

Abstract
The mitochondrial serine protease HtrA2 is a human homolog of the Escherichia coli Deg-proteins exhibiting chaperone and proteolytic roles. HtrA2 is involved in both apoptotic regulation via its ability to degrade inhibitor-of-apoptosis proteins (IAPs), as well as in cellular maintenance as part of the cellular protein quality control machinery, by preventing the possible toxic accumulation of aggregated proteins. In this study, we use advanced solution NMR spectroscopy methods combined with biophysical characterization and biochemical assays to elucidate the crucial role of the substrate recognizing PDZ domain. This domain regulates the protease activity of HtrA2 by triggering an intricate allosteric network involving the regulatory loops of the protease domain. We further show that divalent metal ions can both positively and negatively modulate the activity of HtrA2, leading to a refined model of HtrA2 regulation within the apoptotic pathway.
Similar content being viewed by others


Introduction
To ensure a functional proteome, cells in all kingdoms of life rely on a variety of different unfoldases and proteases to avoid the accumulation of unfolded proteins and possibly toxic protein aggregates1,2,3,4. Whereas the functional details for the ATP-dependent systems residing in the cytosolic compartments of the cells were subject to intense studies in recent years and became well understood, the functional and structural details of protein quality control by the ATP-independent serine–proteases of the HtrA-family (High-temperature requirement A) remain only partially understood3,5,6,7,8,9. Within humans four different HtrA proteins could be identified: whereas HtrA1, HtrA3, HtrA4 are residing in the cytosol, HtrA2 is mainly located in mitochondria under non-stress conditions10. It could be shown that loss of proteolytic activity of this important class of proteins is directly connected to severe diseases, including arthritis, cancer, familial ischemic cerebral small-vessel disease, and age-related macular degeneration, as well as neurodegenerative diseases such as Parkinson’s and Alzheimer´s disease3,6,8,9.
The human mitochondrial serine protease HtrA2 (Omi), a homolog of the Escherichia coli heat shock proteins DegP, DegQ, and DegS, is primarily residing in the mitochondrial intermembrane space performing protein quality functions11,12,13. The full-length protein consists of 458 residues, of which the mature protein lacks the 133 amino-terminal residues containing a mitochondrial targeting sequence followed by a transmembrane domain. The protein is initially attached to the mitochondrial inner membrane and undergoes subsequent maturation by proteolytic auto-processing of the transmembrane domain upon apoptotic stimuli13. In the resulting membrane-dissociated form, the protein consists of a serine protease domain containing a characteristic catalytic triad (S306, H198, and D228), as well as a carboxy-terminal PDZ domain (PSD-95, DLG, and ZO-1)14, serving as a substrate recognition domain and restricting access to the active site of the enzyme (Fig. 1a, b and Supplementary Fig. 1). Furthermore, several regulatory loops, termed LA, LD, L1, L2, and L3, within the protease domain are modulating the proteolytic activity of HtrA2 possessing either activating or inhibitory roles15. It could also be shown that the mature protein assembles into a homotrimer of ~105 kDa, essential for its proteolytic capabilities16. The role of HtrA2 as a key regulator of the apoptotic stress response has been studied in-depth, revealing its role in initiating the apoptotic cascade by processing Inhibitor of Apoptosis proteins (IAPs)17,18. In addition, HtrA2 shares the main function of proteases within the mitochondrial and cellular protein quality control network: the prevention of accumulation of protein aggregates leading to cellular malfunction8,9. As mitochondrial dysfunction as a consequence of protein accumulation is closely linked to neurodegeneration, a possible prominent neuroprotective function of HtrA2 has been hypothesized19,20. This view is further supported by the experimental observation that mutations in the HTRA2 gene cause hereditary tremors which can progress into Parkinson’s disease as well as that mice carrying a loss-of-function variant of HtrA2 develop Parkinson-like symptoms21,22. In addition, HtrA2 has also been reported to co-localize within Lewy bodies and has been shown to be able to reduce the propensity of Parkinson-related α-synuclein seeding whilst also contributing to the removal of existing α-synuclein aggregates23,24.

a Scheme of the used constructs indicating the domain structure of mature human HtrA2. Residues of the catalytic triad and important regulatory loops are labeled. b Crystal structure of the proteolytically active HtrA2 trimer (PDB-ID: 5M3N) focusing on the protease-PDZ-domain interface of a single subunit (complete trimer: Supplementary Fig. 1). Different domains (PDZ domain: green; protease domain: blue) and the catalytic triad composed of H198, D228, and S306 are indicated (gold). c 2D [15N, 1H]-NMR spectrum of the [U-15N,13C]-HtrA2-PDZ domain. The sequence-specific resonance assignment based on triple-resonance experiments is shown. d Secondary structure elements of the HtrA2-PDZ domain in solution (green). The secondary structure elements of the HtrA2-PDZ domain within the full-length crystal structure (PDB-ID: 5M3N) are indicated in gray. e Generalized order parameter (S2) reporting on sub-nanosecond motions plotted on the HtrA2-PDZ structure. The backbone amide moieties of the HtrA2-PDZ domain are shown as spheres and the S2 values are indicated by the yellow-to-blue gradient. f Analysis of 15N-backbone relaxation data with the Lipari–Szabo model-free approach. The generalized order parameter S2 reports on pico- to nanosecond motions, the chemical exchange contributions Rex indicating micro- to millisecond motions, and the rotational correlation time τc. Broken line represents the average value of 8.2 ns. Error bars indicate the standard fitting error obtained from the nonlinear least-squares minimization. g The amide moieties of the HtrA2-PDZ domain are shown as spheres. The calculated Rex is indicated by the yellow-to-red gradient. Source data are provided as a Source Data File.
Despite the importance of HtrA2 in apoptosis regulation and its likely connection to neurodegeneration its functional details remain still largely elusive. Recently, it could be shown that membrane-dissociated HtrA2, prior to activation, is in an equilibrium between trimer and hexamer employing a methyl-TROSY NMR spectroscopy approach25. Remarkably, HtrA2 thus uses a similar sequence motif within the PDZ domain as its bacterial homolog DegP, which could be identified as part of a temperature-dependent activation switch for the bacterial protein25,26. Together with several available high-resolution X-ray structures16,27,28, a picture emerges indicating an important role for the PDZ domain in the activation of HtrA2 proteolytic activity.
Here we set out to specify the role of the HtrA2-PDZ domain for the modulation of HtrA2 proteolytic function. By combining solution NMR spectroscopy, biochemistry, and fluorescence cleavage assays, we show that the inherent dynamics of the PDZ domain play a crucial role for regulating the proteolytic function of the full-length protein triggering an activation cascade of the regulatory loops embedded within the protease domain employing an extended allosteric network. Furthermore, we identify divalent metal ions as important functional modulators, positive or negative, of HtrA2 proteolysis by modulating its activation through targeting the PDZ domain. After initially focusing on the isolated PDZ domain, we establish our observations within the full-length protein exploiting the methyl-TROSY approach, especially suited for large molecular complexes29. By combining this powerful technique with paramagnetic relaxation enhancement (PRE) experiments, we were able to identify a previously unknown role of the amino-terminal helix α1 in the activation cascade of HtrA2. Together with recent observations of regulation by transient oligomerization25 and interprotomer cooperativity30, our results of divalent ion modulated PDZ dynamics as well as the analysis of methyl NOE patterns and dynamics in full-length HtrA2 lead to a refined picture of the individual components underlying HtrA2 regulation, highlighting important aspects of regulating cellular protein quality control to prevent uncontrolled apoptosis.
Results
Isolated HtrA2-PDZ domain in solution
To investigate the structural plasticity and inherent dynamics of the HtrA2-PDZ domain, a construct consisting of the isolated HtrA2-PDZ domain (residues 349–458) was designed (Fig. 1a and Supplementary Fig. 1a). We obtained a nearly complete (97%) assignment of the PDZ domain by sequence-specific backbone and side-chain resonance assignment experiments (Fig. 1c and Supplementary Fig. 2a). Comparison of the secondary structural elements of the HtrA2-PDZ domain in solution as derived from the Cα and Cβ combined secondary shifts shows a large similarity to the secondary structure elements observed in the crystal structure of the HtrA2-PDZ domain and indicates overall a stably folded domain (Fig. 1d and Supplementary Fig. 2b, c)16. The main differences occur in strand β14, which in solution, indicated by the secondary chemical shift analysis (Fig. 1d and Supplementary Fig. 2b), is slightly shifted, and based on the extent of the obtained secondary chemical shift values appears to be only partially populated (~50%) (Fig. 1d). In addition, this segment shows broadened signals in the [15N, 1H]-NMR spectrum although the non-solvent-exchangeable side-chain carbon resonances could be completely assigned (Supplemental Fig. S2d). This behavior could point to either elevated solvent exchange under the used conditions and/or dynamics on the millisecond timescale for this region of the protein, indicative of either structural heterogeneity and/or exchange processes on the micro- to millisecond NMR timescale31. This observation is further supported by the diminished signal intensity in the [15N, 1H]-NMR spectrum for residues within adjacent strand β15 (Supplemental Fig. S2e), pointing to a similar process and suggesting that the interaction between these two strands is only transiently formed. This can be readily explained by the fact that within the full-length protein the strand β14 locks the PDZ domain onto the protease domain rendering the HtrA2 protease inactive and that these interdomain contacts are broken upon activation25,32. Further, we determined the average rotational correlation time (τc) for the PDZ domain to 8.2 ns (Fig. 1f), which is in line with the expected theoretical τc value for globular proteins of this molecular weight (~12.1 kDa).
To further assess the overall stability of the PDZ domain we characterized the methyl–methyl NOE networks (Supplementary Fig. 3) within this domain. Since methyl-bearing residues are well distributed throughout the entire domain, we could thereby acquire an overview of the stabilizing intradomain contacts. We observed that the methyl–methyl NOEs identified are in line with the PDZ domain, exhibiting an overall stable fold in agreement with the HtrA2 X-ray structures previously reported16,27. For instance, structural elements that are in close vicinity in the crystal structure such as β16 and β17 or β17 and β18 show several methyl–methyl NOE contacts, whereas α6 and α7 show only a limited number of NOEs to adjacent β-strands (Supplementary Fig. 3b, c). Interestingly, we could observe that the α5 helix displayed no methyl–methyl NOEs to any other structural element within the PDZ domain, indicating that this part of the PDZ domain might not stably stack against the other structural elements of the domain (Supplementary Fig. 3c, f). Having a more detailed look into the amide as well as the carbon NOEs of the α5 helix revealed that this segment is well stabilized within itself forming a short helix perfectly in line with the observed secondary chemical shifts (Supplementary Fig. 3d, e). In contrast, only a limited number of weak NOE contacts to other segments of the domain could be identified, indicating its rather loose attachment leading us to hypothesize that this helix could be an important trigger within the PDZ domain possibly involved in the activation cascade of HtrA2.
Dynamics of the isolated HtrA2-PDZ domain
In order to elucidate the dynamic properties of the PDZ domain, we analyzed the backbone dynamics of the HtrA2-PDZ domain at a concentration of 500 µM by standard NMR relaxation experiments33. The 15N{1H}-NOE (hetNOE) and 15N longitudinal R1 relaxation rates report on fast motions on the pico- to nanosecond timescale. For HtrA2-PDZ, we observed a stable hetNOE ratio profile, on average 0.8, with a sharp decrease towards the carboxy-terminus, suggesting that the HtrA2-PDZ domain is a stably folded domain with increased flexibility at the carboxy-terminus, which is devoid of stable secondary structure elements. The R1 rate constants show a similar tendency as the hetNOE data (Supplementary Fig. 4a). We also explored the motions in the micro- to millisecond timescale by measuring the 15N transverse relaxation rates R2β and R2 as derived from the R1ρ rates (R2(R1ρ)). The R2(R1ρ) rate constants of HtrA2-PDZ, reporting on motions on the lower microsecond timescale (the used spin-lock radio frequency field of 2000 Hz levels out all exchange contribution (Rex) much slower than 80 µs33), indicate a similar behavior to that observed within the hetNOE and R1 rate measurements, and we observed mainly planar R2(R1ρ) rate constants (Supplementary Fig. 4b). The R2β rate constants, however, show a notable increase in the regions between α5–β15 and β17–β18, suggesting dynamics within these segments on the higher micro- to millisecond timescale, pointing to a possible conformational exchange occurring in these loop regions (Supplementary Fig. 4b). To rule out any contribution of a large anisotropic diffusion tensor to the observed behavior, we also plotted the R1•R2β values alongside the R2β rate constants, showing the same trend as the R2β rates and thus no indication of such a contribution (Supplementary Fig. 4b). Next, we analyzed the backbone relaxation data with the Lipari–Szabo model-free approach using an axially symmetric diffusion tensor (Supplementary Fig. 4c, d) to obtain the generalized order parameters (S2) and the chemical exchange contributions (Rex)34,35. We observed generalized order parameter values with an average of 0.9 ± 0.1 (Fig. 1e, f), indicating structural rigidity for the whole domain, including the loosely attached helix α5 on the pico- to nanosecond timescale. Nevertheless, we observed increased Rex terms for residues in the β15 preceding linker region, as well as in α6 and the region involving α7 and the linker region between α7 and β17, pointing to the presence of chemical exchange contributions as initially observed by the enhanced R2β rates (Fig. 1f, g).
To quantify these exchange contributions, we next employed a BEST TROSY 15N Carr-Purcell-Meiboom-Gill (CPMG) relaxation dispersion (RD) experiment36. The obtained relaxation dispersion profiles provide insight into protein dynamics, where non-flat profiles indicate micro- to millisecond dynamics and flat profiles their absence (Supplementary Fig. 4e, f). The CPMG data recorded at 298 K revealed micro- to millisecond dynamics within the amino-terminus, the α5 helix and the adjacent loop connecting to β15, as well as α7 and its preceding loop (Fig. 2a, b). By transferring the sequence-specific resonance assignment to lower temperatures (283 K) we were then able to compare this data to the initial CPMG data recorded at 298 K. At lower temperature, we observed dynamics in addition also within α7 and the β17–β18 loop, to a larger extent compared to our observations at 298 K and additionally evidenced as line-broadening (Fig. 2a and Supplementary Fig. 4e–h). Quantitative global analysis of the dispersion data showed that this motion occurred on 0.98 ± 0.005 ms (283 K) as well as 0.42 ± 0.004 ms (298 K) timescale (Fig. 2c). At the same time the population of the minor state was also slightly modulated by the temperature, decreasing from 16.1 to 15.2% (Fig. 2c).

a ΔR2eff values for the backbone amide groups, obtained from the difference of R2eff at the lowest and highest CPMG frequency υCPMG, at the indicated temperatures. Residues exchange broadened are highlighted by purple bars; unassigned residues indicated by an asterisk (*). Error bars indicate the standard fitting error obtained from the nonlinear least-squares minimization of one experiment (n = 1). b Amplitude of the CPMG relaxation dispersion profiles ΔR2eff at 21.1 T of an [U-15N,13C]–PDZ sample at 298 K. c Rate constants of the dynamic process (τex) and the relative populations of PDZ states obtained from a global fit of the CPMG relaxation dispersion data. Error bars indicate the standard fitting error obtained from the nonlinear least-squares minimization of one experiment (n = 1). d, e Local methyl group dynamics on the pico- to nanosecond timescale probed by methyl single-quantum (SQ) and triple-quantum (TQ) relaxation experiments showing the product of the local order parameter and the overall tumbling constant, S2axis•τC. Measurements were performed on an [U-2H, Ile-δ1-13CH3, Leu, Val-13CH3, Ala-13CH3, Thr-γ2-13CH3]–PDZ sample at 298 K. Methyl groups (spheres) and the obtained S2axis•τC-values (yellow-to-blue gradient) (d). S2axis•τC-values plotted against the HtrA2-PDZ amino acid sequence (e). Error bars indicate the standard fitting error obtained from the nonlinear least-squares minimization of one experiment (n = 1). f Amplitude of the CPMG relaxation dispersion profiles ΔR2eff at 16.4 T. Source data are provided as a Source Data File.
Methyl side-chain dynamics reveal enhanced motions within HtrA2-PDZ
To derive the origin of the local exchange contributions identified by the backbone relaxation analysis, we exploited the increased sensitivity of the methyl groups to gain insight into the side-chain dynamics of the HtrA2-PDZ. Using an ALVIT-labeled HtrA2-PDZ sample we first determined the product of the side-chain order parameters and the correlation time of the overall molecular tumbling (S2axis•τC), reporting on the extent of the amplitude of motions on the pico- to nanosecond NMR timescale (Fig. 2d, e). Overall, the obtained values indicated the stable fold of the domain averaging to S2axis•τC = 4.3 ± 0.5 ns for residues residing in the core of the domain, comparable to values obtained for the PDZ domains of bacterial DegP26. Looking more specifically at the local distributions of the S2axis•τC values, we observed that these values indicated increased flexibility for residues in the α5-β15-α6 region as well as in α7 and the carboxy-terminus (Fig. 2d, e). As we had already obtained indications of specific line-broadening in a 2D [13C,1H]-NMR spectrum for some methyl resonances, e.g., I373 and V364 (Supplementary Fig. 2a), we used a multiple quantum (MQ) CPMG relaxation dispersion experiment37. The obtained CPMG relaxation dispersion profiles report on the micro- to millisecond dynamics and showed increased rate constants for residues in the α5 to β15 region as well as for residues in the β16/α7 with a 16.6 ± 0.4% minor state population on a 1.19 ± 0.07 ms timescale (Fig. 2f and Supplementary Fig. 5a, b). The obtained data for both, the protein amide backbone as well as the methyl groups, indicate that the PDZ domain is experiencing micro- to millisecond dynamics, indicative of the same underlying process. These dynamics are manifested within the α5 helix and the adjacent linker region connecting α5 and β15, pointing to a possible importance of these inherent local dynamics for the regulation of HtrA2 activation and its protease activity.
Oligomerization properties of the HtrA2-PDZ domain
Given the recently identified hexameric inactive form of HtrA225, we next assessed a potential transient oligomerization of the isolated HtrA2-PDZ domain by size exclusion chromatography coupled with multi-angle light-scattering (SEC-MALS) experiments using a range of different HtrA2-PDZ concentrations. Although the accessible range of protein concentrations was limited by solubility, MALS data for dimerization can be fitted with constrained values for the titration end point masses and lower limits of KD values can be obtained from such solubility-limited datasets38. Under the chosen experimental conditions, the HtrA2-PDZ domain showed only a minute tendency to dimerize with an estimated lower limit of a KD of 13 ± 1.65 mM (Supplementary Fig. 5c, d). This weak transient interaction is about three orders of magnitude weaker than the recently reported trimer-to-hexamer equilibrium for the full-length protein by NMR spectroscopy with ~20 µM25. This difference can be likely attributed to the presumably cooperative nature of the interaction between the three PDZ:PDZ interfaces within the hexameric species25.
Modulation of the PDZ domain by divalent ions facilitates HtrA2s proteolytic activity
We hypothesized that due to HtrA2s role in apoptotic signaling its proteolytic function might be modulated by divalent cations, such as Ca2+, as bacterial HtrA proteins have previously been shown to be modulated by divalent ions39,40. Upon initially adding Mg2+ and Ca2+ ions to the HtrA2-PDZ NMR buffer, we were able to detect localized chemical shift changes within the PDZ domain, indicative of an ability of the domain to bind divalent ions. A detailed analysis of the resulting chemical shift perturbations for each residue revealed that the regions with the most pronounced chemical shift changes were located within the loop region between α5 and β15, as well as the loop region preceding α7. We then extended our analysis by also testing Cu2+ and Zn2+, previously reported as inhibitors for bacterial HtrAs39,40, detecting similar chemical shift perturbations towards HtrA2-PDZ (Supplementary Fig. 6a–d). Upon addition of CuCl2 as well as ZnSO4 we also observed a signal attenuation in the NMR spectra to about 60% and 80%, respectively. Whereas this effect can be attributed for ZnSO4 to a possible oligomerization, in case of CuCl2 it is likely caused by enhanced relaxation due to the presence of paramagnetic Cu2+ leading to paramagnetic relaxation enhancement (PRE)41. Therefore, the observed effect in the presence of Cu2+ reflects rather the solvent accessibility of the different amide moieties as the effect was most pronounced for the loop regions in general (Supplementary Fig. 6c). Coupled with our observations of enhanced dynamics on the micro- to millisecond timescale of both the side-chain methyl groups and the backbone in the regions of HtrA2-PDZ where the divalent metal ions bind, we hypothesized that these inherent dynamics might play a functional role in the modulation of the protease function of HtrA2 by divalent cations. Using the MQ CPMG on the methyl groups for HtrA2-PDZ samples supplemented with either CaCl2 or ZnSO4, we observed however only minor modulations of the inherent dynamics compared to the HtrA2-PDZ in the absence of divalent cations (Supplementary Fig. 6e–h). We next used the NMR line-shape analysis tool TITAN to determine the metal affinity to the HtrA2-PDZ domain from titration series42. We were able to determine KD values for Ca2+ and Zn2+ in the lower millimolar range (4.1 ± 0.5 mM and 1.6 ± 0.3 mM for Ca2+ and Zn2+, respectively) and the dissociation rate koff to 84.3 ± 29 s−1, and 52.8 ± 15.3 s−1 for Ca2+ and Zn2+, respectively (Supplementary Figs. 7 and 8). We were not able to reliably fit the data from the Mg2+ and Cu2+ titration series.
To assess the potential functional role of the divalent cations, we subsequently tested the proteolytic activity of mature HtrA2 toward β-casein in the presence of either Mg2+, Ca2+, Cu2+, or Zn2+. The activity of HtrA2 toward β-casein was increased when Ca2+ was supplemented, and to a lesser extent also in the presence of Mg2+, compared to the activity of the protease in the standard assay buffer (Fig. 3a, b and Supplementary Fig. 9). To rule out any effects of the divalent ions on the β-casein directly, we also performed controls in the absence of HtrA2, resulting in no cleavage, which clearly attributes the observed enhancement of HtrA2 to a modulation of its proteolytic activity by Ca2+ and Mg2+ (Supplementary Fig. 10). In contrast, the addition of Cu2+ and Zn2+ strongly inhibited HtrA2 protease activity (Fig. 3a, b and Supplementary Fig. 9). The observed inhibitory effect of Zn2+ and Cu2+ agree with previously reported effects of these ions on related HtrA proteins39,40.

a Proteolytic assay using β-casein shows that Mg2+ and Ca2+ enhanced the proteolytic activity of HtrA2, while supplementation of Zn2+ and Cu2+ strongly inhibited proteolytic activity. Cleavage assays were done as biological triplicates, yielding similar results (Supplementary Fig. 9). b Comparison of the proteolytic efficiency of HtrA2 towards β-casein without or with divalent metal ions. Cleavage is expressed as percentage of β-casein cleaved after 40 minutes of incubation with HtrA2. Bar colors correspond to the color assigned to each metal ion in (a). c Scheme of the fluorescence assay using an activating peptide and a reporter peptide. d Fluorescence assay showing activity of HtrA2 with an HtrA2 activating peptide (DD-PDZOpt, 50 µM) using varying reporter peptide concentrations as indicated. Data is representative of three replicate experiments. e Analysis of the catalytic efficiency (kcat/KM) of the assay shown in (d) in the absence or presence of divalent metal ions as indicated. f Determination of the apparent KD (KD,app) of the activator peptide using varying peptide concentrations. n.d. indicates not determined. Values are the averages of n = 3 individual repeats, and error bars indicate the SD of these replicates. g Fluorescence assay showing activity of HtrA2 using varying HtrA2 activating peptide concentrations as indicated with a fixed reporter peptide concentration (20 µM). Data are representative of three replicate experiments. h Analysis of the catalytic efficiency (kcat/KM) of the assay shown in (g) with or without divalent metal ions. Source data are provided as a Source Data File.
Previously, a hexameric species of HtrA2 has been characterized as a closed inactive state with reduced substrate affinity compared to the canonical trimeric species of HtrA225. Based on this previous finding and the signal attenuation observed when titrating ZnSO4 toward HtrA2-PDZ, we reasoned that the different divalent ions might influence the oligomeric equilibrium of HtrA2. Therefore, we performed SEC-MALS experiments with the HtrA2-PDZ domain in the presence of the four metal ions used in the proteolytic assay. While the addition of Mg2+, Cu2+, and Ca2+ did not result in a notably increased dimerization rate of the HtrA2-PDZ domain compared to the non-supplemented buffer, the addition of Zn2+ markedly increased the propensity ~50-fold with which the HtrA2-PDZ domains dimerized (Supplementary Fig. 11 and Supplementary Table 1).
Structural basis of the interaction between HtrA2-PDZ and an activating peptide
It has been previously demonstrated that HtrA2 can be activated by the interaction between its PDZ domain and target substrates or custom-made peptides designed for optimized interaction with the HtrA2-PDZ domain, similar to bacterial DegS and DegP43,44,45. We looked more closely at this activation step by analyzing the interaction between the isolated HtrA2-PDZ domain and a customized activator peptide, DD-PDZOpt25. We probed the interaction via titration experiments focusing on the backbone using 2D [15N, 1H]-NMR spectra, as well as by an ALVIT methyl-labeled PDZ sample by recording 2D [13C, 1H]-NMR spectra (Supplementary Fig. 12a–f). While we observed only minor chemical shift changes on the backbone amide moieties, we were able to see the effects of the activating peptide binding more pronounced on the methyl groups. We detected significant chemical shift changes in the regions of β14, β15, and α7 (Supplementary Fig. 12e, f), which correlates well with previous observations of a HtrA2 peptide binding cleft comprising these regions of the PDZ domain25,46. To obtain detailed insight, we then used TITAN42 to analyze the titration data of HtrA2-PDZ and DD-PDZOpt. Employing a two-state ligand binding model, we determined the dissociation constant KD to 3.18 ± 0.9 µM, with a dissociation rate koff of 48.5 ± 2.9 s−1 (Supplementary Fig. 12g), in good agreement with a reported KD of 7.5 µM between wild-type HtrA2 and a related 13-residue activating peptide47. Based on the obtained CSPs we docked the DD-PDZOpt peptide onto the isolated PDZ domain by HADDOCK48,49 (Supplementary Fig. 12h and Supplementary Table 2), revealing a virtual identical arrangement as previously determined by X-ray crystallography for a related activating peptide (PDB-ID: 2PZD; Supplementary Fig. 12i). Finally, we investigated if the binding influences the transient dimerization of the HtrA2-PDZ domain. The SEC-MALS analysis resulted in a similar dimerization constant, 10.3 ± 2.5 mM, as observed for the isolated PDZ, ruling out any contribution to the monomer–dimer equilibrium (Supplementary Fig. 11i, j).
Dissecting the role of divalent ions in a coupled enzymatic assay
Next, we assessed the proteolytic efficiency of HtrA2 in coupled enzymatic fluorescence-based cleavage assays of the H2Opt peptide, a self-quenching fluorescent substrate peptide, in the presence of the activating peptide DD-PDZOpt. In accordance with previous studies25,44,47, we observed that the proteolytic efficiency of HtrA2 towards the fluorescent substrate was greatly enhanced when the activating peptide was present (Fig. 3g). We extracted kinetic parameters for wild-type HtrA2 using two variants of the fluorescence assay: either using a fixed H2Opt concentration and varying the concentration of DD-PDZOpt or alternatively using a fixed concentration of DD-PDZOpt and varying the concentration of the H2Opt peptide. Based on the assay with varying substrate (H2Opt) concentration, we determined a kcat/Km of ~85 M−1 s−1 in standard assay buffer devoid of additional metal ions (Fig. 3d, Supplementary Fig. 13, and Supplementary Table 3). It should be noted that we used here a sub-saturating concentration of DD-PDZOpt (50 μM) that is not sufficient to fully activate HtrA2, for the purpose of analyzing the activation of HtrA2 in the presence of low concentrations of activating peptide. The addition of 2 mM CaCl2 to the assay resulted in a doubling of the vmax value and a decrease of the KM value of about 50% compared to the HtrA2 activity in our standard assay buffer (no metals supplemented), indicating that the enzyme affinity to the substrate is notably enhanced in the presence of Ca2+ and as a result, Ca2+ binding increased the kcat/kM value about fourfold (Fig. 3e). Addition of Mg2+ lowered the KM value of HtrA2 to about 25% of that in standard assay buffer, but the vmax was markedly decreased, resulting in a kcat/kM increase of almost twofold (Fig. 3e). Performing the experiment with 2 mM ZnSO4 or 2 mM CuCl2 supplemented showed, as expected from the β-casein assay, no detectable protease activity (Fig. 3e and Supplementary Fig. 13).
Having established the effect of calcium ions as an uncompetitive activator, as it increases the velocity of the reaction and at the same time decreases the KM, we wanted to clarify if it affects the activation and/or the proteolysis itself. To assess this question, we next used a fixed concentration of substrate (H2Opt) and varying concentrations of activating peptide DD-PDZOpt to extract kinetic parameters under variable concentrations of the activating peptide. We extracted the apparent KD,app and vmax from the experimental data observing that Ca2+ also in this instance lowered the KD,app but that the vmax remained at about the same rate as in standard assay buffer, and kcat/KM of Ca2+-bound HtrA2 increased ~13-fold compared to HtrA2 in standard assay buffer (Supplementary Fig. 14 and Supplementary Table 4). Mg2+-bound HtrA2 had a decrease in KD,app to about 50% of HtrA2 in standard assay buffer but a lowered vmax and as such a kcat/KM of about 50% compared to HtrA2 in standard buffer. The addition of CuCl2 would inhibit HtrA2 but the inhibition could be relieved by concentrations of DD-PDZOpt above ~100 μM (Supplementary Fig. 14d), while Zn2+ addition completely abolished HtrA2 activity at all concentrations of activating peptide tested. It has previously been suggested for other HtrA proteins that Zn2+ may increase oligomer stabilization and Zn2+ ions have been found to bind to the hexameric form of Synechocystis HtrA homolog A39,50. In light of the SEC-MALS results indicating that the isolated PDZ domain is prone to dimerize in the presence of Zn2+ but not in the presence of any other of the tested ions as well as the binding preference of activating peptides to trimeric rather than hexameric HtrA2, this provides a possible explanation for our observations in the proteolytic assays: HtrA2 potentially forms hexamers via the PDZ domain in the presence of Zn2+, preventing efficient binding of the activating peptide and thus keeping HtrA2 in an inactive state. Our experimental data with Cu2+ showed that the inhibition caused by Cu2+ could be relieved by increasing concentrations of DD-PDZOpt (Supplementary Fig. 14), pointing to competitive inhibition by Cu2+ with the activating peptide, in line with our SEC-MALS data showing no indication that the copper ions influence the trimer-to-hexamer equilibrium of HtrA2.
Elucidating the allosteric regulation of HtrA2S306A using methyl-TROSY
For expanding our characterizations also to the full-length protein, we used the catalytically inactive trimeric HtrA2S306A variant to suppress any eventual self-cleavage at the high protein concentrations needed for NMR studies25. By using previously reported assignments for ILVMproR (referring to methyl group labeling of Ile-δ1, Leu-δ1 (proR), Val-γ1 (proR), and Met-ε) HtrA2S306A (see refs. 25,30) as well as our PDZ side-chain assignment, we were able to complete the sequence-specific assignment to ~92% of the methyl groups of a MALVIproS (referring to methyl group labeling of Met-ε, Ala-β, Leu-δ2 (proS), Val-γ2 (proS), and Ile-δ1) labeled HtrA2S306A (Supplementary Fig. 15). We then used this sample to monitor the binding between full-length mature trimeric HtrA2S306A (Supplementary Fig. 1b) and DD-PDZOpt to eventually elucidate the distinct steps underlying its allosteric activation. Also here, we observed significant chemical shift changes as well as line-broadening of residues located in the β14 and β15 strands of the PDZ domain where the peptide binding cleft is located, as well as in the β11 and β12 strands of the proteolytic domain (Fig. 4a–c and Supplementary Fig. 16a). These regions are in close contact in the closed HtrA2 conformation, where the proteolytic domain stacks against the PDZ domain, restricting access to the catalytic site16,25. We observed significant chemical shift changes within the regulatory loops, pointing to an activation cascade of L2-L3-LD-L1 leading to a correct arrangement of the catalytic triad upon binding of the activating peptide to the PDZ domain (Fig. 5a and Supplementary Fig. 16b, c)27. In addition to the CSPs, line-broadening of residues in helix α1 of the protease domain was observed in a similar manner as for the PDZ-domain protease domain interface, which is in general indicative for either conformational heterogeneity and/or micro- to millisecond dynamics. As previous studies confirmed that the effects in the case of the interdomain interface reflect on the detachment of the PDZ domain25, the observation here for helix α1 could suggest that also this helix experiences dynamical changes upon substrate binding, pointing to a similar detachment. The effect on α1 is likely triggered by rearrangements in the LD loop and strand β7 harboring the central M260, stacking closely to helix α1.

a 2D [13C, 1H]-NMR spectra showing titration of DD-PDZOpt toward an 80 μM sample of MALVIproS-labeled HtrA2S306A. Arrows indicate large CSPs. b Upper panel shows residues belonging to the protease domain (blue) or the PDZ domain (green) showing significant CSPs (>1 SD or 2 SD indicated by the broken lines) and line-broadening indicative of PDZ–protease domain separation. Lower panel shows the impact of the titration of DD-PDZOpt on the different regulatory loops of HtrA2 modulating its proteolytic function. Asterisks mark unassigned residues. c Significant CSPs and broadened residues following DD-PDZOpt titration highlighted onto the HtrA2 monomer. Color scheme used for annotating the methyl groups is analogous to (b). d Rotameric states of isoleucine and methionine residues as determined by the methyl 13C shifts in the closed inactive state of HtrA2 (PDB-ID: 5M3N). The catalytic triad residues are shown in gold. e Schematic view of the trans and gauche (−) rotameric states of the isoleucine side-chain. f Changes in rotamer states between the holo and apo form of HtrA2S306A. Residues belonging to the protease domain (blue), residues belonging to the PDZ domain (green) are indicated. g Rotameric changes as seen in (f) plotted onto the structure of HtrA2 (PDB-ID:5M3N). Red spheres show residues in a more gauche (−) conformation while blue spheres show residues in a more trans conformation. The catalytic triad residues are shown in gold. Source data are provided as a Source Data File.

a Flareplot representation visualizing the changes, based on the altered methyl NOE interaction patterns upon DD-PDZOpt interaction (see individual representations of the apo and holo-state in Supplementary Fig. 16). Purple lines represent proposed allosteric coupling of the activation loops, whereas the broken lines represent the putative coupled detachment of helix α1. Yellow lines represent altered NOE contacts only observed in the presence of the activating peptide in line with local structural rearrangements required for the allosteric coupling. b Model of the allosteric modulation of the HtrA2 monomer upon DD-PDZOpt binding involving distinct regulatory steps, leading to the activation of the proteolytic function (for details see main text).
The local structural arrangement of amino acids side-chains can be crucial in substrate binding events with side-chain rotamer changes commonly occurring during substrate binding leading to rearrangements and possible allosteric regulation51. We therefore compared the methyl side-chain rotamer states, extracted from the isoleucine and methionine 13C chemical shifts, of the apo form of HtrA2S306A and its holo form bound to the activating peptide DD-PDZOpt (Fig. 4d–g). Particularly at the domain interface several methyl groups are in an exchange between trans and gauche (−) rotameric states in the closed apo HtrA2 state (Fig. 4d, e). Upon activation, we observed several residues whose rotameric states changed more towards the gauche (−) rotamer at the interdomain interface, e.g., I362, M366, I373, and I397 (Fig. 4f, g). Furthermore, two isoleucine residues in the proximity of the active site, I164 and I166, had an ~20% increased gauche (−) population, pointing to local subtle structural adaptations underlying HtrA2 activation (Fig. 4f, g).
To obtain a more detailed picture of the functional consequences of DD-PDZOpt binding to the PDZ domain, we next characterized the methyl–methyl NOEs within the MALVIproS-HtrA2S306A in the absence of the activating peptide (Supplementary Fig. 16b). The identified NOEs were consistent with the previously reported crystal structures of HtrA2 [16,27] and revealed a network of contacts stabilizing the regulatory loops within the protease domain, whereas especially the L2 loop was stabilized via characteristic interdomain NOEs to β14 as reported before25 (Supplementary Fig. 1e). In addition, we could identify additional NOEs between helix α5 and the L2 loop as well as between β15 and the LA loop (Supplementary Fig. 16b, d). We then analyzed the NOE patterns in the peptide-bound state of MALVIproS-HtrA2S306A. As expected from the CSPs we also see altered NOE patterns, particularly at the amino-terminus as well as in the vicinity of the regulatory loops (Supplementary Fig. 16c). Looking particularly at I150 we can observe that this residue is involved in an intricate NOE network within the apo state, which is largely diminished in the peptide-bound state (Supplementary Fig. 16d), which together with the previously discussed line-broadening of several residues involved in a stabilizing NOE network of helix α1 point to structural changes in this region upon activation. In line with the earlier observation of positive cooperativity upon activator peptide binding30, we also observed changes in the interprotomer NOEs, particularly around the crucial I270, I274, and I150 residues, suggesting local rearrangements at the monomer interfaces (Supplementary Fig. 16d). Other regions of changes in NOE patterns were in the vicinity of the regulatory loops suggesting together with the observed subtle adaptations of the side-chain rotamers a switch from the inactive to active conformation around the catalytic site (Supplementary Fig. 16c). Importantly, we also could detect highly similar NOE patterns for several methyl groups experiencing only minute changes, as highlighted for a local cluster around I333 (Supplementary Fig. 16c, e). This observation rules out that we were detecting a reduced number of NOE cross peaks simply due to sensitivity issues.
In summary, based on the observed methyl CSPs throughout the HtrA2 protein in combination with the altered NOE networks (Fig. 5a), we were able to identify distinctive steps underlying HtrA2 activation (Fig. 5b). Binding of the activator peptide leads to a transition to the HtrA2 open state by detaching the PDZ domain from the protease domain. Particularly, the release of the loop L2 from this domain enables the triggering of the rearrangement cascade L2-L3-LD-L1. This allosteric signaling in the final step leads to perturbations of the amino-terminal α1 helix, and whilst the observed data might indicate a detachment of the α1 helix, based on the observed broadening of the methyl resonances and weakened NOE contacts for I150 no definite conclusions can be drawn, yet.
To also obtain insight into the dynamical adaptations and to probe the relative domain motions, we used the S2axis•τC of the MALVIproS-HtrA2S306A apo state and the peptide-bound MALVIproS-HtrA2S306A. In the apo state we obtained values of 38 ± 19 ns and 37 ± 24 ns for the protease and PDZ domain (Supplementary Fig. 17a, c), respectively, in good agreement with a previous study25 showing that the domains are tightly coupled in the apo state. Analyzing HtrA2S306A in the presence of the DD-PDZOpt peptide yielded decreased values of 29 ± 20 ns and 24 ± 21 ns for the protease and PDZ domain, respectively (Supplementary Fig. 17b, d). It must be noted that due to line-broadening and signal overlap fewer resonances could be analyzed compared to the apo state thus the difference is slightly less pronounced if the same residues are compared: 34 ± 13 ns and 35 ± 19 ns for protease domain and PDZ in the apo state in comparison to 29 ± 20 ns and 24 ± 21 ns in the peptide-bound state (Supplementary Fig. 17d). Nevertheless, the obtained values are in line with the decoupling of the PDZ domain as evidenced by the on average lower values for this domain. Although these values indicate an enhanced flexibility of this domain, values around 6.5 ns, as obtained for the isolated PDZ domain (Fig. 2d, e), would be expected for a completely decoupled movement, pointing to a still restricted mobility after detachment of the PDZ.
Next, we quantified the micro- to millisecond dynamics of MALVIproS-HtrA2S306A by measuring MQ CPMG relaxation rates at 25 and 750 Hz to assess the differences in dynamics of the apo state and the peptide-bound state (Supplementary Fig. 17e–g). On average, we observe ΔR2eff values of 8.5 s−1, with helix α1, parts of the regulatory loops in particular the LA loop, and helix α5 showing the largest extent of conformational exchange (Supplementary Fig. 17e). This observation suggests that parts involved in the activation cascade could experience micro- to millisecond dynamics in the apo state in line with our initial hypothesis of an important trigger function of helix α5 within the PDZ as well as the suggested role of helix α1 in the activation cascade (Fig. 5b).
Looking at the MQ rates at 750 Hz, we obtained an average value of 31.8 ± 14.6 s−1 for the apo state in line with previous results25, whereas using only a subset of values for the apo state and the peptide-bound state for direct comparison yielded values of 27 ± 14.7 s−1 and 16.4 ± 7.8 s−1, respectively. As under these high CPMG pulsing values (750 Hz) only minimal contributions from the exchange between different states with lifetimes larger than ~1.3 ms can be expected, this marked difference for the peptide-bound state indicate a decrease on the pico- to nanosecond timescale dynamics in good agreement with the same trend of the S2axis•τC values.
We reasoned that structural frustration could be the reason for the observation of conformational exchange contributions within the apo state of HtrA2 as it has lately been suggested to be a potential underlying cause for local microsecond dynamics26,52. The analysis of the local structural frustration53 highlighted several hot spots of highly frustrated regions such as the helices α1 and α5 besides the LA loop. These regions matched well with the identified segments experiencing micro- to millisecond conformational exchange in the apo state as detected by the multi-quantum CPMG experiment (Supplementary Fig. 17h–j).
Divalent cations are not sufficient for HtrA2 activation
Having established the potential activation cascade of HtrA2 upon DD-PDZOpt binding, we wondered if the interaction with divalent cations would already be sufficient to trigger the first steps of this activation, namely the domain separation. Titrating the different divalent ions towards MALVIproS-HtrA2S306A led to localized signal attenuations for all four tested ions but no significant CSPs (Supplementary Fig. 18a, b). Whereas in the case of CaCl2 and MgSO4 no clear indication of the domain detachment was observable, the global signal reduction upon ZnSO4 addition pointed to the formation of a larger HtrA2 species. Addressing this possible oligomerization by SEC-MALS revealed a multimerization KD ranging from 0.5 to 1 mM for the absence and presence of CaCl2, respectively, in agreement with previous observations25 (Supplementary Fig. 18c–f and Supplementary Table 5). The KD could not be determined in the presence of ZnSO4, but the data for the isolated PDZ domain as well as the observed strong signal attenuation in the titration of MALVIproS-HtrA2S306A suggest the formation of a multimeric species or even aggregates under these conditions.
To map the binding site(s) of calcium more precisely within full-length mature HtrA2S306A, we used gadolinium (Gd2+) which can substitute calcium in Ca2+ binding sites and whose strong paramagnetic relaxation enhancement (PRE) effect enables mapping of Ca2+ binding sites54,55. We observed extensive line-broadening of residues 412–430 (comprising the β16 and α7 regions) and residues 329–369 (β14) (Supplementary Fig. 19a, b). Using MIB2, a web-based tool for the prediction of metal binding sites in proteins, on the wild-type HtrA2 structure, we identified potential calcium-binding sites56. These sites include as a top hit a region in the α7 helix and the preceding loop region of the PDZ domain comprising residues Q419, Q422, N423, E425, and D426 which correlates well with our gadolinium titration data (Supplementary Fig. 19c). Employing the same in silico approach for identifying theoretical copper binding sites, revealed two potential binding sites partially overlapping with the activator peptide binding groove, in line with our experimental observation that Cu2+-ions can be displaced by increasing the activator peptide concentration (Supplementary Fig. 19d, e).
Thus, Ca2+ promotes stronger binding of activating peptide/substrate to the PDZ domain, leading to enhanced activation of HtrA2 once in the presence of substrate. This hypothesis is based on (i) our titration data of Ca2+ towards full-length HtrA2 did not reveal any of the chemical shift perturbations associated with HtrA2 activation as seen upon the addition of the activating peptide DD-PDZOpt and (ii) our kinetic data from the fluorescence assays suggest that addition of Ca2+ leads to an increased affinity to the substrate as evidenced by a reduced KD value.
PRE-measurements suggest enhanced mobility of helix α1 upon activation
To further clarify the potential detachment of helix α1 and the contribution of Ca2+ to the HtrA2 proteolytic activity, we constructed a single cysteine mutant, HtrA2S306A, S145C, enabling the measurement of PREs by side-specific attachment of a paramagnetic spin label (MTSL). We then compared the PRE effects on HtrA2 in sample buffer (no metals), in buffer containing CaCl2 or after the addition of the activating DD-PDZOpt peptide (Fig. 6a–c). We initially used the apo form of HtrA2 to map the basal mobility of helix α1. We observed mainly localized signal attenuations for a subset of resonances, which is consistent with an attached helix α1 as the measured S2axis•τc values of methyl-bearing residues 150–160 are en par with the average values observed for the complete protein (Supplementary Fig. 20). Upon closer inspection we mainly observed a PRE-dependent line-broadening for I150 and other residues in the direct vicinity of the MTSL-labeling site besides residues at the outside of the interdomain interface such as I274 and I295 and solvent exposed methyl groups of helix α5 in the PDZ domain (Fig. 6d). These localized effects point to the basal degree of flexibility likely due to movements of the unstructured part of helix α1 preceding I150 thus confirming the closed conformation of HtrA2. Upon addition of Ca2+ ions we could observe a more pronounced PRE-induced line-broadening compared to the initial experiment with apo HtrA2 (Fig. 6b, e and Supplementary Fig. 20). We reason that this observation is due to a slight destabilization of the domain interface leading to partial opening as evidenced by additional PRE-dependent line-broadening for helix α5 and the L2 loop, as several residues partially buried at the interdomain interface show PRE effects, e.g., I333 and I373 (Fig. 6e). These more pronounced PRE effects thus clarify the molecular basis of the observed effect of calcium-binding to the PDZ domain: shifting HtrA2 toward a pre-open state, enabling easier access for the activating peptide to its binding site buried at the PDZ:protease domain interface resulting in the observed reduction in dissociation constant (Fig. 3f).

a–c 2D [13C,1H]-NMR of the Ile-region of MTSL-spin-labeled MALVIproS-HtrA2S306A, S145C in the apo state (blue, a), in the presence of 25 mM CaCl2 (dark blue, b), and in the presence of 520 µM DD-PDZOpt (yellow, c) in the oxidized state and after addition of 5 mM ascorbic acid in the spin label reduced state (magenta). d–f PRE effect of a spin label on MALVIproS-HtrA2S306A, S145C. The PRE intensities detected for the different HtrA2 states (d–f) are indicated by the color gradient on the methyl groups depicted as spheres (PDB-ID: 5M3N). Insets illustrate the degree of mobility of the helix α1 under the different conditions.
The addition of the activating peptide to HtrA2 leads to notably larger PRE effects. We observed in this state PRE-dependent line-broadening also in extended patches of the PDZ domain, in the activator peptide binding cleft (β13–β14), as well as in the protease domain, mainly located to β4–β5 region comprising the LA loop, in close vicinity to the catalytic triad and in the now opened interdomain interface. Thus, the PRE data support the conclusions drawn from our metal titration experiments (Fig. 6c and Supplementary Fig. 20b), which indicate that calcium-binding alone is not sufficient for HtrA2 transition into an active state. Furthermore, our PRE data clearly shows that helix α1 is affected when HtrA2 transitions into an active state upon binding an activating peptide (Fig. 6c, f and Supplementary Fig. 20). Our PRE data shows that the PDZ domain is notably affected when a saturating amount of activating peptide is titrated to HtrA2, evidenced by the large signal attenuations. This suggests that the PDZ domain in the activated state of HtrA2 is spatially closer to the paramagnetic spin label than in the closed conformation. Since the peptide binding cleft, which is closely stacked to the protease domain in the closed conformation, shows among the largest signal attenuations changes (Supplementary Fig. 20c), it suggests that the PDZ domain could shift its position relative to the HtrA2 protease domain converting the protein from a closed inactive to an open conformation. Although our data suggests an increased degree of mobility for helix α1, also the conformational flexibility of the undocked PDZ likely contributes to the observed enhanced PRE effects. This is in line with a splayed outwards conformation of the PDZ towards the amino-terminus that was recently demonstrated for HtrA2 when bound to its natural substrate Apollon and has also been observed previously for a bacterial HtrA protein57,58,59. Although the observed effects are perfectly in line with a proposed model of activation upon detachment of the PDZ from the protease domain, based on recent observations by the Kay group of a more pronounced CSP of the characteristic I150 methyl resonance upon substrate binding to the active site60, the open state characterized here most likely represents a still catalytically inactive state and only upon substrate binding to the catalytic site the open fully active state can be populated.
Discussion
It has previously been demonstrated that the proteolytic activity of HtrA2 is modulated both by binding of activating peptides to its regulatory PDZ domain, as well as by its propensity to form inhibitory hexamers25,44,61. Here, we show that HtrA2 activity can be additionally modulated by divalent cations via binding to the PDZ domain in the proximity of helices α5 and α7, where metals such as zinc have inhibitory effects in contrast to magnesium and especially calcium leading to an enhanced HtrA2 activity (Fig. 3 and Supplementary Figs. 13 and 14). Our data suggest that binding of metals to the PDZ domain also modulates the dynamics of helix α5 of the PDZ domain, an important structural element docking the PDZ on top of the protease domain in the non-activated state and potentially acting as a trigger for the domain separation. Calcium ions support the activation of HtrA2 without being themselves sufficient for this important initial step of the activation cascade, as the addition of CaCl2 alone is not resulting in the characteristic CSPs associated with activation of HtrA2 seen in the presence of an activating peptide and the PREs indicate only a partial opening of the binding cleft (Figs. 4a–c and 6 and Supplementary Figs. 18 and 20). Another important feature in this still partially closed complex is the stabilization facilitated by the β14 and β15 in the PDZ domain harboring important methyl-bearing residues locking the closed conformation in solution as identified by us and the Kay lab25. Among these residues particularly I393 appears to be a crucial contact point as on the one hand we observe a large change in its rotameric state upon domain separation and on the other hand as mutations in the directly adjacent helix α6, such as G399S or R405W, impair HtrA2 activation completely supposedly freezing HtrA2 in the inactive state22,62.
The identified basis of the activation of HtrA2 as a multistep event starts with substrate recognition via the PDZ domain, triggering the separation of the PDZ domain and the protease domain, subsequently leading to a rearrangement of the regulatory loops within the protease domain to allow access to the active site (Fig. 5b). A previously not characterized role could be identified for the amino-terminal helix α1 by a local structural rearrangement leading to a possible detachment as evidenced by the extended PRE effects observed for the activator bound HtrA2 (Fig. 6). Even though the exact functional consequences of this feature remain elusive, several previous observations underline the importance of this amino-terminal element: (i) at the far-amino-terminus is an IAP-interacting motif through which supposedly HtrA2 function can be over-activated upon IAP-binding43,44, (ii) the amino-terminus together with helix α1 are involved in inter-subunit communication and therefore important for the positive cooperativity of HtrA216,30, (iii) some Parkinson’s disease associated mutations as well as a putative phosphorylation site reside within this helix63,64.
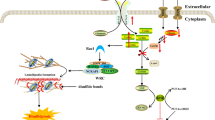
The influence of divalent metal ions on the activity of HtrA2 raises the question of which biological mechanisms these metals might be involved in. In the case of calcium, it is established that calcium levels are rapidly increasing during apoptosis and that calcium can accumulate both in the cytosol and inside the mitochondria, either as controlled calcium signaling or due to pathological conditions of the cell65,66. Therefore, an involvement of Ca2+ in the HtrA2-mediated progression of apoptosis by enhancing the proteolytic activity of HtrA2 against inhibitor-of-apoptosis proteins, such as the well-known HtrA2 target XIAP, seems highly likely13,67. Similarly, the inhibitory effect of zinc on HtrA2 activity might also influence the role of HtrA2 in apoptosis. Zinc is well-known for its ability to suppress apoptosis, and it has been observed in breast and pancreatic cancers that malignant cells accumulate zinc68,69. Interestingly, zinc homeostasis is also known to be affected in prostate cancers where both too high and too low levels of zinc may increase the risk of developing prostate cancer. Surprisingly, inhibition of HtrA2 has also been shown to induce apoptosis in prostate cancer cell lines in a Zn2+-dependent manner together with zinc chelators and a putative drug candidate against prostate cancer, but the exact mechanism remains unclear70. Based on our findings in this study, we thus propose a refined mechanism of how HtrA2 activity is modulated by Ca2+ and Zn2+ ions (Fig. 7).

In its membrane-attached form, HtrA2 performs protein quality control functions within the mitochondrial intermembrane space (IMS). Upon apoptosis-inducing signals HtrA2 matures, leading to the cleaving of its amino-terminus and secretion from the mitochondrion into the cytoplasm. In the presence of elevated Zn2+ levels, the equilibrium of HtrA2 is shifted towards the hexameric inactive state, impairing apoptotic signaling. Under normal apoptotic conditions, the release of HtrA2 from the mitochondrion is coupled to an increase of the cytosolic Ca2+concentration, which further activates the proteolytic function of HtrA2, leading to the cleavage of IAPs and the unleashing of the proapoptotic caspase function, triggering apoptosis. The following crystal structures were used: full-length HtrA2 (PDB-ID: 5M3N), caspase-9:XIAP complex (PDB-ID: 1NW9) as well as the caspase-9 dimer (PDB-ID: 1JXQ).
Methods
Protein preparation
A construct for full-length mature human HtrA2133–458 without the active site serine (S306A) (pET21b-HtrA2) was purchased from GenScript optimized for Escherichia coli codon usage. All other protein constructs were made employing restriction-free cloning71 (Supplementary Tables 6 and 7). Constructs were expressed in the E. coli BL21 (λDE3) strain as amino-terminally SUMO-His6-tagged proteins in a pET28b vector or carboxy-terminally His6-tagged proteins in a pET21b vector (Supplementary Table 6). Cells were grown at 310 K in 1 L medium containing either 50 µg/ml kanamycin (pET28b) or 100 µg/ml ampicillin (pET21b) until an OD600 0.6–0.8 was reached, induced with 0.4 mM IPTG and the protein was expressed at 298 K for an additional 16–18 h. Cells were harvested by centrifugation at 277 K, 4500×g, 20 min. Cells were subsequently resuspended in 25 ml of purification buffer, 50 mM HEPES pH 7.3, 0.5 M NaCl for HtrA2-PDZ or 50 mM HEPES pH 8, 0.5 M NaCl for full-length HtrA2S306A, wild-type HtrA2, and HtrA2S306A, S145C, supplemented with 5 mM imidazole. Prior to cell lysis, ½ cOmplete EDTA-free protease inhibitor (Roche), 100 units of DNase (ArcticZymes) and MgCl2 to a final concentration of 5 mM were added. Protease inhibitor was omitted when purifying wild-type HtrA2. Cells were lysed by three passes through an Emulsiflex (Avestin), and cell debris was removed by centrifugation at 20,000×g, 277 K, for 45 min. Proteins were purified with a Ni2+-HisTrap HP (GE Healthcare) column using purification buffer and eluted with 500 mM imidazole. The protein fractions were isolated and dialyzed against purification buffer together with human SENP1 (Addgene #16356) for removal of the SUMO-His6-tag at 277 K overnight72. Cleaved SUMO-tag and residual SENP1 were separated from the proteins by running the solution over a Ni2+-HisTrap HP column. Collected flow-through and wash fractions of cleaved protein were concentrated using Vivaspin centrifugal concentrations (MWCO: 5000, 10,000, or 50,000; Sartorius) before being applied to gel filtration columns (Superdex 75 increase or Superdex 200 increase, GE Healthcare) equilibrated with PBS pH 7.4 (sample buffer for HtrA2-PDZ), 25 mM Tris-HCl pH 8, 0.1 M NaCl, 1 mM TCEP (sample buffer for full-length HtrA2S306A and wild-type HtrA2) or 25 mM Tris-HCl pH 8, 0.1 M NaCl, 10 mM DTT for HtrA2S145C.
Isotope labeling
Isotope labeling of proteins was achieved by expressing proteins in M9 minimal media with the relevant isotopes supplemented to the media (H2O supplemented with (15NH4)Cl and D-(U-13C)-glucose for uniform double labeling [U-13C,15N], or D2O supplemented with (15NH4)Cl yielding [U-2H,15N]-labeled proteins and with (15NH4)Cl and D-(U-2H,13C)-glucose yielding [U-2H,13C,15N]-labeled proteins73. For specific methyl group labeling of ALVIT-PDZ, D2O-based M9 media supplemented with (15NH4)Cl, D-(U-2H,12C)-glucose, 2% of Bioexpress rich medium, 50 mg/L 2-ketobutyric acid-4-13C,3,3-d2 sodium salt hydrate (isoleucine), 85 mg/L 2-keto-3-(methyl-d3)-butyric acid-4-13C4, 3-d sodium salt (valine/leucine), 50 mg/L [U-2H, γ2-13CH3] threonine and 100 mg/L d5-glycine (threonine), and 50 mg/L 2-[2H], 3-[13C] L-alanine (alanine) were added 1 h prior to induction74,75,76. For MALVIproS–labeled HtrA2S306A, we supplemented the medium with (15NH4)Cl, D-(12C,2H)-glucose, 2% (v/v) Bioexpress cell growth media (98% [U-2H,15N]), 50 mg/L 2-ketobutyric acid-4-13C,3,3-d2 sodium salt hydrate (isoleucine), 50 mg/L [U-2H,13CH3] methionine, 50 mg/L 2-[2H], 3-[13C] L-alanine, and 4 vials/L DLAM-LVproS-kit (2-(13C)-methyl-4-(D3)-acetolactate (valine/leucine proS methyl group only) added 1 h prior to induction77. For specific LV-HtrA2S306A, the medium was supplemented with (15NH4)Cl, D-(13C,2H)-glucose 2-keto-3-(methyl-d3)-butyric acid-1,2,3,4-13C4, 3-2H added 1 h prior to induction. For V-HtrA2S306A, the medium was supplemented with (15NH4)Cl, D-(13C,2H)-glucose, 85 mg/L 2-keto-3-(methyl-d3)-butyric acid-4-13C4, 3-d sodium salt and 40 mg/L of L-leucine-d10 added 1 h prior induction78.
Bioexpress, threonine, alanine, and glycine were purchased from Cambridge Isotope Laboratories. The DLAM-LVproS-kit was purchased from NMR-Bio and all other isotopes from Sigma Aldrich/Merck.
NMR spectroscopy
NMR experiments were performed at 298 K if not stated otherwise on Bruker Avance III HD 700-, 800-, and 900-MHz spectrometers running Topspin 3.6 (Bruker Biospin), equipped with cryogenically cooled triple-resonance probes. Sequence-specific assignments of the backbone resonances were made using 2D [15N,1H]-TROSY-HSQC, 3D HNCO, 3D HNCA and 3D HNCACB as well as 3D CBCACONH experiments for [U-15N,13C] HtrA2-PDZ79,80. Aliphatic side-chain resonance assignments for HtrA2-PDZ were achieved by 3D (H)C(CCO)NH, 3D H(CCCO)NH, and based on 2D [13C, 1H]-HSQC spectra with/without constant time (CT) version by 3D H(C)CH-TOCSY and 3D (H)CCH-TOCSY experiments on [U-15N,13C] HtrA2-PDZ80. In addition, the following NOESY-type experiments with the indicated mixing times were performed: 3D 1Hall-1Hamide-15Namide SOFAST NOESY with 200-ms mixing time on a [U-15N,13C] HtrA2-PDZ sample81 as well as a 3D 13Cmethyl-13Cmethyl-1Hmethyl HMQC NOESY and a 1Hmethyl-13Cmethyl-1Hmethyl NOESY with 600 and 120 ms mixing times on an ALVIT-PDZ sample82,83, respectively. NMR data were processed using the mddNMR2.6 and NMRPipe software packages84,85. Resonance assignments and spectral analyses were done in CARA86. Secondary chemical shifts were calculated relative to the random coil values by the POTENCI algorithm87. Further, a weighting function with weights 1-2-1 for residues (i-1)-i-(i + 1) was applied to the raw data38,88. The chemical shift changes of the amide moiety or the methyl groups were calculated as follows:
For the quantitative analysis of signal intensities, the amplitudes were corrected by differences in the 1H-90° pulse length, the number of scans, and the dilution factor89.
Methyl group assignments
Based on the determined sequence-specific assignment of the methyl groups of the HtrA2-PDZ reported here together with the recently reported ILVMproR-methyl assignment for HtrA2S306A[25], we transferred the assignments and subsequently confirmed as well as completed the MALVIproS-methyl groups of HtrA2S306A by an experimental approach using V-HtrA2S306A and LV-HtrA2S306A samples to identify leucine and valine residues unambiguously, an HMBC-HMQC experiment of the LV-HtrA2S306A to connect leucine/valine geminal pairs, and a 3D 13CMethyl−13CMethyl1HMethyl SOFAST NOESY experiment of the MALVIproS-HtrA2S306A with mixing times of 50 ms and 300 ms, respectively81,90. This approach yielded the following large degree of assignment (~92%): Alaβ (21/30), Ileδ1 (22/22), Leuδ2 (35/37), Metε (5/5), and Valγ2 (37/37). Methyl NOE interaction patterns for ALVIT-PDZ and MALVIproS-HtrA2S306A, respectively, were subsequently generated and visualized using Flareplot (https://gpcrviz.github.io/flareplot/).
Analysis of the rotameric equilibria of the methyl groups
We used the obtained Ile (δ1) and Met (ε) 13C chemical shifts to deduce the rotameric equilibria of the Ile χ2, and Met χ3 angles, employing a previously outlined approach91,92. As the 13C chemical shifts of methyl groups are reportedly directly dependent on the side-chain rotamer, this approach yields insight into the rotameric state of the different methyl groups93,94,95,96. The population of the trans rotameric state (ptrans) for each residue was calculated according to the chemical shift values of the methyl 13C signals (δobs: ppm)93,94 using Eqs. (3–4):
The equations yielded ptrans values ranging from 1 (all trans) to 0 (all gauche (−), respectively. In the analysis residues with values ranging between 0.75 and 1 were considered to be in the trans conformation and 0–0.25 as to be in the gauche (−) conformation.
NMR backbone dynamics
For the analysis of the dynamic properties of HtrA2-PDZ, the following relaxation experiments were measured: 15N{1H}-NOE, R1(15N), R1ρ(15N) and TROSY for rotational correlation times (TRACT) for determining R2α(15N) and R2β(15N)97,98. Steady-state heteronuclear 2D 15N{1H}-NOE experiments were recorded using a 5 s 1H-saturation time for the NOE experiment and the equivalent recovery time for the reference experiment, each preceded by an additional 2.0 s recovery time. R1 measurements were performed with delays of 400, 600, 800, 1200, 1600, 1800, and 2400 ms. R1ρ data measurements were performed by recording delays of 0, 15, 25, 35, 45, 55, and 65 ms. The TROSY (R2β) and were determined by recording relaxation delay points such as 4, 10, 20, 30, 40 ms as well as 0.4, 2, 4, 8,16 ms, respectively. Nonlinear least square fits of relaxation data were done with PINT99 using an in-house analysis pipeline, as recently described100. R2(R1ρ) (15N) values were derived from R1ρ using Eq. (5):
with \({{{{{\rm{\theta }}}}}}=\,{\tan }^{-1}\left({{{{{\rm{\omega }}}}}}/\Omega \right)\), where ω is the spin-lock field strength (2 kHz) and Ω is the offset from the 15N carrier frequency88,101.
Error bars for R1(15N), R1ρ (15N), R2α(15N), and R2β(15N) were calculated by a Monte Carlo simulation embedded within PINT99, and for R2(R1ρ) (15N) by error propagation. Error bars for the 15N{1H}-NOE were calculated from the spectral noise. Analysis of the obtained relaxation rates was performed with Tensor2 on the NMRbox web server102,103. Residues with an R2(R1ρ)/R1 rate quotient above 30 were removed from the analysis with Tensor2 and a prolate axially symmetric diffusion tensor was used to fit the relaxation data to the HtrA2-PDZ structure (PDB-ID: 1LCY). To quantify the observed exchange contributions additionally, a BEST TROSY 15N CPMG experiment using constant time relaxation periods (T) of 40 ms employing CPMG frequencies ranging from 25 to 1500 Hz were used at magnetic field strengths of 16.4 T (700 MHz proton frequency) and 21.1 T (900 MHz proton frequency)36. R2,eff, the effective transverse relaxation rate was calculated according to the following equation:
where I (or I0) are the intensities with and without the presence of a constant time relaxation interval of duration T, during which a variable number of 15N 180° pulses are applied leading to νCPMG = 1/(2δ), where δ is the time between successive pulses. Data were processed in nmrPipe66. Dispersion data were obtained using an in-house extension of PINT 2.199, where the respective intensities for each data point at both fields were extracted separately, and subsequently the effective transverse relaxation rates were fitted globally to a two-site exchange model.
NMR side-chain dynamics
Experiments were performed on a [U-2H, Ile-δ1−13CH3, Leu,Val-13CH3, Ala-13CH3, Thr-γ2-13CH3]]–PDZ (ALVIT-PDZ) sample, with stochastic labeling one methyl isotopomer of the valine and leucine groups, at a temperature of 298 K in 99.9% D2O based NMR buffer104. Side-chain methyl order parameters (S2axis*τC) were determined by cross-correlated relaxation experiments105,106. Single- (SQ) and triple-quantum (TQ) 1H–13C experiments were collected at a series of delay times at a concentration of 500 µM. Ratios of the peak intensities were determined in PINT99 and subsequently fitted by in-house written routines for thirteen values ranging between 2 and 45 ms using the following equation where T is the relaxation delay time and δ a factor to account for coupling due to relaxation with external protons:
S2axis*τC values were determined using Eq. (5) from the fitted \({{{{{\rm{\eta }}}}}}\) values and the separately determined rotational correlation times adjusted for the change in viscosity in 100% D2O (τc)107, for the two different states at 298 K:
where µ0 is the vacuum permittivity constant, γH the gyromagnetic ratio of the proton spin, rHH is the distance between pairs of methyl protons (1.813 Å), S2axis is the generalized order parameter describing the amplitude of motion of the methyl threefold axis, Θaxis,HH is the angle between the methyl symmetry axis and a vector between a pair of methyl protons (90°), and P2(x) = ½ (3x2-1). Finally, the product of the methyl order parameter and the overall correlation time constant, S2axis•τC, was determined. Multiple quantum (MQ) methyl relaxation dispersion experiments were recorded as a series of 2D datasets using constant time relaxation periods (T) of 40 ms (16.4 T (700 MHz)/18.8 T (800 MHz)) and CPMG (Carr-Purcell-Meiboom-Gill) frequencies ranging from 25 to 750 Hz37. Data were processed in nmrPipe66, and peak intensities were extracted with PINT99. Dispersion data were fitted numerically to a two-site exchange model using the program ChemEx (available at https://github.com/gbouvignies/chemex/releases).
Generation of the structural model
The calculation of the structural model between the HtrA2-PDZ and the DD-PDZOpt peptide was based on docking the peptide using the obtained chemical shift perturbation data. For the docking, the HADDOCK web server was used48,49. For the PDZ structure, the chain A of the crystal structure of the HtrA2-PDZ (PDB-ID: 2PZD) was used, whereas for DD-PDZOpt (DDGQYYFV) an unfolded peptide was generated by using XPLOR-NIH108. The active residues were defined as residues bearing methyl groups with a chemical shift change of more than two standard deviations corrected to zero109 in the titration of DD-PDZOpt to ALVIT-PDZ (Supplementary Fig. 12f) as well as all peptide residues. Passive residues were automatically defined by HADDOCK, and standard docking parameters were used. Haddock clustered 173 out of 200 calculated structures into 11 clusters (Supplementary Table 2).
Spin labeling of HtrA2S145C
HtrA2S145C was purified as described above and stored in 20 mM Tris-HCl pH 8, 0.1 M NaCl supplemented with 10 mM DTT. Spin labeling of HtrA2S145C with MTSL (1-Oxyl-2,2,5,5-tetramethyl-Δ3-pyrroline-3-methyl)-methanethiosulfonate; Toronto Research Chemicals) was done according to published protocols88,110. The protein was buffer exchanged into sample buffer without DTT immediately prior to spin labeling using a Zeba spin desalting column 7 kDA MWCO (ThermoFisher). A ten-fold molar excess of MTSL dissolved in acetonitrile was added to the protein solution and the mixture was incubated overnight at room temperature in the dark. Unreacted MTSL was subsequently removed by exchanging the buffer to sample buffer (20 mM Tris-HCl pH 8, 0.1 M NaCl). The spin label was reduced by adding ascorbate dissolved in sample buffer (500 mM) directly into the NMR tube to a final concentration of 5 mM.
Paramagnetic relaxation enhancement (PRE)
A 2D [13C,1H]-NMR spectrum of spin-labeled MALVIproS-HtrA2S145C, S306A was initially measured in the paramagnetic state, and after the addition of 5 mM final concentration of ascorbate to the sample, the diamagnetic reference was measured. The intensities of well-resolved methyl resonances were extracted at 40 °C with PINT99. The experiments were also performed in the presence of 5 mM CaCl2 or DD-PDZOpt.
SEC-MALS
SEC-MALS experiments were performed using a Superdex Increase 200 10/300 GL column (GE Healthcare) on an Agilent 1260 HPLC Infinity II at RT (~297 K). HtrA2-PDZ experiments were run in PBS pH 7.4 or in HBS (20 mM HEPES, 150 mM NaCl) pH 7.4 when divalent metal ions were supplemented. Experiments with HtrA2S306A were run in HBS pH 7.4. Protein elution was monitored by three detectors in series, namely, an Agilent multi-wavelength absorbance detector (absorbance at 280 nm and 254 nm), a Wyatt miniDAWN TREOS multi-angle light-scattering (MALS) detector, and a Wyatt Optilab rEX differential refractive index (dRI) detector. The column was pre-equilibrated overnight in the running buffer to obtain stable baseline signals from the detectors before data collection. Molar mass, elution concentration, and mass distributions of the samples were calculated using the ASTRA 7.1.3 software (Wyatt Technology). A BSA solution (2–4 mg/ml), purchased from Sigma Aldrich and directly used without further purification, was used to calibrate inter-detector delay volumes, band-broadening corrections, and light-scattering detector normalization using standard protocols within ASTRA 7.1.3. To assess the oligomerization state of HtrA2-PDZ and HtrA2S306A, increasing elution concentrations of the proteins were used to fit the dissociation constant (KD) assuming a fast monomer–dimer equilibrium:
where M is the molecular mass of the monomer and [M] the molar concentration of the sample (in terms of monomer) as it passes through the MALS detector after eluting form the column111. The concentration was obtained by the absorbance signal after band-broadening correction, using the MALS detector as the reference instrument. In all, 95% confidence intervals were determined from the fitting error in GraphPad Prism version 9.1.0.
Gadolinium titration
To identify the calcium-binding sites in full-length HtrA2 we resorted to exploiting the relaxation enhancement effect of paramagnetic gadolinium (Gd3+)54. Gd3+ can be readily substituted into Ca2+ binding sites, thus providing a detailed picture of the interacting residues55,112. We titrated increasing amounts of gadolinium(III)-trifluoromethanesulfonate (Gd(OTF)3; (Merck)) to MALVIproS-HtrA2S306A and measured at each step a 2D [13C,1H]-NMR spectrum. Data were processed in nmrPipe66 and peak intensities were extracted with PINT99.
Cleavage assay
Overall, 1 mg/mL β-casein (Sigma Aldrich) was incubated with 623 nM (monomer concentration) of HtrA2 at 310 K in assay buffer (20 mM HEPES pH 7.4) supplemented with 2 mM of either MgSO4, CaCl2, CuCl2, or ZnSO4, to a total volume of 101 μL. Samples were taken at 10-, 20-, 40- and 60-min intervals and the cleavage patterns were analyzed by SDS-PAGE using 4–20% MiniProtean® TGXTM (Bio-Rad) gels stained with SimplyBlue SafeStain (Thermo Scientific). Quantification analysis of band intensities was made using the Image Lab software (Bio-Rad).
Fluorescence cleavage assays
The cleavage assay with varying DD-PDZOpt concentrations was performed using 20 µM of the fluorescent substrate peptide H2Opt (Mca-IRRVSYSF{Lys(Dnp)}KK (GenScript)) with 510 nM wild-type HtrA2 (monomeric concentration) in 20 mM HEPES pH 7.4 supplemented with 1 mM EDTA and increasing activator peptide concentrations (DD-PDZOpt: DDGQYYFV (GenScript)) at 313 K. For assays including 2 mM divalent metal ions, EDTA was omitted from the buffer. The reactions were monitored using a FLUOstar Optima plate reader using λex: 320 nm and λem: 405 nm. The concentration of cleaved product from fluorescence intensity was determined by calculating the fluorescence intensity difference between an internal control of uncleaved H2Opt and the maximum fluorescence intensity obtained at complete peptide cleavage during the assay, which could then be used to determine the amount of cleaved peptide. Catalytic rates were calculated by extraction of initial reaction rates from the fluorescence assay curves. Calculations of the steady-state kinetic parameters vmax and Km were performed by fitting the data to the Michaelis–Menten equation (substrate vs. reaction rate) with GraphPad Prism version 9.1.0 (Eq. (10)):
where \(v\) denotes the reaction velocity, and \(\left[{peptide}\right]\) denotes the DD-PDZOpt concentration. Following a previously outlined approach25, we fitted the cleavage rate of H2Opt as a function of the DD-PDZOpt concentration (5, 10, 25, 50, 75, 100, 150, 200, and 300 μM) to a standard one-site binding model approximating the total peptide concentration by the free peptide concentration:
where \(\left[{DD}-{PDZOpt}\right]\) denotes the activating peptide concentration, k the cleavage rate, k0 the basal substrate cleavage rate and kmax the maximum cleavage rate in the fully peptide-bound form, KD,app is the apparent dissociation constant.
The assay with varying substrate concentration was carried out using 510 nM of wild-type HtrA2 (monomeric concentration), 50 μM DD-PDZOpt and five concentrations of the H2Opt peptide (0.5, 5, 10, 20, and 30 μM). Buffer and temperature conditions as well as plate reader settings were as stated above for the activation peptide concentration variation assay. Calculations of the steady-state kinetic parameters vmax and Km were determined using the webtool InterferENZY113. The kcat value was calculated as shown in Eq. (12).
where \(\left[{HtrA}2\right]\) denotes the trimeric concentration of HtrA2 used in the kinetic assay. All variants of the fluorescence cleavage assays were run in triplicates yielding similar results.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data needed to evaluate the conclusions in the paper are present in the manuscript and/or the Supplementary Materials. Uncropped gel images are provided in a Source Data file. The sequence-specific NMR resonance assignment for HtrA2-PDZ was deposited in the BioMagResBank (www.bmrb.wisc.edu) under accession code 51320 The NMR data used for the relaxation analysis, chemical shift perturbations, and NOE networks have been tabulated and are available alongside the fluorescence data and the structural model generated by HADDOCK on Mendeley data [https://doi.org/10.17632/fgpcmp2kkg.1]. Structure coordinates used in this study are available for the RCSB Protein Data Bank (https://www.rcsb.org) under accession codes 1JXG, 1LCY, 1NW9, 2PZD, and 5M3N. Source data are provided with this paper.
References
Hartl, F. U., Bracher, A. & Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332 (2011).
Burmann, B. M. & Hiller, S. Chaperones and chaperone-substrate complexes: dynamic playgrounds for NMR spectroscopists. Prog. Nucl. Magn. Reson. Spectrosc. 86–87, 41–64 (2015).
Clausen, T., Kaiser, M., Huber, R. & Ehrmann, M. HTRA proteases: regulated proteolysis in protein quality control. Nat. Rev. Mol. Cell Biol. 12, 152–162 (2011).
Hiller, S. & Burmann, B. M. Chaperone–client complexes: a dynamic liaison. J. Magn. Reson. 289, 142–155 (2018).
Merdanovic, M., Clausen, T., Kaiser, M., Huber, R. & Ehrmann, M. Protein quality control in the bacterial periplasm. Annu. Rev. Microbiol. 65, 149–168 (2011).
Vande Walle, L., Lamkanfi, M. & Vandenabeele, P. The mitochondrial serine protease HtrA2/Omi: an overview. Cell. Death Differ. 15, 453–460 (2008).
Sawa, J., Heuck, A., Ehrmann, M. & Clausen, T. Molecular transformers in the cell: lessons learned from the DegP protease-chaperone. Curr. Opin. Struct. Biol. 20, 253–258 (2010).
Quirós, P. M., Langer, T. & López-Otín, C. New roles for mitochondrial proteases in health, ageing and disease. Nat. Rev. Mol. Cell Biol. 16, 345–359 (2015).
Deshwal, S., Fiedler, K. U. & Langer, T. Mitochondrial proteases: multifaceted regulators of mitochondrial plasticity. Annu. Rev. Biochem. 89, 501–528 (2020).
Zurawa-Janicka, D., Skorko-Glonek, J. & Lipinska, B. HtrA proteins as targets in therapy of cancer and other diseases. Expert Opin. Ther. Targets 14, 665–679 (2010).
Faccio, L. et al. Characterization of a novel human serine protease that has extensive homology to bacterial heat shock endoprotease HtrA and is regulated by kidney ischemia. J. Biol. Chem. 275, 2581–2588 (2000).
Gray, C. W. et al. Characterization of human HtrA2, a novel serine protease involved in the mammalian cellular stress response. Eur. J. Biochem. 267, 5699–5710 (2000).
Suzuki, Y. et al. A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol. Cell 8, 613–621 (2001).
Kennedy, M. Origin of PDZ (DHR, GLGF) domains. Trends Biochem. Sci. 20, 350 (1995).
Clausen, T., Southan, C. & Ehrmann, M. The HtrA family of proteases: implications for protein composition and cell fate. Mol. Cell. 10, 443–455 (2002).
Li, W. et al. Structural insights into the pro-apoptotic function of mitochondrial serine protease HtrA2/Omi. Nat. Struct. Biol. 9, 436–441 (2002).
Martins, L. M. et al. The serine protease Omi/HtrA2 regulates apoptosis by binding XIAP through a Reaper-like motif. J. Biol. Chem. 277, 439–444 (2002).
Suzuki, Y., Takahashi-Niki, K., Akagi, T., Hashikawa, T. & Takahashi, R. Mitochondrial protease Omi/HtrA2 enhances caspase activation through multiple pathways. Cell Death Differ. 11, 208–216 (2004).
Aspholm, E. E., Matečko-Burmann, I. & Burmann, B. M. Keeping α-synuclein at bay: A more active role of molecular chaperones in preventing mitochondrial interactions and transition to pathological states? Life 10, 289 (2020).
Martins, L. M. et al. Neuroprotective role of the Reaper-related serine protease HtrA2/Omi revealed by targeted deletion in mice. Mol. Cell Biol. 24, 9848–9862 (2004).
Jones, J. M. et al. Loss of Omi mitochondrial protease activity causes the neuromuscular disorder of mnd2 mutant mice. Nature 425, 721–727 (2003).
Unal Gulsuner, H. et al. Mitochondrial serine protease HTRA2 p.G399S in a kindred with essential tremor and Parkinson disease. Proc. Natl. Acad. Sci. USA 111, 18285–18290 (2014).
Strauss, K. M. et al. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson’s disease. Hum. Mol. Genet. 14, 2099–2111 (2005).
Lautenschläger, J. et al. Intramitochondrial proteostasis is directly coupled to α-synuclein and amyloid β1-42 pathologies. J. Biol. Chem. 295, 10138–10152 (2020).
Toyama, Y., Harkness, R. W., Lee, T. Y. T., Maynes, J. T. & Kay, L. E. Oligomeric assembly regulating mitochondrial HtrA2 function as examined by methyl-TROSY NMR. Proc. Natl. Acad. Sci. USA 118, e2025022118 (2021).
Šulskis, D., Thoma, J. & Burmann, B. M. Structural basis of DegP protease temperature-dependent activation. Sci. Adv. 7, eabj1816 (2021).
Merski, M. et al. Molecular motion regulates the activity of the mitochondrial serine protease HtrA2. Cell Death Dis. 8, e3119 (2017).
Wagh, A. R. & Bose, K. Structural basis of inactivation of human counterpart of mouse motor neuron degeneration 2 mutant in serine protease HtrA2. Biosci. Rep. 38, 1–12 (2018).
Rosenzweig, R. & Kay, L. E. Bringing dynamic molecular machines into focus by methyl-TROSY NMR. Annu. Rev. Biochem. 83, 291–315 (2014).
Toyama, Y., Harkness, R. W. & Kay, L. E. Dissecting the role of interprotomer cooperativity in the activation of oligomeric high-temperature requirement A2 protein. Proc. Natl. Acad. Sci. USA 118, e2111257118 (2021).
McConnell, H. M. Reaction rates by nuclear magnetic resonance. J. Chem. Phys. 28, 430–431 (1958).
Walsh, N. P., Alba, B. M., Bose, B., Gross, C. A. & Sauer, R. T. OMP peptide signals initiate the envelope-stress response by activating DegS protease via relief of inhibition mediated by its PDZ domain. Cell 113, 61–71 (2003).
Palmer, A. G., Kroenke, C. D. & Loria, J. P. Nuclear magnetic resonance methods for quantifying microsecond-to-millisecond motions in biological macromolecules. Methods Enzymol. 339, 204–238 (2001).
Lipari, G. & Szabo, A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J. Am. Chem. Soc. 104, 4546–4559 (1982).
Lipari, G. & Szabo, A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 2. Analysis of experimental results. J. Am. Chem. Soc. 104, 4559–4570 (1982).
Franco, R., Gil-Caballero, S., Ayala, I., Favier, A. & Brutscher, B. Probing conformational exchange dynamics in a short-lived protein folding intermediate by real-time relaxation–dispersion NMR. J. Am. Chem. Soc. 139, 1065–1068 (2017).
Korzhnev, D. M., Kloiber, K., Kanelis, V., Tugarinov, V. & Kay, L. E. Probing slow dynamics in high molecular weight proteins by methyl-TROSY NMR spectroscopy: application to a 723-residue enzyme. J. Am. Chem. Soc. 126, 3964–3973 (2004).
Morgado, L., Burmann, B. M., Sharpe, T., Mazur, A. & Hiller, S. The dynamic dimer structure of the chaperone Trigger Factor. Nat. Commun. 8, 1992 (2017).
Bernegger, S. et al. A novel FRET peptide assay reveals efficient Helicobacter pylori HtrA inhibition through zinc and copper binding. Sci. Rep. 10, 10563 (2020).
Russell, T. M., Tang, X., Goldstein, J. M., Bagarozzi, D. & Johnson, B. J. B. The salt-sensitive structure and zinc inhibition of Borrelia burgdorferi protease BbHtrA. Mol. Microbiol. 99, 586–596 (2016).
Miao, Q. et al. Paramagnetic chemical probes for studying biological macromolecules. Chem. Rev. 122, 9571–9642 (2022).
Waudby, C. A., Ramos, A., Cabrita, L. D. & Christodoulou, J. Two-dimensional NMR lineshape analysis. Sci. Rep. 6, 24826 (2016).
Gupta, S. et al. The C-terminal tail of presenilin regulates Omi/HtrA2 protease activity. J. Biol. Chem. 279, 45844–45854 (2004).
Martins, L. M. et al. Binding specificity and regulation of the serine protease and PDZ domains of HtrA2/Omi. J. Biol. Chem. 278, 49417–49427 (2003).
Meltzer, M. et al. Allosteric activation of HtrA protease DegP by stress signals during bacterial protein quality control. Angew. Chem. 47, 1332–1334 (2008).
Zhang, Y., Appleton, B. A., Wu, P., Wiesmann, C. & Sidhu, S. S. Structural and functional analysis of the ligand specificity of the HtrA2/Omi PDZ domain. Protein Sci. 16, 1738–1750 (2007).
Bejugam, P. R. et al. Allosteric regulation of serine protease HtrA2 through novel non-canonical substrate binding pocket. PLoS ONE 8, e55416 (2013).
de Vries, S. J., van Dijk, M. & Bonvin, A. M. J. J. The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 5, 883–897 (2010).
Honorato, R. V. et al. Structural biology in the clouds: the WeNMR-EOSC ecosystem. Front. Mol. Biosci. 8, 729513 (2021).
Dong, W., Wang, J., Niu, G., Zhao, S. & Liu, L. Crystal structure of the zinc-bound HhoA protease from Synechocystis sp. PCC 6803. FEBS Lett. 590, 3435–3442 (2016).
Gaudreault, F., Chartier, M. & Najmanovich, R. Side-chain rotamer changes upon ligand binding: common, crucial, correlate with entropy and rearrange hydrogen bonding. Bioinformatics 28, 423–430 (2012).
Troussicot, L., Vallet, A., Molin, M., Burmann, B. M. & Schanda, P. Disulfide-bond-induced structural frustration and dynamic disorder in a peroxiredoxin from MAS NMR. J. Am. Chem. Soc. 145, 10700–10711 (2023).
Parra, R. G. et al. Protein Frustratometer 2: a tool to localize energetic frustration in protein molecules, now with electrostatics. Nucleic Acids Res. 44, W356–W360 (2016).
Ravera, E., Cerofolini, L., Fragai, M., Parigi, G. & Luchinat, C. Characterization of lanthanoid-binding proteins using NMR spectroscopy. Methods Enzym. 651, 103–137 (2021).
Bertini, I., Gelis, I., Katsaros, N., Luchinat, C. & Provenzani, A. Tuning the affinity for lanthanides of calcium binding proteins. Biochemistry 42, 8011–8021 (2003).
Lu, C. H. et al. MIB2: metal ion-binding site prediction and modelling server. Bioinformatics 38, 4428–4429 (2022).
Hunkeler, M., Jin, C. Y. & Fischer, E. S. Structures of BIRC6-client complexes provide a mechanism of SMAC-mediated release of caspases. Science 379, 1105–1111 (2023).
MohamedMohaideen, N. N. et al. Structure and function of the virulence-associated high-temperature requirement A of Mycobacterium tuberculosis. Biochemistry 47, 6092–6102 (2008).
Ehrmann, J. F. et al. Structural basis for regulation of apoptosis and autophagy by the BIRC6/SMAC complex. Science 379, 1117–1123 (2023).
Toyama, Y., Harkness, R. W. & Kay, L. E. Structural basis of protein substrate processing by human mitochondrial high-temperature requirement A2 protease. Proc. Natl. Acad. Sci. USA 119, e2203172119 (2022).
Singh, N., D’Souza, A., Cholleti, A., Sastry, G. M. & Bose, K. Dual regulatory switch confers tighter control on HtrA2 proteolytic activity. FEBS J. 281, 2456–2470 (2014).
Bogaerts, V. et al. Genetic variability in the mitochondrial serine protease HTRA2 contributes to risk for Parkinson disease. Hum. Mutat. 29, 832–840 (2008).
Fitzgerald, J. C. et al. Phosphorylation of HtrA2 by cyclin-dependent kinase-5 is important for mitochondrial function. Cell Death Differ. 19, 257–266 (2012).
Plun-Favreau, H. et al. The mitochondrial protease HtrA2 is regulated by Parkinson’s disease-associated kinase PINK1. Nat. Cell. Biol. 9, 1243–1252 (2007).
Rizzuto, R. et al. Calcium and apoptosis: facts and hypotheses. Oncogene 22, 8619–8627 (2003).
Orrenius, S., Zhivotovsky, B. & Nicotera, P. Regulation of cell death: the calcium-apoptosis link. Nat. Rev. Mol. Cell. Biol. 4, 552–565 (2003).
Yang, Q.-H., Church-Hajduk, R., Ren, J., Newton, M. L. & Du, C. Omi/HtrA2 catalytic cleavage of inhibitor of apoptosis (IAP) irreversibly inactivates IAPs and facilitates caspase activity in apoptosis. Genes Dev. 17, 1487–1496 (2003).
Truong-Tran, A. Q., Carter, J., Ruffin, R. E. & Zalewski, P. D. The role of zinc in caspase activation and apoptotic cell death. BioMetals 14, 315–330 (2001).
Franklin, R. B. & Costello, L. C. The important role of the apoptotic effects of zinc in the development of cancers. J. Cell. Biochem. 106, 750–757 (2009).
Gmeiner, W. H., Boyacioglu, O., Stuart, C. H., Jennings-Gee, J. & Balaji, K. C. The cytotoxic and pro-apoptotic activities of the novel fluoropyrimidine F10 towards prostate cancer cells are enhanced by Zn2+-chelation and inhibiting the serine protease Omi/HtrA2. Prostate 75, 360–369 (2015).
Geiser, M., Cèbe, R., Drewello, D. & Schmitz, R. Integration of PCR fragments at any specific site within cloning vectors without the use of restriction enzymes and DNA ligase. Biotechniques 31, 88–92 (2001).
Mikolajczyk, J. et al. Small Ubiquitin-related Modifier (SUMO)-specific proteases: Profiling the specificities and activities of human SENPs. J. Biol. Chem. 282, 26217–26224 (2007).
Sambrook, J. & Russell, D. Molecular Cloning: A Laboratory Manual (Cold Spring Harbor Laboratory Press, 2000).
Callon, M., Burmann, B. M. & Hiller, S. Structural mapping of a chaperone-substrate interaction surface. Angew. Chem. 53, 5069–5072 (2014).
Goto, N. K., Gardner, K. H., Mueller, G. A., Willis, R. C. & Kay, L. E. A robust and cost-effective method for the production of Val, Leu, Ile (δ1) methyl-protonated 15N-, 13C-, 2H-labeled proteins. J. Biomol. NMR 13, 369–374 (1999).
Tzeng, S.-Ru, Pai, M.-T. & Kalodimos, C. NMR studies of large protein systems. Methods Mol. Biol. 831, 133–140 (2012).
Gans, P. et al. Stereospecific isotopic labeling of methyl groups for NMR spectroscopic studies of high-molecular-weight proteins. Angew. Chem. 49, 1958–1962 (2010).
Mas, G., Crublet, E., Hamelin, O., Gans, P. & Boisbouvier, J. Specific labeling and assignment strategies of valine methyl groups for NMR studies of high molecular weight proteins. J. Biomol. NMR 57, 251–262 (2013).
Pervushin, K., Riek, R., Wider, G. & Wüthrich, K. Relaxation by mutual cancellation of dipole–dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc. Natl. Acad. Sci. USA 94, 12366–12371 (1997).
Sattler, M., Schleucher, J. & Griesinger, C. Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog. Nucl. Magn. Reson. Spectrosc. 34, 93–158 (1999).
Rossi, P., Xia, Y., Khanra, N., Veglia, G. & Kalodimos, C. G. 15N and 13C-SOFAST-HMQC editing enhances 3D-NOESY sensitivity in highly deuterated, selectively [1H,13C]-labeled proteins. J. Biomol. NMR 66, 259–271 (2016).
Kerfah, R. et al. Scrambling free combinatorial labeling of alanine-β, isoleucine-δ1, leucine-proS and valine-proS methyl groups for the detection of long range NOEs. J. Biomol. NMR 61, 73–82 (2015).
Tugarinov, V., Choy, W. Y., Orekhov, V. Y. & Kay, L. E. Solution NMR-derived global fold of a monomeric 82-kDa enzyme. Proc. Natl. Acad. Sci. USA 102, 622–627 (2005).
Jaravine, V., Ibraghimov, I. & Orekhov, V. Y. Removal of a time barrier for high-resolution multidimensional NMR spectroscopy. Nat. Methods 3, 605–607 (2006).
Delaglio, F. et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes*. J. Biomol. NMR 6, 277–293 (1995).
Keller, R. L. J. The Computer Aided Resonance Assignment Tutorial (Cantina Verlag, Goldau, 2004).
Nielsen, J. T. & Mulder, F. A. A. POTENCI: prediction of temperature, neighbor and pH-corrected chemical shifts for intrinsically disordered proteins. J. Biomol. NMR 70, 141–165 (2018).
Burmann, B. M., Wang, C. & Hiller, S. Conformation and dynamics of the periplasmic membrane-protein-chaperone complexes OmpX-Skp and tOmpA-Skp. Nat. Struct. Mol. Biol. 20, 1265–1272 (2013).
Wider, G. & Dreier, L. Measuring protein concentrations by NMR spectroscopy. J. Am. Chem. Soc. 128, 2571–2576 (2006).
Siemons, L., Mackenzie, H. W., Shukla, V. K. & Hansen, D. F. Intra-residue methyl–methyl correlations for valine and leucine residues in large proteins from a 3D-HMBC-HMQC experiment. J. Biomol. NMR 73, 749–757 (2019).
Takeuchi, K., Tokunaga, Y., Imai, M., Takahashi, H. & Shimada, I. Dynamic multidrug recognition by multidrug transcriptional repressor LmrR. Sci. Rep. 4, 6922 (2015).
Lidman, J., Sallova, Y., Matečko-Burmann, I. & Burmann, B. M. Structure and dynamics of the mitochondrial DNA-compaction factor Abf2 from S. cerevisiae. J. Struct. Biol. 215, 108008 (2023).
Butterfoss, G. L. et al. Conformational dependence of 13C shielding and coupling constants for methionine methyl groups. J. Biomol. Nmr. 48, 31–47 (2010).
Hansen, D. F., Neudecker, P. & Kay, L. E. Determination of isoleucine side-chain conformations in ground and excited states of proteins from chemical shifts. J. Am. Chem. Soc. 132, 7589–7591 (2010).
Hansen, D. F., Neudecker, P., Vallurupalli, P., Mulder, F. A. A. & Kay, L. E. Determination of Leu side-chain conformations in excited protein states by NMR relaxation dispersion. J. Am. Chem. Soc. 132, 42–43 (2010).
London, R. E., Wingad, B. D. & Mueller, G. A. Dependence of amino acid side chain 13C shifts on dihedral angle: Application to conformational analysis. J. Am. Chem. Soc. 130, 11097–11105 (2008).
Lakomek, N.-A., Ying, J. & Bax, A. Measurement of 15N relaxation rates in perdeuterated proteins by TROSY-based methods. J. Biomol. NMR 53, 209–221 (2012).
Lee, D., Hilty, C., Wider, G. & Wüthrich, K. Effective rotational correlation times of proteins from NMR relaxation interference. J. Magn. Res. 178, 72–76 (2006).
Niklasson, M. et al. Comprehensive analysis of NMR data using advanced line shape fitting. J. Biomol. NMR 69, 93–99 (2017).
Kawale, A. A. & Burmann, B. M. Inherent backbone dynamics fine-tune the functional plasticity of Tudor domains. Structure 29, 1253–1265 (2021).
Walton, T. A., Sandoval, C. M., Fowler, C. A., Pardi, A. & Sousa, M. C. The cavity-chaperone Skp protects its substrate from aggregation but allows independent folding of substrate domains. Proc. Natl. Acad. Sci. USA 106, 1772–1777 (2009).
Maciejewski, M. W. et al. NMRbox: a resource for biomolecular NMR computation. Biophys. J. 112, 1529–1534 (2017).
Dosset, P., Hus, J. C., Blackledge, M. & Marion, D. Efficient analysis of macromolecular rotational diffusion from heteronuclear relaxation data. J. Biomol. NMR 16, 23–28 (2000).
Tugarinov, V. & Kay, L. E. An isotope labeling strategy for methyl TROSY spectroscopy. J. Biomol. NMR 28, 165–172 (2004).
Sun, H., Kay, L. E. & Tugarinov, V. An optimized relaxation-based coherence transfer NMR experiment for the measurement of side-chain order in methyl-protonated, highly deuterated proteins. J. Phys. Chem. B 115, 14878–14884 (2011).
Weinhäupl, K. et al. Structural basis of membrane protein chaperoning through the mitochondrial intermembrane space. Cell 175, 1365–1379 (2018).
Mizukoshi, Y. et al. Improvement of ligand affinity and thermodynamic properties by NMR-based evaluation of local dynamics and surface complementarity in the receptor-bound state. Angew. Chem. 55, 14606–14609 (2016).
Schwieters, C. D., Kuszewski, J. J., Tjandra, N. & Marius Clore, G. The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 160, 65–73 (2003).
Schumann, F. H. et al. Combined chemical shift changes and amino acid specific chemical shift mapping of protein–protein interactions. J. Biomol. NMR 39, 275–289 (2007).
Burmann, B. M., Scheckenhofer, U., Schweimer, K. & Rösch, P. Domain interactions of the transcription–translation coupling factor Escherichia coli NusG are intermolecular and transient. Biochem. J. 435, 783–789 (2011).
Benfield, C. T. O. et al. Mapping the IκB kinase β (IKKβ)-binding interface of the B14 protein, a vaccinia virus inhibitor of IKKβ-mediated activation of nuclear factor κB. J. Biol. Chem. 286, 20727–20735 (2011).
Squier, T. C., Bigelow, D. J., Fernandez-Belda, F. J., deMeis, L. & Inesi, G. Calcium and lanthanide binding in the sarcoplasmic reticulum ATPase. J. Biol. Chem. 265, 13713–13720 (1990).
Pinto, M. F. et al. InterferENZY: a web-based tool for enzymatic assay validation and standardized kinetic analysis. J. Mol. Biol. 433, 166613 (2021).
Acknowledgements
The Swedish NMR Centre of the University of Gothenburg is acknowledged for spectrometer time. The authors thank H. Fremlén for help with graphic design. B.M.B. gratefully acknowledges an EMBO Young Investigator Fellowship as well as funding from the Swedish Research Council (Starting Grant 2016-04721; Consolidator Grant 2020-00466), the Swedish Cancer Foundation (2019-0415 and 2022-2490), and the Knut och Alice Wallenberg Foundation through a Wallenberg Academy Fellowship (2016.0163 and 2020.0300) as well as through the Wallenberg Centre for Molecular and Translational Medicine, University of Gothenburg, Sweden. This study made use of NMRbox: National Center for Biomolecular NMR Data Processing and Analysis, a Biomedical Technology Research Resource (BTRR), which is supported by NIH grant P41GM111135 (NIGMS).
Funding
Open access funding provided by University of Gothenburg.
Author information
Authors and Affiliations
Contributions
B.M.B. conceived the study and designed the experiments together with E.E.A. E.E.A. performed all experimental work. E.E.A., J.L., and B.M.B. analyzed and discussed the data. E.E.A., J.L., and B.M.B. wrote jointly the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Sandra Macedo-Ribeiro, Paul Schanda and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Aspholm, E.E., Lidman, J. & Burmann, B.M. Structural basis of substrate recognition and allosteric activation of the proapoptotic mitochondrial HtrA2 protease. Nat Commun 15, 4592 (2024). https://doi.org/10.1038/s41467-024-48997-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-48997-5
- Springer Nature Limited