
Abstract
Introduction
Acute lung injury and acute respiratory distress syndrome are common complications in patients with coronavirus disease 2019 (COVID-19). Poor outcomes in patients with COVID-19 are associated with cytokine release syndrome. Binding of interleukin-8 (CXCL8/IL-8) to its chemokine receptors, CXCR1/2, may mediate this inflammatory process. The aim of this clinical trial was to determine if CXCR1/2 blockade with reparixin can improve clinical outcomes in hospitalized patients with severe COVID-19 pneumonia. The dose and safety of reparixin have been investigated in clinical trials of patients with metastatic breast cancer.
Methods
This was a phase 2, open-label, multicenter, randomized study in hospitalized adult patients with severe COVID-19 pneumonia from May 5, 2020 until November 27, 2020. Patients were randomized 2:1 to receive 1200 mg reparixin orally three times daily or standard of care (SOC) for up to 21 days. The primary endpoint was defined as a composite of clinical events: use of supplemental oxygen, need for mechanical ventilation, intensive care unit admission, and/or use of rescue medication.
Results
Fifty-five patients were enrolled between reparixin (n = 36) and SOC (n = 19). The rate of clinical events was statistically significantly lower in the reparixin group compared with the SOC group (16.7% [95% CI 6.4–32.8%] vs. 42.1% [95% CI 20.3–66.5%], P = 0.02). The sensitivity analysis based on the Cox regression model provided an adjusted hazard ratio of 0.33 with statistical significance lower than 0.05 (95% CI 0.11–0.99; P = 0.047). Reparixin treatment appeared to be well tolerated.
Conclusion
In patients with severe COVID-19, reparixin led to an improvement in clinical outcomes when compared with the SOC. A larger phase 3 clinical study is needed to confirm these results.
Trial Registration
EudraCT identifier, 2020-001645-40; registered May 6, 2020 (retrospectively registered), and clinicaltrials.gov (NCT04794803) on March 8, 2021.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
Acute lung injury and acute respiratory distress syndrome are common complications in patients with COVID-19. |
Severe respiratory disease associated with COVID-19 has been linked to cytokine release syndrome, and blocking the interaction between the chemokine CXCL8 and its receptors CXCR1/2 has the potential to dampen the inflammatory process and improve patient outcomes. |
This study is the first to investigate the safety and efficacy of a CXCR1/2 inhibitor in hospitalized patients with severe COVID-19 pneumonia. |
What was learned from the study? |
Treatment with reparixin led to improved clinical outcomes in patients with severe COVID-19 pneumonia compared with the standard of care. |
Given the ongoing COVID-19 pandemic and continued hospitalization of patients with severe respiratory complications from hyperinflammation, these findings may assist clinicians with identifying alternative treatment options for this patient population. |
Introduction
Coronavirus disease 2019 (COVID-19), which results from severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection, is responsible for over 5.6 million deaths globally as of February 2022 [1]. Respiratory complications, including acute respiratory distress syndrome (ARDS) and acute lung injury (ALI), have been reported in an estimated one-third of patients hospitalized with COVID-19, with patients often requiring supplemental oxygen, mechanical ventilation, or even extracorporeal membrane oxygenation (ECMO) [2]. Lung edema, endothelial injury, and epithelial injury, characteristic features of ARDS and ALI, are accompanied by an influx of neutrophils into the interstitial and bronchoalveolar space, aiding in COVID-19 tissue damage and the hyperactivation of the immune system related to the development of cytokine release syndrome (CRS) [3].
Neutrophilia, high neutrophil-to-lymphocyte ratio, or elevated levels of neutrophil extracellular traps (NETs) have been reported as indicators of severe respiratory disease and poor outcomes in patients with COVID-19 [4,5,6]. Neutrophil activators (interleukin-8 and granulocyte colony-stimulating factor) and effectors (resistin, lipocalin-2, and hepatocyte growth factor) have been reported as early biomarkers in patients with severe COVID-19, including high levels of CXCL8 transcripts in bronchoalveolar lavage fluid [7, 8]. Moreover, lung autopsies from patients with COVID-19 have shown the presence of neutrophilic mucositis (neutrophil infiltration in pulmonary capillaries with extravasation to alveolar space) [9, 10].
Numerous studies support interleukin-8 (CXCL8/IL-8) receptors C-X-C chemokine receptor type 1 (CXCR1) and C-X-C chemokine receptor type 2 (CXCR2) as potential therapeutic targets in ALI and ARDS [11,12,13,14,15]. In ALI, allergen-induced inflammation, and particulate matter-induced pulmonary fibrosis mouse models, depletion of neutrophils aided in reduction of lung injury [2, 3, 13,14,15]. Moreover, lung injury was attenuated with inhibition of CXCL8 activity in various animal models of ALI, and the inhibition or knockout of CXCR2 receptors diminished neutrophil influx into the lung [10, 16,17,18,19].
Reparixin is being investigated across disease states, including metastatic breast cancer and COVID-19, on the basis of its noncompetitive allosteric inhibition of signals from the IL-8 receptors, CXCR1/CXCR2 [20]. In vitro and preclinical small animal studies have demonstrated that binding of reparixin to CXCR1/CXCR2 can prevent leukocyte recruitment and activation of inflammation. Action of reparixin includes inhibition of neutrophil stimulation and downstream effects of neutrophils, such as NETosis, the formation of NETs, during an inflammatory response [21]. The dose, safety, and pharmacokinetics of reparixin have been investigated in clinical studies of patients with metastatic breast cancer. In patients with breast cancer, there was minimal to no impact on baseline neutrophil counts in patients who received reparixin [22, 23].
Owing to its anti-inflammatory properties, reparixin has the potential to be a COVID-19 treatment option to attenuate or prevent CRS. Reparixin analogues have been shown to attenuate lung infections caused by influenza A virus and Streptococcus pneumoniae in vivo [24]. Additionally, a published case series demonstrated that four hospitalized patients with critical COVID-19 who were treated with reparixin were alive at the 3-month follow-up visit [25]. Together, these studies provided the rationale behind this phase 2, multicenter, open-label, randomized, clinical trial. The aim was to assess the efficacy and safety of reparixin compared with the available treatments considered as standard of care (SOC) in hospitalized patients with severe COVID-19 pneumonia.
Methods
Study Design
This was an open-label, randomized, controlled, multicenter phase 2 study to evaluate the efficacy and safety of reparixin in hospitalized adult patients with severe COVID-19 pneumonia. The definition of severe COVID-19 pneumonia was based upon the definition of severe COVID-19 illness described by the National Institutes of Health (NIH) [26]. This study was conducted at three study sites in Italy and one in Brazil from May 5, 2020 until November 27, 2020. At study entry, all patients gave written informed consent. The protocol and protocol amendment, together with all required clinical trial documentation, were approved by the independent ethics committee (IEC) of each investigational study site before the trial was initiated. The membership of each IEC was also obtained. The central ethics committee for this study was EC IRCSS Istituto Nazionale Per Le Malattie Infettive (approval number 86/2020). The study complied with the tenets of the Declaration of Helsinki, International Conference of Harmonization Tripartite Guidelines for Good Clinical Practice, current international and national regulations, the study protocol, and respective standard operating procedures of the participating sites, sponsor, and contract research organization. The study protocol was registered as EudraCT: 2020-001645-40, Registered May 6, 2020 (retrospectively registered). The first patient was enrolled on May 5, 2020. The study was also registered on clinicaltrials.gov (NCT04794803) on March 8, 2021. Registration of this trial by the authority was delayed by one day because of administrative issues caused by the COVID-19 pandemic.
Participants
Eligible patients included hospitalized adults (aged 18–90 years old) with polymerase chain reaction-confirmed COVID-19 diagnosis and severe pneumonia. Enrolled patients had respiratory distress [respiratory rate ≥ 30 breaths/minute without oxygen)] and/or partial arterial oxygen pressure (PaO2)/fraction of inspiration O2 (FiO2) > 100 to < 300 mmHg (1 mmHg = 0.133 kPa). Lung involvement was confirmed via chest imaging, which the investigators determined was consistent with pneumonia. Patients were also required to have elevated inflammatory markers with at least one of the following: lactate dehydrogenase > normal range, C-reactive protein ≥ 100 mg/L, or IL-6 ≥ 40 pg/mL, serum ferritin ≥ 900 ng/mL, serum crosslinked fibrin > 20 μg/mL.
Patients were excluded if they could not provide informed consent, had severe hepatic dysfunction (Child–Pugh score ≥ C, or aspartate aminotransferase > 5 times the upper limit), severe renal dysfunction (estimated glomerular filtration rate ≤ 30 mL/min/1.73 m2), or received continuous renal replacement therapy, hemodialysis, or peritoneal dialysis. Patients with hypersensitivity to ibuprofen or to more than one non-steroidal anti-inflammatory drug (NSAID) or more than one sulfonamide medication (hypersensitivity to sulfanilamide antibiotics alone, e.g., sulfamethoxazole, did not qualify for exclusion) were excluded. Patients with severe, active bleeding such as hemoptysis, gastrointestinal bleeding, central nervous system bleeding, and nosebleeds within one month before enrollment were excluded. Patients at risk for complications from NSAID use were excluded because one of reparixin’s metabolites is ibuprofen. Additionally, pregnant and lactating women and those planning to get pregnant were excluded. Those participating in other interventional clinical trials were not considered suitable for this study. At the time of enrollment, patients not in a clinical condition compatible with the oral administration of the study drug were excluded. At study entry, all patients provided written informed consent.
Randomization and Masking
Consented patients enrolled in the study were randomly assigned to their treatment in a 2:1 fashion between reparixin and SOC, using an Interactive Response System (IRS). The randomization list was created by an independent statistician not involved in conducting the study. Randomization was stratified by site to ensure balanced assignment across treatment groups. A stratified permuted block randomization list was generated with a computer procedure, randomizing an excess of patients to allow competitive recruitment within each center. Each randomized patient was allocated with a randomization number according to the stratified randomization list. Given the open-label nature of the trial and stratification by site, random block sizes were adopted in the randomization list to avoid forecasted treatment assignments by investigators. The master randomization list was kept confidential other than to those for which it was strictly required (i.e., independent statistician who created the list and IRS provider personnel). Mis-randomization events were recorded as a major deviation and reported in the final study report. Randomization codes were not reused in case of patient dropouts.
Procedures
The study intervention was 1200 mg reparixin tablets three times daily by mouth, with treatment duration lasting at least 7 days. If patients in the reparixin group improved, treatment could be prolonged at the discretion of the investigator until there was clinical improvement or for up to a total of 21 days. After randomization, if the patient was unable or unwilling to take oral tablets, the reparixin dose was crushed and used to create a suspension that was administered via a nasogastric tube. The control group was SOC, which was defined as any medication used to treat COVID-19 pneumonia at the time of the study. As a result of the nature of the COVID-19 pandemic, SOC was expected to evolve over time and, thus, was not prespecified. This group was populated prospectively and offered a rescue medication in case of worsening clinical status (e.g., need for intensive care unit [ICU] admission and/or mechanical ventilation). In the case of worsening clinical status, patients in both treatment groups were offered rescue medication, which was based on their physicians’ judgements without any constraint from the sponsor and could include intravenously administered reparixin in either the treatment or SOC group. As a result of the evolving understanding of novel treatments being tested during the COVID-19 pandemic, the sponsor did not place any restriction on the selection of the rescue medication. The overall study duration was a maximum of 1 month, including up to 21 days of treatment and up to 7 ± 3 days of follow-up.
Patients were assessed at screening, enrollment, day 1, day 2, and day 7 post-randomization, at the end of treatment (EOT), and at the end of study (EOS; 7 ± 3 days after ending treatment) for concomitant medications, clinical severity, dyspnea, oxygen treatment, mechanical ventilation, ICU admission, and respiratory parameters (FiO2, PaO2). All laboratory and clinical parameters, including radiography imaging assessments, were collected according to the local procedures of the participating clinical sites.
Outcomes
The primary objective was to determine the efficacy of reparixin compared with SOC in adult patients with severe COVID-19 pneumonia, as measured by the time-to-event analysis of the composite endpoint of clinical events. The composite endpoint was defined as patients requiring at least one of the following at any time during treatment or follow-up: supplemental oxygen, based on deterioration of the PaO2/FiO2 ratio by at least one-third (− 33.3%) from baseline PaO2/FiO2, mechanical ventilation use, admission to ICU, and/or use of a rescue medication for any reason. Supplemental oxygen was defined as requiring nasal cannula or invasive mechanical ventilation including high flow nasal cannula, bilevel-positive airway pressure, continuous positive airway pressure, or non-rebreather mask.
Secondary endpoints included changes in clinical severity score defined as the time to clinical improvement of two points from the time of randomization on a seven-category ordinal scale recommended by the World Health Organization or live discharge from the hospital, whichever occurred first. The seven-category ordinal scale consisted of the following: (1) not hospitalized, with resumption of normal activities; (2) not hospitalized, but unable to resume normal activities; (3) hospitalized, not requiring supplemental oxygen; (4) hospitalized, requiring supplemental oxygen; (5) hospitalized, requiring high-flow oxygen therapy, non-invasive mechanical ventilation, or both; (6) hospitalized, requiring ECMO, invasive mechanical ventilation, or both; and (7) death [27]. Other secondary endpoints included dyspnea severity (measured using the Likert scale by patients grading their breathing at the time of assessment compared with when they first started treatment on a scale of − 3 to 3 as follows: “0” = no change, “1” = minimally better, “2” = moderately better, “3” = markedly better, “− 1” = minimally worse, “− 2” = moderately worse, “− 3” = markedly worse”, and changes in the PaO2/FiO2 ratio.
Adverse events (AEs) were defined as any untoward medical occurrence in a patient, which resulted in an unfavorable and unintended sign (including an abnormal laboratory finding), symptom, or disease temporally associated with the use of the treatment, regardless of the relation to the medicinal product. Treatment emergent adverse events (TEAEs) were defined as AEs that started at or after the first administration of study treatment. AEs were graded as mild (acceptable discomfort), moderate (disturbing discomfort), or severe (unacceptable discomfort). Serious adverse events (SAE) were defined as any untoward medical occurrence that at any dose resulted in death or was life-threatening, required prolongation of existing hospitalization, resulted in persistent or significant disability or incapacity, or was medically significant.
Statistical Analysis
A target sample size of 48 was determined expecting a 1.5-fold improvement of the median time to the primary endpoint (the composite event) and assuming an exponential distribution to provide 80% power to show superiority of reparixin compared with SOC using a one-sided log-rank test at a significance level of 0.025.
The primary endpoint was analyzed along with the single components by a binomial response rate at each time point (only the first event was considered). The Clopper–Pearson method was used to estimate the two-sided 95% confidence intervals (CIs). Primary analysis on time to the composite endpoint was performed using Kaplan–Meier methodology, and the one-sided log-rank test was used to test for differences between treatment groups. The time to event for each single component of the primary endpoint was performed separately to adjust for the presence of possible competing risks, and the presence of other competing events were treated as censored. A multivariate Cox proportional hazard regression model was used to adjust for age (≤ 65 vs. > 65 years old), gender (male vs. female), and clinical severity score (based on seven-category ordinal scale recommended by the World Health Organization) at baseline as qualitative covariates. Changes from baseline in clinical severity score, dyspnea (Likert scale), and PaO2/FiO2 ratio were analyzed by a two-sided Fisher’s exact test. A Shapiro–Wilk test and visual inspection (QQ plot) were used to assess the normality of the quantitative variables. Median (interquartile range) and non-parametric tests were used in cases when normality was not met. Statistical tests for interaction were performed to decide the need for further investigation of subgroups by means of a Cox regression model.
Randomized patients who received at least one dose of treatment were included in the primary, secondary, and safety analyses. All statistical analyses and data processing were performed using the Statistical Analysis Systems (SAS®) Software (release 9.4). Final statistical analyses were performed by CROS NT S.r.l. (Verona, Italy) following sponsor approval of the statistical analysis plan and database lock. As a result of the open-label nature of this trial, an independent data monitoring committee provided timely recommendations regarding trial adaptations and data analyses.
Data Management
Data were collected through an electronic case report form and subsequently entered into a validated database which was used to produce the final statistical analyses. Once all data were entered into the database and all outstanding queries were solved, the clinical coding and the SAE reconciliation was performed; a data review meeting was held to review major and minor deviations and to define the study populations. A database lock occurred once queries were resolved.
Results
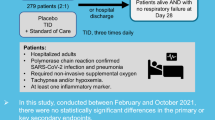
Fifty-six patients were screened, enrolled, and randomized from May 5, 2020 and followed until November 27, 2020. One patient who was randomized to the reparixin group did not take at least one dose of the assigned treatment and was excluded from the full analysis and safety analysis sets (Fig. 1). Baseline characteristics of the 55 enrolled patients in the full analysis set were similar between the reparixin (n = 36) and SOC (n = 19) groups and are displayed in Table 1. The average age was 60.6 ± 13.5 years for the reparixin group and 63.6 ± 14.2 years for the SOC group, with 61.1% (n = 22/36) and 57.9% (n = 11/19) of patients aged 65 years or older in the reparixin and SOC groups, respectively. The majority of study participants were male (72.2% [n = 26/36] vs. 84.2% [n = 16/19], reparixin and SOC groups, respectively) and had at least one concomitant disease (77.8% [n = 28/36] vs. 84.2% [n = 16/19], reparixin and SOC groups, respectively). Patients enrolled in the SOC group received remdesivir, corticosteroids, anticoagulants, and/or antibiotics. IL-6 receptor antagonists, such as tocilizumab, were not utilized in any patient in the SOC group because of the time period of this study. Baseline laboratory values were similar in both groups and are displayed in Table 2. The median number of treatment days in the reparixin group was 7 (Q1–Q3 = 6–7), with 62% of patients receiving treatment for more than 7 days.

Flowchart of the enrolled patients. Both the full analysis and safety sets consisted of all randomized patients who received at least one dose of reparixin. The full analysis was analyzed according to the intent-to-treat (ITT) principle and was used to determine efficacy results. The safety set was used to determine safety results
The Kaplan–Meier analysis of the primary endpoint of time to composite event demonstrated that reparixin statistically significantly prolonged the time to achieving at least one clinical event as compared with SOC (P = 0.02) (Fig. 2). Additionally, the proportion of patients with at least one composite event at the EOS was statistically significantly lower in the reparixin group compared with the SOC group (16.7% [95% CI 6.4–32.8%] vs. 42.1% [95% CI 20.3–66.5%]) (Table 3). The sensitivity analysis based on the Cox regression model provided an adjusted hazard ratio of 0.33 after controlling for covariates, with statistical significance lower than 0.05 (95% CI 0.11–0.99; P = 0.047). No statistically significant effects were observed for age (P = 0.20), gender (P = 0.54), and clinical severity score (P = 0.62), and the proportional hazard assumption was met (P = 0.16) (Table 3). After controlling for additional covariates within a post hoc analysis including obesity, diabetes mellitus, and hypertension, the hazard ratio (HR) and the statistical significance lower than 0.05 were maintained with reparixin [HR 0.32 (95% CI 0.11–0.95), P = 0.04] for the primary composite endpoint (Supplementary Material) as well as for adjusting for each clinical trial site (HR 0.30 [95% CI 0.10–0.97], P = 0.04).

Primary endpoint analysis: time to composite endpoint of clinical events. The Kaplan–Meier estimate shows that reparixin prolonged the time to achieving at least one clinical event as compared with the placebo group
No patients required rescue medication for any reason in the reparixin group (0.0%), as compared with 26.3% (n = 5/19) of patients in the SOC group (P = 0.001). Additionally, five patients in the SOC group were switched to rescue medications. For all of these patients, intravenously administered reparixin was used as the rescue therapy by the investigator. There was a trend favoring reparixin in requiring supplemental oxygen based on PaO2/FiO2 worsening (reparixin 13.9%, n = 5/36 vs. SOC 26.3%, n = 5/19; P = 0.20), requiring invasive mechanical ventilation (reparixin 2.8%, n = 1/36 vs. SOC 5.3%, n = 1/19; P = 0.30), and admission to the ICU (reparixin 2.8%, n = 1/36 vs. 5.3%, n = 1/19; P = 0.56) (Table 3).
The proportion of patients with improvement in clinical severity score of at least two points in the reparixin group demonstrated no statistically significant difference when compared with those in the SOC group at any time point up to week 1 (Table 3). The proportion of patients with improvement in dyspnea severity, as measured by the Likert scale, also demonstrated no statistically significant difference in the reparixin group when compared with those in the SOC group at any time point up to week 1 (Table 3). Worsening respiratory status, defined as a decrease of PaO2/FiO2 of at least one-third from baseline, was reported in several patients in both groups at various post-baseline time points. The proportion of patients with worsening respiratory status in the reparixin group when compared with the SOC group was not statistically significantly different at day 1 (7.4%, n = 2/27 vs. 14.3%, n = 2/14; P = 0.60), day 2 (12.9%, n = 4/31 vs. 20.0%, n = 3/15; P = 0.67), or EOT (0.0%, n = 0/29 vs. 8.3%, n = 1/13; P = 0.29) but was statistically significantly different at day 7 (0.0%, n = 0/26 vs. 21.4%, n = 3/14; P = 0.04) (Table 3). Notably, no patient in the reparixin group had worsening respiratory status from week 1 onwards.
Three patients (8.3%) in the reparixin group and five patients (26.3%) in the SOC group reported at least one TEAE (Table 4). None of the TEAEs were related to treatment and none led to temporary or permanent discontinuation of treatment. Severe TEAEs were reported in two patients (5.6%) in the reparixin group and in four patients (21.1%) in the SOC group. Serious AEs were reported in two patients (5.6%) in the reparixin group and in four patients (21.1%) in the SOC group. All serious AEs consisted of respiratory failure. Death occurred in one patient (2.8%) in the reparixin group and in three patients (15.8%) in the SOC group and was due to respiratory insufficiency/failure in all cases.
Discussion
In this phase 2 study, patients with severe COVID-19 pneumonia who received 1200 mg reparixin orally three times daily for up to 21 days had a lower incidence of the primary composite clinical outcome of supplemental oxygen, based on deterioration of the PaO2/FiO2 ratio by at least one-third (− 33.3%) from baseline PaO2/FiO2, mechanical ventilation use, admission to ICU, and/or use of a rescue medication for any reason, relative to those who received SOC. To our knowledge, this is the first study to test the safety and efficacy of a CXCR1/2 inhibitor in hospitalized patients with severe COVID-19 pneumonia.
A growing body of evidence shows that elevated IL-8 is upregulated early and remains elevated during the disease course of COVID-19 [10, 28]. These elevated levels of IL-8 have been significantly associated with increased mortality and greater disease severity in patients, indicating that IL-8 plays a role in disease progression [10, 28,29,30]. Considering these data, it is possible to speculate that decreased neutrophil recruitment to the lungs due to the CXCR1/2 blockade likely contributed to the improved clinical outcomes seen in this study by mitigating the severe and uncontrolled hyperinflammatory disease manifestations.
Previous studies on CXCL8 receptors CXCR1 and CXCR2 have identified them as potential therapeutic targets in ALI and ARDS. Preliminary data on four hospitalized patients with COVID-19 pneumonia and treated with reparixin under compassionate use procedures have been reported [25]. Patients had a clinical indication for mechanical ventilation and were treated with reparixin IV infusion (2.772 mg/kg), which started within 1 week of hospital admission and continued for 5 days. As of the publication of the report (3 months after hospital admission), all four patients were alive. While reparixin treatment was ongoing, the clinicians observed an improvement or at least stabilization of inflammatory markers (C-reactive protein, procalcitonin, and ferritin) and tissue damage markers (lactate dehydrogenase, aspartate, and alanine aminotransferase), a finding that was also seen during this phase 2 study utilizing the oral formulation of reparixin. The IV formulation of reparixin was used in compassionate use cases for critical patients with COVID-19 that required invasive mechanical ventilation compared to the current phase 2 study that used reparixin’s oral formulation in severe patients with COVID-19 requiring oxygen support or non-invasive ventilation. Most of these patients were treated outside of the ICU where oral medications are preferred. Additionally, the safety profile of the oral formulation of reparixin has already been well established in patients with breast cancer.
Reparixin was well tolerated in all patients, with no AEs related to the drug. When orally administered reparixin was studied in women with HER2 (also known as ERBB2)-negative breast cancer, the most common side effects (i.e., gastrointestinal disorders) were mild (grade ≤ 2) with no drug-related serious events [22, 31]. This tolerable safety profile was maintained when reparixin was investigated in combination with paclitaxel in a phase 2 randomized, double-blind, placebo-controlled clinical trial assessing reparixin or placebo in combination with paclitaxel in patients with metastatic triple-negative breast cancer. Vomiting, headache, anemia, and rash were the most common (> 10% incidence) side effects reported with reparixin [23]. Interestingly, a significantly lower frequency of cancer-related fatigue was noted in patients receiving reparixin compared with placebo [18%, n = 11 vs. 43.3%, n = 26, P = 0.003] [23]. This has been hypothesized to be due to the role of IL-8 in fatigue syndromes as reported in several studies, and may be of additional importance in patients with COVID-19. Importantly, within this immunocompromised patient population, neither neutropenia nor a sustained decrease in absolute neutrophil count was noted during these investigations, which aligns with the novel allosteric mechanism of action of reparixin.
As reparixin is a small molecule, its use could be applicable to global healthcare systems and to reduce pressure on healthcare systems. Moreover, the efficacy of reparixin is unlikely to be affected by any emergent SARS-CoV-2 variant, and it could represent an important resource for managing any crises determined by new variants awaiting vaccine implementation. Furthermore, reparixin represents a possible option for hospitalized patients requiring nasal cannula support where there is a current lack of recommended immunomodulators or within the severe population as resources and therapeutic shortages continue to be an issue [32].
There are limitations to our study. First, this was designed as an open-label study since a placebo-controlled group was not practical at the time of study design. This study was conducted at the beginning of the COVID-19 pandemic and for ethical reasons the sponsor was requested to have the investigators unmasked to the treatment the patients were receiving. The expected degree of real bias in an open-label study for a new disease such as COVID-19 pneumonia is still unknown. Second, given the severe state of disease experienced by some patients, rescue medications were allowed without restriction. While this was necessary in a small number of cases, their use may have reduced the significance of the difference in treatment response. Third, this relatively small trial consisted of 56 patients and was limited to three sites in Northern Italy and a single site in Brazil, thus lacking a sample that would be applicable to more widespread populations and also reducing the power to detect hazard ratio differences when controlling for baseline factors. Fourth, randomized patients who received at least one dose of reparixin were included in all analyses, which may not have been sufficient for patients to achieve steady state or therapeutic levels based on available pharmacokinetic data. Fifth, while the primary endpoint did not depend on patient input, the secondary endpoint of dyspnea severity was measured by the Likert scale, in which a patient grades their own breathing. Therefore, patients who were ventilated were unable to provide their input and were excluded from this analysis. Furthermore, CRP levels at baseline were low in both groups and the patients would not have qualified as having severe pneumonia based on this measure alone. Nonetheless, heterogeneity in the definition for severe pneumonia exists, and the clinical profile of patients in this study is consistent with that of severe pneumonia: all patients had a respiratory rate ≥ 30 breaths/min and 90% were receiving oxygen.
Additionally, the sensitivity and specificity of the different biomarkers, alongside their different cutoffs, led to the use of several alternative indicators of systemic inflammation: LDH, CRP, IL-6, serum ferritin, or serum cross-linked fibrin. Thus, inflammation status was not defined using a single marker, such as CRP (e.g., CRP > 100 mg/L often indicates a bacterial infection). Lastly, at the time of this study neither SOC treatments nor a regulatory guideline for severe patients was defined. During the time this study was conducted in 2020, corticosteroids were considered the standard of care with limited use of immunomodulators, such as interleukin-6 (IL-6) inhibitors or Janus kinase (JAK) inhibitors, which are now used to target inflammation and recommended by current guidelines in patients with rapidly increasing oxygen needs and systemic inflammation in addition to corticosteroids [32]. However, these various limitations will be addressed in a subsequent, larger phase 3 study.
Conclusions
Preclinical and clinical data support the use of CXCR1/2 blockade to dampen innate immune responses for improved clinical outcomes. In the present study, the administration of reparixin in patients with severe COVID-19 pneumonia improved clinical outcomes and may facilitate respiratory recovery relative to those receiving SOC. On the basis of these results, a larger phase 3 clinical study is needed to understand and confirm the role of reparixin in improving the clinical management of patients with respiratory distress related to severe COVID-19 pneumonia.
References
Weekly epidemiological update on COVID-19—1 February 2022. http://www.who.int. https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19---1-february-2022. Accessed 4 Feb 2022.
Tzotzos SJ, Fischer B, Fischer H, Zeitlinger M. Incidence of ARDS and outcomes in hospitalized patients with COVID-19: a global literature survey. Crit Care. 2020;24(1):516. https://doi.org/10.1186/s13054-020-03240-7.
Cavalcante-Silva LHA, Carvalho DCM, de Lima ÉA, et al. Neutrophils and COVID-19: the road so far. Int Immunopharmacol. 2021;90:107233.
Singh K, Mittal S, Gollapudi S, Butzmann A, Kumar J, Ohgami RS. A meta-analysis of SARS-CoV-2 patients identifies the combinatorial significance of d-dimer, C-reactive protein, lymphocyte, and neutrophil values as a predictor of disease severity. Int J Lab Hematol. 2020;43(2):324–8.
Liu J, Liu Y, Xiang P, et al. Neutrophil-to-lymphocyte ratio predicts critical illness patients with 2019 coronavirus disease in the early stage. J Transl Med 2020;18(1):206.
Veras FP, Pontelli MC, Silva CM, et al. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J Exp Med. 2020;217(12):e20201129. https://doi.org/10.1084/jem.20201129.
Meizlish ML, Pine AB, Bishai JD, et al. A neutrophil activation signature predicts critical illness and mortality in COVID-19. Blood Adv. 2021;5(5):1164–77.
Xiong Y, Liu Y, Cao L, et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg Microbes Infect. 2020;9(1):761–70.
Buja LM, Wolf DA, Zhao B, et al. The emerging spectrum of cardiopulmonary pathology of the coronavirus disease 2019 (COVID-19): report of 3 autopsies from Houston, Texas, and review of autopsy findings from other United States cities. Cardiovasc Pathol. 2020;48:107233.
Masso-Silva JA, Moshensky A, Lam MTY, et al. Increased IL-8, neutrophil activation phenotypes and NETosis in critically ill COVID-19 patients. SSRN Electron J. 2020. https://doi.org/10.2139/ssrn.3705291.
Puneet P, Moochhala S, Bhatia M. Chemokines in acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol. 2005;288(1):L3-15.
Keane MP, Donnelly SC, Belperio JA, et al. Imbalance in the expression of CXC chemokines correlates with bronchoalveolar lavage fluid angiogenic activity and procollagen levels in acute respiratory distress syndrome. J Immunol. 2002;169(11):6515–21.
Zarbock A, Allegretti M, Ley K. Therapeutic inhibition of CXCR2 by reparixin attenuates acute lung injury in mice. Br J Pharmacol. 2008;155(3):357–64.
Hosoki K, Rajarathnam K, Sur S. Attenuation of murine allergic airway inflammation with a CXCR 1/ CXCR 2 chemokine receptor inhibitor. Clin Exp Allergy. 2018;49(1):130–2.
Cheng I-Y, Liu C-C, Lin J-H, et al. Particulate matter increases the severity of bleomycin-induced pulmonary fibrosis through KC-mediated neutrophil chemotaxis. Int J Mol Sci. 2019;21(1):227.
Yang X-D, Corvalan JRF, Wang P, Roy CM-N, Davis CG. Fully human anti-interleukin-8 monoclonal antibodies: potential therapeutics for the treatment of inflammatory disease states. J Leukoc Biol. 1999;66(3):401–10.
Auten RL, Richardson RM, White JR, Mason SN, Vozzelli MA, Whorton MH. Nonpeptide CXCR2 antagonist prevents neutrophil accumulation in hyperoxia-exposed newborn rats. J Pharmacol Exp Ther. 2001;299(1):90–5.
Belperio JA, Keane MP, Burdick MD, et al. CXCR2/CXCR2 ligand biology during lung transplant ischemia-reperfusion injury. J Immunol. 2005;175(10):6931–9.
Gonçalves A-S, Appelberg R. The involvement of the chemokine receptor CXCR2 in neutrophil recruitment in LPS-induced inflammation and in Mycobacterium avium infection. Scand J Immunol. 2002;55(6):585–91.
Bertini R, Allegretti M, Bizzarri C, et al. Noncompetitive allosteric inhibitors of the inflammatory chemokine receptors CXCR1 and CXCR2: prevention of reperfusion injury. Proc Natl Acad Sci. 2004;101(32):11791–6.
Cheng OZ, Palaniyar N. NET balancing: a problem in inflammatory lung diseases. Front Immunol. 2013;4:1. https://doi.org/10.3389/fimmu.2013.00001.
Schott AF, Goldstein LJ, Cristofanilli M, et al. Phase Ib pilot study to evaluate reparixin in combination with weekly paclitaxel in patients with HER-2-negative metastatic breast cancer. Clin Cancer Res. 2017;23(18):5358–65.
Goldstein LJ, Mansutti M, Levy C, et al. A randomized, placebo-controlled phase 2 study of paclitaxel in combination with reparixin compared to paclitaxel alone as front-line therapy for metastatic triple-negative breast cancer (fRida). Breast Cancer Res Treat. 2021;190(2):265–75.
Tavares LP, Garcia CC, Machado MG, et al. CXCR1/2 antagonism is protective during influenza and post-influenza pneumococcal infection. Front Immunol. 2017;13:8.
Piemonti L, Landoni G. COVID-19 and islet transplantation: different twins. Am J Transplant. 2020;20(11):2983–8.
Clinical Spectrum. COVID-19 treatment guidelines. 2021. https://www.covid19treatmentguidelines.nih.gov/overview/clinical-spectrum/. Accessed 3 Feb 2022.
COVID-19 Therapeutic Trial Synopsis. http://www.who.int. https://www.who.int/publications/i/item/covid-19-therapeutic-trial-synopsis. Accessed 6 June 2021.
Liu L, Chen H-G, Li Y, et al. Temporal profiles of antibody responses, cytokines, and survival of COVID-19 patients: a retrospective cohort. Engineering. 2021;7(7):958–65.
Li H, Zhang J, Fang C, et al. The prognostic value of IL-8 for the death of severe or critical patients with COVID-19. Medicine. 2021;100(11):e23656.
Del Valle DM, Kim-Schulze S, Huang H-H, et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat Med. 2020;26(10):1636–43.
Goldstein LJ, Perez RP, Yardley D, et al. A window-of-opportunity trial of the CXCR1/2 inhibitor reparixin in operable HER-2-negative breast cancer. Breast Cancer Res. 2020;22(1):4.
COVID-19 Treatment Guidelines Panel. Coronavirus Disease 2019 (COVID-19) Treatment Guidelines. NIH. https://files.covid19treatmentguidelines.nih.gov/guidelines/covid19treatmentguidelines.pdf.
Acknowledgements
The authors thank all of the patients for their participation in this study.
Funding
This work was supported by Dompé Farmaceutici SpA, Milan, Italy. Dompé Farmaceutici SpA participated in the design and conduct of the study; management, analysis, and interpretation of the data; preparation and review of the manuscript; and provided funding for the journal’s Rapid Service Fee.
Medical Writing, Editorial, and Other Assistance
Medical writing support was provided by Meraki Consulting and funded by Dompé Farmaceutici SpA.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
GL, LP, MA, NP, and FM conceived and designed the study; GL, LP, AdM, PG, and AZ collected the study data; MA and FM supervised the study; GG and FM analyzed the data; MD was responsible for project administration for the study; NP drafted the manuscript; GL, LP, AdM, PG, AZ, EB, MA, GG, EG, NP, MD, GP, and FM reviewed and edited the manuscript. All authors read and approved the final version of the manuscript.
Disclosures
Marcello Allegretti, ChemD; Giovanni Goisis, MSc; Elizabeth M. Gavioli, PharmD; Neal Patel, PharmD; Maria DePizzol, PhD; Georgea Pasedis, PharmD; and Flavio Mantelli, MD are Dompé Farmaceutici SpA employees. Giovanni Landoni, MD; Lorenzo Piemonti, MD; Antonella d’Arminio Monforte, MD; Paolo Grossi, MD; and Alberto Zangrillo, MD have received grant support from Dompé Farmaceutici SpA. Enrico Bucci, PhD has no competing interests to declare.
Compliance with Ethics Guidelines
At study entry, all patients gave a written informed consent. The protocol and protocol amendment, together with all required clinical trial documentation, were approved by the independent ethics committee (IEC) of each investigational study site before the trial was initiated. The membership of each IEC was also obtained. The central ethics committee for this study was EC IRCSS Istituto Nazionale Per Le Malattie Infettive (approval number 86/2020). The study complied with the tenets of the Declaration of Helsinki, International Conference of Harmonization Tripartite Guidelines for Good Clinical Practice, current international and national regulations, the study protocol, and respective standard operating procedures of the participating sites, sponsor, and contract research organization.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Landoni, G., Piemonti, L., Monforte, A.d. et al. A Multicenter Phase 2 Randomized Controlled Study on the Efficacy and Safety of Reparixin in the Treatment of Hospitalized Patients with COVID-19 Pneumonia. Infect Dis Ther 11, 1559–1574 (2022). https://doi.org/10.1007/s40121-022-00644-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40121-022-00644-6