
Abstract
Understanding the stability and aggregation of nanoparticles in aqueous milieu is critical for assessing their behavior in the natural and engineered environmental systems and establishing their threat to human and ecosystems health. In this study, the colloidal stability and aggregation kinetics of nanocrystal quantum dots (QDs) —CdSe/ZnS QDs—were thoroughly explored under a wide range of aqueous environmental conditions. The z-average hydrodynamic diameters (z-avg. HDs) and zeta potential (ξ potential) of CdSe/ZnS QDs were measured in monovalent electrolyte (NaCl) and divalent electrolyte (CaCl2) solutions in both the absence and presence of natural organic matter (NOM)—Suwannee River natural organic matter, SRNOM to assess the dynamic growth of these nanoaggregate-QD-complexes, and the evaluation of their colloidal stability. Results show that CaCl2 was more effective to destabilize the QDs compared to NaCl at similar concentrations. An increase in NaCl concentration from 0.01 to 3.5 M increased the z-avg. HD of QD aggregates from 61.4 nm to 107.2 nm. The aggregation rates of QDs increased from 0.007 to 0.042 nm·s−1 with an increase in ionic strength from 0.5 to 3.5 M NaCl solutions, respectively. In the presence of Na+ cations, the aggregation of QDs was limited as steric forces generated by the original surface coating of QDs prevailed. In the presence of CaCl2, the aggregation of QDs was observed at a low concentration of CaCl2 (0.0001 M) with a z-avg. HD of 74.2 nm that significantly increased when the CaCl2 was higher than 0.002 M. Larger sizes of QD aggregates were observed at each level of CaCl2 concentration in suspensions of 0.002–0.1 M, as the z-avg. HDs of QDs increased from 125.1 to 560.4 nm, respectively. In the case of CaCl2, an increase in aggregation rates occurred from 0.035 to 0.865 nm·s−1 with an increase in ionic strength from 0.0001 M to 0.004 M, respectively. With Ca2+ cations, the aggregation of QDs was enhanced due to the bridging effects from the formation of complexes between Ca2+ cations in solution and the carboxyl group located on the surface coating of QDs. In the presence of SRNOM, the aggregation of QDs was enhanced in both monovalent and divalent electrolyte solutions. The degree of aggregation formation between QDs through cation-NOM bridges was superior for Ca2+ cations compared to Na+ cations. The presence of SRNOM resulted in a small increase in the size of the QD aggregates for each of NaCl concentrations tested (i.e., 0.01 to 3.5 M, except 0.1 M), and induced a monodispersed and narrower size distribution of QDs suspended in the monovalent electrolyte NaCl concentrations. In the presence of SRNOM, the aggregation rates of QDs increased from 0.01 to 0.024 nm 1 with the increase of NaCl concentrations from 0.01 to 2 M, respectively. The presence of SRNOM in QDs suspended in divalent electrolyte CaCl2 solutions enhanced the aggregation of QDs, resulting in the increase of z-avg. HDs of QDs by approximately 19.3%, 42.1%, 13.8%, 1.5%, and 24.8%, at CaCl2 concentrations of 0.002, 0.003, 0.005, 0.01, and 0.1 M, respectively. In the case of CaCl2, an increase in aggregation rates occurred from 0.035 to 0.865 nm·s−1 with an increase in ionic strength from 0.0001 to 0.004 M, respectively. Our findings demonstrated the colloidal stability of QDs and cations-NOM-QD nanoparticle complexes under a broad spectrum of conditions encountered in the natural and engineered environment, indicating and the potential risks from these nanoparticles in terms of human and ecosystem health.
Similar content being viewed by others


1 Introduction
Among the various types of nanoparticles, quantum dots (QDs), which are semiconductor nanoscale crystals, encompass a wide range of areas of application owing to their optical and electronic properties [1]. Their capacity of fluorescence in different spectral regions, with a superior excitation enhancement, high quantum yield, and elevated photobleaching threshold, makes them suitable for potential applications in biomedical imaging [2, 3], biomarkers and biosensors [4,5,6,7], site-specific gene medical treatment and site-specific drug delivery or targeting [8, 9], diagnosis and therapy [10, 11], nanophotocatalysis [12,13,14,15], and luminescence based-probes [16, 17]. In addition, QDs are used in various applications in electronics industries such as solar cells [18,19,20,21], visual and display technologies such as light emitting diodes [22,23,24], and information technologies such as quantum information processing (QIP) for storing, processing and communicating data [25].
The large amount of production and diversity of applications of nanoparticles greatly augments the probability of their discharge into the natural and engineered environmental systems. The concurrent risk to environmental health and public safety from such releases has generated much concern worldwide about their fate and transport in the environment after release and potential toxicity following exposure [26,27,28,29,30,31,32].
At present, a consistent body of studies on the fate, behavior, and effects of nanoparticles in the environment indicates a clear correlation between the mobility and retention of nanoparticles in various media, including soil and water, and their physical and chemical properties and the physical, chemical, and biological factors and processes in the environment. The physical and chemical properties of nanoparticles and the physical, chemical, and biological conditions and processes in the environment that affect the nanoparticles’ transport behavior and fate encompass adsorption, aggregation/stabilization, dissolution, and transformation [33]. The mobility of nanoparticles in soil and water is governed by their aggregation, deposition kinetics, and straining. Although nanoparticles may aggregate to form larger colloidal size particles [34], these nanoaggregates have fractal structures [35] and impact the surface interaction processes; consequently the nanoparticles size and geometry are of environmental significance [36]. Concerning the QD nanomaterials, the presence of a shell, surface modification, and resistance to aggregation significantly enhances their mobility in sand columns [37,38,39] and in other media [26, 40, 41]. Environmental factors influencing the fate and transport characteristics of nanoparticles, including QDs, include: water chemistry (e.g., pH, electrolyte type, ionic strength, organic matter, and surfactant) [39, 42,43,44,45,46,47,48]; natural and engineered porous media materials (e.g., sand and soil) [47, 49]; degree of saturation (e.g., unsaturated and saturated) [45, 50, 51]; hydraulic properties of porous media materials (e.g., flow velocity and hydraulic conductivity); fluid flow in porous media materials (e.g., saturated flow, unsaturated flow, and preferential flow phenomena such as unstable fingered flow) [42, 50, 52, 53]; and porous media geometrical characteristics (e.g., grain size distribution and grain shape irregularity) [45].
Like transport, the bioavailability and toxicity of nanoparticles are influenced by their physico-chemical characteristics, particularly the size and surface properties that determine the nanoparticle aggregation state and stability behavior. Stability (i.e., the resistance to aggregation) has also been evaluated and is believed to play a pivotal role in determining toxicity [26, 54, 55]. Research on the toxicity of QD nanoparticles on the ecosystem has involved investigations of microorganisms, fungi, aquatic and terrestrial plants (e.g., algae, Arabidopsis thaliana), invertebrates (e.g., mussels), and vertebrates (e.g., crustaceans, fish, mice, and primates) [29, 56,57,58,59,60] at the cellular, subcellular, and molecular scales [61]. Moreover, with an accumulation of evidence, the adverse effect of QD nanoparticles on the ecosystem is now beyond dispute. For instance, growth inhibition in bacteria [62] and protozoa [63]; physical changes in cellular structure decreasing catalase activities of algae [28, 64]; DNA damage-causing immunotoxin effects mediated in mussel cells [65, 66]; damage to the hematopoietic organ and hematocytes of invertebrate (e.g., Daphnia magna) organisms [67,68,69,70]; reduced the phagocytic activity of phagocytes of fish [71]; and aberrations including malformations of the placenta and fetus in mice [72, 73] have all been observed following exposure to QD nanoparticles.
The potential exposure routes for human to QDs are contaminated environmental media, workplace, and therapeutic or diagnostic administration for medicinal purposes [56]. The adverse effect of QDs on human cells (e.g., lung, kidney, liver, skin, breast, intestine, vein, and prostate) [72, 74,75,76,77,78,79] is also now well recognized. Cytotoxicity in human cells has been observed following exposure to QDs in many in vitro studies. For instance, mercapto-undecanoic acid (MUA) modified CdSe QDs caused bronchial epithelial cell apoptosis, change in gene level response, and generation of reactive oxygen species [77].
Considering the significant influence of stability on the QD nanoparticles’ fate and transport [49, 80, 81], bioavailability [82,83,84], and toxicity [26, 61, 85,86,87], it is of immense importance to understand their stability and aggregation behavior in the natural and engineered environmental systems, and thus to evaluate the interaction mechanisms and to model and forecast the fate, transport, transformation, and toxicity of nanoparticles in these environmental systems [26, 88].
Several studies have revealed that nanoparticles exhibit different properties upon aggregation [89, 90]. Aggregation kinetics are controlled by particle properties—size [91,92,93,94], shape [91, 93], chemical composition, surface charge, and surface modification [39, 92, 93, 95]. Aggregation kinetics are also influenced by environmental chemistries such as pH levels [42, 96,97,98,99,100], ionic strength [96,97,98, 101,102,103,104] and electrolyte types [97, 105,106,107], and natural organic matter (NOM) [36, 106, 108,109,110,111,112].
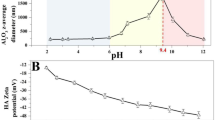
The role of pH in the stability behavior and aggregation kinetics of nanoparticles is well known in terms of modifying the surface charge of nanoparticles and disrupting the electrostatic energy barriers [26, 42, 96, 97]. The variation of surface charge of the nanoparticles at different pH levels is caused by the change in the protonation-deprotonation equilibria of the functional groups of capping agent located at the surface of the nanoparticles. Rapid aggregation of nanoparticles occurs at the point zero charge (pHPZC) because of the decrease of repulsive electrostatic interactions between the surfaces of nanoparticles, whereas stabilization of nanoparticles occurs at both higher and lower pH conditions due to the increasing repulsive electrostatic interactions between the surfaces of nanoparticles [26, 40, 97, 113, 114].
Among the factors that may affect nanoparticles’ stability, the effects of ionic strength and electrolyte types have been the subject of substantive studies. Electrolytes in the environment can cause the compression of electric double layers (EDLs) of nanoparticles, resulting in their destabilization as a result of the decrease of the electrostatic repulsion energy barrier [50, 85, 96, 97, 103, 115]. Compared to the monovalent electrolytes (e.g., NaCl and KCl), divalent electrolytes (e.g., CaCl2 and MgCl2) have a more pronounced effect on the destabilization of the nanoparticles in suspension by cause of the high efficiency in screening the EDLs and the bridging effect through the complexation of divalent cations [105, 111, 115,116,117].
NOM existing in the natural and engineered environmental systems can alter the chemical processes and affect the aggregation of nanoparticles through various mechanisms depending upon the properties of nanoparticle and the environmental chemistry [100, 105, 106, 118, 119]. The various mechanisms involved in the interactions between nanoparticles and NOM include hydrophobic interactions, electrostatic interactions, steric interactions, hydrogen bonding, and bridging. In monovalent electrolyte and low concentrations of divalent electrolyte solutions at higher pH well above the pKa values of NOM, the presence of NOM has been found to enhance the stability of nanoparticles. The enhanced stability is caused by the electrostatic repulsions from the strongly negative surface of nanoparticles due to either the completely deprotonated functional groups or steric repulsion. In a higher concentration divalent electrolyte suspension, NOM is thought to enhance the aggregation of nanoparticles due to the bridging interactions [111, 115].
The potential for synergistic interaction between electrolytes and NOM is thus important for understanding the mechanisms of QD nanoparticle aggregation for the purpose of predicting their environmental fate, transport, and toxicity. However, the interaction among QD nanoparticles and electrolytes and NOM that results in a variation of colloidal stability and aggregation kinetics is complex. Indeed, only a few studies have sought to explore the reasons that tie aggregation heterogeneity to the complex interactions of electrostatic and steric forces among particles, electrolytes, and NOM in natural surface and subsurface environments [120]. Therefore, it is critical to study the interactions between nanoparticles, the interactions between nanoparticles and electrolytes, the interactions between nanoparticles and electrolyte-NOM systems and their resulting structures for purposes of elucidating their effect on the aggregation and stability of QD nanoparticles in aqueous systems.
The objective of this study is to systematically investigate the effects of ionic strength, monovalent and divalent electrolytes, and NOM upon the aggregation behavior and the mechanisms that control the stability of QD nanoparticles in aqueous systems. To this end, we investigated the dynamic stability of QD nanoparticles as a function of monovalent (NaCl) and divalent (CaCl2) cations concentration in both the absence and presence of NOM. The aggregation kinetics of QD nanoparticles were studied using time-resolved dynamic light scattering (DLS) and electrophoretic light scattering (ELS) for the various nanoparticle-aqueous systems. Linking experimental measurements and theoretical calculations, the critical coagulation concentration (CCC) values for the aggregation and the DLVO energy profiles between QD nanoparticles were defined. The results obtained through this systematic study of QD nanoparticles in aqueous systems contribute to advance the understanding of the water chemistry factors and mechanisms that influence the stability of QD nanoparticle. Such an understanding is critical to assess the environmental and toxicological risks to QD nanoparticles exposure.
The novelty of this research consists of the analysis of the mechanisms governing the interfacial interactions between the surface of QDs in the presence of NOM and different electrolytes (monovalent electrolyte NaCl and divalent electrolyte CaCl2), the description and measurement of the dynamic growth of these nanoaggregate-QD-complexes, and the evaluation of their colloidal stability in aqueous environment.
2 Materials and methods
2.1 CdSe/ZnS quantum dots synthesis
The synthesis of water-dispersible CdSe/ZnS QDs that include a core of cadmium selenium (CdSe) and a protecting shell of zinc sulfide (ZnS) was achieved following fabrication processes reported in the research literature [50, 121,122,123]. The QD ligand shell is made of tetradecylphosphonic acid (TDPA) and is covered by 40% octylamine-adjusted poly(acrylic acid) (OA-PAA) [124, 125]. The synthesized CdSe/ZnS core–shell nanoscale crystals consist of the following elements and dimensions: the core of CdSe/ZnS QD has a diameter of 2.3 nm and is encapsulated by a shell with a layer thickness of 1.5 nm, yielding CdSe/ZnS QDs that have a particle size of 5.3 nm in diameter. These synthesized QDs have a quantum yield that is evaluated at 30% [122]. These QDs formed have an emission wavelength that is measured at 630 nm. Transmission Electron Microscopy (TEM) characterization of the CdSe core of QDs are displayed in micrographs (Fig. 1). Knowledge of the properties of nanoscale crystal structured materials, such as QDs, at the atomic level can be accomplished by utilizing atomic number contrast scanning transmission electron microscopy (Z-STEM) [126, 127]. The Z-STEM imaging of the structural details of CdSe/ZnS core/shell nanoscale crystals are presented in the research of McBride et al. (2006). The synthesized water-soluble QDs in this study had an original concentration of 1.38 × 10–5 M and a z-average hydrodynamic diameter (z-avg. HD) of 46.8 nm. Additional dilution of the original concentration of the QDs suspension was performed to achieve a concentration of 5 × 10–7 M utilizing ultrapure deionized water (diH2O) with a resistivity of 18.2 MΩ·cm, obtained using a laboratory water purification system (Millipore Corporation, Billerica, MA) to prepare the stock suspension of QDs. This QDs stock suspension was then stored in the dark at 4 °C.

TEM micrographs of a characteristic sample of core CdSe quantum dots. QDs utilized in the experiments were of a smaller average size (\(\sim\) 2.3 nm). TEM micrographs of core CdSe QDs were acquired employing the TEM JEOL JEM-3010. The JEOL JEM-3010 is a 300 kV transmission electron microscope instrumented with a LaB6 electron source and equipped with an ultra-high resolution pole piece (Cs = 0.6 mm). The JEOL JEM-3010 has a resolution of 0.14 nm lattice and 0.17 nm point-to-point
2.2 Suwannee River natural organic matter
Suwannee River natural organic matter (SRNOM, RO Isolation 2R101N) was acquired from the International Humic Substances Society (IHSS), St. Paul, MN and used as a model to mimic NOM in aqueous systems. The stock solution for SRNOM was made by mixing 50 mg of SRNOM into 250 mL of ultrapure diH2O. The SRNOM solution was then stirred for 24 h in darkness and subject to filtration using a 0.2 μm Polyethersulfone (PES) membrane filter under vacuum (VWR, Randor, PA), after which the solution was stored in the dark at 4 °C. The initial pH of the SRNOM stock solution was 3.45 ± 0.01. The total organic carbon (TOC) content of the SRNOM stock solution quantified by a TOC analyzer (TOC-V CSH, Shimadzu, Kyoto, Japan) was 82.68 ± 2.20 mg/L.
2.3 Electrolyte-Suwannee River natural organic matter-Quantum dots-based suspensions preparation
Sodium chloride (BDH®, ACS Grade) and calcium chloride dihydrate (Fisher Chemical®, ACS Grade) were used for the ionic strength and electrolyte type dependent QD aggregation experiments. Different concentrations of background electrolyte solutions were prepared by dissolving electrolytes in ultrapure diH2O and then stirred for 1 h. All the electrolyte-QD suspensions were prepared by mixing 1.8 mL background electrolyte solutions and 0.2 mL QDs stock suspension in a BI-SCP plastic sample cuvette (10 W × 10L × 52H mm, 3.5 mL), resulting in a QD concentration of 5 × 10–8 M. Before incorporation, both the QD stock suspension and background solution were subjected to filtration utilizing a syringe filter unit (Millex®, MilliporeSigma, Burlington, MA) with a 100 nm pore size membrane to eliminate contaminations. The total ionic strengths of the QD suspensions were 0.001–3.5 M for sodium chloride and 0.0003–0.3 M for calcium chloride as calculated using Eq. (1) [128, 129]:
where \(I\) is the ionic strength of solution (M), \({c}_{i}\) is \(i\) th species molar concentration (M), and \({Z}_{i}\) is the charge number of the \(i\) th species.
For the experiments involving electrolyte-NOM-QD-based suspensions, a fixed amount of SRNOM stock solution was added to ionic strength-modified electrolyte solutions to attain SRNOM concentrations of 10 mg/L. All background electrolyte-NOM solutions were titrated with 0.1 M sodium hydroxide (Sigma-Aldrich, Reagent Grade) or 0.1 M hydrochloric acid (BDH®, ACS Grade) to pH of 7.00 ± 0.01. The measurements of the pH values of the solutions were performed using an Orion 720A pH/Ion Meter (Thermo Fisher Scientific, Waltham, MA). A total of 42 solutions were created for experimentation including diH2O, two types of electrolytes — monovalent electrolyte NaCl at concentrations ranging from 0.001 to 3.5 M, and divalent electrolyte CaCl2 at concentrations ranging from 0.0001 to 0.1 M—in both the absence and presence of SRNOM at 10 mg/L at pH 7 (Table 1).
2.4 Particle size and zeta potential measurements
The z-avg. HD of the QD nanoaggregates suspended in a wide range of ionic strengths and electrolytes in both the absence and presence of SRNOM were quantified by the time-resolved dynamic light scattering (DLS) method using a NanoBrook 90Plus Zeta Particle Size Analyzer (Brookhaven Instruments Corporation, Holtsville, NY). Immediately after mixing, the electrolyte-NOM-QD suspensions were agitated manually for 10 s to achieve a well-mixed and homogeneous suspension for z-avg. HD measurements at a 90° scattering angle. The z-avg. HD measurements data were obtained at 10 s intervals for approximately ~ 11 min to obtain the aggregation kinetics of QDs. The z-avg. HD were reported as average values and included standard deviations. All measurements were conducted at 25 °C.
The NanoBrook 90Plus Zeta Particle Size Analyzer was also utilized to measure the zeta potential (ξ potential) of QDs under various ionic strengths and electrolytes in both the absence and presence of SRNOM. Regarding particle size analysis, immediately after mixing, the electrolyte-NOM-QD suspensions placed in a BI-SCP plastic cell were agitated manually for 10 s, and the ξ potential was measured using electrophoretic light scattering (ELS). Three ELS measurements (10 cycles per measurement) were made for each sample within ~ 11 min. ξ potentials were reported as average values and included standard deviations. The temperature was maintained at 25 °C during all the measurements. The pH value at the pHPZC for the QDs was measured between pH 1.5 and 3.5 [130].
2.5 Aggregation kinetics
The quantitative analysis of the aggregation kinetics of QDs was determined by the aggregation rate constant\({(k}_{11})\), that was correlated to the rate of increase of the z-avg. HDs \((d{d}_{z-h}\left(t\right))\) of the aggregates with time \(t\) measured by DLS, and \({k}_{11}\), which is expressed as follows [105, 131,132,133]:
where, \({N}_{o}\) is the initial particle concentration.
The aggregation rate was computed using a linear least-squares (LLS) regression analysis of the measured z-avg. HD data during a ~ 11 min aggregation stage [111]. The aggregation kinetics of the colloidal systems were characterized by the aggregation attachment efficiencies \((\alpha )\) as a function of the experimental electrolyte types and concentrations, and in both the absence and presence of SRNOM. The attachment efficiency was then computed by normalizing the aggregation rate constant \({(k}_{11})\) acquired under reaction-limited aggregation (RLA) regimes to the aggregation rate constant \({{((k}_{11})}_{fast})\) taken under the diffusion limited aggregation (DLA) regimes and \(\alpha\), which is expressed as follows [105, 131, 132]:
The RLA and DLA regimes take place at ion concentrations below and above the CCC, respectively. Two lines of attachment efficiencies dependent upon the ion concentrations under RLA and DLA regimes were then extrapolated by linear functions with their intersections generating the corresponding CCC values [105, 111, 131, 134]. To compute the attachment efficiency with SRNOM, \({k}_{11}\) obtained in the presence of SRNOM was normalized to \({{(k}_{11})}_{fast}\) acquired in the absence of SRNOM [105, 135].
2.6 Potential energy of interaction between the particles
Classical DLVO theory can be utilized to forecast the stability of (nano)particles in aqueous solutions by studying the interaction energy between particles. As stated in this theory, the two main forces between particles are the van der Waals attraction and the EDL repulsion [136, 137]. The sum of these two forces is then used to determine either a repulsive or attractive net interaction between the particles [106, 138]. The EDL repulsion \(({\varnothing }_{EDL})\) energy can be calculated and expressed as follows:
In the case of \(\kappa r>5\), the Hogg-Healy-Fuerstenau (HHF) formula is used [139]:
In the case of \(\kappa r<5\), the linear superposition approximation (LSA) expression is used [140]:
The van der Waals attraction (\({\varnothing }_{vdW})\) energy can be calculated using the following expression [141]:
The sum of these two energies, known as the total potential energy, is used to determine if the net interaction between two particles is either repulsive or attractive. It is expressed as follows:
where \(r\) is the radius of particles (m), \(\zeta\) is the zeta potential (V), \(\varepsilon\) is the permittivity of water, which is \(6.95\times {10}^{-10} {(C}^{2}{J}^{-1}{m}^{-1})\)[98], \(H\) is the separation distance between two particles (m), and \(A\) is the Hamaker constant of particles in water [142]. A value of \(6.216\times {10}^{-20} J\) was used for \(A\) [143, 144]. Further, \(e\) is the charge of an electron \(\left(1.60\times {10}^{-19} C\right)\), \({k}_{B}\) is the Boltzmann constant \(\left(1.38\times {10}^{-23} J{K}^{-1}\right)\), \(T\) is the absolute temperature, and \({k}_{B}T\) is thermal energy (at room temperature \(25 ^\circ \mathrm{C}\), \(1 {k}_{B}T=4.11\times {10}^{-21} J\)).
Furthermore, \(\kappa\) is the inverse of the Debye length \(({m}^{-1})\) [145, 146] and is expressed as:
where, \({N}_{A}\) is the Avogadro number \(\left(6.02\times {10}^{23} {mol}^{-1}\right)\), \({Z}_{i}\) is the valency of the \({i}^{th}\) ion, and \({c}_{i}^{\infty }\) is the bulk concentration of ion species \(i\), expressed in \({mol L}^{-1}\). At 25 °C, \(\kappa\) is calculated as a function of the electrolyte concentration \(C\) with units of \(M\) [147,148,149,150], and is expressed as follows:
For 1:1 electrolyte,
For 2:1 electrolyte,
Finally, \(Y\) is the scaled effective surface potential (or the asymptotic constant) [140] and is expressed as follows:
3 Results
3.1 Characterization of quantum dot nanoparticles at pH 7 in diH2O
The z-avg. HD of QDs dispersed in diH2O at the neutral pH of 7 was measured by DLS prior to investigating the effect of the electrolyte type and concentration, in both the absence and presence of SRNOM on their aggregation kinetics. The QDs were well dispersed, with a z-avg. HD of approximately 57.5 nm. The aggregates’ size distribution indicated that 9%, 48%, 36%, and 7% of the QD aggregates were in the ranges of 21–40, 41–60, 61–80, and 81–100 nm, respectively (Figure S1).
The surface charge measurements indicated that the QDs with a ZnS shell and OA-PAA surface modification used in this study were negatively charged in a diH2O suspension at a pH of 7. The average ξ potential of QDs in this condition was approximately − 3.99 mV (Table 2).
3.2 Effect of monovalent and divalent electrolytes on quantum dot nanoparticles
NaCl as a monovalent electrolyte and CaCl2 as a divalent electrolyte were added to QD samples, both with suspensions at pH 7.0 to determine their effect on QD stability and aggregation. No significant change in QDs aggregate size was observed in NaCl concentrations from 0.001 to 0.875 M (except for 0.1 M). However, the effects of the monovalent electrolyte on the QD nanoparticle aggregation significantly increased when the NaCl concentration was higher than 1 M (Fig. 2a). Larger sizes of QD aggregates were observed at each level of ionic strength in suspensions of 1–3.5 M, with the z-avg. HDs of QDs increasing from 57.5 nm (diH2O, pH 7) to approximately 102.3 nm (3.5 M), respectively (Table 2). Monodispersed and narrow size distribution of QDs were in suspensions with NaCl concentrations varying between 0.001 and 3.5 M (Fig. 3a, b).

DLS measurements of the z-average hydrodynamic diameter (z-avg. HD) of QDs suspended in aqueous chloride salt solutions as a function of the types and concentrations of electrolytes and the absence and presence of SRNOM at 10 mg/L: a Monovalent electrolyte NaCl in both the absence and presence of the SRNOM at 10 mg/L, b Divalent electrolyte CaCl2 in both the absence and presence of the SRNOM at 10 mg/L

Particle size distribution of QDs in suspensions as a function of NaCl (0.001–3.5 M) and the presence of SRNOM at 10 mg/L: a NaCl-QD systems within 0–100 s; b NaCl-QD systems at last 100 s; c NaCl-SRNOM-QD systems within 0–100 s; d NaCl-SRNOM-QD systems at last 100 s
In the presence of divalent electrolyte CaCl2, the aggregation of QD nanoparticles was detected at a low concentration of CaCl2 (0.0001 M) with a z-avg. HD of 74.2 nm that significantly increased when the CaCl2 was higher than 0.002 M. Larger sizes of QD aggregates were observed at each level of CaCl2 concentration in suspensions of 0.002–0.1 M, as the z-avg. HDs of QDs increased from 125.1 nm to 560.4 nm, respectively (Fig. 2b). Monodispersed and narrow size distribution of QDs were observed in suspensions with CaCl2 concentrations varying between 0.0001 and 0.002 M (Fig. 4a, b). Both poly-dispersed and larger size distribution of QDs were present in suspensions with CaCl2 concentrations varying between 0.002 and 0.1 M (Fig. 4a, b).

Particle size distribution of QDs in suspensions as a function of CaCl2 (0.001–0.25 M) and the presence of SRNOM at 10 mg/L: a CaCl2-QD systems within 0–100 s; b CaCl2-QD systems at last 100 s; c CaCl2-SRNOM-QD systems within 0–100 s; and d CaCl2-SRNOM-QD systems at last 100 s
The measured ξ potentials of QDs at different ionic strength solutions for monovalent and divalent electrolytes are shown in Table 2, and in Figs. 5a and c. These data indicated both an increase of the ξ potential (i.e., becoming less negative and sometimes slightly positive) and a decrease in magnitude of the ξ potential of QDs, with increasing ionic strength of both monovalent and divalent electrolyte concentrations. Significant differences were observed for the concentrations of monovalent and divalent electrolytes at which a reduction in the magnitude of the ξ potential was observed. The values of the ξ potential of QDs were approximately − 15.03 mV at a NaCl concentration of 0.001 M, which increased rapidly to approximately − 1.89 mV once the ionic strength increased beyond 0.875 M.

Zeta potential (ξ potential) variation of QDs suspended in aqueous chloride salt solutions as a function of the types and concentrations of electrolytes and the absence and presence of SRNOM at 10 mg/L: a Monovalent electrolyte NaCl, b Monovalent electrolyte NaCl in the presence of SRNOM at 10 mg/L, c Divalent electrolyte CaCl2, and d Divalent electrolyte CaCl2 in the presence of SRNOM at 10 mg/L
The effect of the divalent electrolyte CaCl2 on the ξ potential of QD nanoparticles was similar to that of the monovalent electrolyte. The values of the ξ potential of QDs were approximately − 10.77 mV at a CaCl2 concentration of 0.0001 M and increased rapidly to values near 0 mV and − 7 mV once the CaCl2 concentration increased beyond 0.03 M. However, compared to the monovalent electrolyte concentrations, at lower concentrations the presence of divalent electrolyte in suspensions enhanced both the increase of the ξ potential of the QD nanoparticles (i.e., becoming less negative) and also the decrease in the magnitude of the ξ potential of those particles. In addition, as the NaCl concentration increases in the NaCl-QDs system, the ξ potential of QDs decreased, as illustrated by the trend (linear) regression analysis (Fig. 5a), although large standard deviations are observed for ξ potential values for high concentrations of NaCl at about and above 1 M. Similarly, as the CaCl2 concentration increases in the CaCl2-QDs system, the ξ potential of QDs decreased, as illustrated by the trend (linear) regression analysis (Fig. 5c), although large standard deviations are reported for ξ potential values for high concentrations of CaCl2 at about and above 0.1 M. The Pearson’s correlation coefficients for the ξ potential values of QDs and the ionic strength values of NaCl-QDs system and the ξ potential values of QDs and the ionic strength values of CaCl2-QD system are 0.21 and 0.63, respectively.
3.3 Effect of Suwannee River natural organic matter on quantum dot nanoparticles in monovalent and divalent electrolyte solutions
The stability of QDs was also investigated as a function of both the monovalent and divalent electrolyte concentrations in the presence SRNOM at 10 mg/L. Here, an increase in the monovalent electrolyte NaCl concentration from 0.01 to 3.5 M increased the z-avg. HD of QD aggregates from 61.4 nm to 107.2 nm. The presence of SRNOM resulted in a small increase in the size of the QD aggregates for each of NaCl concentrations tested (i.e., 0.01 to 3.5 M, except 0.1 M), and induced a monodispersed and narrower size distribution of QDs suspended in the monovalent electrolyte NaCl concentrations (Fig. 3c, d).
Also in the presence of SRNOM, the effect of the divalent electrolyte CaCl2 on the z-avg. HDs of QDs in suspensions was concentration-dependent (Fig. 2b), and an increase in the divalent electrolyte CaCl2 concentration from 0.001 to 0.25 M increased the z-avg. HD of QD aggregates from 51.9 nm to 692.9 nm. The presence of SRNOM in QDs suspended in divalent electrolyte CaCl2 solutions enhanced the aggregation of QDs, resulting in the increase of z-avg. HDs of QDs by approximately 19.3%, 42.1%, 13.8%, 1.5%, and 24.8%, at CaCl2 concentrations of 0.002, 0.003, 0.005, 0.01, and 0.1 M (Table 3), respectively. A monodispersed and narrow size distribution of QDs was observed in suspensions in the presence of 10 mg/L SRNOM with CaCl2 concentrations varying between 0.001 and 0.002 M (Fig. 4c, d). However, a poly-dispersed and larger size distribution of QDs was observed in suspensions with CaCl2 concentrations, varying between 0.0025 and 0.25 M (Fig. 4c, d).
The influence of electrolyte type and concentration on the surface charge properties of QDs in the presence of 10 mg/L SRNOM was characterized as shown in Figs. 5b and d. The presence of SRNOM did not show a measurable difference in ξ potentials of QDs at each electrolyte concentration (both for monovalent electrolyte NaCl and divalent electrolyte CaCl2) compared to the absence of SRNOM due to the overlap of the standard deviations. However, as the NaCl concentration increases in the NaCl-SRNOM-QDs system, the ξ potential of QDs decreased, as illustrated by the trend (linear) regression analysis (Fig. 5b), although large standard deviations are observed for ξ potential values for high concentrations of NaCl at about and above 1 M. Similarly, as the CaCl2 concentration increases in the CaCl2-SRNOM-QDs system, the ξ potential of QDs decreased, as illustrated by the trend (linear) regression analysis (Fig. 5d), although large standard deviations are reported for ξ potential values for high concentrations of CaCl2 at about and above 0.1 M. The Pearson’s correlation coefficients for the ξ potential values of QDs and the ionic strength values of NaCl-SRNOM-QDs system and the ξ potential values of QDs and the ionic strength values of CaCl2-SRNOM-QD system are 0.45 and 0.61, respectively.
3.4 Effect of monovalent and divalent electrolytes on aggregation kinetics of quantum dot nanoparticles
To study the effects of electrolytes type and ionic strength on the aggregation kinetics of QDs, the z-avg. HDs of QD nanoparticles were measured at different electrolyte concentrations over a period of ~ 11 min (Figs. 6 and 7). The type and concentration of the electrolytes significantly affected both the aggregation kinetics and the z-avg. HDs. An increase in the electrolyte concentration resulted in a significant increase in the aggregation rates in most of the systems analyzed (Fig. 8). The CaCl2 systems produced a higher set of aggregation rates than NaCl within the ranges of electrolyte concentrations analyzed. The aggregation rates of the NaCl increased from 0.007 to 0.042 nm·s−1 with an increase in ionic strength from 0.5 to 3.5 M, respectively. The size of the QD aggregates in NaCl was observed as nearly constant with time at low electrolyte concentrations (0.001 to 0.875 M), with a slight increase observed at higher electrolyte concentrations (2.5 to 3.5 M) (Table 2 and Fig. 6). QD aggregation was observed in all suspensions with the addition of CaCl2, as the z-avg. HD of QDs increased with time (Table 2 and Fig. 7). Higher aggregation rates of QD nanoparticles were measured in the presence of the divalent electrolyte compared to the monovalent electrolyte for the same ionic strength. In the case of CaCl2, an increase in aggregation rates occurred from 0.035 to 0.865 nm·s−1 with an increase in ionic strength from 0.0001 to 0.004 M, respectively. The aggregation process was almost constant and independent of the CaCl2 concentration again when the CaCl2 concentration was higher than 0.004 M (Fig. 8).

The growth of the z-average hydrodynamic diameter (z-avg. HD) of QDs suspended in aqueous chloride salt solutions of representative monovalent electrolyte NaCl concentrations and the absence and presence of SRNOM at 10 mg/L: a 0.01 M, b 0.1 M, c 2 M, d 2.5 M, e 3 M, and f 3.5 M

The growth of the z-average hydrodynamic diameter (z-avg. HDs) of QDs suspended in aqueous chloride salt solutions of representative divalent electrolyte CaCl2 concentrations and the absence and presence of SRNOM at 10 mg/L: a 0.001 M, b 0.002 M, c 0.003 M, d 0.005 M, e 0.01 M, and f 0.05 M

Aggregation rates of QDs suspended in aqueous chloride salt solutions as a function of the types and concentrations of electrolytes and the absence and presence of the SRNOM at 10 mg/L: a Monovalent electrolyte NaCl in both the absence and presence of the SRNOM at 10 mg/L, b Divalent electrolyte CaCl2 in both the absence and presence of the SRNOM at 10 mg/L
3.5 Effect of Suwannee River natural organic matter on aggregation kinetics of quantum dot nanoparticles in monovalent and divalent electrolytes solutions
To examine the effects of 10 mg/L of SRNOM on the aggregation kinetics of QDs in the presence of different types of electrolytes and ionic strengths, the z-avg. HDs of QDs were measured over a period of ~ 11 min (Fig. 6 and 7). The presence of SRNOM had significant effects on the aggregation rates of QDs in suspensions with different concentrations of NaCl and CaCl2 (Fig. 8). The aggregation profiles of QDs as a function of time in suspensions at different monovalent electrolyte NaCl and divalent electrolyte CaCl2 concentrations in the presence of 10 mg/L SRNOM are shown in Figs. 6 and 7, respectively.
In the presence of SRNOM, the size of the QD aggregates in suspensions under low NaCl concentration conditions (i.e., 0.01 to 2 M) increased slightly with time (Fig. 6). Therefore, in the presence of SRNOM, the aggregation rates of QDs increased from 0.01 to 0.024 nm s−1 with the increase of NaCl concentrations from 0.01 to 2 M, respectively (Fig. 8a). The presence of 10 mg/L SRNOM enhanced the aggregation rates of QDs significantly compared to the absence of SRNOM in the monovalent electrolyte solutions under low NaCl concentration conditions (i.e., 0.01 to 2 M). Under high NaCl concentration conditions (i.e., 2.5 to 3.5 M), the aggregation rates of QDs in suspensions in the presence of SRNOM increased significantly with the increase of the NaCl concentrations.
In the presence of SRNOM, the z-avg. HDs of QDs increased rapidly with time once the concentration of CaCl2 in suspension was above 0.001 M, which resulted in a rapid aggregation of QDs (Fig. 7). The aggregation rates of QDs were highly correlated with the concentration of CaCl2 in the regimes of 0.001 to 0.003 M (Fig. 8b). QD aggregation rates increased from 0.012 to 0.800 nm∙s−1 as CaCl2 concentration increased from 0.001 to 0.003 M (Fig. 8), respectively. The aggregation rate was then almost constant and independent of the CaCl2 concentration when the CaCl2 concentrations were higher than 0.003 M (Fig. 8b). As also observed in the absence of SRNOM, the CaCl2 systems produced higher aggregation rates as the NaCl at the same electrolyte concentration in the presence of 10 mg/L SRNOM. As an overall trend, the presence of SRNOM in the divalent electrolyte CaCl2 solutions enhanced the aggregation rates of QDs under all CaCl2 concentration conditions examined in this study.
4 Discussion
4.1 Stability of quantum dot nanoparticles in diH2O at pH 7
The water-soluble QDs with a concentration of 5 × 10–8 M diluted by diH2O at pH 7 utilized in this study were well dispersed, with a z-avg. HD of 57.5 nm. Compared to the synthesized water-soluble QDs at concentration of 1.38 × 10–5 M with a z-avg. HD of 46.8 nm, the slight increase in size to 57.5 nm when diluted in diH2O is a strong indicator of their stability. The size of the QDs diluted in diH2O is smaller than the results in previous studies which are summarized in Table 4. For example, the diameters of TGA-CdTe QDs, SLN-CdSe QDs, and F-68/SDS/CTAB-CdSe QDs dispersed in diH2O were 60–100 nm [151], 92.3 nm [152], and 159/178/266 nm [153], respectively. Therefore, the spontaneous aggregation and the formation of QD nanoaggregates did not occur when the QDs were diluted in diH2O, which differed from other studies on metal-based nanoparticles, such as ZnO, in which nanoparticles easily formed large aggregates when diluted in water [154].
The stability of the CdSe QDs in water is determined by both the surface modification and the surface charge [93, 153]. The measurements of the ξ potential of the nanoparticles suspended in aqueous solutions are used to assess changes in the surface charges. When the ξ potential values are higher than \(\pm\) 30 mV, the charged particles may provide sufficient electrostatic repulsive forces to stabilize the particles in suspension by forming an EDL [134, 155]. In this study, the QDs suspended in diH2O with a neutral pH had a negative charge because of the deprotonation of the poly(acrylic acid) which contained carboxyl functionalities and which was used in the QD surface coatings. This finding is in agreement with previous reports indicating a negatively charged nanocrystal CdSe/ZnS QD coated with MUA (i.e., QD-COOH)[156]. Regarding the negative sign of the charged surface, the ξ potential of QDs was lower (ca. − 3.99 mV) than the ξ potential that was reported previously. For example, the ξ potentials of poly(acrylic acid) modified CdSe/ZnS QDs in suspensions at pH 7.0 were − 45.8 mV [38] and − 53.9 mV [123]. Therefore, the limited surface charge may prove insufficient for generating an electrostatic repulsive barrier between the QDs to prevent aggregation.
To examine the interaction between particles, the total potential energy was calculated based on the classical DLVO theory of the stability of colloids. The results of the DLVO interaction net potential energy profile are presented in Fig. 9. The total potential energy is described essentially by the van der Waals attractive interaction between particles at distance less than 20 nm. Though the net total potential energy was dominated by the electrostatic repulsive interaction between particles at distances of more than 20 nm, the energy barrier was negligible with potential energies as low as 0.25 \({k}_{B}T\). This limited repulsive energy barrier was not sufficient for maintaining QD dispersal in water as compared with the required minimum repulsive energy of 15 \({k}_{B}T\) [157]. Although the z-avg. HDs of QDs remained constant with time in diH2O at pH 7.0, the contribution of the electrostatic interactions was minor, thus inferring a dominant role of the steric repulsive force in ensuring the stability of QDs under this condition.

Total potential energy versus inter-particle distance according to DLVO theory, showing the influence of neutral (pH 7) diH2O for QDs suspended in aqueous solution. Calculation of DLVO theory includes the contributions of the van der Waals and electric double layer (EDL) interactions. The net energy (Net) shown is the sum of EDL repulsion and van der Waals attraction between two approaching particles
4.2 Stability and aggregation kinetics of quantum dot nanoparticles as a function of both ionic strength and electrolyte type
In this study, divalent (Ca2+) electrolyte was more effective in destabilizing the QDs compared to monovalent (Na+) electrolyte. The decrease in magnitude of the ξ potentials of QDs was more pronounced with CaCl2 than with NaCl, revealing the effect of ion valence on the surface charge of QDs. The results of the z-avg. HDs of QDs measured in systems with monovalent electrolyte (NaCl) concentrations varying between 0.001 and 3.5 M were between 50.1 and 122.5 nm. These results are consistent with previous findings of PAA modified QDs dispersed in monovalent electrolyte solutions. The diameter of PAA-OA-QDs (CdSe/ZnS) suspended in 0.003 M NaCl solution was 30.8 nm [38], and the PAA-QDs (CdTe/CdS) and the ODA-QDs (CdSe/ZnS) suspended in monovalent electrolyte KCl solutions with concentrations varying between 0.0001 and 0.1 M were 72–114 nm and 76–175 nm, respectively [47]. When QDs were suspended in 0.0001–0.1 M divalent electrolyte CaCl2 solutions, the QD diameters ranged from 52.5 to 685.0 nm, findings which again are corroborated by previous results on the influence of CaCl2 on QD diameters ranging from 86 to 674 nm [47, 158].
As modelled by the classical DLVO theory, the QDs were less stable when suspended in monovalent and divalent electrolyte solutions compared to diH2O, the results of which are corroborated by observed increases in the diameters of QD nanoaggregates and the enhanced aggregation rates. The attachment efficiency which is a function of particle size was also computed for different monovalent electrolyte NaCl and divalent electrolyte CaCl2 concentrations (Fig. 10). At low NaCl concentrations (0.001 to 2 M), the increase in the electrolyte concentrations did not affect the aggregation rate of QDs which resulted in an attachment efficiency (α) equal to zero. At high NaCl concentrations (2.5, 3, and 3.5 M), however, each of these electrolyte concentrations induced a high aggregation rate and a high attachment efficiency. The attachment efficiency profile of QDs for the electrolyte NaCl with concentrations ranging from 0.001 to 3.5 M was atypical for the two regimes—RLA and DLA regimes—that are consistent with the DLVO colloidal theory. The attachment efficiency profile of QDs indicated the absence of distinct DLA regimes for the QDs dispersed in suspensions with monovalent electrolyte NaCl, thus compounding the difficulty of determining the CCC for NaCl. The lack of CCC in the electrolyte NaCl concentration ranges from 0.001 to 3.5 M may indicate the absence of a sufficient level of electrolytes needed to eliminate the existing energy barrier preventing aggregation between the negatively charged QDs. Similar findings detailing the failure to develop CCCs in nanoparticles suspended in electrolyte solutions are also available for both polyvinylpyrrolidone (PVP)-Ag nanoparticles and branched polyethyleneimine (BPEI)-Ag nanoparticles suspended in NaCl solutions [159]. The aggregation kinetics of PVP-Ag and BPEI-Ag nanoparticles were not conforming to the DLVO theory, and the nanoparticles did not undergo aggregation in spite of being exposed to high electrolyte concentrations. Further, an increase in the NaCl electrolyte concentrations to 8 M for PVP-Ag nanoparticles and 1 M for BPEI-Ag nanoparticles failed to create the RLA or fast aggregation, indicating the absence of CCC in both the PVP-Ag nanoparticles and the BPEI-Ag nanoparticles at the investigated ionic strength values.

Attachment efficiency of QDs suspended in aqueous chloride salt solutions as a function of the types and concentrations of electrolytes and the absence and presence of SRNOM at 10 mg/L: a Monovalent electrolyte NaCl in both the absence and presence of SRNOM at 10 mg/L, b Divalent electrolyte CaCl2 in both the absence and presence of the SRNOM at 10 mg/L
The aggregation kinetics of QDs in the divalent electrolyte CaCl2 revealed the existence of both RLA and DLA regimes. Within the RLA regime at low CaCl2 concentrations (i.e., 0.0001 to 0.004 M), the attachment efficiency increased with an increase in CaCl2 concentrations. Under these conditions, the increase in the electrolyte concentration elevated the degree of charge screening of QDs (i.e., becoming less negative) and hence reduced the energy barrier to aggregation, consequently causing a faster rate of aggregation. Once the electrolyte concentration reached and exceeded the CCC, the sufficient presence of electrolyte in the solution, induced a complete compression of the EDL of QDs to eliminate the energy barrier between the particles. Under such DLA conditions, QDs undergo a random walk due to Brownian motion to cluster and form QD nanoaggregates. The attachment efficiencies reached a maximum value of 1 at the CCC and were independent of the CaCl2 concentration once past the CCC. As determined from the attachment efficiency profile, the CCC for CaCl2 was 0.004 M in this study.
The results of the aggregation kinetics of QDs indicated that the divalent electrolyte CaCl2 was more efficient in destabilizing the QDs than the monovalent electrolyte NaCl, a finding that is consistent with the results reported previously for other nanoparticles. Specifically, the CCC of NaCl and CaCl2 were 0.12 M and 0.0048 M, respectively, for fullerene nanoparticles [131], 0.04 M and 0.002 M, respectively, for silver nanoparticles at pH 7.0 [133], and 0.122 M and 0.0022 M, respectively, for citrate coated silver nanoparticles [111]. In the divalent electrolyte systems, the CaCl2 was reported to cause aggregation from the bridging effects between the particles [160, 161] and a screening of the surface charge of the particles [116, 162, 163]. The adsorption of Ca2+ is hypothesized as the catalyst behind the formation of interparticle bridging flocculation. As previously mentioned, QDs have a negatively charged surface. Thus, when using CaCl2 as the electrolyte, 2 mol of Ca2+ is added, which was double that of the per mol electrolyte used in NaCl. The divalent ions in the electrolyte were observed to promote a greater aggregation of QDs than their monovalent counterparts as they placed a greater level of compression on the EDLs, and increased the neutralization of the surface charge of QDs as counter-ions. Hence, screening of the surface charge resulted in a reduction of the electrostatic force of repulsion between the particles.
The DLVO theory is useful in analyzing the interaction energies of QD nanoaggregates suspended in an electrolyte solution. The effects of type and concentration of electrolytes on the DLVO calculations for QD nanoaggregates are shown in the resulting potential energy profiles in Figs. 11 and 12. Here, a demonstrable decrease of the energy barrier is evident with a diminishing electrostatic repulsion and a more significant van der Waals attraction with an increase in the concentration of both monovalent and divalent electrolytes.

Total potential energy versus inter-particle distance according to DLVO theory, showing the influence of representative monovalent electrolyte NaCl concentrations and the absence and presence of SRNOM at 10 mg/L for QDs suspended in aqueous chloride salt solutions: a 0.01 M, b 0.1 M, c 2 M, d 2.5 M, e 3 M, and f 3.5 M. Calculation of DLVO theory includes the contributions of the van der Waals and electric double layer (EDL) interactions. The net energy (Net) shown is the sum of EDL repulsion and van der Waals attraction between two approaching particles

Total potential energy versus inter-particle distance according to DLVO theory, showing the influence of representative divalent electrolyte CaCl2 concentrations and the absence and presence of the SRNOM at 10 mg/L for QDs suspended in aqueous chloride salt solutions: a 0.001 M, b 0.002 M, c 0.003 M, d 0.005 M, e 0.01 M, and f 0.05 M. Calculation of DLVO theory includes the contributions of the van der Waals and electric double layer (EDL) interactions. The net energy (Net) shown is the sum of EDL repulsion and van der Waals attraction between two approaching particles
As indicated in the potential energy profile, the introduction of a low concentration of NaCl (0.001 M) resulted in a significant compression of the EDL such that the electrostatic repulsive energy was rather small (< 10 \({K}_{B}T\)) and that the net energy between particles was attractive when the distance between particles was less than 5 nm. An increase in NaCl concentration caused a rapid decrease in the energy barriers between particles. For instance, the repulsive energy was less than 5 \({K}_{B}T\) in suspension with a NaCl concentration of 0.01 M and an EDL that was completely compressed, which eliminated the repulsive energy barrier when the NaCl concentration exceeded 0.5 M. For CaCl2, the attractive energy was the dominating contributor for the net potential energy of all electrolyte concentrations investigated. The correlation between the electrostatic interaction energy and the CaCl2 concentration was positive only at the low electrolyte concentration range. For example, the net potential energy between QDs at the separation distance of 50 nm increased from 0.01 \({K}_{B}T\) to 3.09 \({K}_{B}T\) when the CaCl2 concentration increased from 0.001 M to 0.005 M, which was independent of the electrolyte concentrations either at or above CCC. The results of the aggregation kinetics of QDs in the CaCl2 solutions support this finding.
4.3 Stability and aggregation kinetics of quantum dot nanoparticles as a function of ionic strength, electrolyte type, and Suwannee River natural organic matter
The presence of SRNOM destabilized the QDs at all NaCl concentrations examined (i.e., 0.01 to 3.5 M). The addition of SRNOM failed to produce appreciable effects on the calculated net potential energy (Fig. 11), and to influence the aggregation extent and rates by changing the energy barriers between QDs. The results suggested a governing role of the steric interactions between the nanoparticles, the electrolytes, and the SRNOM in determining the colloidal stability and aggregation of QDs under monovalent electrolyte conditions. Monovalent cations are known to form bridges between molecules [164,165,166] with the subsequent bridging interactions resulting in nanoparticle aggregation. For example, AuNPs coated with MUA (11-mercaptoundecanoic acid) strengthened the bridging interaction between monovalent cations and the carboxylate groups on two interacting nanoparticles. This interaction, which was stronger than the van der Waals attractions, accounted for the aggregation of like-charged nanoparticles [166]. In this study, the formation of single rod-like complexes [29, 166] occurred between the SRNOM molecules and the monovalent cations, which brought QDs together by adsorbing concurrently onto at least two QDs. This bridging effect may be rational explanation for the destabilization of QDs by the SRNOM in the monovalent electrolyte.
As shown in Fig. 10, the enhanced aggregation of QDs by the SRNOM was also observed in the divalent electrolyte system with the van der Waals attraction predominant in the interaction between QDs (Fig. 12). The increase in the magnitude of the net potential energy between QDs was obtained by the SRNOM in the divalent electrolyte system. Specifically, CaCl2 concentrations of 0.003 and 0.005 M respectively caused an increase in the net potential energy between QDs at a separation distance of 10 nm from 13.30 to 20.41 KBT and from 26.63 to 30.32 KBT in the presence of SRNOM. The increased net potential energy may account for the observed enhancement of aggregation of QDs by the SRNOM. Furthermore, the CaCl2 may enhance the aggregation of QDs through the mechanism of divalent cation bridging. The interaction between divalent cation and SRNOM resulted in the formation of macromolecule aggregates of SRNOM through calcium complexation, aggregates that subsequently bridge the QDs and cause the enhanced destabilization. These bridging phenomena are also proposed to explain the enhanced aggregation of fullerene nanoparticles by HA [105], hematite nanoparticles by alginate [167], zinc oxide by NOM [34], citrate-coated silver nanoparticles by HA [111], graphene oxide nanomaterials by NOM [168], titanium oxide by EPS [169], and copper nanoparticles by DOC [170]. The degree of aggregation formation between QDs through cation-SRNOM bridges was superior for Ca2+ cations compared to Na+ cations due to more effective and strong interactions of Ca2+ cations with SRNOM than Na+ cations.
5 Conclusion
The environmental behavior and toxicological risks of nanoparticles are related to their physico-chemical properties, particularly their surface coating properties, and the chemistry of the environment encountered. Although it is impossible to characterize the behavior of nanoparticles in all possible chemical scenarios found in the environment, our study aimed at contributing to the understanding of QDs in the presence of salts and organic matter commonly encountered in the terrestrial and aquatic environment. Results demonstrate that both the types and concentrations of monovalent (Na+) and divalent (Ca2+) electrolytes will significantly influence the particle size distribution, the stability behavior, and aggregation kinetics of QD nanoparticles suspended in these aqueous systems. The formation of QD nanoaggregates was controlled by the valency and concentration of cations. The increase in the cation concentrations induced a decrease in the electrostatic repulsion as a result of the enhancement of the compression of the EDL. Divalent cations generated larger QD nanoaggregates than monovalent cations at the same concentrations. Under monovalent (Na+) solutions, the aggregation of QDs was limited in that the steric forces generated by the original surface coating of QDs prevailed, whereas under divalent (Ca2+) solutions, the aggregation of QDs was enhanced due to the bridging formations between the Ca2+ in solution and the carboxyl group located in the surface coating of QDs. The QD suspended in divalent (Ca2+) solutions remained stable at low Ca2+ concentrations (< 0.002 M) or formed nanoaggregates of smaller sizes compared to QDs in high Ca2+ concentrations ranging from 0.004 M to 0.1 M.
The changes in solution chemistry exhibited a substantial effect upon the interactions between QDs and SRNOM. In the presence of SRNOM, QDs suspended in the monovalent (NaCl) and divalent (CaCl2) electrolyte solutions formed electrolyte-SRNOM-QDs’ complexes with higher aggregation rates compared to the absence of SRNOM. Despite similarities in the effect of the SRNOM on the general aggregation trends of QDs between the monovalent and divalent electrolytes, each behaved differently in terms of the mechanisms that accounted for the enhanced aggregation. In the monovalent electrolyte solutions, the destabilization induced by the SRNOM was attributed to the bridging effect caused by the single rod-like complexes of monovalent (Na+) cation and the SRNOM. In the divalent electrolyte solutions, the destabilization by the SRNOM was imputed to the bridging effect of the SRNOM aggregates produced through the calcium complexation between Ca2+ cations in solution and the carboxyl group on the SRNOM. The resulting aggregation between QDs through cation-NOM bridges was superior for Ca2+ cations compared to Na+ cations due to more effective and strong interactions of Ca2+ cations with NOM than Na+ cations.
Equations predicting the attachment efficiency indicated the presence of both the RLA and DLA regimes only for the divalent (Ca2+) electrolyte solution in both the absence and presence of SRNOM. The experimental CCCs of the QDs at pH 7.0 were 0.004 M and 0.003 for the divalent (Ca2+) electrolytes in both the absence and presence of SRNOM, respectively. The absence of a distinct DLA for the monovalent (Na+) electrolyte solution did not allow to determine the CCC of the systems in both the absence and presence of SRNOM. It was also determined that although classical DLVO theory could simulate the QD nanoparticles behavior, certain physico-chemical properties of QD nanoparticles manifested in non-DLVO behavior in systems involving nanoparticles, organic ligands coating, NOM, and salts. Examples comprise the utilization of polymer and organic ligand coatings to alter the surfaces of QD nanoparticles in the presence of monovalent electrolytes, which caused steric stabilization and a reduction in nanoparticle aggregation. Surface chemistry was also determined as most important in controlling the stability of QD nanoparticles in aqueous systems.
Although SRNOM was selected to represent NOM in aquatic systems in our study, it prevented an assessment of the effects of the fractions of NOM such as humic acids and fulvic acids. Therefore, the influence of these NOM fractionations on the interfacial interactions between metal ions and QD nanoparticles, and the mechanism of aggregation of QD nanoparticles should be further evaluated in future studies.
Thus, elucidating the stabilization/aggregation, interaction, and transformation of QD nanoparticles in aqueous systems of different chemistries will allow the forecasting of exact QD nanoparticle characteristics, aggregation states, and stability behavior under a broad range of environmental settings. Aggregation and sedimentation of nanoparticles that are released in the natural and engineered environmental systems have critical effects on the fate and transport, reactivity, bioavailability, and toxicity of these nanoparticles. Consequently, although the risk from QD nanoparticles suspended in water with salts and NOM under relevant environmental conditions may be lessened as nanoparticles are inclined to aggregation, sedimentation and/or adsorption onto the surfaces of media encountered, QDs released in water with high salt concentrations and NOM may threaten ecosystems and public health as they may settle in the sediments of aquatic systems. Such knowledge is essential for evaluating the environmental and toxicological risks of exposure to QD nanoparticles.
References
Vasudevan D, Gaddam RR, Trinchi A, Cole I (2015) Core–shell quantum dots: Properties and applications. J Alloy Compd 636:395–404. https://doi.org/10.1016/j.jallcom.2015.02.102
Alivisatos P (2004) The use of nanocrystals in biological detection. Nat Biotechnol 22(1):47–52. https://doi.org/10.1038/nbt927
Smith AM, Duan H, Mohs AM, Nie S (2008) Bioconjugated quantum dots for in vivo molecular and cellular imaging. Adv Drug Deliv Rev 60(11):1226–1240. https://doi.org/10.1016/j.addr.2008.03.015
Geissler D, Charbonniere LJ, Ziessel RF, Butlin NG, Lohmannsroben HG, Hildebrandt N (2010) Quantum dot biosensors for ultrasensitive multiplexed diagnostics. Angewandte Chemie-International Edition 49(8):1396–1401. https://doi.org/10.1002/anie.200906399
Reiss P, Protière M, Li L (2009) Core/shell semiconductor nanocrystals. Small 5(2):154–168. https://doi.org/10.1002/smll.200800841
Yang YL, Mao GB, Ji XH, He ZK (2020) DNA-templated quantum dots and their applications in biosensors, bioimaging, and therapy. J Mater Chem 8(1):9–17. https://doi.org/10.1039/c9tb01870k
Yu TY, Wei DM, Li Z, Pan LJ, Zhang ZL, Tian ZQ, Liu ZH (2020) Target-modulated sensitization of upconversion luminescence by NIR-emissive quantum dots: A new strategy to construct upconversion biosensors. Chem Commun 56(13):1976–1979. https://doi.org/10.1039/c9cc09220j
Li D, Li G, Guo W, Li P, Wang E, Wang J (2008) Glutathione-mediated release of functional plasmid DNA from positively charged quantum dots. Biomaterials 29(18):2776–2782. https://doi.org/10.1016/j.biomaterials.2008.03.007
Qi L, Gao X (2008) Emerging application of quantum dots for drug delivery and therapy. Expert Opin Drug Deliv 5(3):263–267. https://doi.org/10.1517/17425247.5.3.263
Bagalkot V, Zhang L, Levy-Nissenbaum E, Jon S, Kantoff PW, Langer R, Farokhzad OC (2008) Quantum dot−aptamer conjugates for synchronous cancer imaging, therapy, and sensing of drug delivery based on bi-fluorescence resonance energy transfer. Nano Lett 7(10):3065–3070. https://doi.org/10.1021/nl071546n
Yezhelyev MV, Gao X, Xing Y, Al-Hajj A, Nie S, O’Regan RM (2006) Emerging use of nanoparticles in diagnosis and treatment of breast cancer. Lancet Oncol 7(8):657–667. https://doi.org/10.1016/S1470-2045(06)70793-8
Rajabi HR, Khani O, Shamsipur M, Vatanpour V (2013) High-performance pure and Fe3+-ion doped ZnS quantum dots as green nanophotocatalysts for the removal of malachite green under UV-light irradiation. J Hazard Mater 250:370–378. https://doi.org/10.1016/j.jhazmat.2013.02.007
Shamsipur M, Rajabi HR, Khani O (2013) Pure and Fe3+-doped ZnS quantum dots as novel and efficient nanophotocatalysts: Synthesis, characterization and use for decolorization of Victoria blue R. Mater Sci Semicond Process 16(4):1154–1161. https://doi.org/10.1016/j.mssp.2013.02.010
Rajabi HR, Farsi M (2015) Effect of transition metal ion doping on the photocatalytic activity of ZnS quantum dots: Synthesis, characterization, and application for dye decolorization. J Mol Catal A Chem 399:53–61. https://doi.org/10.1016/j.molcata.2015.01.029
Rajabi HR, Shahrezaei F, Farsi M (2016) Zinc sulfide quantum dots as powerful and efficient nanophotocatalysts for the removal of industrial pollutant. J Mater Sci Mater Electron 27(9):9297–9305. https://doi.org/10.1007/s10854-016-4969-4
Shamsipur M, Rajabi HR (2014) Pure zinc sulfide quantum dot as highly selective luminescent probe for determination of hazardous cyanide ion. Mater Sci Eng C Mater Biol Appl 36:139–145. https://doi.org/10.1016/j.msec.2013.12.001
Rajabi HR, Shamsipur M, Khosravi AA, Khani O, Yousefi MH (2013) Selective spectrofluorimetric determination of sulfide ion using manganese doped ZnS quantum dots as luminescent probe. Spectrochim Acta Part A Mol Biomol Spectrosc 107:256–262. https://doi.org/10.1016/j.saa.2013.01.045
Hetsch F, Xu X, Wang H, Kershaw SV, Rogach AL (2011) Semiconductor nanocrystal quantum dots as solar cell components and photosensitizers: Material, charge transfer, and separation aspects of some device topologies. J Phys Chem Lett 2(15):1879–1887. https://doi.org/10.1021/jz200802j
Kamat PV (2008) Quantum dot solar cells. Semiconductor nanocrystals as light harvesters. J Phys Chem C 112(48):18737–18753. https://doi.org/10.1021/jp806791s
Becker-Koch D, Albaladejo-Siguan M, Lami V, Paulus F, Xiang HY, Chen ZY, Vaynzof Y (2020) Ligand dependent oxidation dictates the performance evolution of high efficiency PbS quantum dot solar cells. Sustain Energy Fuels 4(1):108–115. https://doi.org/10.1039/c9se00602h
Jean J (2020) Getting high with quantum dot solar cells. Nat Energy 5(1):10–11. https://doi.org/10.1038/s41560-019-0534-8
Anikeeva PO, Halpert JE, Bawendi MG, Bulović V (2009) Quantum dot light-emitting devices with electroluminescence tunable over the entire visible spectrum. Nano Lett 9(7):2532–2536. https://doi.org/10.1021/nl9002969
Qian L, Zheng Y, Xue J, Holloway PH (2011) Stable and efficient quantum-dot light-emitting diodes based on solution-processed multilayer structures. Nat Photonics 5(9):543–548. https://doi.org/10.1038/nphoton.2011.171
Zhao J, Bardecker JA, Munro AM, Liu MS, Niu Y, Ding IK, Luo J, Chen B, Jen AKY, Ginger DS (2006) Efficient CdSe/CdS quantum dot light-emitting diodes using a thermally polymerized hole transport layer. Nano Lett 6(3):463–467. https://doi.org/10.1021/nl052417e
Wu Y, Li X, Steel D, Gammon D, Sham LJ (2004) Coherent optical control of semiconductor quantum dots for quantum information processing. Physica E 25(2):242–248. https://doi.org/10.1016/j.physe.2004.06.023
Keller AA, Wang H, Zhou D, Lenihan HS, Cherr G, Cardinale BJ, Miller R, Ji Z (2010) Stability and aggregation of metal oxide nanoparticles in natural aqueous matrices. Environ Sci Technol 44(6):1962–1967. https://doi.org/10.1021/es902987d
Li B, Chen Y, Liang W-Z, Mu L, Bridges WC, Jacobson AR, Darnault CJG (2017) Influence of cerium oxide nanoparticles on the soil enzyme activities in a soil-grass microcosm system. Geoderma 299:54–62. https://doi.org/10.1016/j.geoderma.2017.03.027
Morelli E, Cioni P, Posarelli M, Gabellieri E (2012) Chemical stability of CdSe quantum dots in seawater and their effects on a marine microalga. Aquat Toxicol 122–123:153–162. https://doi.org/10.1016/j.aquatox.2012.06.012
Navarro DA, Bisson MA, Aga DS (2012) Investigating uptake of water-dispersible CdSe/ZnS quantum dot nanoparticles by Arabidopsis thaliana plants. J Hazard Mater 211–212:427–435. https://doi.org/10.1016/j.jhazmat.2011.12.012
Tang S, Allagadda V, Chibli H, Nadeau JL, Mayer GD (2013) Comparison of cytotoxicity and expression of metal regulatory genes in zebrafish (Danio rerio) liver cells exposed to cadmium sulfate, zinc sulfate and quantum dots. Metallomics 5(10):1411–1422. https://doi.org/10.1039/C3MT20234H
Tang Y, Han S, Liu H, Chen X, Huang L, Li X, Zhang J (2013) The role of surface chemistry in determining in vivo biodistribution and toxicity of CdSe/ZnS core-shell quantum dots. Biomaterials 34(34):8741–8755. https://doi.org/10.1016/j.biomaterials.2013.07.087
Werlin R, Priester JH, Mielke RE, Krämer S, Jackson S, Stoimenov PK, Stucky GD, Cherr GN, Orias E, Holden PA (2011) Biomagnification of cadmium selenide quantum dots in a simple experimental microbial food chain. Nat Nanotechnol 6(1):65–71. https://doi.org/10.1038/nnano.2010.251
Dale AL, Casman EA, Lowry GV, Lead JR, Viparelli E, Baalousha M (2015) Modeling nanomaterial environmental fate in aquatic systems. Environ Sci Technol 49(5):2587–2593. https://doi.org/10.1021/es505076w
Zhou D, Keller AA (2010) Role of morphology in the aggregation kinetics of ZnO nanoparticles. Water Res 44(9):2948–2956. https://doi.org/10.1016/j.watres.2010.02.025
Kim AY, Berg JC (2000) Fractal aggregation: Scaling of fractal dimension with stability ratio. Langmuir 16(5):2101–2104. https://doi.org/10.1021/la990841n
Thio BJR, Zhou D, Keller AA (2011) Influence of natural organic matter on the aggregation and deposition of titanium dioxide nanoparticles. J Hazard Mater 189(1):556–563. https://doi.org/10.1016/j.jhazmat.2011.02.072
Navarro DA, Banerjee S, Watson DF, Aga DS (2011) Differences in soil mobility and degradability between water-dispersible CdSe and CdSe/ZnS quantum dots. Environ Sci Technol 45(15):6343–6349. https://doi.org/10.1021/es201010f
Wang Y, Becker MD, Colvin VL, Abriola LM, Pennell KD (2014) Influence of residual polymer on nanoparticle deposition in porous media. Environ Sci Technol 48(18):10664–10671. https://doi.org/10.1021/es500523p
Wang YG, Zhu HG, Becker MD, Englehart J, Abriola LM, Colvin VL, Pennell KD (2013) Effect of surface coating composition on quantum dot mobility in porous media. J Nanopart Res 15(8):1805. https://doi.org/10.1007/s11051-013-1805-0
Hotze EM, Phenrat T, Lowry GV (2010) Nanoparticle aggregation: Challenges to understanding transport and reactivity in the environment. J Environ Qual 39(6):1909–1924. https://doi.org/10.2134/jeq2009.0462
Zhang S, Jiang Y, Chen C-S, Spurgin J, Schwehr KA, Quigg A, Chin W-C, Santschi PH (2012) Aggregation, dissolution, and stability of quantum dots in marine environments: Importance of extracellular polymeric substances. Environ Sci Technol 46(16):8764–8772. https://doi.org/10.1021/es301000m
Godinez IG, Darnault CJG (2011) Aggregation and transport of nano-TiO2 in saturated porous media: Effects of pH, surfactants and flow velocity. Water Res 45(2):839–851. https://doi.org/10.1016/j.watres.2010.09.013
Godinez IG, Darnault CJG, Khodadoust AP, Bogdan D (2013) Deposition and release kinetics of nano-TiO2 in saturated porous media: Effects of solution ionic strength and surfactants. Environ Pollut 174:106–113. https://doi.org/10.1016/j.envpol.2012.11.002
Torkzaban S, Kim Y, Mulvihill M, Wan J, Tokunaga TK (2010) Transport and deposition of functionalized CdTe nanoparticles in saturated porous media. J Contam Hydrol 118(3):208–217. https://doi.org/10.1016/j.jconhyd.2010.10.002
Lv X, Gao B, Sun Y, Shi X, Xu H, Wu J (2014) Effects of humic acid and solution chemistry on the retention and transport of cerium dioxide nanoparticles in saturated porous media. Water Air Soil Pollut 225(10):2167. https://doi.org/10.1007/s11270-014-2167-7
Quevedo IR, Olsson ALJ, Tufenkji N (2013) Deposition kinetics of quantum dots and polystyrene latex nanoparticles onto alumina: Role of water chemistry and particle coating. Environ Sci Technol 47(5):2212–2220. https://doi.org/10.1021/es303392v
Quevedo IR, Tufenkji N (2012) Mobility of functionalized quantum dots and a model polystyrene nanoparticle in saturated quartz sand and loamy sand. Environ Sci Technol 46(8):4449–4457. https://doi.org/10.1021/es2045458
Wang M, Gao B, Tang D, Yu C (2018) Concurrent aggregation and transport of graphene oxide in saturated porous media: Roles of temperature, cation type, and electrolyte concentration. Environ Pollut 235:350–357. https://doi.org/10.1016/j.envpol.2017.12.063
Lin D, Tian X, Wu F, Xing B (2010) Fate and transport of engineered nanomaterials in the environment. J Environ Qual 39(6):1896–1908. https://doi.org/10.2134/jeq2009.0423
Uyusur B, Darnault CJG, Snee PT, Kokën E, Jacobson AR, Wells RR (2010) Coupled effects of solution chemistry and hydrodynamics on the mobility and transport of quantum dot nanomaterials in the vadose zone. J Contam Hydrol 118(3):184–198. https://doi.org/10.1016/j.jconhyd.2010.09.013
Sang W, Stoof CR, Zhang W, Morales VL, Gao B, Kay RW, Liu L, Zhang Y, Steenhuis TS (2014) Effect of hydrofracking fluid on colloid transport in the unsaturated zone. Environ Sci Technol 48(14):8266–8274. https://doi.org/10.1021/es501441e
Lecoanet HF, Wiesner MR (2004) Velocity effects on fullerene and oxide nanoparticle deposition in porous media. Environ Sci Technol 38(16):4377–4382. https://doi.org/10.1021/es035354f
Li CY, Hassan A, Palmai M, Xie Y, Snee PT, Powell BA, Murdoch LC, Darnault CJG (2021) Experimental measurements and numerical simulations of the transport and retention of nanocrystal CdSe/ZnS quantum dots in saturated porous media: Effects of pH, organic ligand, and natural organic matter. Environ Sci Pollut Res 28(7):8050–8073. https://doi.org/10.1007/s11356-020-11097-0
Limbach LK, Li Y, Grass RN, Brunner TJ, Hintermann MA, Muller M, Gunther D, Stark WJ (2005) Oxide nanoparticle uptake in human lung fibroblasts: Effects of particle size, agglomeration, and diffusion at low concentrations. Environ Sci Technol 39(23):9370–9376. https://doi.org/10.1021/es051043o
Sager TM, Porter DW, Robinson VA, Lindsley WG, Schwegler-Berry DE, Castranova V (2007) Improved method to disperse nanoparticles for in vitro and in vivo investigation of toxicity. Nanotoxicology 1(2):118–129. https://doi.org/10.1080/17435390701381596
Hardman R (2006) A Toxicologic review of quantum dots: Toxicity depends on physicochemical and environmental factors. Environ Health Perspect 114(2):165–172. https://doi.org/10.1289/ehp.8284
Lovrić J, Bazzi HS, Cuie Y, Fortin GRA, Winnik FM, Maysinger D (2005) Differences in subcellular distribution and toxicity of green and red emitting CdTe quantum dots. J Mol Med 83(5):377–385. https://doi.org/10.1007/s00109-004-0629-x
Navarro E, Baun A, Behra R, Hartmann NB, Filser J, Miao A-J, Quigg A, Santschi PH, Sigg L (2008) Environmental behavior and ecotoxicity of engineered nanoparticles to algae, plants, and fungi. Ecotoxicology 17(5):372–386. https://doi.org/10.1007/s10646-008-0214-0
Yang Y, Wang J, Zhu HG, Colvin VL, Alvarez PJ (2012) Relative susceptibility and transcriptional response of nitrogen cycling bacteria to quantum dots. Environ Sci Technol 46(6):3433–3441. https://doi.org/10.1021/es203485f
Yang Y, Mathieu JM, Chattopadhyay S, Miller JT, Wu T, Shibata T, Guo W, Alvarez PJJ (2012) Defense mechanisms of Pseudomonas aeruginosa PAO1 against quantum dots and their released heavy metals. ACS Nano 6(7):6091–6098. https://doi.org/10.1021/nn3011619
Auffan M, Rose J, Wiesner MR, Bottero J-Y (2009) Chemical stability of metallic nanoparticles: A parameter controlling their potential cellular toxicity in vitro. Environ Pollut 157(4):1127–1133. https://doi.org/10.1016/j.envpol.2008.10.002
Wang L, Zheng H, Long Y, Gao M, Hao J, Du J, Mao X, Zhou D (2010) Rapid determination of the toxicity of quantum dots with luminous bacteria. J Hazard Mater 177(1):1134–1137. https://doi.org/10.1016/j.jhazmat.2009.12.001
Xiao Q, Huang S, Su W, Li P, Liang Z, Ou J, Ma J, Liu Y (2012) Evaluate the potential environmental toxicity of quantum dots on ciliated protozoa by microcalorimetry. Thermochim Acta 547:62–69. https://doi.org/10.1016/j.tca.2012.08.013
Bouldin JL, Ingle TM, Sengupta A, Alexander R, Hannigan RE, Buchanan RA (2008) Aqueous toxicity and food chain transfer of quantum DotsTM in freshwater algae and Ceriodaphnia dubia. Environ Toxicol Chem 27(9):1958–1963. https://doi.org/10.1897/07-637.1
Katsumiti A, Gilliland D, Arostegui I, Cajaraville MP (2014) Cytotoxicity and cellular mechanisms involved in the toxicity of CdS quantum dots in hemocytes and gill cells of the mussel Mytilus galloprovincialis. Aquat Toxicol 153:39–52. https://doi.org/10.1016/j.aquatox.2014.02.003
Rocha TL, Gomes T, Cardoso C, Letendre J, Pinheiro JP, Sousa VS, Teixeira MR, Bebianno MJ (2014) Immunocytotoxicity, cytogenotoxicity and genotoxicity of cadmium-based quantum dots in the marine mussel Mytilus galloprovincialis. Mar Environ Res 101:29–37. https://doi.org/10.1016/j.marenvres.2014.07.009
Ingle TM, Alexander R, Bouldin J, Buchanan RA (2008) Absorption of semiconductor nanocrystals by the aquatic invertebrate Ceriodaphnia dubia. Bull Environ Contam Toxicol 81(3):249–252. https://doi.org/10.1007/s00128-008-9481-y
Kominkova M, Michalek P, Moulick A, Nemcova B, Zitka O, Kopel P, Beklova M, Adam V, Kizek R (2014) Biosynthesis of quantum dots (CdTe) and its effect on Eisenia fetida and Escherichia coli. Chromatographia 77(21):1441–1449. https://doi.org/10.1007/s10337-014-2775-9
Lewinski NA, Zhu H, Jo H-J, Pham D, Kamath RR, Ouyang CR, Vulpe CD, Colvin VL, Drezek RA (2010) Quantification of water solubilized CdSe/ZnS quantum dots in Daphnia magna. Environ Sci Technol 44(5):1841–1846. https://doi.org/10.1021/es902728a
Liu T, Xing R, Zhou YF, Zhang J, Su YY, Zhang KQ, He Y, Sima YH, Xu SQ (2014) Hematopoiesis toxicity induced by CdTe quantum dots determined in an invertebrate model organism. Biomaterials 35(9):2942–2951. https://doi.org/10.1016/j.biomaterials.2013.12.007
Tang S, Cai Q, Chibli H, Allagadda V, Nadeau JL, Mayer GD (2013) Cadmium sulfate and CdTe-quantum dots alter DNA repair in zebrafish (Danio rerio) liver cells. Toxicol Appl Pharmacol 272(2):443–452. https://doi.org/10.1016/j.taap.2013.06.004
Jatana S, Callahan LM, Pentland AP, DeLouise LA (2016) Impact of cosmetic lotions on nanoparticle penetration through ex vivo C57BL/6 hairless mouse and human skin: A comparison study. Cosmetics 3(1):6. https://doi.org/10.3390/cosmetics3010006
Li X, Yang X, Yuwen L, Yang W, Weng L, Teng Z, Wang L (2016) Evaluation of toxic effects of CdTe quantum dots on the reproductive system in adult male mice. Biomaterials 96:24–32. https://doi.org/10.1016/j.biomaterials.2016.04.014
Chen M, Yin H, Bai P, Miao P, Deng X, Xu Y, Hu J, Yin J (2016) ABC transporters affect the elimination and toxicity of CdTe quantum dots in liver and kidney cells. Toxicol Appl Pharmacol 303:11–20. https://doi.org/10.1016/j.taap.2016.04.017
Michalet X, Pinaud FF, Bentolila LA, Tsay JM, Doose S, Li JJ, Sundaresan G, Wu AM, Gambhir SS, Weiss S (2005) Quantum dots for live cells, in vivo imaging, and diagnostics. Science 307(5709):538. https://doi.org/10.1126/science.1104274
Naderi S, Zare H, Taghavinia N, Irajizad A, Aghaei M, Panjehpour M (2018) Cadmium telluride quantum dots induce apoptosis in human breast cancer cell lines. Toxicol Ind Health 34(5):339–352. https://doi.org/10.1177/0748233718763517
Nagy A, Steinbrück A, Gao J, Doggett N, Hollingsworth JA, Iyer R (2012) Comprehensive analysis of the effects of CdSe quantum dot size, surface charge, and functionalization on primary human lung cells. ACS Nano 6(6):4748–4762. https://doi.org/10.1021/nn204886b
Singh BR, Singh BN, Khan W, Singh HB, Naqvi AH (2012) ROS-mediated apoptotic cell death in prostate cancer LNCaP cells induced by biosurfactant stabilized CdS quantum dots. Biomaterials 33(23):5753–5767. https://doi.org/10.1016/j.biomaterials.2012.04.045
Yan M, Zhang Y, Qin H, Liu K, Guo M, Ge Y, Xu M, Sun Y, Zheng X (2016) Cytotoxicity of CdTe quantum dots in human umbilical vein endothelial cells: The involvement of cellular uptake and induction of pro-apoptotic endoplasmic reticulum stress. Int J Nanomed 11:529–542. https://doi.org/10.2147/IJN.S93591
Lundqvist M, Stigler J, Elia G, Lynch I, Cedervall T, Dawson KA (2008) Nanoparticle size and surface properties determine the protein corona with possible implications for biological impacts. Proc Natl Acad Sci 105(38):14265. https://doi.org/10.1073/pnas.0805135105
Tourinho PS, van Gestel CAM, Lofts S, Svendsen C, Soares AMVM, Loureiro S (2012) Metal-based nanoparticles in soil: Fate, behavior, and effects on soil invertebrates. Environ Toxicol Chem 31(8):1679–1692. https://doi.org/10.1002/etc.1880
Baun A, Hartmann NB, Grieger K, Kusk KO (2008) Ecotoxicity of engineered nanoparticles to aquatic invertebrates: A brief review and recommendations for future toxicity testing. Ecotoxicology 17(5):387–395. https://doi.org/10.1007/s10646-008-0208-y
Colvin VL (2003) The potential environmental impact of engineered nanomaterials. Nat Biotechnol 21(10):1166–1170. https://doi.org/10.1038/nbt875
Oberdörster G, Maynard A, Donaldson K, Castranova V, Fitzpatrick J, Ausman K, Carter J, Karn B, Kreyling W, Lai D, Olin S, Monteiro-Riviere N, Warheit D, Yang H (2005) Principles for characterizing the potential human health effects from exposure to nanomaterials: Elements of a screening strategy. Part Fibre Toxicol 2(1):8. https://doi.org/10.1186/1743-8977-2-8
Jiang J, Oberdörster G, Biswas P (2009) Characterization of size, surface charge, and agglomeration state of nanoparticle dispersions for toxicological studies. J Nanopart Res 11(1):77–89. https://doi.org/10.1007/s11051-008-9446-4
Levard C, Hotze EM, Lowry GV, Brown GE (2012) Environmental transformations of silver nanoparticles: Impact on stability and toxicity. Environ Sci Technol 46(13):6900–6914. https://doi.org/10.1021/es2037405
Li MH, Pokhrel S, Jin X, Madler L, Damoiseaux R, Hoek EMV (2011) Stability, bioavailability, and bacterial toxicity of ZnO and iron-doped ZnO nanoparticles in aquatic media. Environ Sci Technol 45(2):755–761. https://doi.org/10.1021/es102266g
Afrooz ARMN, Khan IA, Hussain SM, Saleh NB (2013) Mechanistic heteroaggregation of gold nanoparticles in a wide range of solution chemistry. Environ Sci Technol 47(4):1853–1860. https://doi.org/10.1021/es3032709
Peng C, Zhang W, Gao H, Li Y, Tong X, Li K, Zhu X, Wang Y, Chen Y (2017) Behavior and potential impacts of metal-based engineered nanoparticles in aquatic environments. Nanomaterials. https://doi.org/10.3390/nano7010021
Bathi JR, Moazeni F, Upadhyayula VKK, Chowdhury I, Palchoudhury S, Potts GE, Gadhamshetty V (2021) Behavior of engineered nanoparticles in aquatic environmental samples: Current status and challenges. Sci Total Environ 793. ARTN 148560 https://doi.org/10.1016/j.scitotenv.2021.148560
Albanese A, Tang PS, Chan WCW (2012) The effect of nanoparticle size, shape, and surface chemistry on biological systems. Annu Rev Biomed Eng 14(1):1–16. https://doi.org/10.1146/annurev-bioeng-071811-150124
Liu JF, Legros S, Ma GB, Veinot JGC, von der Kammer F, Hofmann T (2012) Influence of surface functionalization and particle size on the aggregation kinetics of engineered nanoparticles. Chemosphere 87(8):918–924. https://doi.org/10.1016/j.chemosphere.2012.01.045
Mulvihill MJ, Habas SE, Jen-La Plante I, Wan J, Mokari T (2010) Influence of size, shape, and surface coating on the stability of aqueous suspensions of CdSe nanoparticles. Chem Mater 22(18):5251–5257. https://doi.org/10.1021/cm101262s
Wiese GR, Healy TW (1970) Effect of particle size on colloid stability. Trans Faraday Soc 66:490–499. https://doi.org/10.1039/TF9706600490
Baalousha M, Manciulea A, Cumberland S, Kendall K, Lead JR (2008) Aggregation and surface properties of iron oxide nanoparticles: Influence of pH and natural organic matter. Environ Toxicol Chem 27(9):1875–1882. https://doi.org/10.1897/07-559.1
El Badawy AM, Luxton TP, Silva RG, Scheckel KG, Suidan MT, Tolaymat TM (2010) Impact of environmental conditions (pH, Ionic strength, and electrolyte type) on the surface charge and aggregation of silver nanoparticles suspensions. Environ Sci Technol 44(4):1260–1266. https://doi.org/10.1021/es902240k
French RA, Jacobson AR, Kim B, Isley SL, Penn RL, Baveye PC (2009) Influence of ionic strength, pH, and cation valence on aggregation kinetics of titanium dioxide nanoparticles. Environ Sci Technol 43(5):1354–1359. https://doi.org/10.1021/es802628n
Hu J-D, Zevi Y, Kou X-M, Xiao J, Wang X-J, Jin Y (2010) Effect of dissolved organic matter on the stability of magnetite nanoparticles under different pH and ionic strength conditions. Sci Total Environ 408(16):3477–3489. https://doi.org/10.1016/j.scitotenv.2010.03.033
Oriekhova O, Stoll S (2016) Effects of pH and fulvic acids concentration on the stability of fulvic acids–cerium (IV) oxide nanoparticle complexes. Chemosphere 144:131–137. https://doi.org/10.1016/j.chemosphere.2015.08.057
Zhu M, Wang H, Keller AA, Wang T, Li F (2014) The effect of humic acid on the aggregation of titanium dioxide nanoparticles under different pH and ionic strengths. Sci Total Environ 487:375–380. https://doi.org/10.1016/j.scitotenv.2014.04.036
Lee J, Bartelt-Hunt SL, Li Y, Gilrein EJ (2016) The influence of ionic strength and organic compounds on nanoparticle TiO2 (n-TiO2) aggregation. Chemosphere 154:187–193. https://doi.org/10.1016/j.chemosphere.2016.03.059
Mylon SE, Chen KL, Elimelech M (2004) Influence of natural organic matter and ionic composition on the kinetics and structure of hematite colloid aggregation: Implications to iron depletion in estuaries. Langmuir 20(21):9000–9006. https://doi.org/10.1021/la049153g
Van Hoecke K, De Schamphelaere KAC, Van der Meeren P, Smagghe G, Janssen CR (2011) Aggregation and ecotoxicity of CeO2 nanoparticles in synthetic and natural waters with variable pH, organic matter concentration and ionic strength. Environ Pollut 159(4):970–976. https://doi.org/10.1016/j.envpol.2010.12.010
Zhao J, Li Y, Wang XJ, Xia XH, Shang EX, Ali J (2021) Ionic-strength-dependent effect of suspended sediment on the aggregation, dissolution and settling of silver nanoparticles. Environ Pollut 279. ARTN 116926 https://doi.org/10.1016/j.envpol.2021.116926
Chen KL, Elimelech M (2007) Influence of humic acid on the aggregation kinetics of fullerene (C60) nanoparticles in monovalent and divalent electrolyte solutions. J Colloid Interface Sci 309(1):126–134. https://doi.org/10.1016/j.jcis.2007.01.074
Zhang Y, Chen Y, Westerhoff P, Crittenden J (2009) Impact of natural organic matter and divalent cations on the stability of aqueous nanoparticles. Water Res 43(17):4249–4257. https://doi.org/10.1016/j.watres.2009.06.005
Cao TC, Elimelech M (2021) Colloidal stability of cellulose nanocrystals in aqueous solutions containing monovalent, divalent, and trivalent inorganic salts. J Colloid Interface Sci 584:456–463. https://doi.org/10.1016/j.jcis.2020.09.117
Furman O, Usenko S, Lau BLT (2013) Relative importance of the humic and fulvic fractions of natural organic matter in the aggregation and deposition of silver nanoparticles. Environ Sci Technol 47(3):1349–1356. https://doi.org/10.1021/es303275g
Navarro DAG, Watson DF, Aga DS, Banerjee S (2009) Natural organic matter-mediated phase transfer of quantum dots in the aquatic environment. Environ Sci Technol 43(3):677–682. https://doi.org/10.1021/es8017623
Slaveykova VI, Startchev K (2009) Effect of natural organic matter and green microalga on carboxyl-polyethylene glycol coated CdSe/ZnS quantum dots stability and transformations under freshwater conditions. Environ Pollut 157(12):3445–3450. https://doi.org/10.1016/j.envpol.2009.06.017
Baalousha M, Nur Y, Römer I, Tejamaya M, Lead JR (2013) Effect of monovalent and divalent cations, anions and fulvic acid on aggregation of citrate-coated silver nanoparticles. Sci Total Environ 454–455:119–131. https://doi.org/10.1016/j.scitotenv.2013.02.093
Gallego-Urrea JA, Holmberg JP, Hassellov M (2014) Influence of different types of natural organic matter on titania nanoparticle stability: Effects of counter ion concentration and pH. Environ Sci-Nano 1(2):181–189. https://doi.org/10.1039/c3en00106g
Dunphy Guzman KA, Finnegan MP, Banfield JF (2006) Influence of surface potential on aggregation and transport of titania nanoparticles. Environ Sci Technol 40(24):7688–7693. https://doi.org/10.1021/es060847g
Nowack B, Bucheli TD (2007) Occurrence, behavior and effects of nanoparticles in the environment. Environ Pollut 150(1):5–22. https://doi.org/10.1016/j.envpol.2007.06.006
Akaighe N, Depner SW, Banerjee S, Sharma VK, Sohn M (2012) The effects of monovalent and divalent cations on the stability of silver nanoparticles formed from direct reduction of silver ions by Suwannee River humic acid/natural organic matter. Sci Total Environ 441:277–289. https://doi.org/10.1016/j.scitotenv.2012.09.055
Huynh KA, Chen KL (2011) Aggregation kinetics of citrate and polyvinylpyrrolidone coated silver nanoparticles in monovalent and divalent electrolyte solutions. Environ Sci Technol 45(13):5564–5571. https://doi.org/10.1021/es200157h
Quevedo IR, Tufenkji N (2009) Influence of solution chemistry on the deposition and detachment kinetics of a CdTe quantum dot examined using a quartz crystal microbalance. Environ Sci Technol 43(9):3176–3182. https://doi.org/10.1021/es803388u
Chen KL, Elimelech M (2008) Interaction of fullerene (C60) nanoparticles with humic acid and alginate coated silica surfaces: Measurements, mechanisms, and environmental implications. Environ Sci Technol 42(20):7607–7614. https://doi.org/10.1021/es8012062
Franchi A, O’Melia CR (2003) Effects of natural organic matter and solution chemistry on the deposition and reentrainment of colloids in porous media. Environ Sci Technol 37(6):1122–1129. https://doi.org/10.1021/es015566h
Jones EH, Su C (2014) Transport and retention of zinc oxide nanoparticles in porous media: Effects of natural organic matter versus natural organic ligands at circumneutral pH. J Hazard Mater 275:79–88. https://doi.org/10.1016/j.jhazmat.2014.04.058
Snee PT, Chan Y, Nocera DG, Bawendi MG (2005) Whispering-gallery-mode lasing from a semiconductor nanocrystal/microsphere resonator composite. Adv Mater 17(9):1131–1136. https://doi.org/10.1002/adma.200401571
Snee PT, Tyrakowski CM, Page LE, Isovic A, Jawaid AM (2011) Quantifying quantum dots through Förster resonant energy transfer. The Journal of Physical Chemistry C 115(40):19578–19582. https://doi.org/10.1021/jp205833q
Uyuşur B, Snee PT, Li C, Darnault CJG (2016) Quantitative imaging and in situ concentration measurements of quantum dot nanomaterials in variably saturated porous media. J Nanomater 2016:10. https://doi.org/10.1155/2016/8237029
Liu D, Snee PT (2011) Water-soluble semiconductor nanocrystals cap exchanged with metalated ligands. ACS Nano 5(1):546–550. https://doi.org/10.1021/nn1025934
Pouya S, Koochesfahani M, Snee P, Bawendi M, Nocera D (2005) Single quantum dot (QD) imaging of fluid flow near surfaces. Exp Fluids 39(4):784–786. https://doi.org/10.1007/s00348-005-0004-x
McBride J, Treadway J, Feldman LC, Pennycook SJ, Rosenthal SJ (2006) Structural basis for near unity quantum yield core/shell nanostructures. Nano Lett 6(7):1496–1501. https://doi.org/10.1021/nl060993k
Nellist PD, Chisholm MF, Dellby N, Krivanek OL, Murfitt MF, Szilagyi ZS, Lupini AR, Borisevich A, Sides WH, Pennycook SJ (2004) Direct sub-angstrom imaging of a crystal lattice. Science 305(5691):1741–1741. https://doi.org/10.1126/science.1100965
Conway JR, Adeleye AS, Gardea-Torresdey J, Keller AA (2015) Aggregation, dissolution, and transformation of copper nanoparticles in natural waters. Environ Sci Technol 49(5):2749–2756. https://doi.org/10.1021/es504918q
Solomon T (2001) The definition and unit of ionic strength. J Chem Educ 78(12):1691. https://doi.org/10.1021/ed078p1691
Li C, Hassan A, Palmai M, Snee PT, Baveye PC, Darnault CJG (2020) Colloidal stability and aggregation kinetics of nanocrystal CdSe/ZnS quantum dots in aqueous systems: Effects of pH and organic ligands. J Nanoparticle Res 22(11). ARTN 349 https://doi.org/10.1007/s11051-020-05080-6
Chen KL, Elimelech M (2006) Aggregation and deposition kinetics of fullerene (C60) nanoparticles. Langmuir 22(26):10994–11001. https://doi.org/10.1021/la062072v
Chen KL, Elimelech M (2009) Relating colloidal stability of fullerene (C60) nanoparticles to nanoparticle charge and electrokinetic properties. Environ Sci Technol 43(19):7270–7276. https://doi.org/10.1021/es900185p
Li XA, Lenhart JJ, Walker HW (2010) Dissolution-accompanied aggregation kinetics of silver nanoparticles. Langmuir 26(22):16690–16698. https://doi.org/10.1021/la101768n
Nur Y, Lead JR, Baalousha M (2015) Evaluation of charge and agglomeration behavior of TiO2 nanoparticles in ecotoxicological media. Sci Total Environ 535:45–53. https://doi.org/10.1016/j.scitotenv.2014.11.057
Metreveli G, Philippe A, Schaumann GE (2015) Disaggregation of silver nanoparticle homoaggregates in a river water matrix. Sci Total Environ 535:35–44. https://doi.org/10.1016/j.scitotenv.2014.11.058
Derjaguin B, Landau L (1941) The theory of stability of highly charged lyophobic sols and coalescence of highly charged particles in electrolyte solutions. Acta Physicochim URSS 14(633–52):58
Verwey EJW, Overbeek JTG (1955) Theorie der stabilität lyophober kolloide. Kolloid-Zeitschrift 141(1):44–45. https://doi.org/10.1007/BF01511752
Behrens SH, Christl DI, Emmerzael R, Schurtenberger P, Borkovec M (2000) Charging and aggregation properties of carboxyl latex particles: Experiments versus DLVO theory. Langmuir 16(6):2566–2575. https://doi.org/10.1021/la991154z
Hogg R, Healy TW, Fuerstenau DW (1966) Mutual coagulation of colloidal dispersions. Trans Faraday Soc 62:1638–1651. https://doi.org/10.1039/TF9666201638
Ohshima H (1995) Effective surface potential and double-layer interaction of colloidal particles. J Colloid Interface Sci 174(1):45–52. https://doi.org/10.1006/jcis.1995.1362
Hamaker HC (1937) The London—van der Waals attraction between spherical particles. Physica 4(10):1058–1072. https://doi.org/10.1016/S0031-8914(37)80203-7
He YT, Wan JM, Tokunaga T (2008) Kinetic stability of hematite nanoparticles: The effect of particle sizes. J Nanopart Res 10(2):321–332. https://doi.org/10.1007/s11051-007-9255-1
Noh M, Kim T, Lee H, Kim C-K, Joo S-W, Lee K (2010) Fluorescence quenching caused by aggregation of water-soluble CdSe quantum dots. Colloids Surf, A 359(1):39–44. https://doi.org/10.1016/j.colsurfa.2010.01.059
Rabani E (2001) An interatomic pair potential for cadmium selenide. J Chem Phys 116(1):258–262. https://doi.org/10.1063/1.1424321
Blum L, Hoeye JS (1977) Mean spherical model for asymmetric electrolytes. 2. Thermodynamic properties and the pair correlation function. J Phys Chem 81(13):1311–1316. https://doi.org/10.1021/j100528a019
Kohonen MM, Karaman ME, Pashley RM (2000) Debye length in multivalent electrolyte solutions. Langmuir 16(13):5749–5753. https://doi.org/10.1021/la991621c
Butt H-J (1991) Measuring electrostatic, van der Waals, and hydration forces in electrolyte solutions with an atomic force microscope. Biophys J 60(6):1438–1444. https://doi.org/10.1016/S0006-3495(91)82180-4
Israelachvili JN (1985) Intermolecular and surface forces: With applications to colloidal and biological systems. Academic Press, Cambridge
Israelachvili JN (2011) Intermolecular and surface forces. Revised third edition. Academic Press
Israelachvili JN, Adams GE (1978) Measurement of forces between two mica surfaces in aqueous electrolyte solutions in the range 0–100 nm. J Chem Soc Faraday Trans 1: Phys Chem Condens Phases 74:975–1001. https://doi.org/10.1039/F19787400975
Zhang Y, Chen Y, Westerhoff P, Crittenden JC (2007) Stability and removal of water soluble CdTe quantum dots in water. Environ Sci Technol 42(1):321–325. https://doi.org/10.1021/es0714991
Liu W, He Z, Liang J, Zhu Y, Xu H, Yang X (2008) Preparation and characterization of novel fluorescent nanocomposite particles: CdSe/ZnS core-shell quantum dots loaded solid lipid nanoparticles. J Biomed Mater Res Part A 84(4):1018–1025. https://doi.org/10.1002/jbm.a.31205
Guo G, Liu W, Liang J, He Z, Xu H, Yang X (2007) Probing the cytotoxicity of CdSe quantum dots with surface modification. Mater Lett 61(8):1641–1644. https://doi.org/10.1016/j.matlet.2006.07.105
Peng Y-H, Tso C-P, Tsai Y-C, Zhuang C-M, Shih Y-H (2015) The effect of electrolytes on the aggregation kinetics of three different ZnO nanoparticles in water. Sci Total Environ 530–531:183–190. https://doi.org/10.1016/j.scitotenv.2015.05.059
Honary S, Zahir F (2013) Effect of zeta potential on the properties of nano-drug delivery systems—a review (Part 1). Trop J Pharm Res 12(2):255–264. https://doi.org/10.4314/tjpr.v12i2.19
Hoshino A, Fujioka K, Oku T, Suga M, Sasaki YF, Ohta T, Yasuhara M, Suzuki K, Yamamoto K (2004) Physicochemical properties and cellular toxicity of nanocrystal quantum dots depend on their surface modification. Nano Lett 4(11):2163–2169. https://doi.org/10.1021/nl048715d
Uchikawa H, Hanehara S, Sawaki D (1997) The role of steric repulsive force in the dispersion of cement particles in fresh paste prepared with organic admixture. Cem Concr Res 27(1):37–50. https://doi.org/10.1016/S0008-8846(96)00207-4
Torkzaban S, Bradford SA, Wan J, Tokunaga T, Masoudih A (2013) Release of quantum dot nanoparticles in porous media: Role of cation exchange and aging time. Environ Sci Technol 47(20):11528–11536. https://doi.org/10.1021/es402075f
El Badawy AM, Scheckel KG, Suidan M, Tolaymat T (2012) The impact of stabilization mechanism on the aggregation kinetics of silver nanoparticles. Sci Total Environ 429:325–331. https://doi.org/10.1016/j.scitotenv.2012.03.041
Gao J, Youn S, Hovsepyan A, Llaneza VL, Wang Y, Bitton G, Bonzongo J-CJ (2009) Dispersion and toxicity of selected manufactured nanomaterials in natural river water samples: effects of water chemical composition. Environ Sci Technol 43(9):3322–3328. https://doi.org/10.1021/es803315v
Kirby GH, Harris DJ, Li Q, Lewis JA (2004) Poly(acrylic acid)–poly(ethylene oxide) comb polymer effects on BaTiO3 nanoparticle suspension stability. J Am Ceram Soc 87(2):181–186. https://doi.org/10.1111/j.1551-2916.2004.00181.x
Domingos RF, Peyrot C, Wilkinson KJ (2010) Aggregation of titanium dioxide nanoparticles: Role of calcium and phosphate. Environ Chem 7(1):61–66. https://doi.org/10.1071/EN09110
Topuz E, Traber J, Sigg L, Talinli I (2015) Agglomeration of Ag and TiO2 nanoparticles in surface and wastewater: Role of calcium ions and of organic carbon fractions. Environ Pollut 204:313–323. https://doi.org/10.1016/j.envpol.2015.05.034
Braunschweig H, Dewhurst RD, Hupp F, Schneider C (2014) Silver(I) and thallium(I) cations as unsupported bridges between two metal bases. Chem Commun 50(99):15685–15688. https://doi.org/10.1039/C4CC08508F
Kara S, Arda E, Pekcan Ö (2007) Monovalent and divalent cation eeffects on phase transitions of ι-carrageenan. J Bioact Compat Polym 22(1):42–61. https://doi.org/10.1177/0883911506073361
Wang DW, Tejerina B, Lagzi I, Kowalczyk B, Grzybowski BA (2011) Bridging interactions and selective nanoparticle aggregation mediated by monovalent cations. ACS Nano 5(1):530–536. https://doi.org/10.1021/nn1025252
Chen KL, Mylon SE, Elimelech M (2006) Aggregation kinetics of alginate-coated hematite nanoparticles in monovalent and divalent electrolytes. Environ Sci Technol 40(5):1516–1523. https://doi.org/10.1021/es0518068
Chowdhury I, Mansukhani ND, Guiney LM, Hersam MC, Bouchard D (2015) Aggregation and stability of reduced graphene oxide: complex roles of divalent cations, pH, and natural organic matter. Environ Sci Technol 49(18):10886–10893. https://doi.org/10.1021/acs.est.5b01866
Lin D, Story SD, Walker SL, Huang QY, Cai P (2016) Influence of extracellular polymeric substances on the aggregation kinetics of TiO2 nanoparticles. Water Res 104:381–388. https://doi.org/10.1016/j.watres.2016.08.044
Xiao Y, Vijver MG, Peijnenburg WJGM (2018) Impact of water chemistry on the behavior and fate of copper nanoparticles. Environ Pollut 234:684–691. https://doi.org/10.1016/j.envpol.2017.12.015
Acknowledgements
We wish to convey our appreciation to Clemson University for supporting this research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, C., Hassan, A., Palmai, M. et al. Colloidal stability and aggregation kinetics of nanocrystal CdSe/ZnS quantum dots in aqueous systems: Effects of ionic strength, electrolyte type, and natural organic matter. SN Appl. Sci. 4, 101 (2022). https://doi.org/10.1007/s42452-022-04948-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-022-04948-7