
Abstract
Sunflower (Helianthus annuus L.) production is challenged by different biotic and abiotic stresses, among which downy mildew (DM) is a severe biotic stress that is detrimental to sunflower yield and quality in many sunflower-growing regions worldwide. Resistance against its infestation in sunflower is commonly regulated by single dominant genes. Pl17 and Pl19 are two broad-spectrum DM resistance genes that have been previously mapped to a gene cluster spanning a 3.2 Mb region at the upper end of sunflower chromosome 4. Using a whole-genome resequencing approach combined with a reference sequence-based chromosome walking strategy and high-density mapping populations, we narrowed down Pl17 to a 15-kb region flanked by SNP markers C4_5711524 and SPB0001. A prospective candidate gene HanXRQChr04g0095641 for Pl17 was identified, encoding a typical TNL resistance gene protein. Pl19 was delimited to a 35-kb region and was approximately 1 Mb away from Pl17, flanked by SNP markers C4_6676629 and C4_6711381. The only gene present within the delineated Pl19 locus in the reference genome, HanXRQChr04g0095951, was predicted to encode an RNA methyltransferase family protein. Six and eight SNP markers diagnostic for Pl17 and Pl19, respectively, were identified upon evaluation of 96 diverse sunflower lines, providing a very useful tool for marker-assisted selection in sunflower breeding programs.
Similar content being viewed by others


Introduction
Sunflower (Helianthus annuus L.) is native to North America and is an important economic crop worldwide because of its edible seeds (confectionary sunflower) and oil products (oilseed sunflower), as well as for ornamental purposes. Like other crops, sunflower production is often hindered by many factors, including biotic (e.g., diseases, pests, birds, etc.) and abiotic stresses (e.g., drought, waterlogging, and salinity). Among the diseases, downy mildew (DM), incited by the oomycete Plasmopara halstedii (Farl.) Berlese & de Toni, is one of the most severe biotic factors affecting sunflower production worldwide, particularly in Europe and North America1,2. Typical symptoms of DM infections include leaf chlorosis, seedling dwarfing, and white sporulation on the underside of leaves (https://www.ag.ndsu.edu/extensionentomology/recent-publications-main/publications/A-1331-sunflower-production-field-guide). Seedling death is commonly seen upon DM infection. Despite some survival, the infected plants display stunted growth and typically yield little or no seeds. Generally, it is publicly accepted that the deployment of disease resistance in crops is the most effective and environmentally sound means of controlling disease infestation. The identification of resistance is prerequisite for sunflower breeding against pathogen infection.
Resistance (R) against downy mildew in sunflower is, in most cases, governed by a single dominant gene (designated as Pl), although some partial and quantitative resistance have also been reported3,4. To date, a total of 36 Pl genes, Pl1–Pl35, and PlArg, have been reported from the DM resistance pool in cultivated sunflower and its wild relatives (Supplementary Table S1). However, DM resistance is often rendered ineffective by the rapid genetic changes in the pathogen populations due to the coevolution between the pathogen and sunflower host5,6. Some of the Pl genes that have been widely used to combat DM infection in sunflower have already been ineffective against new races of P. halstedii, such as Pl6 and Pl77,8,9. A recent survey showed that only the PlArg, Pl15, Pl17, Pl18, and Pl33 genes remained effectively resistant against a total of 185 P. halstedii isolates collected from North Dakota, South Dakota and Nebraska sunflower production regions in the United States when a total of twelve known DM R genes were tested, including Pl1, Pl2, Pl5, Pl6, Pl13, Pl15–Pl18, Pl21, Pl33, and PlArg10.
Intensive breeding efforts in sunflower have narrowed down the genetic variability of the sunflower genome, resulting in a constant need to identify and deploy new agronomically important genes. There are 53 wild sunflower species belonging to the Helianthus genus, which are invaluable reservoirs of agronomically desirable genes11,12,13. The oil maintainer line HA 458 (PI 655009) is resistant to all North American P. halstedii races identified thus far10,14. It harbors the DM R gene, Pl17, originating from the wild H. annuus L. accession PI 468435. The other DM R gene, Pl19, was also identified from the wild H. annuus L. accession PI 43541415. Both Pl17 and Pl19 genes were previously mapped to sunflower chromosome 4 corresponding to linkage group 4 in a similar position16,17. Recently, four additional novel DM R genes, Pl27–Pl29 and Pl33, were identified in proximity to Pl17 and Pl19 on chromosome 418,19, while Pl17, Pl19, and Pl33 were in an interval spanning a physical distance of approximately 3.2 Mb when the flanking markers were positioned on chromosome 4 pseudomolecules of the HA412-HO genome sequence16,17,19.
The three DM R genes, Pl17, Pl19, and Pl33, were highly effective toward the most predominant and virulent races of P. halstedii and have not been widely used for commercial sunflower production. The broad-spectrum DM resistance and similar position of these R genes make it infeasible to select individuals harboring respective R gene based on phenotyping. Diagnostic molecular markers would provide a timely and accurate selection tool for sunflower breeding programs and would be developed with the advancement of rapidly developing sequencing technology combined with the single nucleotide polymorphism (SNP) genotyping system.
The publicly available genomic resources of the two assembled and annotated genome reference sequences of HA412-HO and XRQ in sunflower are powerful tools to study the genetic basis of agronomically important traits, to utilize sequence information for marker development, and to dissect the trait-governing genes genetically and molecularly20. Whole-genome resequencing can be utilized for efficiently identifying SNP, insertion and deletion (InDel), structure variation (SV), and copy number variation (CNV) in a massively parallel manner. The extremely high distribution of SNPs in the genomes of all organism makes it a powerful genetic tool for population genetics studies and marker-trait association analyses. However, current use of PCR-based approaches for genotyping of individual SNPs of special interest is still limited by accuracy, throughput, simplicity, and operational costs. An innovative SNP genotyping method has been developed in our laboratory, which adapts to multiple platforms and throughputs, allowing a PCR-based technology to genotype individual SNPs16,21,22. In the current study, we report the use of reference sequence-based chromosome walking toward the target genes, Pl17 and Pl19, identify candidate genes, and develop user-friendly SNP markers diagnostic for Pl17 and Pl19.
Results
Saturation and fine mapping of Pl17
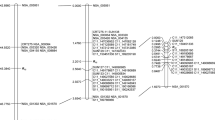
Two strategies were adopted for marker development. At first, the genome sequence of chromosome 4 was extracted from the HA412-HO reference assembly from 3,621,089 to 6,852,749 bp and the XRQ assembly from 5,662,479 to 5,707,598 bp, which covers the Pl17 and Pl19 loci reported in previous studies16,17. A total of 101 pairs of primers, including 40 STSs and 61 SSRs were screened for polymorphisms between the parents HA 458 (Pl17) and HA 234. Polymorphic markers were further used to genotype 186 F2 individuals of HA 234 × HA 458. Five markers were mapped around the Pl17 locus (Table 1), reducing the Pl17 gene interval from 2.9 cM between SFW04052 and ORS963 to 1.3 cM between SUN232 and ORS963 (Fig. 1a,b).

Pl17 genetic maps. (a) Pl17 basic map (Qi et al. 2015); (b) Pl17 saturation map; (c) Pl17 fine map; (d) physical position of the Pl17 candidate gene. The diagnostic marker for Pl17 is shown in bold. *SSR/STS markers.
To further narrow down the Pl17 gene region, a total of 80 SNPs was identified from the HA458-WGS1 (27 SNPs) and HA458-WGS2 (53 SNPs) in the targeted gene interval. Ten contigs were identified from the HA458-WGS1, which fell in the Pl17 interval and were used as queries to align against the HA412-HO and XRQ references with BLASTn. A total of 27 SNP markers (SPB0001–SPB0027) were selected between the HA458-WGS1 and the XRQ sequence (Supplementary Table S2). Another 53 SNP markers were selected based on SNPs/InDels between the HA458-WGS2 and the two references, with 16 from the 69.3 kb segment (5,951,824–6,021,131 bp) of the HA412-HO assembly and 37 from the 138.1 kb segment (5,600,607–5,738,736 bp) of the XRQ assembly. Of 80 SNP markers screened, 12 showed polymorphisms between HA 234 and HA 458 and were used to genotype the F2 population. Linkage analysis indicated that ten SNP markers co-segregated with Pl17 and one (SPB0007) was proximal to Pl17 at a 0.3 cM genetic distance (Fig. 1b).
Fine mapping of Pl17 was performed to dissect the SNP marker cluster co-segregating with Pl17 and to increase the map resolution. The two previously reported Pl17 flanking markers, SNP marker SFW04052 and SSR marker ORS963 covering a 2.9 cM interval (Fig. 1a), were used to genotype the 3,008 F3 individuals from the selected F3 families that were heterozygous for Pl17. One hundred and three recombinants were identified and advanced into the next generation. The SSR marker SUN232 identified from saturation mapping was closer to the Pl17 locus than SFW04052 and was then used to screen the 103 recombinants (Fig. 1b). Twenty-two of them were found to have recombination events in the target interval of 1.3 cM flanked by the SSR markers SUN232 (0.5 cM) and ORS963 (0.8 cM), and their advanced generation was inoculated with P. halstedii race 734 for the resistance test.
The 12 polymorphic SNP markers mapped to the Pl17 interval between markers SUN232 and ORS963 using the 186 F2 individuals were further used to genotype 22 recombinants identified from 3,008 F3 individuals. As a result, Pl17 was placed in a 0.0665 cM interval at the upper end of chromosome 4, flanked by markers C4_5711524 (0.0332 cM) and SPB0001 (0.0333 cM) (Fig. 1c). Most of the markers were physically in accordance with their genetic positions, although five SPB SNPs had a reversed order in both the HA412-HO and XRQ assemblies compared with their genetic positions (Table 2). The flanking markers C4_5711524 and SPB0001 delimited Pl17 to a 15 kb interval on the XRQ genome assembly.
Saturation and fine mapping of Pl19
One hundred and one SSR and STS markers previously used in the Pl17 saturation mapping were also used to genotype the two parents of the Pl19 population, CONFSCLB1 and PI 435414. In addition, 56 SSRs were identified from the 296.4 kb sequence of XRQ from 6,238,999 to 6,535,440 bp on chromosome 4. Of 157 SSR and STS markers tested, 11 showed polymorphisms between the parents and were further used to genotype the BC1F2 population (Table 1). Linkage analysis of marker-trait associations indicated that all SSR markers mapped distal to Pl19 (Fig. 2a,b).

Pl19 genetic maps. (a) Pl19 basic map (Zhang et al. 2017); (b) Pl19 saturation map; (c) Pl19 fine map; (d) physical position of the Pl19 candidate gene. The diagnostic marker for Pl19 is shown in bold. *SSR/STS markers.
Based on the physical positions of the newly mapped SSR marker SUN461, which was located from 7,383,392–7,383,599 bp in HA412-HO and 6,413,020–6,413,227 bp in XRQ, 104 SNPs were selected from a 308.4 kb region (7,690,106–7,998,497 bp) of the HA412-HO sequence, and 168 SNPs were selected from a 398.7 kb region (6,400,728–6,799,385 bp) of the XRQ sequence. Of 272 SNP markers tested in CONFSCLB1 and HA-DM5, 66 were polymorphic and were used to genotype the 139 BC1F2 individuals derived from the cross of CONFSCLB1 × PI 435414 (Pl19). Total of 35 SNPs were mapped around Pl19, with four SNPs designed from the HA412-HO assembly and 31 designed from the XRQ assembly. A total of 37 co-segregating markers, including eight SSR and 29 SNP markers, were mapped to a 0.7 cM genetic distance distal to Pl19 (Fig. 2b).
To further fine map Pl19, the SSR marker SUN391 and the SNP marker SFW02206 were used as the flanking markers to screen the 2,256 BC1F3 individuals selected from the BC1F3 families heterozygous for Pl19. A total of 77 BC1F3 individuals with recombination events close to the Pl19 gene were identified and advanced to the next generation. Of 77 recombinants, 23 with recombination events occurred in the proximity to the Pl19 region, and their families (30 seedlings per family) were tested with P. halstedii race 734. Of 35 mapped SNP markers, 15 were selected for further genotyping of the 77 recombinants to increase the map resolution. The Pl19 gene was placed in the 0.2216 cM interval, flanked by SNP markers C4_6676629 (0.0443 cM) and C4_6711381 (0.1773 cM) (Fig. 2c). This genetic region corresponds to a 35 kb segment in the XRQ assembly (Table 3).
Collinearity of SNPs between the two reference genome assemblies
In the present study, two reference genomes, HA412-HO and XRQ, were used for SNP marker development. Most SNPs from either HA412-HO or XRQ had a collinear order in both genome assemblies (Tables 2 and 3). However, of 104 SNPs selected from HA412-HO in a region of 308. 4 kb for Pl19, only four SNP markers were mapped to the Pl19 region. A search for these SNP positions in the XRQ genome assembly revealed that 30 SNPs residing in a 57.9 kb segment (7,690,106–7,748,049 bp) of HA412-HO were aligned to a 7.2 Mb segment (147,656,203–154,868,683 bp) of XRQ, which is outside the Pl19 region (Supplementary Table S3). The remaining 74 SNPs in a 236.2 kb region (7,762,321–7,998,497 bp) were aligned to a corresponding region of 518.1 kb (6,458,182–6,976,291 bp) in the XRQ assembly.
Identification of candidate genes for Pl17 and Pl19
Most SNP markers mapped around the Pl17 locus were physically between 5,676,065 to 5,711,324 bp on chromosome 4 of the XRQ assembly (Table 2). The genetic positions of those markers were generally in accordance with their physical positions, although there was some conflict. The 104 kb genomic sequence of XRQ was analyzed from 5,670,000 to 5,780,000 bp on chromosome 4 encompassing the newly identified SNP markers from the XRQ sequence (https://www.heliagene.org/HanXRQ-SUNRISE/). Four putative genes were found in the corresponding genomic region (Table 4). One defense-associated gene HanXRQChr04g0095641 at nucleotide positions from 5,672,715 to 5,705,044 bp with a length of 32.329 kb had the typical TNL motif of the resistance gene model, encoding the full-length Toll/interleukin-1-receptor, nucleotide-binding site, and leucine-rich repeat. Moreover, all 12 polymorphic SNP markers identified from the fine mapping were in this 32.329 kb region, supporting its candidacy for Pl17 (Fig. 1d).
Pl19 was located between marker C4_6676629 and C4_6711381, and the good collinearity of the genetic and physical positions of markers in this region suggested the presence of Pl19 in the interval from 6,676,629–6,711,381 bp on chromosome 4 of the XRQ assembly (Table 3). A 120-kb genomic sequence on XRQ chromosome 4 was analyzed from 6,640,000 to 6,760,000 bp, which covers newly identified SNP markers for Pl19 (Table 3). Three putative genes were discovered, with one candidate gene HanXRQChr04g0095951 falling into the interval of 6,676,629–6,711,381 bp, which was predicted as a probable RNA methyltransferase family protein (Table 4, Fig. 2d).
Development of diagnostic markers for Pl17 and Pl19
Currently, three different DM resistance genes, Pl17, Pl19, and Pl33, have been mapped to a similar position on sunflower chromosome 416,17,19. The 12 and 35 SNP markers mapped to Pl17 and Pl19, respectively, were first tested in three resistant lines, HA 458 (Pl17), HA-DM5 (Pl19), and TX16R (Pl33), and three susceptible lines, HA 234, CONFSCLB1, and HA 434. Seven and 17 SNP markers mapped to Pl17 and Pl19, respectively, showed unique PCR pattern in HA 458 and HA-DM5 each, in contrast to the two other resistant and susceptible lines. These markers were further genotyped in an evaluation panel with 96 selected sunflower lines (Supplementary Table S4) to determine their specificity in the sunflower population and to assess their potential in marker-assisted selection for Pl17 and Pl19.
Six of the seven SNP markers, C4_5696413, C4_5705018, C4_5705841, C4_5709499, SPB0001, and SPB0007, could differentiate Pl17 from other reported Pl genes, including PlArg, Pl1–Pl3, Pl6–Pl13, Pl15–Pl21, Pl33, and Pl34 in the selected sunflower lines (Fig. 3). HA 458 (Pl17 donor line) and those sunflower lines introgressed with the Pl17 gene, including HA-DM3, HA-BSR2 to HA-BSR4, and HA-BSR6 to BA-BSR8, showed unique Pl17 SNP marker alleles, distinguishing them from other sunflower lines (Fig. 3). The SNP marker C4_5696413 also amplified a fragment with a similar size to the Pl17 allele in HA 291 (lane 3 in Fig. 3a). Sunflower line HOLS 1 showed a heterozygous pattern in all six diagnostic SNP markers for Pl17 (lane 95 in Fig. 3).

The polymerase chain reaction (PCR) amplification pattern of 96 selected sunflower lines with Pl17 diagnostic single nucleotide polymorphism (SNP) markers. (a) PCR amplification pattern with SNP marker C4_5696413. (b) PCR amplification pattern with SNP marker SPB0001. Names and pedigrees of 96 selected sunflower lines (lanes) are listed in Supplementary Table S4. Lane 30: HA 458, lane 33: HA-DM3, lane 88: HA-BSR2, lane 89: HA-BSR3, lane 90: HA-BSR4, lane 92: HA-BSR6, lane 93: HA-BSR7, and lane 94: HA-BSR8, all of which have the Pl17 gene and show the Pl17 marker allele.
Of 17 SNP markers tested in the evaluation panel, eight, C4_6401756, C4_6407910, C4_6647557, C4_6656705, C4_6666835; C4_6675662, C4_6676629, and S4_7964876, could differentiate Pl19 from other reported Pl genes in the selected sunflower lines. HA-DM5 was the only sunflower line carrying the Pl19 gene in the 96-line evaluation panel and had a unique PCR pattern of Pl19 marker alleles compared with the remaining 95 lines (Fig. 4). These Pl17 and Pl19 unique markers are of essential utility in sunflower breeding to assist selection for these two genes.

The PCR amplification pattern of 96 selected sunflower lines with Pl19 diagnostic SNP markers. (a) PCR amplification pattern with SNP marker C4_6666835, and b: PCR amplification pattern with SNP marker S4_7964876. Names and pedigrees of 96 selected sunflower lines (lanes) are listed in Supplementary Table S4. Lane 35: HA-DM5 with the Pl19 gene.
Discussion
Like other crops, DM R genes in sunflower are organized in clusters in the sunflower genome, such as in chromosomes 1 (cluster 1, PlArg, Pl23, Pl24, and Pl35; cluster 2, Pl13, Pl14, Pl16, and Pl25), 4 (Pl17, Pl19, Pl27–Pl29, and Pl33), 8 (Pl1, Pl2, Pl6, Pl7, Pl15, and Pl20), and 13 (Pl5, Pl8, Pl21, Pl22, Pl31, Pl32, and Pl34) (Supplementary Table S1)23,24,25,26,27,28. Distinguishing genes from a cluster can be achieved through traditional allelic analysis, polymorphic marker analysis, resistance specificity to different pathotypes, and the presence or absence of host reactions to pathogen effectors. Our previous studies have indicated that Pl17 and Pl19 are different but closely linked genes on sunflower chromosome 4 (data for allelic analysis not shown). Common markers ORS963 and NSA_003564 are downstream of Pl17 but upstream of Pl1916,17. Both genes are delimited in an interval of 3.2 Mb on chromosome 4 of the HA412-HO assembly, at which time the XRQ reference was not available. Using a sequence-based chromosome walking strategy toward the target gene in this study, Pl17 was refined into an interval of 15 kb at a position from 5,696,076–5,711,324 bp on chromosome 4 in the XRQ assembly. In contrast, Pl19 was precisely mapped to an interval of 35 kb at a position from 6,676,429–6,711,781 bp in the XRQ assembly, approximately 1 Mb apart from Pl17. A recently reported DM R gene, Pl33, is located in an interval of 1.56 Mb from 4,208,180–5,766,419 bp on chromosome 4 in the XRQ assembly19. Marker analysis among the three gene donors suggested that Pl33 is different from Pl17 and Pl19 (Figs 3 and 4).
Pecrix et al. (2018) reported mapping of the three new DM R genes, Pl27–Pl29, to the upper end of sunflower chromosome 4, a similar region to that of Pl17 and Pl1918. Both Pl27 and Pl28 originate from a wild perennial sunflower H. tomentosus (synonym H. tuberrosus), while Pl29 is derived from the same source as Pl17, Pl19, and Pl33 of the wild annual sunflower H. annuus. The Pl17 donor accession PI 468435 was collected from Idaho, USA, and both accessions of PI 435414 for Pl19 and Texas-16 for Pl33 were collected in Texas, USA, while Pl29 accession Wyoming 358 was collected from Wyoming, USA16,17,18,19. The flanking markers place Pl27 in an interval between 2.18–6.40 Mb, Pl28 between 6.62–8.42 Mb, and Pl29 between 6.93–7.07 Mb in the XRQ assembly, respectively18. Three genes fall within a small region between Pl17 and Pl19 on chromosome 4 with Pl29 close to Pl19. Further fine mapping of Pl27–Pl29 or cloning of Pl17 and Pl19 will elucidate the genetic architecture and evolutionary mechanisms underlying this gene cluster.
The sunflower genome is approximately 3.6 Gb in size with more than 80% highly repetitive sequences. The assembly of a large and complex genome with a high level of repetitive sequences remains a challenge in the community, but longer read length, higher genome coverage, and more sophisticated bioinformatics would reduce this difficulty and provide more accurate results. The HA412-HO whole-genome sequence was assembled from Illumina reads (100 bp) and 454 Roche reads (400–1,000 bp), while the XRQ whole-genome sequence was assembled from PacBio sequencing data with an average read length of 10.3 kb20. High quality genome assembly is crucial for reference sequence-based chromosome walking to anchor a specific region for the target gene. In the present study, comparison of mapped SNP positions between two assemblies revealed the coincidence of their positions in the two reference genomes of most SNPs. However, when searching the positions of 104 SNPs derived from HA412-HO for Pl19 in the XRQ genome assembly, 30 SNPs located in a 57.9 kb segment between 7,690,106 and 7,748,049 bp were found to align to a 7.2 Mb segment between 147,656,203 and 154,868,683 bp in XRQ (Supplementary Table S3), and none of them was mapped to the Pl19 region. This finding complicates the use of the reference genome for chromosome walking.
The two sunflower reference sequences provide alternative opportunities for SNP discovery. In the current study, SNPs from the XRQ genome showed more polymorphisms than those from HA412-HO. A total of 120 SNPs from HA412-HO were used for Pl17 (16 SNPs) and Pl19 (104 SNPs) fine mapping, and only four were mapped (3.3%). In contrast, of 232 SNPs from XRQ tested for Pl17 (64 SNPs) and Pl19 (168 SNPs), 43 were mapped (18.5%). Considering its assembly from very long PacBio reads, the XRQ genome sequence can be used as the first choice in sequence-based chromosome walking aiming for fine mapping and gene cloning in the sunflower community, while the HA412-HO genome provides a useful comparison to the XRQ genome and a second selection of SNP markers.
In the prior five years, 18 new DM R genes (Pl17–Pl20, Pl22–Pl35) have been identified and mapped with a total of 36 DM R genes in sunflower1,16,17,18,19,29,30,31,32. Despite this great progress, none of the DM R genes has been cloned in sunflower to date. The R genes cloned from other crops indicate that most R genes encode proteins with nucleotide binding and leucine-rich repeat domains (NLRs)33,34. The putative candidate gene HanXRQChr04g0095641 identified from the reference genome of XRQ for Pl17 belongs to this class. A preliminary expression analysis suggested it is potentially a Pl17 gene with the expected kinetics in cotyledons and roots between susceptible and resistant parents in chronological order (data not shown). EMS-induced mutation was performed in a large population of HA 458 seeds and advanced into the M2 generation. DM testing of the M2 population is currently underway to screen for mutants showing susceptible phenotypes. The sequences of the candidate gene HanXRQChr04g0095641 will be further evaluated and compared between wild type and mutants. These studies will provide a foundation to facilitate our efforts of cloning Pl17 in the future.
The 35 kb region of the XRQ reference genome harboring Pl19 contains only one annotated gene, HanXRQChr04g0095951, predicted as a probable RNA methyltransferase family protein. RNA methylation and its role in human diseases have been reported, however, genes with similar annotation have not thus far been implicated in disease resistance in plants35,36. Genomic regions harboring plant disease resistance genes are often complex, exhibiting structural variations between resistant and susceptible genotypes37. Thus, it is possible that the Pl19 gene is absent from the available sunflower reference assemblies. Alternatively, the single gene identified at the Pl19 locus in the XRQ assembly may be indicative of a novel resistance mechanism. Similarly, although a more conventional prospective candidate gene was identified for Pl17, the gene conferring resistance may also be absent from the reference assembly. Future work on the cloning of Pl17 and Pl19 will be required to distinguish between these possibilities and elucidate the genetic basis of the broad-spectrum disease resistance.
Downy mildew remains the major disease threat to sunflower production because of its high-level ability to develop new virulence and its worldwide distribution. Two prerequisites are essential to the use of host resistance in breeding programs, i.e., a resistance resource and diagnostic markers. Both Pl17 and Pl19 show broad-spectrum resistance to all known isolates of P. halstedii10,17,18. Because of their biallelic nature, SNP markers show fewer polymorphisms in the breeding population in nature, especially if the marker is not closely linked to the target gene. In the current study, we applied a whole-genome resequencing approach combined with reference sequence-based chromosome walking to narrow down the gene intervals and develop diagnostic SNP markers for Pl17 and Pl19, respectively. Six diagnostic SNP markers for Pl17 spanned a physical distance of 15 kb in the XRQ genome within the candidate gene HanXRQChr04g0095641. Two diagnostic SNP markers, C4_6675662 and C4_6672629, closest to Pl19 were in a 35-kb interval of Pl19 within the candidate gene HanXRQChr04g0095951. The high-density maps and diagnostic SNP markers for Pl17 and Pl19 developed in this study provide useful tools to accelerate the transfer of these genes to elite sunflower lines in breeding programs, as well as facilitate pyramiding of these genes with other broadly effective Pl genes for durable DM control38.
Methods
Mapping populations and evaluation panel
The initial F2 mapping population for Pl17 was created from a cross between HA 234 and HA 458 with 186 individuals. HA 458 (PI 655009) is an oilseed maintainer line that is resistant to all North American P. halstedii races identified thus far. HA 234 is an oilseed sunflower maintainer line that is susceptible to DM. The DM resistance gene Pl17 in HA 458 was previously mapped to sunflower chromosome 416. This F2 population was used for saturation mapping of additional markers in the present study. For fine mapping, recombinants were screened from 3,008 F3 individuals selected from the previously characterized F2:3 families heterozygous for Pl17. Each selected heterozygous F3 family equates to a segregating F2 population.
Saturation mapping of the DM R gene Pl19 was performed in the BC1F2 population developed from the cross of cytoplasmic male sterile (CMS) CONFSCLB1 and PI 435414 with 139 F2 individuals, which was previously used for the initial mapping of Pl1917. PI 435414, which is resistant to DM, is a wild H. annuus accession that was collected from Paris, Texas, U.S. in 1978. CONFSCLB1 is a confectionary maintainer line that is susceptible to DM. For fine mapping, recombinants were screened from 2,256 BC1F3 individuals selected from the previously characterized BC1F2:3 families heterozygous for Pl19. In our follow-up breeding program, Pl19 was successfully introgressed from wild PI 435414 into confectionary sunflower, named HA-DM5 (PI 687025), which was used for whole-genome resequencing to fine map the Pl19 gene.
The evaluation panel consisted of 96 sunflower inbred lines with diverse origins, including 24 and 17 lines harboring different DM and rust R genes, respectively (Supplementary Table S4). This panel was used to identify diagnostic DNA markers in marker-assisted selection for Pl17 and Pl19, respectively.
SSR and STS marker identification
Previous genetic mapping studies have placed both Pl17 and Pl19 on sunflower chromosome 4 in an interval between 3,621,089 and 6,852,749 bp16,17. This stretch of 3.2 Mb genomic sequence covering both loci was extracted from the HA412-HO (https://www.heliagene.org/HA412.v1.1.bronze.20141015/) and XRQ reference genomes (GenBank accession GCA_002127325.1), respectively. The type and distribution of simple sequence repeats (SSRs) were analyzed using GRAMENE Ssrtool (http://archive.gramene.org/db/markers/ssrtool), and those repeated no less than five times were utilized for primer design. Sequence-tagged sites (STSs) were also analyzed within this 3.2 Mb sequence of the HA412-HO reference. A total of 157 pairs of primers, including 40 STSs and 117 SSRs (40 STSs and 55 SSRs were from the HA412-HO sequence and 62 SSRs from the XRQ sequence), were designed for amplification.
Resequencing and SNP marker identification
Initially, HA 458 whole genome sequence (named HA458-WGS1 with a low genome coverage) was provided by Dr. Loren Rieseberg of the University of British Columbia, Canada, and aligned with the reference genome XRQ (https://www.heliagene.org/HanXRQ-SUNRISE/) around the Pl17 region to identify SNPs and InDels. Subsequently, HA 458 (HA458-WGS2) and HA-DM5 (released germplasm with Pl19) were sequenced at 40 and 35 × depth, respectively, on the Illumina HiSeq sequencing platform at CD Genomics Inc. according to their protocols. Briefly, quality DNA samples were used for library construction using CoVaris S/E210 for fragmentation, and qualified libraries for each gene were pooled for sequencing. Raw reads resulting from sequencing process were filtered to remove reads containing adaptors, reads with >1% ambiguous bases, and reads with low quality (greater than 50% bases less than 15 Q score). A total of 961,980,260 (98.95%) clean reads were obtained from HA 458 sequencing, where 952,508,154 (99.02% mapping rate) and 954,743,565 (99.25%) reads could be mapped to the HA412-HO and XRQ reference genomes, respectively. All SNPs and InDels were identified using the mapped reads and annotated with ANNOVAR software. HA-DM5 was also whole-genome resequenced at CD Genomics Inc. with the same protocols and on the same platforms. The SNP markers were named with prefix C4 or S4 followed by a number representing the physical position of the SNPs along chromosome 4 of each reference genome assembly. C4 represents the SNP from the XRQ reference, while prefix S4 represents the SNP from the HA412-HO reference.
Genotyping of PCR-based markers and linkage analysis
SSR and STS primers were designed using the Primer 3 program (Table 1)39,40. For SNP genotyping, primers were designed as described by Qi et al. (2015)16 and Long et al.21 based on SNP flanking sequences (Supplementary Tables S2, S5). Polymerase chain reaction (PCR) for SSR and STS was performed as described by Qi et al. (2011)41, while SNP PCR was conducted as described by Qi et al. (2016)32. PCR products were visualized by gel electrophoresis on a 6.5% polyacrylamide gel using an IR2 4300/4200 DNA analyzer (LI-COR, Lincoln, NE, USA).
Genotyping data for each marker was first assessed for goodness of fit to the Mendelian segregation ratio (1:3 for dominant and 1:2:1 for codominant) using the Chi-square (χ2) test. Those fitted markers were linkage analyzed with phenotyping data using JoinMap 4.1 software42. Regression mapping algorithm and Kosambi’s mapping function were chosen. The cutoffs of linkage analysis among markers were set at a likelihood of odds (LOD) ≥3.0 and maximum genetic distance ≤50 centimorgans (cM).
Downy mildew resistance evaluation
The P. halstedii isolate of race 734 was chosen to test seedlings of the recombinants selected from the fine mapping populations for resistance to DM, together with their respective parents, HA 234 and HA 458 for Pl17, and CONFSCLB1 and HA-DM5 for Pl19, using the whole seedling immersion method as described by Gulya et al.43 and Qi et al. (2015)16. Race 734 was first identified in 2009 in North America and overcame the Pl6 and Pl7 genes8. The seedling was considered susceptible (S) if sporulation was observed on cotyledons and true leaves and resistant (R) if no sporulation was observed. A total of approximately 30 seedlings from each recombinant were inoculated with the P. halstedii isolate of race 734 and evaluated. The recombinants were classified as homozygous resistant if none of the seedlings exhibited sporulation, segregating if some seedlings showed sporulation on cotyledons and true leaves, and homozygous susceptible if all seedlings showed sporulation on cotyledons and true leaves, which represented the genotypes of DM resistance in each recombinant.
Ethical standards
The experiments were performed in compliance with the current laws of the USA.
References
Ma, G. J., Markell, S. G., Song, Q. J. & Qi, L. L. Genotyping-by-sequencing targeting of a novel downy mildew resistance gene Pl 20 from wild Helianthus argophyllus for sunflower (Helianthus annuus L.). Theor Appl Genet 130, 1519–1529 (2017).
Vear, F. Breeding disease-resistant sunflowers. CAB Reviews 12, 1–11 (2017).
Vear, F. et al. Origins of major genes for downy mildew resistance in sunflower. In: Proceedings of 17 th International Sunflower Conference. 125–130 (2008).
Vincourt, P. et al. Consensus mapping of major resistance genes and independent QTL for quantitative resistance to sunflower downy mildew. Theor Appl Genet 125, 909–920 (2012).
Viranyi, F., Gulya, T. J. & Tourieille, D. L. Recent changes in the pathogenic variability of Plasmopara halstedii (sunflower downy mildew) populations from different continents. Helia 38, 149–162 (2015).
Trojanová, Z., Sedlářová, M., Gulya, T. J. & Lebeda, A. Methodology of virulence screening and race characterization of Plasmopara halstedii, and resistance evaluation in sunflower-a review. Plant Pathol 66, 171–185 (2017).
Gulya, T. J., Due, B., & Hutter, M. New virulent races of downy mildew: distribution, status of DM resistant hybrids, and USDA sources of resistance to races that overcome the Pl 6 gene. In: Proceedings of 32 nd Sunflower Research Forum., http://www.sunflowernsa.com/uploads/16/gulya1_virulentraces_10.pdf (2010).
Gulya, T. J., Markell, S., McMullen, M., Harveson, B. & Osborne, L. New virulent races of downy mildew: distribution, status of DM resistant hybrids, and USDA sources of resistance. In: Proceedings of 33 rd Sunflower Research Forum., http://www.sunflowernsa.com/uploads/17/gulya_virulentracesdownymildew.pdf (2011).
Ahmed, S., de Labrouhe, D. T. & Delmotte, F. Emerging virulence arising from hybridization facilitated by multiple introductions of the sunflower downy mildew pathogen Plasmopara halstedii. Fungal Genet Biol 49, 847–855 (2012).
Gilley, M., Misar, C., Gulya, T., & Markell, S. Prevalence and virulence of Plasmopara halstedii (downy mildew) in sunflowers. In: Proceeding 38 th Sunflower Research Forum, http://www.sunflowernsa.com/uploads/research/1277/Prevalence.Downey_Gilley.etal_2016.rev.pdf (2016).
Schilling, E. E. “Helianthus,” in Flora of North America Committee, Vol. 21, ed Flora of North America Editorial Committee 141–169 (New York, NY; Oxford: Oxford University Press, 2006).
Stebbins, J. C., Winchell, C. J. & Constable, J. V. H. Helianthus winteri (Asteraceae), a new perennial species from the southern Sierra Nevad a foothills, California. Aliso 31, 19–24, https://doi.org/10.5642/aliso.20133101.04 (2013).
Seiler, G. J., Qi, L. L. & Marek, L. F. Utilization of sunflower crop wild relatives for cultivated sunflower improvement. Crop Sci 57, 1083–1101 (2017).
Hulke, B. S., Miller, J. F., Gulya, T. J. & Vick, B. A. Registration of the oilseed sunflower genetic stocks HA 458, HA 459, and HA 460 possessing genes for resistance to downy mildew. J Plant Reg 4, 93–97 (2010).
Gulya, T. J. Evaluation of wild annual Heliathus species for resistance to downy mildew and Sclerotinia stalk rot. In: Proceeding 27 th Sunflower Research Forum., http://www.sunflowernsa.com/uploads/research/265/Gulya_WildHelianthus_studies_05.pdf (2005).
Qi, L. L., Long, Y. M., Jan, C. C., Ma, G. J. & Gulya, T. J. Pl 17 is a novel gene independent of known downy mildew resistance genes in the cultivated sunflower (Helianthus annuus L.). Theor Appl Genet 128, 757–767 (2015).
Zhang, Z. W., Ma, G. J., Zhao, J., Markell, S. G. & Qi, L. L. Discovery and introgression of the wild sunflower-derived novel downy mildew resistance gene Pl 19 in confection sunflower (Helianthus annuus L.). Theor Appl Genet 130, 29–39 (2017).
Pecrix, Y. et al. Ten broad spectrum resistances to downy mildew physically mapped on the sunflower genome. Front Plant Sci 9, 1780 (2018).
Liu, Z. et al. Molecular mapping of the downy mildew and rust resistance genes in a sunflower germplasm line TX16R. Mol Breed 39, 19 (2019).
Badouin, H. et al. The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution. Nature 546, 148–152 (2017).
Long, Y. M. et al. An innovative SNP genotyping method adapting to multiple platforms and throughputs. Theor Appl Genet 130, 597–607 (2017).
Ma, G. et al. Cloning and characterization of the homoeologous genes for the Rec8-like meiotic cohesion in polyploid wheat. BMC Plant Biol 18, 224 (2018).
Meyers, B. C. et al. The major resistance gene cluster in lettuce is highly duplicated and spans several megabases. Plant Cell 10, 1817–1832 (1998).
Michelmore, R. W. & Meyers, B. C. Cluster of resistance genes in plants evolve by divergent selection and a birth-and-death process. Genome Res 8, 1113–1130 (1998).
Wei, F. et al. The Mla (powdery mildew) resistance cluster is associated with three NBS-LRR gene families and suppressed recombination within a 240-kb DNA interval on chromosome 5S (1HS) of barley. Genetics 153, 1929–1949 (1999).
Hulbert, S. H., Webb, C. A., Smith, S. M. & Sun, Q. Resistance gene complexes: evolution and utilization. Ann Rev Phytopathol 39, 285–312 (2001).
Kong, L. R., Cambron, S. E. & Ohm, H. W. Hessian fly resistance genes H16 and H17 are mapped to a resistance gene cluster in the distal region of chromosome 1AS in wheat. Mol Breed 21, 183–194 (2008).
Ashfield, T. et al. Evolution of a complex disease resistance gene cluster in diploid Phaseolus and tetraploid Glycine. Plant Physiol 159, 336–354 (2012).
Pecrix, Y. et al. Sunflower resistance to multiple downy mildew pathotypes revealed by recognition of conserved effectors of the oomycete Plasmopara halstedii. Plant J 97, 730–748 (2018).
Talukder, Z. I., Ma, G., Hulke, B. S., Jan, C. C. & Qi, L. L. Linkage mapping and genome-wide association studies of the Rf gene cluster in sunflower (Helianthus annuus L.) and their distribution in world sunflower collections. Front Genet 10, 216, https://doi.org/10.3389/fgene.2019.00216 (2019).
Qi, L. L., Ma, G. J., Li, X. H., & Seiler, G. J. Diversification of the downy mildew resistance gene pool by introgression of a new gene, Pl 35, from wild Helianthus argophyllus into oilseed and confection sunflowers (Helianthus annuus L.). Theor Appl Genet https://doi.org/10.1007/s00122-019-03370-9 (2019).
Qi, L. L., Foley, M. E., Cai, X. W. & Gulya, T. J. Genetics and mapping of a novel downy mildew resistance gene, Pl 18, introgressed from wild Helianthus argophyllus into cultivated sunflower (Helianthus annuus L.). Theor Appl Genet 129, 741–752 (2016).
Liu, J., Liu, X., Dai, L. & Wang, G. Recent progress in elucidating the structure, function and evolution of disease resistance genes in plants. J Genet Genom 34, 765–776 (2007).
Sekhwal, M. K. et al. Disease resistance gene analogs (RGAs) in plants. Int J Mol Sci 16, 19248–19290 (2015).
Blanco, S. & Frye, M. Role of RNA methyltransferases in tissue renewal and pathology. Curr Opin Cell Biol 31, 1–7 (2014).
Bohnsack, K. E., Höbartner, C. & Bohnsack, M. T. Eukaryotic 5-methylcytosine (m5C) RNA methyltransferases: mechanisms, cellular functions, and links to disease. Gene 10, 102 (2019).
van Wersch, S. & Li, X. Stronger when together: clustering of plant NLR disease resistance genes. Trends Plant Sci 24, 688–699 (2019).
Ma, G. J., Seiler, G. J., Markell, S. G. & Qi, L. L. Registration of three confection sunflower germplasm, HA-DM2, HA-DM3, and HA-DM4, resistant to downy mildew and rust. J Plant Reg 13, 103–108 (2019).
Koressaar, T. & Remm, M. Enhancements and modifications of primer design program Primer3. Bioinformatics 23, 1289–1291 (2007).
Untergasser, A. et al. Primer3 – new capabilities and interfaces. Nucleic Acids Res 40, e115 (2012).
Qi, L. L., Hulke, B. S., Vick, B. A. & Gulya, T. J. Molecular mapping of the rust resistance gene R4 to a large NBS-LRR cluster on linkage group 13 of sunflower. Theor Appl Genet 123, 351–358 (2011).
Van Ooijen, J. W. JoinMap® 4, Software for the calculation of genetic linkage maps in experimental populations. Kyazma BV, Wageningen, Netherlands (2006).
Gulya, T. J. et al. Metalaxyl resistance in sunflower downy mildew in North America. In: Proceeding 21st Sunflower Research Forum. (1999).
Acknowledgements
The authors would like to thank Angelia Hogness for technical assistance. This project was supported by the USDA-AMS Specialty Crop Block Grant Programs 16-SCBGPND-0029 and AM170100XXXXG05 and the USDA-ARS CRIS Project No. 3060-2100-043-00D. The mention of trade names or commercial products in this report is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the US Department of Agriculture. The USDA is an equal opportunity provider and employer.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: L.L.Q., G.J.M. Performed the experiments: G.J.M., L.L.Q., Z.W.Z. Analyzed data: G.J.M., L.L.Q., Q.J.S., W.R.U., J.D.F., X.H.L. Wrote the paper: G.J.M., L.L.Q. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ma, G., Song, Q., Underwood, W.R. et al. Molecular dissection of resistance gene cluster and candidate gene identification of Pl17 and Pl19 in sunflower by whole-genome resequencing. Sci Rep 9, 14974 (2019). https://doi.org/10.1038/s41598-019-50394-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-50394-8
- Springer Nature Limited
This article is cited by
-
QTL Mapping and Genetic Map for the Ornamental Sunflower in China
Plant Molecular Biology Reporter (2024)
-
Whole-genome sequencing enables molecular dissection and candidate gene identification of the rust resistance gene R12 in sunflower (Helianthus annuus L.)
Theoretical and Applied Genetics (2023)
-
Introgression and targeting of the Pl37 and Pl38 genes for downy mildew resistance from wild Helianthus annuus and H. praecox into cultivated sunflower (Helianthus annuus L.)
Theoretical and Applied Genetics (2023)
-
Genetic and physiological characterization of sunflower resistance provided by the wild-derived OrDeb2 gene against highly virulent races of Orobanche cumana Wallr
Theoretical and Applied Genetics (2022)
-
Characterization and mapping of a downy mildew resistance gene, Pl36, in sunflower (Helianthus annuus L.)
Molecular Breeding (2022)