
Abstract
Adult-onset Still’s disease (AOSD) is a multisystemic complex disorder clinically characterised by episodes of spiking fever, evanescent rash, polyarthritis or diffuse arthralgias; multiorgan involvement may develop according to the hyper-inflammatory extent. The pathogenesis of AOSD is not completely recognised. The central role of macrophage activation, which results in T helper 1 (Th1) cell cytokine activation, is well established. Pro-inflammatory cytokines such as interleukin (IL)-1, IL-6 and IL-18 play a fundamental role in disease onset and progression. The disease may develop in both children and adults with overlapping clinical features, and although several subsets depending on the clinical manifestations and the cytokines expressed have been identified, the dichotomy between systemic juvenile idiopathic arthritis (sJIA) and AOSD nowadays has been overcome, and the pathology is considered a disease continuum between ages. Various therapeutic approaches have been evaluated thus far, and different compounds are under assessment for AOSD treatment. Historically, glucocorticoids have been employed for treating systemic manifestations of Still’s disease, while conventional synthetic disease modifying anti-rheumatic drugs (csDMARDs) demonstrated efficacy in controlling the articular manifestations. Currently, biological (b) DMARDs are widely employed; IL-1 inhibitors such as anakinra and canakinumab have proven to have high efficacy and an excellent safety profile and the anti-IL-6 tocilizumab is approved for sJIA, with several trials and longitudinal studies confirming its efficacy and safety. Moreover, in the light of the ‘window of opportunity’, new evidence showed that the earlier these treatments are initiated, the sooner clinical inactivity can be achieved. Other treatment options are being considered since several molecules involved in the disease pathophysiology can be targeted through various mechanisms. This review will provide a broad overview of AOSD pathophysiology, insights into specific organ manifestations and the currently available treatments with the identification of potential therapeutic targets involved in AOSD pathogenesis will be outlined.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
AOSD is a complex multisystemic autoinflammatory disease for which diagnosis is often challenging and based on the exclusion of mimickers such as infections and haematological malignancies. |
In AOSD, both innate and adaptive immunity are upregulated, contributing to the clinical manifestations presented. |
Therapeutic options include glucocorticoids, csDMARDS and bDMARDs; however, new targets are being evaluated and should be considered in the future. |
Among bDMARDs, IL-1 and IL-6 inhibitors proved high evidence of efficacy and safety in both children and adults, to date. |
1 Introduction: Clinical Aspects of a Unique Disease
Adult-onset Still’s disease (AOSD) is a multisystem autoinflammatory disease clinically characterised by recurrent episodes of spiking fever (> 39–40°C), pink–salmon transient skin rash, and the presence of arthralgia or polyarthritis. Laboratory examinations usually show an increase in white blood cell counts (WBC), with neutrophilia [1]. An elevation in inflammatory markers such as ferritin and C-reactive protein (CRP) along with transaminases is common. Still’s disease occurs in both children and adults, and for years, systemic juvenile idiopathic arthritis (sJIA) and AOSD have been considered as two separate entities. Recent studies underscored that both sJIA and AOSD represent the same disease. The theory of the disease continuum between children and adults has been proposed by Nirmala and colleagues, who observed similarities in the gene expression of patients with sJIA and AOSD following canakinumab treatment [2], and the expression of certain pro-inflammatory cytokines was also found to be similar between children and adults [3]. However, recent evidence pointed out that there are some differences in the clinical presentation of Still’s disease in children, adults and the elderly. A multicentre study including adults and children observed that the frequency of fever, rash, myalgia, weight loss and sore throat was higher in patients with adult onset, while laboratory findings were similar amongst the groups, except for an increase in transaminases and neutrophilia that was more present in adults [4]. Conversely, patients with elderly onset (≥ 65 years) had less skin rash, sore throat, myalgia, hepatosplenomegaly and overall Pouchot’s score compared with adults (< 65 years)[5]. Furthermore, different subsets of AOSD were recently identified depending on clinical/laboratory features and on the basis of the different expressions of pro-inflammatory cytokines [6]: a first phenotype was recognised in younger patients clinically presenting with fever, skin rash and arthritis at the onset of the disease, accompanied by a remarkable increase in ferritin, and thus indicating a phenotype mostly driven by an IL-1β response [7]; the second subset included patients with high CRP and liver enzymes with hepatomegaly, implying a predominant role of IL-6 [8]; and a third pattern is instead characterised by a high systemic score and the recurrence of life-threatening complications with liver and lung involvement, and a role for interferon (IFN)-γ was therefore speculated; and finally, a fourth phenotype was associated with less fever and CRP, but with a prevalence of arthritis, and thus with a predominant role exerted by tumour necrosis factor (TNF)-α; indeed, a hyper-expression of some genes of the TNF pathway was observed in this group [6]. AOSD was historically subdivided into three different phenotypes depending on the evolution over time: the systemic pattern characterised by recurrent episodes of inflammation, the monocyclic pattern that presents only once in a lifetime and the chronic articular pattern that is predominantly characterised by joint involvement.
The diagnosis of sJIA/AOSD is essentially clinical and based on the exclusion of potential mimics such as infections, malignancies, in particular hematologic conditions such as lymphoma and other autoimmune/autoinflammatory disorders that may present with overlapping clinical and biological characteristics, that is, vasculitis, periodic fevers or the newly described vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic (VEXAS) syndrome [9]. Several sets of classification criteria have been proposed thus far, with Yamaguchi’s and Fautrel’s being the most employed [10, 11], however, at present, there are no diagnostic criteria available. To facilitate the diagnostic process, a new algorithm fixed on points-based score has been recently proposed, aiming to be an appropriate diagnostic tool for clinical practice and research [12]. The clinimetric score known as modified Pouchot’s score (mPS) [13], is uncommonly employed to evaluate disease activity, however, a recent study comparing the mPS and the systemic manifestation score (mSMS), demonstrated that both mSMS and mPS can assess disease severity, with a higher discriminative ability of mPS to identify patients with high disease severity compared with mSMS. However, there are some limitations in the application of both the scores, and mPS is not able to assess the change of disease severity in patients with serious complications [14]; for this reason, a European Alliance of Associations for Rheumatology (EULAR) ongoing initiative (DAVID project) is currently working on the development and validation of an EULAR disease activity score.
The treatment of AOSD varies according to the pattern and the clinical manifestations exhibited by patients; generally, the first-line therapy relies on the employment of traditional drugs such as non-steroidal anti-inflammatory drugs (NSAIDs) and glucocorticoids (GCs); most of the data on the efficacy of conventional synthetic (cs) disease-modifying anti-rheumatic drugs (DMARDs) come from longitudinal studies, and csDMARDs have proven to be more effective at controlling articular manifestations. The use of biological (b) DMARDs instead represents a valid option for treating both systemic and articular manifestations, providing satisfactory results in terms of safety and efficacy, despite data derived mostly from clinical trials involving children with sJIA [1]. Nowadays, new therapies are being evaluated, and several drugs are being assessed through clinical trials both in children and in adults. In this review, we will provide new insights into AOSD pathophysiology, describing the therapeutic strategies in use and the possible employable compounds for this complex multisystemic disease. An online literature search was conducted in Medline via PubMed and Embase via Ovid. The search strategy included synonyms of medical subject headings (MeSH)/Emtree and free terms of “adult-onset Still’s disease”, along with eligible drugs in use, approved and evaluated in randomised controlled clinical trials (RCTs), longitudinal observational prospective (LOP) and retrospective studies (LOR). For new drugs and compounds under evaluation in AOSD, we conducted an online search on clinicatrials.gov considering the studies listed (recruiting, completed/terminated and withdrawn).
2 The Initial Step Towards the Development of the Disease is the Interaction Between Genetic and Environmental Factors
AOSD pathophysiology is not clearly understood. Indeed, as for the majority of autoimmune and autoinflammatory disorders, an association between environmental factors and a genetic predisposition is conceivable. A single gene responsible for AOSD has not been identified, but several studies demonstrated a genetic susceptibility to AOSD and sJIA. Associations with polymorphisms in the genes encoding human leukocyte antigen (HLA), including HLA-Bw35, HLA-B17, HLA-B18, HLA-B35, HLA-DR4, HLA-DR2, HLA-DRw6 and HLA-DRB1, were noticed in the past [15,16,17,18,19]. Several studies have observed that AOSD can be strongly related to HLA-DRB1-12 and HLA-DRB1-15, but a negative correlation with HLA-DR1 and HLA-DRB1-04 21 was also reported [18]. In this context, it is important to know that subjects carrying the HLA-DRB1*15:01, or those carrying other alleles of the HLA-DRB1*15 family, may be at risk of inhibitor-triggered reactions. This is of relevance, since recent evidence observed that drug rash with eosinophilia and systemic symptoms (DRESS)-type reactions might occur in patients treated with IL-1 or IL-6 inhibitors, and are strongly associated with common HLA-DRB1*15 haplotypes [20]. Therefore, a consideration of HLA typing and vigilance for serious reactions to these drugs are suggested, and clinicians should be aware of the occurrence of these events. Regarding other genetical (non-HLA) possible associations, variations in genes involved in other systemic autoinflammatory diseases such as MEFV, responsible for Familial Mediterranean Fever, TNFRSF1A, responsible for TNF receptor-associated periodic syndrome (TRAPS) [21, 22] and NOD2, related to granulomatous diseases, were observed in patients with AOSD, despite the fact that a causative role or direct association between those variants and AOSD has not been confirmed [23, 24]. Furthermore, mutations in LACC1 gene, responsible for monogenic juvenile arthritis, were found to be associated with the systemic form of JIA [25]. A recent study demonstrated that the expression of mRNAs of molecules involved in the interferon STING pathway (i.e. CGAS, NLRP4, PKDC, STING1, XRCC5), derived from peripheral blood mononuclear cells (PBMCs) of subjects with AOSD were significantly higher than in healthy subjects [26]. In particular, NLRP4 expression and IFN-γ resulted increased in patients with AOSD complicated by macrophage activation syndrome (MAS) or secondary hemophagocytic lymphohystiocitosis (sHLH), suggesting that additional mechanisms involving NLRP4 might be responsible for this life-threatening complication. Regarding MAS, IL-18 is one of the pivotal biomarkers highly expressed during episodes. A study reported by Sugiura et al. [27] investigated the genetic polymorphisms in the human IL-18 gene and observed that seven single nucleotide polymorphisms and a single 9-base pair (bp) insertion were more frequently present in AOSD patients compared with subjects with rheumatoid arthritis (RA). Similarly, Chen et al. found a functional association between IL-18 gene-607 promoter polymorphisms and the disease course in Chinese patients with AOSD. This genotype, with a low IL-18 level, was probably related to a protective factor against both AOSD severity and progression to chronic arthritis [28]. Genetic variants near colony-stimulating factor 1 (CSF1) are associated with AOSD and the rs11102024 T allele is linked to higher macrophage (M)-CSF levels. Similarly, polymorphisms in the migration inhibitory factor (MIF) gene have been reported to influence plasma MIF levels in AOSD and can be associated with disease susceptibility or clinical presentation [29]. The associations between genetic polymorphisms of NLRP3 and its components with AOSD susceptibility and outcomes were recently investigated. A single-nucleotide polymorphism (SNP) rs11672725 of the CARD8 gene encoding for CARD, a negative regulator of NLRP3, was significantly associated with AOSD. In fact, patients carrying the rs11672725CC genotype and C allele had lower CARD8 levels and were predisposed to the development of a systemic pattern with recurrence of hyperinflammation [30]. Recently, SNPs of the autophagy-related 16-like 1 (ATG16L1) gene were reported to have a link with different AOSD phenotypes. Indeed, patients with the AA/CC/TT haplotype of ATG16L1 had lower mRNA expression of light chain (LC)3-II, reflected by autophagosome formation. This finding was clinically associated with a higher proportion of skin rash and systemic AOSD features compared with other haplotypes [31]. An association between neutrophil extracellular trap (NET) formation and a new genetic susceptibility factor was also investigated. Indeed, plasma levels of leukocyte immunoglobulin-like receptor A3 (LIR-A3), encoded by LILRA3 gene, were found to be higher in patients with AOSD compared with healthy controls; LIR-A3 is able to interfere with certain inhibitory mechanisms and in turn amplify the NETosis process and promote inflammation in AOSD [32].
These results are of remarkable interest, especially in the perspective of early intervention and treatment of specific disease manifestations [33]. If genetic susceptibility demonstrates a relevant influence in AOSD pathogenesis, it is well known that infectious triggers, including viruses (Parvovirus B19, Epstein-Barr virus, Cytomegalovirus) and bacteria (Yersinia and Mycoplasma spp.), may contribute to the onset and evolution of AOSD. In this context, seasonality was described in both sJIA [34] and AOSD [35]; however, to date the role of external factors in AOSD has not been established and a specific environmental trigger has not been identified.
3 Pathophysiology: Main Player and Side Character: The Role of Innate and Adaptive Immunity
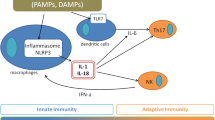
The pathophysiology of AOSD is known to be based on an imbalance between innate and adaptive immunity and is characterised by a substantial increase in pro-inflammatory cytokines that contribute to disease progression (see Fig. 1). A pivotal role is played by cells of innate immunity, mostly macrophages and neutrophils; Toll-like receptors (TLR) are widely expressed on the surface of dendritic cells (DCs), neutrophils, natural killer (NK) cells, macrophages, mast cells, B cells, T cells and other non-immune cells [36]. Their function is to sense and transfer the danger signals, namely damage-associated molecular patterns (DAMPs), and pathogen-associated molecular patterns (PAMPs), into the intracellular signalling pathways of different immune cells. Neutrophil activation is fundamental in AOSD pathogenesis, since neutrophils are responsible for the initiation and development of inflammation through the release of a wide variety of granular enzymes and antimicrobial proteins [37]. During Still’s disease flares, more than 80% of patients may develop neutrophilic leucocytosis. In addition, increasing evidence suggests that the release of NETs plays a key role in activating macrophages and perpetuating the overproduction of several pro-inflammatory cytokines through the activation of NLRP3 [38]. In turn, the activation of NLRP3 stimulates the production of IL-1β and IL-18 through caspase-1. Similarly, after sensing DAMPs and PAMPs, macrophages may recruit the MYD88 adapter and activate the NFkB intracellular pathway, inducing the release of pro-inflammatory mediators. In addition, the increase in macrophage activity can be reflected by an upregulation of MIF, M-CSF and of soluble receptor (s)CD163, the latter notably related to hyperferritinemia. In AOSD, NK cells may also be activated, and during MAS episodes their levels can be decreased in favour of an expansion of macrophages and T cells. Indeed, previous studies reported an elevation of sCD25, the soluble receptor of IL-2, potentially suggesting the activation and proliferation of CD4+ T cells [17, 39]. The Th1 polarisation of CD4+ T cells may be reflected by IL-4 increase and by the infiltration of IL-4-producing T cells in skin, sera and synovial tissues of patients with AOSD [39]. Several studies have shown that after innate immunity, adapted immunity is upregulated in AOSD, supporting the evidence that both immunological counterparts are involved in disease pathogenesis and progression [40, 41]. The role of adaptive immunity in Still’s disease has recently been investigated, particularly in relation to the newly described life-threatening complication that may involve the lungs of those affected. Indeed, pulmonary alveolar proteinosis (PAP) is a condition characterised by the deposition of proteinaceous material within the alveolar airspace. This reaction was supposed to be caused by the employment of IL-1 (i) or IL-6 receptor (R)-i inhibitors, however, the association with these anti-cytokines is still under observation and highly debated. Indeed, PAP cases have also been reported in patients with sJIA not treated with IL-1i or IL-6i, and several factors, including the recurrence of MAS, young age and 21 trisomy, have been identified as possible risk factors for PAP. The pathophysiological mechanisms proposed for PAP development in AOSD include several hypotheses, ranging from an infectious agent acting in a context of HLA susceptibility to a disease caused by an altered antigen presentation (DRESS hypothesis) or an altered cytokine milieu (cytokine plasticity hypotheses). In the DRESS hypothesis, IL-1 and IL-6 inhibitors, or their excipients, might drive pathogenic T cell responses as antigens or by altering the antigen presentation to CD4+ T cells, leading to Th2 polarisation and activating CD8+ T cells. In the cytokine plasticity hypothesis, instead, the elevation of IL-1 and IL-6 during the disease is responsible for Th17 skewing in CD4+ Th and Treg cells. By targeting IL-1 or IL-6, these cells are converted to IFN-γ-producing Th1 cells and/or IL-4-producing Th2 cells, in particular CD4+ T cells, recognising HLA-DRB1*15:XX-presented antigens (exogenous or endogenous) [20, 42]. Moreover, a high expression of chemokine (C-X-C motif) ligands (CXCL) 9, CXCL10 and IL-18 was observed in the bronchoalveolar lavage fluid of sJIA-interstitial lung disease (ILD), reflecting IFN-γ and T-cell activity. Although the proposed mechanisms appear valid to explain ILD occurrence in sJIA, these hypotheses should be further investigated with larger cohorts of children and adults. In fact, PAP has been widely described in sJIA, but has been observed in only two adults to date [43, 44]; however, these findings strengthen the concept that adaptive immunity is involved not only in Still’s disease onset and progression, but also into specific manifestations. The final result of the immunological imbalance is the massive release of pro-inflammatory cytokines and the exuberant production of mediators that may amplify and perpetuate, including IL-1β, IL-6, IL-8, IL-17, TNF-α and IFN-γ; when the production is uncontrolled, the exacerbation of this inflammatory upregulation may provoke the so-called cytokine storm [45]. The concept of cytokine storm has been extensively mentioned in the last years in the context of coronavirus disease 2019 (COVID-19) infection. It is recognised that the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) could induce a dramatic pro-inflammatory cascade, dominated mostly by IL-1, IL-6 and IFN-γ [46]; although AOSD and COVID-19 have different pathogenesis, the hyperinflammation provoked by this uncontrolled release of pro-inflammatory mediators is common to both the diseases. That is the reason why several anti-cytokines, in particular anakinra and tocilizumab, commonly employed in autoimmune and autoinflammatory diseases, have been used to contrast the hyperinflammation caused by COVID-19 [47, 48]. In addition, the experience of the pandemic has opened the way to other modalities of drug administration which are uncommon in adults affected by systemic autoinflammatory diseases, showing high proof of safety and easy handling even in critical patients [49, 50].

Pathophysiology of Still’s disease, activation of innate and adaptive immunity, possible and confirmed therapeutic targets. Created with BioRender.com. PAMPs pathogen-associated molecular pattern, DAMPs damage-associated molecular pattern, TLR Toll-like receptor, GSDMD gasdermin D, JAK Janus kinase, STAT signal transducers and activators of transcription, GM-CSF granulocyte-macrophage colony-stimulating factor, IgIV intravenous immunoglobulins, IL interleukin, IFN interferon, NET neutrophil extracellular traps, NK natural killer, Th T helper cells
4 Supporting Diagnosis: Old and New Biomarkers and the Role of Imaging Tools
4.1 Biomarkers
Currently, several biomarkers are employed to monitor the course of AOSD, although a specific and measurable marker able to confirm the diagnosis is not available. High ferritin levels are normally observed during flares [51,52,53], however, hyperferritinemia is not unique to AOSD and can be distinctive of the so-called hyperferritinemic syndromes. This is an umbrella term which refers to pathologies characterised by significant increases in ferritin such as MAS, catastrophic anti-phospholipid syndrome, septic shock [54] and severe COVID-19 [55]. Indeed, ferritin dosage has a poor positive predictive value alone without the presence of a supportive clinical background [56]. Ferritin levels usually correlate with the increase of IL-1β, IL-18, TNF-α and IFN-γ, which may contribute to the perpetuation of the cytokine storm through a loop mechanism in which the inflammatory properties of ferritin are exacerbated [57].Therefore, ferritin can be considered a mechanistic biomarker directly involved in the pathogenic mechanism of AOSD and not just a descriptive biomarker [58]. In clinical practice, ferritin is included in the mPS [13] to assess disease activity. Another unspecific biomarker is sCD163, expressed on M2 macrophages. However, sCD163 may also be released in serum after infections, as a reflection of macrophage activation, and therefore it may be considered a possible biomarker useful for prognosis and disease activity assessment [54]. The measurement of glycosylated ferritin (GF) instead is not common in routine clinical practice; normal GF levels are usually greater than 50% of total ferritin levels. Since glycosylation mechanisms cannot keep up with the increase in ferritin production [59], the concentration of GF drops in inflammatory conditions, typically ranging from 20 to 50%. However, GF, like ferritin, is not specific for AOSD and its decrease is exacerbated in MAS, where GF levels are usually very low. GF is nowadays part of the classification criteria proposed by Fautrel [11], for which the cut-off of < 20% has been established. The combination of hyperferritinemia and GF < 20% have increased AOSD diagnosis sensitivity and specificity at 70.5% and 83.2%, respectively [58]. The increase in neutrophil count is another typical hallmark of AOSD. A recent observational study identified the neutrophil-to-lymphocyte ratio (cut-off value of ≥ 4) as a promising biomarker of AOSD with a high sensitivity, and that could replace the neutrophil percentage (≥ 80%) in the classification criteria [60]. Serum calprotectin (a heterodimer of the S100A8 and S100A9 proteins) belongs to the S100 family proteins (S100A8, A9, A12), a group of calcium-binding proteins also called calgranulines. Several studies observed higher levels of calprotectin in AOSD in comparison with subjects with RA [61, 62], although unspecific, high levels of S100 positive correlate not only with increase of CRP, erythrocyte sedimentation rate (ESR) and WBCs, but also with IL-1β and TNF-α. Despite the fact that serum calprotectin is not currently employed in routine clinical practice, it represents a good biomarker for disease activity assessment [63]. Concerning pro-inflammatory cytokines, the IL-1 family is upregulated in acute AOSD. Although IL-1β is not specific to AOSD, it correlates with disease activity and can be predictive of therapeutic response [1]. IL-33 is the ligand of the orphan receptor ST2 [64]; it was found to be higher in AOSD compared with RA. Moreover, IL-33 levels correlated clinically with the mPS, ESR, ferritin and transaminases. IL-18 is involved in both AOSD and MAS pathogenesis and is a potent pro-inflammatory cytokine that may be increased in other autoinflammatory conditions as well [65]. A recent study by Shiga and colleagues investigated 46 patients with AOSD (including 9 patients with AOSD-MAS) and 31 adult patients with non-AOSD-associated MAS. IL-18 levels of patients with AOSD-MAS were significantly higher than those of the non-AOSD MAS group, and they positively correlated with ferritin and soluble interleukin-2 receptor levels. As a result, IL-18 can be used as a diagnostic biomarker to distinguish AOSD with or without MAS from other secondary forms of lymphohystiocitosis [66]. Another Italian study found that IL-18 levels are significantly higher in patients with active AOSD and positively correlated to disease activity, ferritin and ESR levels. Furthermore, IL-18 levels were found to be significantly higher in patients with AOSD than in those with sepsis [67]. Finally, a recent study compared COVID-19 and AOSD, confirming that active AOSD had 68-fold higher IL-18 and five-fold higher ferritin levels than severe COVID-19 [68]. Among other cytokines, IL-37, IL-6 and TNF-α may be upregulated during AOSD flares, despite the lack of specificity. Regarding chemokines, CXCL10 is produced in response to IFN-γ, while CXCL13 is expressed in lymphoid tissues and regulated by TNF-α. CXCL10 may correlate with ferritin and systemic scores, while serum CXCL13 levels correlate mostly with haemoglobin, CRP, ferritin and systemic score. Higher levels of these two chemokines were observed not only in AOSD, but also in other pathologies involving lymphoid tissues such as lymphoma, histiocytic necrotising lymphadenitis, tuberculous lymphadenitis and reactive hyperplasia [69]. Is it noteworthy that several biomarkers may express not only the degree of systemic inflammation, but also organ involvement. CXCL10, CXCL13 and the already mentioned IL-33, for example, correlate with skin manifestations. Lipocalin-2 (LCN2) may instead be increased in hepatic inflammation and was found to be upregulated in AOSD neutrophils by RNA sequencing and confirmed at the mRNA and protein levels. In addition, LCN2 levels correlated with inflammatory markers and systemic score, and higher levels were detected in liver biopsies of patients with AOSD with liver injury, suggesting it as a potential biomarker able to identify liver damage provoked by the inflammation itself [70]. Triggering receptor expressed on myeloid cells-1 (TREM-1) is an amplifier of inflammatory signals expressed on neutrophils and monocytes. In a recent study on 108 adult patients, higher levels of sTREM-1 were observed in comparison with patients with RA and controls; in addition, serum levels of sTREM-1 correlated with systemic score, ferritin, leucocyte count, CRP, IL-1β and IL-6 [71]. The already mentioned TLRs respond to danger signalling sensing DAMPs and PAMPs and transmit the signal resulting in an increased expression of pro-inflammatory chemokines and cytokines. Recent evidence proved that the expression of TLR1, TLR4, TLR7 and TLR9 was higher in lymph nodes of patients with AOSD than in those with T-cell lymphoma and reactive lymphadenopathy. TLR2 and TLR9 in particular were mostly related to skin inflammation in AOSD, while higher circulating TLR2-positive cells were more increased in patients with AOSD and arthritis compared with those without arthritis [72]. Finally, elevated advanced glycation end products (AGE) and decreased soluble receptors for AGE (sRAGE) were detected in subjects with AOSD, and the associations of AGEs and sRAGE levels with disease activity estimate their involvement in AOSD pathogenesis [73]. MicroRNAs (miRNAs) have recently been identified as crucial negative post-transcriptional immune regulators that target mRNA expression involved in both innate and adaptive immune responses. Their dysregulation has been observed in different pathological conditions, including inflammatory diseases. Emerging evidence demonstrated that miRNAs are present in stable form in body fluids and are thought to serve as clinical non-invasive biomarkers of disease. Liao et al. found that the upregulation of circulating miR-134 positively correlated with AOSD activity and significantly decreased after effective treatment, suggesting it as a potentially novel diagnostic biomarker. MiR-134 may contribute to AOSD pathogenesis through the elevation of free IL-18 levels, downregulating IL-18BP expression [74]. Hu et al. identified a set of plasma miRNAs as potential biomarkers to monitor disease activity and diagnose AOSD. It was indeed observed that five miRNAs (miR-142-5p, miR-101-3p, miR-29a-3p, miR-29c-3p and miR-141-3p) were significantly increased in the plasma of patients with AOSD when compared with healthy controls, by using a model in which a combination of three miRNAs (miR-142-5p, miR-101-3p and miR-29a-3p) distinguished AOSD from healthy controls [75]. Finally, Kamiya et al. found that the levels of miR-223 in sera were significantly elevated in active patients with sJIA and correlated with ESR. In addition, increased levels of miR-146a were found in sera of patients with sJIA, suggesting that both miR-223 and miR-146a have a potential novel value for Still’s disease diagnosis [76].
4.2 Imaging
A specific imaging tool capable of performing or supporting AOSD diagnosis is not currently available. X-rays or ultrasounds have proven to be unhelpful in the diagnostic process and no radiological criteria for AOSD exist. However, it was reported that the employment of 18F-fluoro-2-deoxyglucose (18F-FDG) positron emission tomography (PET) with computed tomography (CT) or magnetic resonance (MR) may support AOSD diagnosis and response to therapy [77]. Although a specific uptake cannot be detected and the differential diagnosis with haematological malignancies is mandatory, subjects with AOSD usually present with higher 18F-FDG uptake than non-AOSD in specific areas, namely bone marrow, spleen and lymph nodes [77]. The qualitative analysis, usually performed by an expert nuclear medicine radiologist, is essential for detecting active areas, whereas the quantitative analysis, through the assessment of standardised uptake values (SUVs), is important to establishing and quantifying hypermetabolic areas. Furthermore, PET-CT/MR has some limitations: it is not specific for AOSD, it cannot be used in routine clinical practice and costs can be high. However, as new radiotracers related to inflammation are currently being evaluated, this imaging technique should be considered in the near future to improve the diagnostic process of AOSD.
5 Therapeutic Strategies and Cutting-Edge Therapies in the Era of Biologics
5.1 Conventional Therapies
The management of AOSD relies on the employment of different therapeutic approaches, and the recommendations for treatment of sJIA/AOSD proposed by EULAR/PReS are still ongoing. Regarding NSAIDs, there is insufficient evidence to prove their use in monotherapy. NSAIDs can be employed when the diagnosis is still not clear. High doses of indomethacin or ibuprofen may control the inflammatory symptoms in a satisfactory way [45], however, there are no clinical trials or longitudinal studies exhibiting data on the efficacy of NSAIDs alone. GCs represent an efficient therapeutic approach and remain a cornerstone in AOSD treatment, especially at high doses [78]. Indeed, the response to both articular and systemic manifestations can be impressive, and the employment of GCs is required when MAS or other life-threatening complications are forthcoming. However, the employment of GCs should be limited in time due to the risk of hypertension, diabetes, cataracts, early-onset osteoporosis and cushingoid habits. Methyl-prednisolone is adequate to control most of the manifestations, while intravenous (IV) dexamethasone should be reserved for patients with multiorgan involvement [79] or MAS, as is done in the HLH-2004 protocol for the treatment of primary HLH [80]. Despite the lack of a defined posology in severe AOSD, the clinical approach should be based on the careful assessment of clinical manifestations and the evaluation of serum biomarkers. Evidence for conventional synthetic disease-modifying anti-rheumatic drugs (csDMARDs) comes primarily from longitudinal studies. Indeed, methotrexate (MTX) and ciclosporin A (CsA) have proven to be effective in limiting the general intake of GCs and in allowing for the achievement of disease inactivity [81, 82], especially on articular manifestations. However, nowadays csDMARDs should be considered if other treatments are not available (e.g. in low-income countries) or in specific cases, and biological (b) DMARDs should be preferred.
5.2 Anti-cytokines
Currently, the anti-cytokines employed for the treatment of AOSD target IL-1, IL-6 and TNF-alpha, which are the pivotal cytokines involved in AOSD pathogenesis. IL-1 inhibitors include anakinra (ANK), canakinumab (CAM) and rilonacept (RIL).
5.2.1 Anakinra
ANK is a non-glycosylated form of the human receptor antagonist (IL-Ra); ANK therefore targets both IL-1α and IL-1β [1]. ANK was approved in 2011 for the treatment of RA and is currently approved by EMA for the treatment of Still’s disease at all ages. ANK has a short half-life (6–8 h), and is usually administered at the posology of 100 mg/day (1–2 mg/kg/day in children) subcutaneously (SC), but higher dosages may be required if clinical manifestations are not sufficiently controlled. In addition, despite not being approved, the IV route may be considered if life-threatening features or MAS occur, or if a severe laboratory hyperinflammation is detected, allowing an increase of up to 400 mg/day [49]. The efficacy of ANK in AOSD was evaluated in several case series (Table 1), although most of the studies derive from children’s cohorts [83]. The only open randomised trial [84] carried out in adults included 12 patients treated with ANK and 10 patients treated with csDMARD (MTX, azathioprine, leflunomide). After 6 months, 50% of the ANK-treated patients achieved clinical inactive disease (CID), compared with 20% of the csDMARD-treated group, but these differences were not statistically significant. Several systematic reviews reported cumulative data on ANK efficacy. An analysis by Vastert et al. [85] included 15 studies on AOSD with a total of 444 patients. Despite the heterogeneity of the studies, the response rates ranged from 50% to 100% after a follow-up of 3 > 12 months. In a systematic review (SR) proposed by Giacomelli et al., of 455 cases of AOSD, 320 patients achieved a complete response (remission rate of roughly 70%) [86], and the SR by Hong et al. highlighted similar results, with an overall remission rate and complete remission of 82% and 67%, respectively [87]. Similar findings were observed in the recent SR published by Fautrel et al., who reported a remission rate of around 70% after ANK employment [88]. Therefore, the use of ANK in Still’s disease demonstrated high efficacy, confirming the drugs’ capability to achieve CID rapidly and prevent the chronicity of the articular damage. During ANK treatment, no major safety concerns were raised. The most common non-serious adverse event (AE) reported in the cohorts was injection site reaction (ISR). In case of infection, the relatively short half-life of ANK allows for prompt modulation; indeed, ANK was employed for the treatment of hyperinflammation following severe sepsis [89] and is currently approved for the treatment of severe COVID-19, exhibiting an excellent safety profile.
5.2.2 Canakinumab
Canakinumab is a fully human monoclonal antibody targeting IL-1β. It is approved by EMA for sJIA/AOSD treatment, and is normally administered subcutaneously at a dosage of 150 mg or 300 mg/4 weeks. Although studies with solid evidence are required to support the efficacy of CAM in AOSD, there are favourable results which describe its efficacy in young patients. Indeed, CAM was evaluated in several RCTs [90, 91], but only one pooled analysis [92] and one RCT [93] were conducted in adults. Feist et al. pooled data from four studies on sJIA. After 3 months, the ACR70 response of adolescents, reflecting an adult population, was 72%, which is comparable to the response observed in children [92]. Despite being prematurely concluded, the double-blind RCT by Kedor et al. compared CAM-treated subjects and a placebo group; the ACR90 response, EULAR response and DAS28 reduction were all significant at 3 months, confirming the efficacy of CAM in adults [93]. Besides these two studies, a recent SR illustrating the efficacy and safety of CAM [94] included 11 case series/reports and 4 observational studies. The authors reported a complete response on articular and systemic manifestations and laboratory values in 69% of the patients treated with canakinumab at the end of the study period, while only 16% had a partial improvement and 15% an inadequate response. Despite only a few cohorts of adult patients having been assessed so far, data are in favour of the employment of CAM. Furthermore, infections of different severity and episodes of MAS were reported in several cohorts, but no major concerns arose after CAM employment (see Table 2).
5.2.3 Rilonacept
Rilonacept (RIL) is an IL-1 trap molecule which is not currently approved in Europe for sJIA/AOSD treatment. Two trials have been proposed in children, both with favourable outcomes of efficacy and safety [95, 96]. In AOSD, data come from a small number of case series in which RIL was used successfully at the dosage of 160 mg/week in refractory forms with a predominant articular involvement [97].
5.2.4 IL-6 inhibitors
IL-6 is another central cytokine involved in AOSD pathogenesis and is often associated with articular manifestations. Tocilizumab (TCZ) competitively inhibits the binding of IL-6 to its receptor (IL-6R), and it is currently approved for the treatment of refractory sJIA and in patients older than 2 years of age; the usual dose is 8 mg/kg once every 2 weeks in patients weighing more than or equal to 30 kg or 12 mg/kg once every 2 weeks in patients weighing less than 30 kg. Tocilizumab is not approved for adults, but several trials have been carried out in children, proving evidence of efficacy [98, 99]. A recent meta-analysis reported by Ma et al. analysed 147 adult patients; the overall partial and complete remission rates were 85.3% (95% CI 69.32–96.88%) and 77.9% (95% CI 57.91–90.04%), respectively, and the remission rate of refractory patients was 87.9% (95% CI 56.53–100.00%), demonstrating high efficacy [100]. The only RCT conducted in adults reported a higher rate of ACR70 response in those receiving TCZ compared with placebo (46% versus 31%) at 3 months. Moreover, in the long-term extension phase, the ACR70 response rate reached 61% at 1 year [101]. As reported in Table 1, a longitudinal observational study reported a decrease of > 2 modified Pouchot score at 6 months in 50% of the patients, and in 64% after 1 year [102], while in another study a DAS28 ≦ 2.6 was achieved by 78% of the patients within 1 year [103]. The efficacy of another IL-6Ra, sarilumab, was assessed only in one case report to date, in a patient refractory to TCZ therapy [104].
IL-6 inhibitors are generally safe, and no major concerns have been raised. Yokota [105] reported the safety data of one of the largest cohorts of children with sJIA treated with TCZ, observing a rate of non-serious and serious infections of 69.8/100 patient-years (PY) and 18.2/100 PY, respectively.
5.2.5 TNF-α inhibitors
A variable efficacy is exhibited by TNF-α inhibitors, such as infliximab, etanercept and adalimumab. TNF-α inhibitors were the first bDMARDs employed in AOSD and current data derive mostly from observational studies and limited case series, but there are no RCTs that support the efficacy and safety of this therapeutic strategy in Still’s disease. Overall, TNF-α inhibitors are well tolerated, although their efficacy appears to be limited. Cavagna et al. first reported the use of infliximab in three patients with AOSD, proving a rapid and sustained efficacy for both articular and systemic symptoms [106]. However, larger cohorts reported mixed results. In the study reported by Fautrel et al. [107], only 25% of the patients treated with infliximab or etanercept achieved CID; similarly, an Italian cohort reported a CID achievement in only 22% of the patients treated at last follow-up.
5.2.6 IL-17 inhibitors
IL-17 increase has been reported in AOSD [56]. Secukinumab is a human monoclonal antibody that binds to IL-17A, and is currently approved for spondyloarthritis (SpA), psoriatic arthritis and cutaneous psoriasis with a favourable safety profile. Only one case of AOSD overlap with SpA was treated with anti- IL-17A, resulting in complete remission of articular manifestations [108].
5.3 Other therapeutic approaches
Other treatment options have been considered over time. Anti-CD20 (rituximab) and anti-CTLA-4 (abatacept) were evaluated in limited case reports or series [109,110,111,112], but did not provide substantial evidence in terms of efficacy; the same applies for intravenous immunoglobulins (IVIg) [113,114,115], but they should be considered in selected cases (e.g. pregnancy). Finally, a recent report on colchicine found clinical improvement in 20 patients with AOSD with serositis features. [116].
5.4 Cutting edge-therapies and proposed treatments
New treatment options are under assessment through clinical trials; in addition, several case series/reports present in the literature are currently evaluating new compounds with various pharmacokinetic and dynamic properties (Table 2).
5.4.1 JAK inhibitors
A few case series and case reports reported the efficacy and safety of different JAK-I in refractory sJIA and AOSD to date [117]. In AOSD, JAK-I employment was described in a French cohort of six patients in which baricitinib was administered to three patients, ruxolitinib to two, and tofacitinib to one, in addition to GCs [118]. Although no one achieved complete remission, the general intake of glucocorticoids was significantly reduced at the last follow-up in almost all patients. In another cohort of 14 cases of refractory AOSD, tofacitinib contributed to achieving disease remission, providing a consistent steroid-sparing effect, especially in those with active polyarthritis [119]. Mixed results were observed with baricitinib in a small cohort [120], however, a recent clinical trial (ChiCTR2200061599) reported a significant reduction in the systemic score and clinical improvement after 4 weeks of treatment with baricitinib in seven cases of refractory AOSD [121]. Overall, the JAK-I-related side effects were minor, with a prevalence of non-serious infections; no thrombotic or cardiovascular events were recorded [117]. Despite the diverse selectivity on Janus kinases, the efficacy of one JAK-I over another in AOSD remains unknown. Due to the modulating effect on a wide variety of cytokines, JAK-I appear to be a promising treatment strategy in AOSD, but larger studies are required to assess the long-term efficacy and safety in refractory AOSD.
5.4.2 IL-18 BP (tadekinig alpha)
Tadekinig alpha is a recombinant human IL-18-binding protein (BP) that binds to high-affinity IL-18, inhibiting the production of several pro-inflammatory cytokines, including TNF-α, IFN-γ and IL-1 [122]. In 23 patients with AOSD, a multicentre open-label dose-escalating trial was conducted. Overall, 44% achieved a reduction of CRP > 70% or normal CRP at 3 months, and joint count reduction ≥ 20%. Side effects were minor, mostly due to injection site reactions and infections, and only one serious event (optic neuropathy) was reported [123]. To date, only one case reported two adult patients treated successfully with IL-18 BP [124].
5.4.3 Emapalumab
IFN-γ is a central cytokine that is usually increased in patients with macrophage activation syndrome complicating sJIA and AOSD [125]. Emapalumab is a fully human monoclonal antibody that neutralises both free- and receptor-bound IFN-γ by inhibiting receptor dimerisation and signalling transduction. The potential efficacy of emapalumab was recently investigated in children with MAS, demonstrating the achievement of remission in particular in those who failed high-dose glucocorticoids [126]; however, the experience of IFN-γ blockade in adults is still limited [127].
5.4.4 Camoteskimab
Camoteskimab (also known as CERC 007, AEVI 007 or MEDI 2338) is a fully human monoclonal antibody which targets IL-18. It is not currently available as a possible treatment option; however, a phase I open-label trial is undergoing in the USA (NCT04752371). In this study, patients will be administered intravenous camoteskimab at a dose of 7 mg/kg, and subsequently, on the basis of efficacy and tolerability outcomes in the first cohort, the other participants will be administered a dose escalation or reduction of camoteskimab to assess the long-term efficacy.
5.4.5 MAS 825
MAS-825 is a bi-specific antibody against IL-1 and IL-18. Several patients with sJIA, MAS and ILD have received the drug for compassionate use. The current trials for MAS-825 are recruiting patients with NLRC4-gain-of-function (GOF), XIAP deficiency, CDC42 mutations (NCT04641442) and severe COVID-19 [128]. Efficacy is also being investigated in other inflammatory conditions such as hidradenitis suppurativa. On the basis of its specificity against two pivotal cytokines of AOSD/MAS, this treatment option should also be evaluated in RCTs on sJIA/AOSD.
5.4.6 RPH-104
RPH-104 is currently under development for the treatment of several inflammatory conditions, including idiopathic recurrent pericarditis, myocardial infarction, familial Mediterranean fever (FMF) and SARS-CoV-2 infection. RPH-104 is a fusion protein that targets IL-1β/IL-1F2-induced signalling. A two-part multicentre RCT (NCT05432960) aimed to compare the efficacy and safety of RPH-104 in AOSD versus placebo, but the study was recently halted due to decision made by sponsor.
5.4.7 APB R3
APB R3 is a long-acting recombinant fusion protein composed of IL-18BP that is fused to an anti-human serum albumin Fab fragment. Research is underway regarding its role in the treatment of AOSD, and evidence is currently available on liver inflammation [129].
5.4.8 NLRP3 Inflammasome Inhibitors
Inflammasome NLRP3 and its components are deeply involved in the pathogenesis of systemic AID. Various compounds can bind NLRP3 and thus prevent the release of IL-1β. Dapansutrile (OLT1177), an orally active beta-sulfonyl nitrile, was recently tested in gouty arthritis as one of the inhibitors targeting NLRP3 protein and assembly. [130]. Dapansutrile is a candidate therapeutic option in AOSD, particularly for treating the subset of patients with a predominance of articular involvement, on the basis of promising results in the resolution of articular manifestations and a favourable safety profile observed in gouty arthritis. Similarly, INF39 is a non-toxic, irreversible, specific inhibitor of the NLRP3 inflammasome that prevents NLRP3 activation, and as a result, dampens the release of IL-1β from macrophages [131]. NLRP3 inhibitors may also have an effect on inflammasome components; for example, MCC950 directly binds to the NACHT domain and inhibits inflammasome assembly by interfering with ASC oligomerisation. For this reason, it was found to be a promising option in reducing IL-1β production in vivo and in mouse models of cryopirinopathies [132]. A similar selective mechanism on NACHT has been attributed to CY09 [133] and to tranilast, an anthranilic acid whose potential anti-inflammatory effect was assessed in COVID-19 and several cardiovascular disorders [134, 135]. Similarly, oridonin, a primary active component of Rabdosia rubescens, may exert anti-inflammatory effects on NLRP3 by blocking the NACHT domain [136]. In cryopyrin-associated autoinflammatory syndromes (CAPS), there are currently ongoing studies with inzomelid (NCT04015076), a selective oral NLRP3 inhibitor, and diacerein, an anthraquinone compound that may downregulate both NLRP3/caspase-1/IL-1β axis or IL-6/pSTAT3 [137]; other compounds targeting caspase-1 are belnacasan (VX-765), which attenuated disease progression in osteoarthritis in preclinical evaluations [138], and endogenous alpha-1 antitrypsin (A1-AT), which exerts anti-inflammatory and anti-apoptotic properties and directly acts on caspase I preventing IL-1β release. Gasdermin D (GSDMD) is a downstream effector of caspase-1 that regulates pyroptosis and the release of IL-1β and IL-18 into the extracellular space [139]. By suppressing GSDMD, the inflammasome-induced pyroptosis is dampened, making Gasdermin D a promising novel target for regulating inflammation [140]. Overall, many compounds are under study and evaluation in in vivo or in vitro models. Despite most of the studies currently being at an early stage, it is possible to conceive their future employment in refractory AOSD.
5.4.9 GM-CSF Inhibitors
GM-CSF may enhance the pro-inflammatory activities of neutrophils and macrophages by inducing the production of NETs, and in turn, upregulating the release of pro-inflammatory cytokines. Mavrilimumab (CAM-3001), an IgG4 monoclonal antibody (mAb), and otilimab (MOR-103), an IgG1 mAb, bind to the GM-CSF-alpha receptor and GM-CSF, respectively. The efficacy of both otilimab and mavrilimumab was already assessed in RA [141,142,143] and the potent anti-inflammatory effect of mavrilimumab has been observed in hyperinflammation following COVID-19 infection [144]; therefore, the employment of these compounds in systemic AOSD is conceivable.
6 Conclusions
The most recent advances, as well as the introduction of biologic drugs, have significantly improved the quality of life of patients with AOSD. On the basis of efficacy and safety data, the inhibition of IL-1 and IL-6 represent the best treatment option in the long term. According to this review, a wide range of new drugs and compounds are presently being studied as possible and efficient targets for AOSD. Furthermore, advances in understanding the physiopathology and genetic aspects will enable the investigation of specific targets relevant for the treatment of specific organ–disease manifestations.
References
Sfriso P, Bindoli S, Galozzi P. Adult-Onset still’s disease: molecular pathophysiology and therapeutic advances. Drugs. 2018;78:1187–95. https://doi.org/10.1007/s40265-018-0956-9.
Nirmala N, Brachat A, Feist E, et al. Gene-expression analysis of adult-onset Still’s disease and systemic juvenile idiopathic arthritis is consistent with a continuum of a single disease entity. Pediatr Rheumatol. 2015;13:50. https://doi.org/10.1186/s12969-015-0047-3.
Kudela H, Drynda S, Lux A, et al. Comparative study of Interleukin-18 (IL-18) serum levels in adult onset Still’s disease (AOSD) and systemic onset juvenile idiopathic arthritis (sJIA) and its use as a biomarker for diagnosis and evaluation of disease activity. BMC Rheumatol. 2019;3:4. https://doi.org/10.1186/s41927-019-0053-z.
Pay S, Türkçapar N, Kalyoncu M, et al. A multicenter study of patients with adult-onset Still’s disease compared with systemic juvenile idiopathic arthritis. Clin Rheumatol. 2006;25:639–44. https://doi.org/10.1007/s10067-005-0138-5.
Li S, Ying S, Bai J, et al. Clinical characteristics and outcome of elderly onset adult-onset Still’s disease: a 10-year retrospective study. J Transl Autoimmun. 2023;6: 100196. https://doi.org/10.1016/j.jtauto.2023.100196.
Ruscitti P, Berardicurti O, Giacomelli R, et al. The clinical heterogeneity of adult onset Still’s disease may underlie different pathogenic mechanisms. Implications for a personalised therapeutic management of these patients. Semin Immunol. 2021;58:101632. https://doi.org/10.1016/j.smim.2022.101632.
Berardicurti O, Conforti A, Iacono D, et al. Dissecting the clinical heterogeneity of adult-onset Still’s disease: results from a multi-dimensional characterization and stratification. Rheumatology. 2021;60:4844–9. https://doi.org/10.1093/rheumatology/keaa904.
Del Giudice M, Gangestad SW. Rethinking IL-6 and CRP: why they are more than inflammatory biomarkers, and why it matters. Brain Behav Immun. 2018;70:61–75. https://doi.org/10.1016/j.bbi.2018.02.013.
Delplanque M, Aouba A, Hirsch P, et al. USAID Associated with myeloid neoplasm and VEXAS syndrome: two differential diagnoses of suspected adult onset still’s disease in elderly patients. J Clin Med. 2021;10:5586. https://doi.org/10.3390/jcm10235586.
Yamaguchi M, Ohta A, Tsunematsu T, et al. Preliminary criteria for classification of adult Still’s disease. J Rheumatol. 1992;19(3):424–30.
Fautrel B, Zing E, Golmard J-L, et al. Proposal for a new set of classification criteria for adult-onset still disease. Medicine (Baltimore). 2002;81:194–200. https://doi.org/10.1097/00005792-200205000-00003.
Daghor-Abbaci K, Ait Hamadouche N, Makhloufi CD, et al. Proposal of a new diagnostic algorithm for adult-onset Still’s disease. Clin Rheumatol. 2023;42:1125–35. https://doi.org/10.1007/s10067-023-06509-8.
Rau M, Schiller M, Krienke S, et al. Clinical manifestations but not cytokine profiles differentiate adult-onset Still’s disease and sepsis. J Rheumatol. 2010;37:2369–76. https://doi.org/10.3899/jrheum.100247.
Zhu D, Meng J, Jia J, et al. Performance of the modified systemic manifestation score for systemic juvenile idiopathic arthritis in adult-onset Still’s disease. Clin Rheumatol. 2023;42:187–95. https://doi.org/10.1007/s10067-022-06340-7.
Terkeltaub R, Esdaile JM, Décary F, et al. HLA—Bw35 and prognosis in adult Still’s disease. Arthritis Rheum. 1981;24:1469–72. https://doi.org/10.1002/art.1780241203.
Wouters JMGW, Reekers P, van de Putte LBA. Adult-onset still’s disease. Disease course and HLA associations. Arthritis Rheum. 1986;29:415–8. https://doi.org/10.1002/art.1780290316.
Fujii T, Nojima T, Yasuoka H, et al. Cytokine and immunogenetic profiles in Japanese patients with adult Still’s disease. Association with chronic articular disease. Rheumatology (Oxford). 2001;40:1398–404. https://doi.org/10.1093/rheumatology/40.12.1398.
Joung CI, Lee HS, Lee SW, et al. Association between HLA-DR B1 and clinical features of adult onset Still’s disease in Korea. Clin Exp Rheumatol. 2003;21:489–92. http://www.ncbi.nlm.nih.gov/pubmed/12942703.
Li S, Zheng S, Tang S, et al. Autoinflammatory pathogenesis and targeted therapy for adult-onset Still’s disease. Clin Rev Allergy Immunol. 2020;58:71–81. https://doi.org/10.1007/s12016-019-08747-8.
Saper VE, Ombrello MJ, Tremoulet AH, et al. Severe delayed hypersensitivity reactions to IL-1 and IL-6 inhibitors link to common HLA-DRB1*15 alleles. Ann Rheum Dis. 2022;81:406–15. https://doi.org/10.1136/annrheumdis-2021-220578.
Kim JJ, Kim J-K, Shim S-C, et al. MEFV gene mutations and their clinical significance in Korean patients with adult-onset Still’s disease. Clin Exp Rheumatol. 2013;31:60–3. http://www.ncbi.nlm.nih.gov/pubmed/24064016.
Cosan F, Emrence Z, Erbag G, et al. The association of TNFRSF1A gene and MEFV gene mutations with adult onset Still’s disease. Rheumatol Int. 2013;33:1675–80. https://doi.org/10.1007/s00296-012-2609-8.
Garcia-Melchor E, Grados D, González-Roca E, et al. CIAS1 and NOD2 genes in adult-onset Still’s disease. J Rheumatol. 2014;41:1566–7. https://doi.org/10.3899/jrheum.131563.
Sighart R, Rech J, Hueber A, et al. Evidence for genetic overlap between adult onset Still’s disease and hereditary periodic fever syndromes. Rheumatol Int. 2018;38:111–20. https://doi.org/10.1007/s00296-017-3885-0.
Wakil SM, Monies DM, Abouelhoda M, et al. Association of a mutation in LACC1 With a monogenic form of systemic juvenile idiopathic arthritis. Arthritis Rheumatol. 2015;67:288–95. https://doi.org/10.1002/art.38877.
Ruscitti P, Berardicurti O, Di Cola I, et al. The hyper-expression of NLRP4 characterizes the occurrence of macrophage activation syndrome assessing STING pathway in adult-onset Still’s disease. Clin Exp Immunol. 2022;208:95–102. https://doi.org/10.1093/cei/uxac014.
Sugiura T, Kawaguchi Y, Harigai M, et al. Association between adult-onset Still’s disease and interleukin-18 gene polymorphisms. Genes Immun. 2002;3:394–9. https://doi.org/10.1038/sj.gene.6363922.
Chen D-Y, Chen Y-M, Chen H-H, et al. Functional association of interleukin 18 gene -607 (C/A) promoter polymorphisms with disease course in Chinese patients with adult-onset Still’s disease. J Rheumatol. 2009;36:2284–9. https://doi.org/10.3899/jrheum.090316.
Wang F-F, Huang X-F, Shen N, et al. A genetic role for macrophage migration inhibitory factor (MIF) in adult-onset Still’s disease. Arthritis Res Ther. 2013;15:R65. https://doi.org/10.1186/ar4239.
Hung W-T, Chen Y-M, Hung S-I, et al. CARD8 SNP rs11672725 Identified as a potential genetic variant for adult-onset Still’s disease. Life. 2021;11:382. https://doi.org/10.3390/life11050382.
Hung W-T, Hung S-I, Chen Y-M, et al. The association of ATG16L1 variations with clinical phenotypes of adult-onset Still’s disease. Genes (Basel). 2021;12:904. https://doi.org/10.3390/genes12060904.
Wang M, Liu M, Jia J, et al. Association of the leukocyte immunoglobulin-like receptor A3 gene with neutrophil activation and disease susceptibility in adult-onset Still’s disease. Arthritis Rheumatol. 2021;73:1033–43. https://doi.org/10.1002/art.41635.
Chen Y-M, Hung W-T, Chang W-C, et al. Genetic association and expression correlation between colony-stimulating factor 1 gene encoding M-CSF and adult-onset Still’s disease. J Immunol Res. 2020;2020:1–11. https://doi.org/10.1155/2020/8640719.
Uziel Y, Pomeranz A, Brik R, et al. Seasonal variation in systemic onset juvenile rheumatoid arthritis in Israel. J Rheumatol. 1999;26(5):1187–9.
Lee JY-Y, Hsu C-K, Liu M-F, et al. Evanescent and persistent pruritic eruptions of adult-onset Still disease: a clinical and pathologic study of 36 patients. Semin Arthritis Rheum. 2012;42:317–26. https://doi.org/10.1016/j.semarthrit.2012.05.003.
Rao S, Tsang LS-L, Zhao M, et al. Adult-onset Still’s disease: a disease at the crossroad of innate immunity and autoimmunity. Front Med. 2022. https://doi.org/10.3389/fmed.2022.881431.
Kim J-W, Ahn M-H, Jung J-Y, et al. An update on the pathogenic role of neutrophils in systemic juvenile idiopathic arthritis and adult-onset Still’s disease. Int J Mol Sci. 2021;22:13038. https://doi.org/10.3390/ijms222313038.
Hu Q, Shi H, Zeng T, et al. Increased neutrophil extracellular traps activate NLRP3 and inflammatory macrophages in adult-onset Still’s disease. Arthritis Res Ther. 2019;21:9. https://doi.org/10.1186/s13075-018-1800-z.
Choi JH, Suh CH, Lee YM, et al. Serum cytokine profiles in patients with adult onset Still’s disease. J Rheumatol. 2003;30(11):2422–7.
Chen D-Y, Chen Y-M, Chen H-H, et al. The associations of circulating CD4 + CD25 high regulatory T cells and TGF-β with disease activity and clinical course in patients with adult-onset Still’s disease. Connect Tissue Res. 2010;51:370–7. https://doi.org/10.3109/03008200903461462.
Zhou L, Ivanov II, Spolski R, et al. IL-6 programs TH-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–74. https://doi.org/10.1038/ni1488.
Binstadt BA, Nigrovic PA. The conundrum of lung disease and drug hypersensitivity-like reactions in systemic juvenile idiopathic arthritis. Arthritis Rheumatol (Hoboken, NJ). 2022;74:1122–31. https://doi.org/10.1002/art.42137.
Ito Y, Nakahara H, Nakajima A. Development of pulmonary alveolar proteinosis in a patient with adult-onset Still disease treated with tocilizumab. J Rheumatol. 2023;50:288–9. https://doi.org/10.3899/jrheum.220128.
Bindoli S, Lococo S, Calabrese F, et al. Pulmonary alveolar proteinosis in an adult patient affected by Still’s disease and recurrent episodes of macrophage activation syndrome. Jt Bone Spine. 2023. https://doi.org/10.1016/j.jbspin.2023.105654.
Galozzi P, Bindoli S, Doria A, et al. Progress in biological therapies for adult-onset Still’s disease. Biol Targets Ther. 2022;16:21–34. https://doi.org/10.2147/BTT.S290329.
Bindoli S, Felicetti M, Sfriso P, et al. The amount of cytokine-release defines different shades of Sars-Cov2 infection. Exp Biol Med Published Online First. 2020. https://doi.org/10.1177/1535370220928964.
Mehta P, McAuley DF, Brown M, et al. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395:1033–4. https://doi.org/10.1016/S0140-6736(20)30628-0.
Perrone F, Piccirillo MC, Ascierto PA, et al. Tocilizumab for patients with COVID-19 pneumonia. The single-arm TOCIVID-19 prospective trial. J Transl Med. 2020. https://doi.org/10.1186/s12967-020-02573-9.
Bindoli S, Galozzi P, Doria A, et al. Intravenous anakinra to curb cytokine storm in adult-onset Still’s disease and in macrophage activation syndrome: a case series. Jt bone spine. 2023;90: 105524. https://doi.org/10.1016/j.jbspin.2023.105524.
Shakoory B, Carcillo JA, Chatham WW, et al. Interleukin-1 receptor blockade is associated with reduced mortality in sepsis patients with features of macrophage activation syndrome. Crit Care Med. 2016;44:275–81. https://doi.org/10.1097/CCM.0000000000001402.
Ota T, Higashi S, Suzuki H, et al. Increased serum ferritin levels in adult Still’s disease. Lancet. 1987;329:562–3. https://doi.org/10.1016/S0140-6736(87)90204-2.
Zandman-Goddard G, Shoenfeld Y. Ferritin in autoimmune diseases. Autoimmun Rev. 2007;6:457–63. https://doi.org/10.1016/j.autrev.2007.01.016.
Loh NK, Lucas M, Fernandez S, et al. Successful treatment of macrophage activation syndrome complicating adult Still disease with anakinra. Intern Med J. 2012;42:1358–62. https://doi.org/10.1111/imj.12002.
Colafrancesco S, Priori R, Alessandri C, et al. The hyperferritinemic syndromes and CD163: a marker of macrophage activation. Isr Med Assoc J. 2014;16:662–3. http://www.ncbi.nlm.nih.gov/pubmed/25438465.
Kaushal K, Kaur H, Sarma P, et al. Serum ferritin as a predictive biomarker in COVID-19. A systematic review, meta-analysis and meta-regression analysis. J Crit Care. 2022;67:172–81. https://doi.org/10.1016/j.jcrc.2021.09.023.
Feist E, Mitrovic S, Fautrel B. Mechanisms, biomarkers and targets for adult-onset Still’s disease. Nat Rev Rheumatol. 2018;14:603–18. https://doi.org/10.1038/s41584-018-0081-x.
Jamilloux Y, Gerfaud-Valentin M, Martinon F, et al. Pathogenesis of adult-onset Still’s disease: new insights from the juvenile counterpart. Immunol Res. 2015;61:53–62. https://doi.org/10.1007/s12026-014-8561-9.
Mitrovic S, Fautrel B. New markers for adult-onset Still’s disease. Jt Bone Spine. 2018;85:285–93. https://doi.org/10.1016/j.jbspin.2017.05.011.
Fautrel B, Le Moël G, Saint-Marcoux B, et al. Diagnostic value of ferritin and glycosylated ferritin in adult onset Still’s disease. J Rheumatol. 2001;28(2):322–9.
Daghor Abbaci K, Ait Hamadouche N, Otmani F, et al. Validation of the neutrophil-to-lymphocyte ratio as a new simple biomarker of adult onset Still’s disease: a STROBE-compliant prospective observational study. Medicine (Baltimore). 2022;101: e29970. https://doi.org/10.1097/MD.0000000000029970.
Kim H-A, Han J, Kim W-J, et al. TLR4 Endogenous ligand S100A8/A9 levels in adult-onset Still’s disease and their association with disease activity and clinical manifestations. Int J Mol Sci. 2016;17:1342. https://doi.org/10.3390/ijms17081342.
Jung S-Y, Park Y-B, Ha Y-J, et al. Serum calprotectin as a marker for disease activity and severity in adult-onset Still’s disease. J Rheumatol. 2010;37:1029–34. https://doi.org/10.3899/jrheum.091120.
Maranini B, Ciancio G, Govoni M. Adult-onset Still’s disease: novel biomarkers of specific subsets, disease activity, and relapsing forms. Int J Mol Sci. 2021;22:13320. https://doi.org/10.3390/ijms222413320.
Han JH, Suh C-H, Jung J-Y, et al. Serum levels of interleukin 33 and soluble ST2 are associated with the extent of disease activity and cutaneous manifestations in patients with active adult-onset Still’s disease. J Rheumatol. 2017;44:740–7. https://doi.org/10.3899/jrheum.170020.
Baggio C, Bindoli S, Guidea I, et al. IL-18 in Autoinflammatory diseases: focus on adult onset Still disease and macrophages activation syndrome. Int J Mol Sci. 2023;24:11125. https://doi.org/10.3390/ijms241311125.
Shiga T, Nozaki Y, Tomita D, et al. Usefulness of interleukin-18 as a diagnostic biomarker to differentiate adult-onset Still’s disease with/without macrophage activation syndrome from other secondary hemophagocytic lymphohistiocytosis in adults. Front Immunol. 2021. https://doi.org/10.3389/fimmu.2021.750114.
Priori R, Colafrancesco S, Alessandri C, et al. Interleukin 18: a biomarker for differential diagnosis between adult-onset Still’s disease and sepsis. J Rheumatol. 2014;41:1118–23. https://doi.org/10.3899/jrheum.130575.
Chen P-K, Lan J-L, Huang P-H, et al. Interleukin-18 is a potential biomarker to discriminate active adult-onset Still’s disease from COVID-19. Front Immunol. 2021. https://doi.org/10.3389/fimmu.2021.719544.
Kim H-A, Kim YH, Jeon YK, et al. Histopathology and expression of the chemokines CXCL10, CXCL13, and CXCR3 and the endogenous TLR-4 ligand S100A8/A9 in lymph nodes of patients with adult-onset Still’s disease. Sci Rep. 2019;9:7517. https://doi.org/10.1038/s41598-019-44032-6.
Jia J, Yang L, Cao Z, et al. Neutrophil-derived lipocalin-2 in adult-onset Still’s disease: a novel biomarker of disease activity and liver damage. Rheumatology. 2021;60:304–15. https://doi.org/10.1093/rheumatology/keaa368.
Wang Z, Chi H, Sun Y, et al. Serum sTREM-1 in adult-onset Still’s disease: a novel biomarker of disease activity and a potential predictor of the chronic course. Rheumatology. 2020;59:3293–302. https://doi.org/10.1093/rheumatology/keaa135.
Han JH, Ahn M-H, Jung J-Y, et al. Elevated expression of TLR2 and its correlation with disease activity and clinical manifestations in adult-onset Still’s disease. Sci Rep. 2022;12:10240. https://doi.org/10.1038/s41598-022-14004-4.
Chen D-Y, Chen Y-M, Lin C-C, et al. The potential role of advanced glycation end products (AGEs) and soluble receptors for AGEs (sRAGE) in the pathogenesis of adult-onset still’s disease. BMC Musculoskelet Disord. 2015;16:111. https://doi.org/10.1186/s12891-015-0569-3.
Liao T-L, Chen Y-M, Hsieh C-W, et al. Upregulation of circulating microRNA-134 in adult-onset Still’s disease and its use as potential biomarker. Sci Rep. 2017;7:4214. https://doi.org/10.1038/s41598-017-04086-w.
Hu Q, Gong W, Gu J, et al. Plasma microRNA profiles as a potential biomarker in differentiating adult-onset Still’s disease from sepsis. Front Immunol. 2019. https://doi.org/10.3389/fimmu.2018.03099.
Kamiya Y, Kawada J, Kawano Y, et al. Serum microRNAs as potential biomarkers of juvenile idiopathic arthritis. Clin Rheumatol. 2015;34:1705–12. https://doi.org/10.1007/s10067-015-2922-1.
Bindoli S, Galozzi P, Magnani F, et al. 18F-fluorodeoxyglucose positron emission tomography and computed tomography with magnetic resonance for diagnosing adult-onset Still’s disease. Front Med. 2020. https://doi.org/10.3389/fmed.2020.544412.
Ruscitti P, Cipriani P, Liakouli V, et al. Managing adult-onset Still’s disease: the effectiveness of high-dosage of corticosteroids as first-line treatment in inducing the clinical remission. Results from an observational study. Medicine (Baltimore). 2019;98: e15123. https://doi.org/10.1097/MD.0000000000015123.
Koizumi R, Yoshito Tsukada H. Treatment of adult Still’s disease with dexamethasone, an alternative to prednisolone. Scand J Rheumatol. 2000;29:396–8. https://doi.org/10.1080/030097400447624.
Henter JI, Samuelsson-Horne AC, Aricò M, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002. https://doi.org/10.1182/blood-2002-01-0172.
Fautrel B, Borget C, Rozenberg S, et al. Corticosteroid sparing effect of low dose methotrexate treatment in adult Still’s disease. J Rheumatol. 1999;26(2):373–8.
Pal P, Giri PP, Sinha R. Cyclosporine in resistant systemic arthritis—a cheaper alternative to biologics. Indian J Pediatr. 2019;86:590–4. https://doi.org/10.1007/s12098-019-02912-9.
Quartier P, Allantaz F, Cimaz R, et al. A multicentre, randomised, double-blind, placebo-controlled trial with the interleukin-1 receptor antagonist anakinra in patients with systemic-onset juvenile idiopathic arthritis (ANAJIS trial). Ann Rheum Dis. 2011;70:747–54. https://doi.org/10.1136/ard.2010.134254.
Nordström D, Knight A, Luukkainen R, et al. Beneficial effect of interleukin 1 inhibition with anakinra in adult-onset Still’s disease. An open, randomized, multicenter study. J Rheumatol. 2012;39:2008–11. https://doi.org/10.3899/jrheum.111549.
Vastert SJ, Jamilloux Y, Quartier P, et al. Anakinra in children and adults with Still’s disease. Rheumatology. 2019;58:vi9-22. https://doi.org/10.1093/rheumatology/kez350.
Giacomelli R, Sota J, Ruscitti P, et al. The treatment of adult-onset Still’s disease with anakinra, a recombinant human IL-1 receptor antagonist: a systematic review of literature. Clin Exp Rheumatol. 2021;39:187–95. https://doi.org/10.55563/clinexprheumatol/fsq5vq.
Hong D, Yang Z, Han S, et al. Interleukin 1 inhibition with anakinra in adult-onset Still disease: a meta-analysis of its efficacy and safety. Drug Des Devel Ther. 2014;8:2345–57. https://doi.org/10.2147/DDDT.S73428.
Fautrel B, Patterson J, Bowe C, et al. Systematic review on the use of biologics in adult-onset still’s disease. Semin Arthritis Rheum. 2023;58: 152139. https://doi.org/10.1016/j.semarthrit.2022.152139.
Shakoory B, Carcillo JA, Chatham WW, et al. Interleukin-1 receptor blockade is associated with reduced mortality in sepsis patients with features of macrophage activation syndrome: reanalysis of a prior phase III trial∗. Crit Care Med Published Online First. 2016. https://doi.org/10.1097/CCM.0000000000001402.
Ruperto N, Brunner HI, Quartier P, et al. Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. N Engl J Med. 2012;367:2396–406. https://doi.org/10.1056/NEJMoa1205099.
Quartier P, Alexeeva E, Constantin T, et al. Tapering canakinumab monotherapy in patients with systemic juvenile idiopathic arthritis in clinical remission: results from a phase IIIb/IV open-label, randomized study. Arthritis Rheumatol. 2021;73:336–46. https://doi.org/10.1002/art.41488.
Feist E, Quartier P, Fautrel B, et al. Efficacy and safety of canakinumab in patients with Still’s disease: exposure-response analysis of pooled systemic juvenile idiopathic arthritis data by age groups. Clin Exp Rheumatol 2018;36(4):668–75.
Kedor C, Listing J, Zernicke J, et al. Canakinumab for treatment of adult-onset Still’s disease to achieve reduction of arthritic manifestation (CONSIDER): phase II, randomised, double-blind, placebo-controlled, multicentre, investigator-initiated trial. Ann Rheum Dis. 2020;79:1090–7. https://doi.org/10.1136/annrheumdis-2020-217155.
Cota-Arce JM, Cota J, De León-Nava MA, et al. Efficacy and safety of canakinumab in the treatment of adult-onset Still’s disease: a systematic review. Semin Arthritis Rheum. 2021;51:1282–90. https://doi.org/10.1016/j.semarthrit.2021.08.007.
Ilowite NT, Prather K, Lokhnygina Y, et al. Randomized, double-blind, placebo-controlled trial of the efficacy and safety of rilonacept in the treatment of systemic juvenile idiopathic arthritis. Arthritis Rheumatol (Hoboken, NJ). 2014;66:2570–9. https://doi.org/10.1002/art.38699.
Lovell DJ, Giannini EH, Reiff AO, et al. Long-term safety and efficacy of rilonacept in patients with systemic juvenile idiopathic arthritis. Arthritis Rheum. 2013;65:2486–96. https://doi.org/10.1002/art.38042.
Petryna O, Cush JJ, Efthimiou P. IL-1 Trap rilonacept in refractory adult onset Still’s disease. Ann Rheum Dis. 2012;71:2056–8. https://doi.org/10.1136/annrheumdis-2012-201409.
De Benedetti F, Brunner HI, Ruperto N, et al. Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. N Engl J Med. 2012;367:2385–95. https://doi.org/10.1056/NEJMoa1112802.
Ruperto N, Brunner HI, Ramanan AV, et al. Subcutaneous dosing regimens of tocilizumab in children with systemic or polyarticular juvenile idiopathic arthritis. Rheumatology. 2021;60:4568–80. https://doi.org/10.1093/rheumatology/keab047.
Ma Y, Wu M, Zhang X, et al. Efficacy and safety of tocilizumab with inhibition of interleukin-6 in adult-onset Still’s disease: a meta-analysis. Mod Rheumatol. 2018;28:849–57. https://doi.org/10.1080/14397595.2017.1416924.
Kaneko Y, Kameda H, Ikeda K, et al. Tocilizumab in patients with adult-onset still’s disease refractory to glucocorticoid treatment: a randomised, double-blind, placebo-controlled phase III trial. Ann Rheum Dis. 2018;77:1720–9. https://doi.org/10.1136/annrheumdis-2018-213920.
Song ST, Kim JJ, Lee S, et al. Efficacy of tocilizumab therapy in Korean patients with adult-onset Still’s disease: a multicentre retrospective study of 22 cases. Clin Exp Rheumatol 2016;34(6 Suppl 102):S64–71.
Tang K-T, Hsieh C-W, Chen H-H, et al. The effectiveness of tocilizumab in treating refractory adult-onset Still’s disease with dichotomous phenotypes: IL-18 is a potential predictor of therapeutic response. Clin Rheumatol. 2022;41:557–66. https://doi.org/10.1007/s10067-021-05921-2.
Simeni Njonnou SR, Soyfoo MS, Vandergheynst F-A. Efficacy of sarilumab in adult-onset Still’s disease as a corticosteroid-sparing agent. Rheumatology. 2019;58:1878–9. https://doi.org/10.1093/rheumatology/kez154.
Yokota S, Itoh Y, Morio T, et al. Tocilizumab in systemic juvenile idiopathic arthritis in a real-world clinical setting: results from 1 year of postmarketing surveillance follow-up of 417 patients in Japan. Ann Rheum Dis. 2016;75:1654–60. https://doi.org/10.1136/annrheumdis-2015-207818.
Cavagna L, Caporali R, Epis O, et al. Infliximab in the treatment of adult Still’s disease refractory to conventional therapy. Clin Exp Rheumatol 2001;19:329–32. http://www.ncbi.nlm.nih.gov/pubmed/11407090.
Fautrel B, Sibilia J, Mariette X, et al. Tumour necrosis factor alpha blocking agents in refractory adult Still’s disease: an observational study of 20 cases. Ann Rheum Dis. 2005;64:262–6. https://doi.org/10.1136/ard.2004.024026.
Mitrovic S, Hassold N, Kamissoko A, et al. Adult-onset Still’s disease or systemic-onset juvenile idiopathic arthritis and spondyloarthritis: overlapping syndrome or phenotype shift? Rheumatology. 2022;61:2535–47. https://doi.org/10.1093/rheumatology/keab726.
Ahmadi-Simab K. Successful treatment of refractory adult onset Still’s disease with rituximab. Ann Rheum Dis. 2006;65:1117–8. https://doi.org/10.1136/ard.2005.047621.
Belfeki N, Smiti Khanfir M, Said F, et al. Successful treatment of refractory adult onset Still’s disease with rituximab. Reumatismo. 2016;68:159–62. https://doi.org/10.4081/reumatismo.2016.888.
Ostrowski RA, Tehrani R, Kadanoff R. Refractory adult-onset Still disease successfully treated with abatacept. JCR J Clin Rheumatol. 2011;17:315–7. https://doi.org/10.1097/RHU.0b013e31822c53ad.
Quartuccio L, Maset M, De Vita S. Efficacy of abatacept in a refractory case of adult-onset Still’s disease. Clin Exp Rheumatol 2010;28(2):265–7.
Permal S, Wechsler B, Cabane J, et al. Traitement de la maladie de Still de l’adulte par immunoglobulines intraveineuses. La Rev Médecine Interne. 1995;16:250–4. https://doi.org/10.1016/0248-8663(96)80703-X.
Vignes S, Wechsler B, Amoura Z, et al. Intravenous immunoglobulin in adult Still’s disease refractory to non-steroidal anti-inflammatory drugs. Clin Exp Rheumatol 1998;16(3):295–8.
Uziel Y, Laxer RM, Schneider R, et al. Intravenous immunoglobulin therapy in systemic onset juvenile rheumatoid arthritis: a followup study. J Rheumatol. 1996;23(5):910–8.
Myachikova V, Moiseeva O, Konradi A, et al. A retrospective analysis of colchicine in combination with NSAIDs therapy in patients with systemic form of adult-onset Still’s disease with serositis. Clin Exp Rheumatol. 2022;40:1474–9. https://doi.org/10.55563/clinexprheumatol/1o41c8.
Boyadzhieva Z, Ruffer N, Burmester G, et al. Effectiveness and safety of JAK inhibitors in autoinflammatory diseases: a systematic review. Front Med. 2022. https://doi.org/10.3389/fmed.2022.930071.
Gillard L, Mitrovic S, Reumaux H, et al. AB0772 JAK inhibitors in refractory adult and childhood onset Still’s disease. Ann Rheum Dis. 2021;80:1412.2-1413. https://doi.org/10.1136/annrheumdis-2021-eular.2210.
Hu Q, Wang M, Jia J, et al. Tofacitinib in refractory adult-onset Still’s disease: 14 cases from a single centre in China. Ann Rheum Dis. 2020;79:842–4. https://doi.org/10.1136/annrheumdis-2019-216699.
Kacar M, Fitton J, Gough AK, et al. Mixed results with baricitinib in biological-resistant adult-onset Still’s disease and undifferentiated systemic autoinflammatory disease. RMD Open. 2020;6: e001246. https://doi.org/10.1136/rmdopen-2020-001246.
Sun Z, Li R, Wang Y, et al. Efficacy of baricitinib in patients with refractory adult-onset Still’s disease. Drugs R D. 2023;23:109–20. https://doi.org/10.1007/s40268-023-00417-7.
Dinarello CA, Novick D, Kim S, et al. Interleukin-18 and IL-18 binding protein. Front Immunol. 2013. https://doi.org/10.3389/fimmu.2013.00289.
Gabay C, Fautrel B, Rech J, et al. Open-label, multicentre, dose-escalating phase II clinical trial on the safety and efficacy of tadekinig alfa (IL-18BP) in adult-onset Still’s disease. Ann Rheum Dis. 2018. https://doi.org/10.1136/annrheumdis-2017-212608.
Kiltz U, Kiefer D, Braun J, et al. Prolonged treatment with Tadekinig alfa in adult-onset Still’s disease. Ann Rheum Dis. 2020;79:e10–e10. https://doi.org/10.1136/annrheumdis-2018-214496.
Bracaglia C, de Graaf K, Pires Marafon D, et al. Elevated circulating levels of interferon-γ and interferon-γ-induced chemokines characterise patients with macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. Ann Rheum Dis. 2017;76:166–72. https://doi.org/10.1136/annrheumdis-2015-209020.
De Benedetti F, Grom AA, Brogan PA, et al. Efficacy and safety of emapalumab in macrophage activation syndrome. Ann Rheum Dis. 2023;82:857–65. https://doi.org/10.1136/ard-2022-223739.
Gabr JB, Liu E, Mian S, et al. Successful treatment of secondary macrophage activation syndrome with emapalumab in a patient with newly diagnosed adult-onset Still’s disease: case report and review of the literature. Ann Transl Med. 2020;8:887–887. https://doi.org/10.21037/atm-20-3127.
Hakim AD, Awili M, O’Neal HR, et al. Efficacy and safety of MAS825 (anti-IL-1β/IL-18) in COVID-19 patients with pneumonia and impaired respiratory function. Clin Exp Immunol. 2023;213:265–75. https://doi.org/10.1093/cei/uxad065.
Kim D-H, Lee K-J, Park J, et al. Disruption of IL-18 signaling via engineered IL-18BP biologics alleviates experimental cholestatic liver disease. Biomed Pharmacother. 2023;167: 115587. https://doi.org/10.1016/j.biopha.2023.115587.
Klück V, Jansen TLTA, Janssen M, et al. Dapansutrile, an oral selective NLRP3 inflammasome inhibitor, for treatment of gout flares: an open-label, dose-adaptive, proof-of-concept, phase 2a trial. Lancet Rheumatol. 2020;2:e270–80. https://doi.org/10.1016/S2665-9913(20)30065-5.
Shi Y, Lv Q, Zheng M, et al. NLRP3 inflammasome inhibitor INF39 attenuated NLRP3 assembly in macrophages. Int Immunopharmacol. 2021;92: 107358. https://doi.org/10.1016/j.intimp.2020.107358.
Coll RC, Robertson AAB, Chae JJ, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21:248–55. https://doi.org/10.1038/nm.3806.
Jiang H, He H, Chen Y, et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J Exp Med. 2017;214:3219–38. https://doi.org/10.1084/jem.20171419.
Jia H, Qi X, Fu L, et al. <scp>NLRP3</scp> inflammasome inhibitor ameliorates ischemic stroke by reprogramming the phenotype of microglia/macrophage in a murine model of distal middle cerebral artery occlusion. Neuropathology. 2022;42:181–9. https://doi.org/10.1111/neup.12802.
Saeedi-Boroujeni A, Mahmoudian-Sani M-R, Nashibi R, et al. Tranilast: a potential anti-inflammatory and NLRP3 inflammasome inhibitor drug for COVID-19. Immunopharmacol Immunotoxicol. 2021;43:247–58. https://doi.org/10.1080/08923973.2021.1925293.
Owona BA, Schluesener HJ. Molecular insight in the multifunctional effects of oridonin. Drugs R D. 2015;15:233–44. https://doi.org/10.1007/s40268-015-0102-z.
Fouad AA, Abdel-Aziz AM, Hamouda AAH. Diacerein downregulates NLRP3/Caspase-1/IL-1β and IL-6/STAT3 pathways of inflammation and apoptosis in a rat model of cadmium testicular toxicity. Biol Trace Elem Res. 2020;195:499–505. https://doi.org/10.1007/s12011-019-01865-6.
Wannamaker W, Davies R, Namchuk M, et al. ( S )-1-(( S )-2-{[1-(4-Amino-3-chloro-phenyl)-methanoyl]-amino}-3,3-dimethyl-butanoyl)-pyrrolidine-2-carboxylic acid ((2 R,3 S )-2-ethoxy-5-oxo-tetrahydro-furan-3-yl)-amide (VX-765), an orally available selective interleukin (IL)-converting enzyme/caspa. J Pharmacol Exp Ther. 2007;321:509–16. https://doi.org/10.1124/jpet.106.111344.
He W, Wan H, Hu L, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015;25:1285–98. https://doi.org/10.1038/cr.2015.139.
Hu JJ, Liu X, Xia S, et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat Immunol. 2020;21:736–45. https://doi.org/10.1038/s41590-020-0669-6.
Burmester GR, McInnes IB, Kremer JM, et al. Mavrilimumab, a fully human granulocyte–macrophage colony-stimulating factor receptor α monoclonal antibody. Arthritis Rheumatol. 2018;70:679–89. https://doi.org/10.1002/art.40420.
Weinblatt ME, McInnes IB, Kremer JM, et al. A randomized phase <scp>II</scp> b study of mavrilimumab and golimumab in rheumatoid arthritis. Arthritis Rheumatol. 2018;70:49–59. https://doi.org/10.1002/art.40323.
Behrens F, Tak PP, Østergaard M, et al. MOR103, a human monoclonal antibody to granulocyte–macrophage colony-stimulating factor, in the treatment of patients with moderate rheumatoid arthritis: results of a phase Ib/IIa randomised, double-blind, placebo-controlled, dose-escalation trial. Ann Rheum Dis. 2015;74:1058–64. https://doi.org/10.1136/annrheumdis-2013-204816.
De Luca G, Cavalli G, Campochiaro C, et al. GM-CSF blockade with mavrilimumab in severe COVID-19 pneumonia and systemic hyperinflammation: a single-centre, prospective cohort study. Lancet Rheumatol. 2020;2:e465–73. https://doi.org/10.1016/S2665-9913(20)30170-3.
Sfriso P, Priori R, Valesini G, et al. Adult-onset Still’s disease: an Italian multicentre retrospective observational study of manifestations and treatments in 245 patients. Clin Rheumatol. 2016;35:1683–9. https://doi.org/10.1007/s10067-016-3308-8.
Campochiaro C, Tomelleri A, Sartorelli S, et al. Drug retention and discontinuation reasons between seven biologics in patients with Takayasu arteritis. Semin Arthritis Rheum. 2020;50:509–14. https://doi.org/10.1016/j.semarthrit.2020.01.005.
Rossi-Semerano L, Fautrel B, Wendling D, et al. Tolerance and efficacy of off-label anti-interleukin-1 treatments in France: a nationwide survey. Orphanet J Rare Dis. 2015;10:19. https://doi.org/10.1186/s13023-015-0228-7.
Lequerré T, Quartier P, Rosellini D, et al. Interleukin-1 receptor antagonist (anakinra) treatment in patients with systemic-onset juvenile idiopathic arthritis or adult onset Still disease: preliminary experience in France. Ann Rheum Dis Published Online First. 2008. https://doi.org/10.1136/ard.2007.076034.
Laskari K, Tzioufas AG, Moutsopoulos HM. Efficacy and long-term follow-up of IL-1R inhibitor anakinra in adults with Still’s disease: a case-series study. Arthritis Res Ther. 2011;13:R91. https://doi.org/10.1186/ar3366.
Giampietro C, Ridene M, Lequerre T, et al. Anakinra in adult-onset Still’s disease: long-term treatment in patients resistant to conventional therapy. Arthritis Care Res (Hoboken). 2013;65:822–6. https://doi.org/10.1002/acr.21901.
Iliou C, Papagoras C, Tsifetaki N, et al. Adult-onset Still’s disease: clinical, serological and therapeutic considerations. Clin Exp Rheumatol. 2013;31(1):47–52.
Vercruysse F, Barnetche T, Lazaro E, et al. Adult-onset Still’s disease biological treatment strategy may depend on the phenotypic dichotomy. Arthritis Res Ther Published Online First. 2019. https://doi.org/10.1186/s13075-019-1838-6.
Colafrancesco S, Priori R, Valesini G, et al. Response to interleukin-1 inhibitors in 140 Italian patients with adult-onset Still’s disease: a multicentre retrospective observational study. Front Pharmacol. 2017. https://doi.org/10.3389/fphar.2017.00369.
Fautrel B. Tumour necrosis factor blocking agents in refractory adult Still’s disease: an observational study of 20 cases. Ann Rheum Dis. 2005;64:262–6. https://doi.org/10.1136/ard.2004.024026.
Ruscitti P, Ursini F, Sota J, et al. The reduction of concomitant glucocorticoids dosage following treatment with IL-1 receptor antagonist in adult onset Still’s disease A systematic review and meta-analysis of observational studies. Ther Adv Musculoskelet Dis. 2020;12:1759720X2093313. https://doi.org/10.1177/1759720X20933133.
Fujii T, Akizuki M, Kameda H, et al. Methotrexate treatment in patients with adult onset Still’s disease–-retrospective study of 13 Japanese cases. Ann Rheum Dis. 1997;56:144–8. https://doi.org/10.1136/ard.56.2.144.
Mitamura M, Tada Y, Koarada S, et al. Cyclosporin A treatment for Japanese patients with severe adult-onset Still’s disease. Mod Rheumatol. 2009;19:57–63. https://doi.org/10.1007/s10165-008-0126-0.
Franchini S, Dagna L, Salvo F, et al. Efficacy of traditional and biologic agents in different clinical phenotypes of adult-onset Still’s disease. Arthritis Rheum. 2010;62:2530–5. https://doi.org/10.1002/art.27532.
Gerfaud-Valentin M, Hot A, Huissoud C, et al. Adult-onset Still’s disease and pregnancy: about ten cases and review of the literature. Rheumatol Int. 2014;34:867–71. https://doi.org/10.1007/s00296-013-2765-5.
Harel M, Fauteux-Daniel S, Rodriguez E, et al. IL-18 binding protein–producing cells attenuate anemia in murine macrophage activation syndrome. J Immunol. 2023;210:1790–803. https://doi.org/10.4049/jimmunol.2300065.
Huang Z, Lee PY, Yao X, et al. Tofacitinib treatment of refractory systemic juvenile idiopathic arthritis. Pediatrics. 2019. https://doi.org/10.1542/peds.2018-2845.
Vojinovic J, Damjanov N, D’Urzo C, et al. Safety and efficacy of an oral histone deacetylase inhibitor in systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2011;63:1452–8. https://doi.org/10.1002/art.30238.
Bader-Meunier B, Hadchouel A, Berteloot L, et al. Effectiveness and safety of ruxolitinib for the treatment of refractory systemic idiopathic juvenile arthritis like associated with interstitial lung disease : a case report. Ann Rheum Dis. 2022;81:e20–e20. https://doi.org/10.1136/annrheumdis-2020-216983.
Chen Q-L, Yin H-R, He Q-Y, et al. Targeting the NLRP3 inflammasome as new therapeutic avenue for inflammatory bowel disease. Biomed Pharmacother. 2021;138: 111442. https://doi.org/10.1016/j.biopha.2021.111442.
Zhang X, Wang Z, Zheng Y, et al. Inhibitors of the NLRP3 inflammasome pathway as promising therapeutic candidates for inflammatory diseases (Review). Int J Mol Med. 2023;51:35. https://doi.org/10.3892/ijmm.2023.5238.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Open access funding provided by Università degli Studi di Padova within the CRUI-CARE Agreement.
Conflicts of Interest
We have no conflicts of interest to declare.
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Availability of Data and Materials
Not applicable.
Code Availability
Not applicable.
Author Contributions
SB carried out the search strategy, retrieved the literature and wrote the manuscript; CB prepared the figure, wrote the manuscript and revised it; AD revised the manuscript; and PS revised and drafted the final version of the manuscript. All authors have read and agreed to the published version of the manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Bindoli, S., Baggio, C., Doria, A. et al. Adult-Onset Still’s Disease (AOSD): Advances in Understanding Pathophysiology, Genetics and Emerging Treatment Options. Drugs 84, 257–274 (2024). https://doi.org/10.1007/s40265-024-01993-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-024-01993-x