Springer, Berlin, Heidelberg, 2010).
Jezierska-Tys, S. & Rutkowska, A. Chemical and enzymatic changes in soil treated with ammonium glufosinate. Journal of Elementology. 19(1) (2014)
Follmer, C. Insights into the role and structure of plant ureases. Phytochemistry. 69(1), 18–28 (2008).
Sun, C., Li, B., Su, Y., Jia, X. & Chen, G. Dynamic succession of soil microbial community during continuous cropping of Astragalus membranaceus Bge. var. mongholicus (Bge.). PeerJ Preprints (2019).
Chen, S. et al. Continuous-cropping tobacco caused variance of chemical properties and structure of bacterial network in soils. Land Degradation & Development 29(11), 4106–4120 (2018).
Cleveland, C. C., Nemergut, D. R., Schmidt, S. K. & Townsend, A. R. Increases in soil respiration following labile carbon additions linked to rapid shifts in soil microbial community composition. Biogeochemistry 82(3), 229–240 (2007).
Högberg, M. N., Yarwood, S. A. & Myrold, D. D. Fungal but not bacterial soil communities recover after termination of decadal nitrogen additions to boreal forest. Soil Biology and Biochemistry 72, 35–43 (2014).
Lei, T. et al. Urease activity and urea hydrolysis rate under coupling effects of moisture content, temperature, and nitrogen application rate. International Journal of Agricultural and Biological Engineering 11(2), 132–138 (2018).
Liang, Y., Li, F., Nong, M., Luo, H. & Zhang, J. H. Microbial activity in paddy soil and water-use efficiency of rice as affected by irrigation method and nitrogen level. Communications in Soil Science and Plant Analysis 47(1), 19–31 (2016).
Konig, C., Kaltvasser, H. & Schiegel, H. G. The formation of urease after use of other nitrogen sources in Hidrogenumonas. Archives of Microbiology 53, 231–241 (1966).
Vlek, P. L. G., Stumpe, J. M. & Byrnes, B. H. Urease activity and inhibition in flooded soil systems. Fertilizer Research 1(3), 191–202 (1980).
Reyon, D. et al. FLASH assembly of TALENs for high-throughput genome editing. Nature Biotechnology 30(5), 460–465 (2012).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nature Methods 7(5), 335–336 (2010).
Edgar, R. C. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nature Methods. 10(10), 996–998 (2013).
Wang, Q., Garrity, G. M., Tiedje, J. M. & Cole, J. R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and Environmental Microbiology 73(16), 5261–5267 (2007).
Acknowledgements
We acknowledge the Shanghai OE Biotech.Co., Ltd for its assistance in original data processing and related bioinformatics analysis. This work was supported by grants from the National Natural Science Foundation of China (Grant Nos. 31571618 and 31771734), the Agricultural Science and Technology Innovation Program (ASTIP), and the National Modern Agro-industry Technology Research System (nycytx-19-E16).
Author information
Authors and Affiliations
Contributions
S.Z. planned and designed the research. Y.W. and X.X. performed experiment. T.L. and H.W. conducted the fieldwork, and prepared the reagents of experiments. Y.Y. and X.C. prepared the soil samples, and S.Z. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, Y., Xu, X., Liu, T. et al. Analysis of bacterial and fungal communities in continuous-cropping ramie (Boehmeria nivea L. Gaud) fields in different areas in China. Sci Rep 10, 3264 (2020). https://doi.org/10.1038/s41598-020-58608-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-58608-0
- Springer Nature Limited
This article is cited by
-
Composition and Diversity of Endophytic Rhizosphere Microbiota in Apple Tree with Different Ages
Molecular Biotechnology (2023)
-
Analysis of rhizosphere bacterial communities of tobacco resistant and non-resistant to bacterial wilt in different regions
Scientific Reports (2022)
-
The Mexican giant maize of Jala landrace harbour plant-growth-promoting rhizospheric and endophytic bacteria
3 Biotech (2021)