
Abstract
Introduction
Polymorphonuclear cell influx into the interstitial and bronchoalveolar spaces is a cardinal feature of severe coronavirus disease 2019 (COVID-19), principally mediated by interleukin-8 (IL-8). We sought to determine whether reparixin, a novel IL-8 pathway inhibitor, could reduce disease progression in patients hospitalized with severe COVID-19 pneumonia.
Methods
In this Phase 3, randomized, double-blind, placebo-controlled, multicenter study, hospitalized adult patients with severe COVID-19 pneumonia were randomized 2:1 to receive oral reparixin 1200 mg three times daily or placebo for up to 21 days or until hospital discharge. The primary endpoint was the proportion of patients alive and free of respiratory failure at Day 28, with key secondary endpoints being the proportion of patients free of respiratory failure at Day 60, incidence of intensive care unit (ICU) admission by Day 28 and time to recovery by Day 28.
Results
Of 279 patients randomized, 182 received at least one dose of reparixin and 88 received placebo. The proportion of patients alive and free of respiratory failure at Day 28 was similar in the two groups {83.5% versus 80.7%; odds ratio 1.63 [95% confidence interval (CI) 0.75, 3.51]; p = 0.216}. There were no statistically significant differences in the key secondary endpoints, but a numerically higher proportion of patients in the reparixin group were alive and free of respiratory failure at Day 60 (88.7% versus 84.6%; p = 0.195), fewer required ICU admissions by Day 28 (15.8% versus 21.7%; p = 0.168), and a higher proportion recovered by Day 28 compared with placebo (81.6% versus 74.9%; p = 0.167). Fewer patients experienced adverse events with reparixin than placebo (45.6% versus 54.5%), most mild or moderate intensity and not related to study treatment.
Conclusions
This trial did not meet the primary efficacy endpoints, yet reparixin showed a trend toward limiting disease progression as an add-on therapy in COVID-19 severe pneumonia and was well tolerated.
Trial Registration
ClinicalTrials.gov: NCT04878055, EudraCT: 2020-005919-51.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Why carry out this study? |
High interleukin-8 (IL-8) levels and neutrophil infiltration in the airways are hallmarks of disease severity of several pulmonary conditions, including COVID-19. |
Given IL-8 is the principal chemoattractant of neutrophils in the lung, a therapy that targets this pathway is of potential interest. |
This study follows a previous Phase 2 study, and was conducted to evaluate the efficacy and safety of reparixin, a noncompetitive allosteric inhibitor of IL-8 receptors, in patients hospitalized with severe COVID-19 pneumonia. |
What was learned from the study? |
This Phase 3 study demonstrated the safety and tolerability of reparixin in a group of patients with hypoxemic respiratory failure due to COVID-19 pneumonia. Despite no significant effect, it identified a trend in preventing disease deterioration necessitating escalation of care, when combined with standard of care. |
Our upcoming studies will build on these encouraging findings by determining the efficacy and safety of reparixin in hospitalized patients with hypoxemic respiratory failure due to pneumonia. |
Digital Features
This article is published with digital features, including a graphical abstract, to facilitate understanding of the article. To view digital features for this article, go to https://doi.org/10.6084/m9.figshare.24087894.
Introduction
Although most patients infected with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) experience mild disease [1], a proportion develops pneumonia and hypoxemic respiratory failure that can lead to fatal outcomes. The immunological phenotype of this severe “coronavirus disease 2019” (COVID-19) is characterized by a high neutrophil-to-lymphocyte ratio that correlates with disease severity [2, 3], predominantly due to a pronounced increase in neutrophil count [4]. Neutrophils mount anti-pathogen responses including the generation of neutrophil extracellular traps (NETosis), degranulation, and de novo production of cytokines and chemokines. These are vital innate immune responses against SARS-CoV-2, as they appear to be the most activated cellular immune responses in COVID-19 [5, 6]. However, these same processes may contribute to lung tissue damage and systemic complications such as thrombosis, acute respiratory distress syndrome, and multisystem inflammatory disease in children [7, 8]. Interleukin-8 [IL-8, also known as chemokine ligand 8 (CXCL8)] is the principal chemoattractant of neutrophils in the lung, with elevated systemic levels associated with poor outcomes in COVID-19 [9, 10].
Reparixin is a noncompetitive allosteric inhibitor of the IL-8 receptors 1 and 2 (CXCR1/CXCR2), which inhibits IL-8 mediated chemotaxis of human neutrophils [11], and their downstream biological effects such as NETosis [12]. In animal models of acute lung injury, reparixin significantly reduced capillary permeability and interstitial/alveolar neutrophil migration, improving gas exchange in both prophylactic and therapeutic approaches [13]. Similar biological and clinical outcomes, in terms of neutrophil infiltration and lung damage, were obtained using reparixin in lung injury due to influenza A virus and Streptococcus pneumoniae [14]. Reparixin also reduced pulmonary fibrosis and improved lung function in a particulate matter mice model [15]. In a mice sepsis model, reparixin decreased neutrophil extracellular traps (NET) formation without impairment of bacterial clearance, improving organ function and decreasing mortality [12].
In the pulmonary clinical setting, the role of reparixin in the prevention of primary graft dysfunction was investigated in a randomized controlled trial that included 114 patients; reparixin, although well tolerated with a good overall safety profile, was unable to show a statistically significant effect of reparixin on functional and clinical outcomes after lung transplantation [16]. In a previous Phase 2 study, completed during the first stages of the pandemic, reparixin in addition to standard of care (SoC) reduced disease progression in patients hospitalized with COVID-19 pneumonia, as determined by the proportion of patients who required supplemental oxygen, mechanical ventilation, intensive care unit (ICU) admission, and/or rescue medication for any reason (16.7% in the reparixin group versus 42.1% in the SoC group; p = 0.02) [17].
To further explore these initial findings, we conducted a larger Phase 3 study to evaluate the efficacy and safety of reparixin in patients hospitalized with severe COVID-19 pneumonia.
Methods
Study Design
This was a randomized, double-blind, placebo-controlled, multicenter study to evaluate the efficacy and safety of reparixin in hospitalized adult patients with severe COVID-19 pneumonia, conducted between February and October 2021. The definition of severe COVID-19 followed the National Institutes of Health (NIH) recommendation, and required the presence of hypoxemia in combination with lung infiltrates and/or tachypnea (Table S1 in the electronic supplementary material) [18]. The protocol and all required clinical trial documentation were approved by the independent ethics committee of each investigational study site before the study was initiated. The central ethics committee for this study was EC IRCSS Istituto Nazionale Per Le Malattie Infettive (approval number 254). The study complied with the tenets of the Declaration of Helsinki and the International Conference of Harmonization Tripartite Guidelines for Good Clinical Practice (ICH/CPMP/135/95), and was registered at EudraCT (2020-005919-51, 19 January 2021) and ClinicalTrials.gov (NCT04878055, 7 May 2021). All patients provided informed consent to participate in the study.
Participants
Eligible patients were hospitalized adults (aged 18–90 years) with polymerase chain reaction confirmed SARS-CoV-2 infection within 10 days of randomization, and radiologically verified pneumonia that required non-invasive supplemental oxygen. Patients had tachypnea (respiratory rate ≥ 24 breaths/min without oxygen) and/or hypoxemia [partial pressure of oxygen (PaO2) to fraction of inspiration O2 (FiO2) ratio 100–300 mmHg or peripheral arterial oxygen saturation (SpO2) ≤ 94% while breathing ambient air]. In addition, patients had at least one of the following inflammatory markers: lactate dehydrogenase above normal range, C-reactive protein ≥ 100 mg/L, IL-6 ≥ 40 pg/mL, serum ferritin ≥ 900 ng/mL, or serum cross-linked fibrin > 20 μg/mL. Patients were excluded if they had moderate/severe hepatic dysfunction (Child–Pugh score B–C, or aspartate aminotransferase > 5 times the upper limit of normal), moderate/severe renal dysfunction (estimated glomerular filtration rate ≤ 50 mL/min/1.73 m2, or were on continuous renal replacement therapy, hemodialysis, or peritoneal dialysis), history of hypersensitivity to ibuprofen (metabolite of reparixin) or to more than one nonsteroidal anti-inflammatory drug or to more than one sulfonamide medication. Patients with severe active bleeding, or a recent history of such, were also excluded. All patients provided written informed consent prior to any study-related procedure. A complete list of the inclusion and exclusion criteria is in Table S1 in the electronic supplementary material.
Procedures
In addition to SoC, patients were randomly assigned 2:1 to reparixin 1200 mg tablets three times daily by mouth or identical placebo (reparixin and placebo tablet composition are in Table S2 in the electronic supplementary material), using an interactive response system, based on a randomization list created by an independent statistician using a computer-generated stratified permuted block scheme. Randomization was stratified by site, sex, and age (< 65 versus ≥ 65 years). After randomization, patients unwilling or unable to swallow could receive tablets crushed and dispersed in water via naso-gastric tube, if already positioned. The duration of treatment was up to 21 days or until hospital discharge if occurring earlier than 21 days. Given the evolving nature of the disease, SoC was not strictly defined, and included any medication used during the study to treat COVID-19 pneumonia. Demographic data, medical history, previous and concomitant medications were collected at screening and/or at baseline. Clinical, laboratory, and hematology assessments were performed at screening, baseline, on Days 3, 7, 14, and 21 and on the day of discharge from hospital (or maximum at Day 28). Follow-up visits, in person or via telephone call, were conducted on Days 60 and 90, unless consent was withdrawn.
Clinical assessments included the 7-point ordinal scale recommended by the World Health Organization (WHO-OS) [19], oxygenation status (SpO2, PaO2, FiO2, and PaO2/FiO2), severity of dyspnea [assessed using a visual analogue scale (VAS) and a 100 mm Likert scale], and use of supplemental oxygen. The seven WHO-OS categories were: (1) not hospitalized, with resumption of normal activities; (2) not hospitalized, but unable to resume normal activities; (3) hospitalized, not requiring supplemental oxygen; (4) hospitalized, requiring supplemental oxygen; (5) hospitalized, requiring high-flow oxygen therapy, non-invasive mechanical ventilation, or both; (6) hospitalized, requiring extracorporeal membrane oxygenation (ECMO), invasive mechanical ventilation, or both; and (7) death. Supplemental oxygen was defined as any oxygen administered through nasal cannula or non-invasive ventilation (NIV), including high-flow nasal cannula, bilevel-positive airway pressure, continuous positive airway pressure, or non-rebreather mask. On Days 60 and 90 information was collected on patients’ general condition, the occurrence of clinically important adverse events, WHO-OS, need for supplemental oxygen or NIV, and new hospitalization and/or ICU admission since hospital discharge.
Outcomes
The primary endpoint was the proportion of patients alive and free of respiratory failure at Day 28, defined as no need for invasive mechanical ventilation or ECMO, or admission to ICU due to worsening respiratory function. The four key secondary endpoints were the proportion of patients alive and free of respiratory failure at Day 60, mortality up to Day 28, the incidence of ICU admission or death up to Day 28, and the time to recovery as defined by reversion to WHO-OS category 1, 2, or 3 up to Day 28. Additional secondary endpoints, which included the proportion of patients alive and free of respiratory failure at fixed time points and other measures of clinical improvement and level of care, are listed in Table S3 in the electronic supplementary material. Safety was assessed throughout the study in terms of the occurrence of adverse events (AEs), and hematology and laboratory evaluations.
Statistical Analysis
With a 2:1 (reparixin:placebo) randomization ratio and a one-sided alpha of 0.025, a total of 264 evaluable patients would have allowed an overall power of 90% to detect a group difference ≥ 20% in the proportion of patients alive and free of respiratory failure at Day 28 in favor of reparixin, assuming that the proportion of patients alive and free of respiratory failure in the placebo group was approximately 60%. The primary and key secondary endpoints were analyzed by a logistic regression model, adjusted by treatment, sex, age group, and presence of concomitant disease as fixed effects, and site as a random effect, with a one-sided test used to evaluate the difference between treatment groups. Time to recovery was analyzed using the cumulative incidence function, with the treatment groups compared by means of a Gray’s test.
The full analysis set (FAS), which comprised all randomized patients who received at least one dose of study drug, was used for all efficacy analyses, with patients analyzed according to treatment allocation. The per-protocol (PP) population, consisting of all patients in the FAS who did not have any major protocol deviations, was used for the sensitivity analysis of the primary endpoint. Safety analyses were performed using the safety population, which was the FAS with patients analyzed according to treatment received. All statistical analyses and data processing were performed using the Statistical Analysis Systems (SAS) Software (release 9.4).
Results
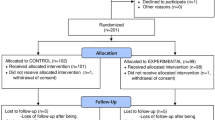
A total of 279 patients were randomized at 14 Italian sites; 270 received at least one dose of study treatment (182 and 88 in the reparixin and placebo groups, respectively), with 221 patients (148 and 73, respectively) completing treatment (Fig. 1). The patients’ baseline characteristics and clinical parameters were similar in the two groups (Table 1). The distribution of WHO-OS scores was similar in the two groups, with all patients having a score of 4 or 5, i.e., requiring supplemental oxygen, with half receiving high-flow supplemental oxygen and/or NIV. All patients had radiological imaging confirming lung involvement, and approximately one-third had ground-glass opacifications.

Patient disposition. The full analysis set and the safety population consisted of all randomized patients who received at least one dose of study medication. The full analysis set was performed according to the intent-to-treat principle and was used for the efficacy analyses. The safety set was used for the safety analyses
A total of 229 patients took at least one medication for COVID-19, most commonly glucocorticoids [219 (84.8%)] and remdesevir [74 (27.4%)]. Few patients received tocilizumab [two (1.1%) in the reparixin group versus none in the placebo group] or anakinra [nine (4.9%) versus two (2.3%)]. Prophylactic anticoagulants were used by 142 patients (78.0%) in the reparixin group and 65 (73.9%) in the placebo group.
The mean (± SD) duration of treatment was 9.5 ± 4.9 days [median 9.0 (range 1, 21 days)] in the reparixin group and 9.6 ± 4.8 days [8.0 (1, 21) days] in the placebo group. Compliance to study medication was similar in the two arms: a median of 97.2% [interquartile range (IQR) 93.3%–100%] in the reparixin group and 97.9% (IQR 94.2%–100%) in the placebo group. Compliance ≥ 80% was reported in 163 patients (89.6%) in the reparixin group and 81 (92.0%) in the placebo group.
For the primary endpoint, 152 patients (83.5%) in the reparixin group and 71 (80.7%) in the placebo group were alive and free of respiratory failure at Day 28 [odds ratio (OR) 1.63; p = 0.216; Table 2 and Fig. 2]. This was confirmed in the various sensitivity analyses, including in the PP population {1.92 [95% confidence interval (CI) 0.77, 4.79]; p = 0.162}, when patients who were in the ICU at baseline were excluded [1.84 (0.81, 4.19); p = 0.148], when only complete cases were considered [1.32 (0.41, 4.24); p = 0.638], or when analyzed by means of multiple imputation under missing at random assumptions [1.70 (0.74, 3.92); p = 0.215].

Proportion of patients alive and free of respiratory failure at each visit throughout the study (full analysis set)
For the key secondary outcomes (Table 2), 141 patients (88.7%) in the reparixin group and 66 (84.6%) in the placebo group were alive and free of respiratory failure at Day 60 (OR 1.77; p = 0.195), with 10 (6.0%) and 7 (8.6%), respectively dying up to Day 28 (0.47; p = 0.170). A total of 141 patients in the reparixin group and 63 in the placebo group recovered by Day 28, i.e., reverted to WHO-OS scores of 1, 2, or 3, corresponding to cumulative incidence functions of 81.6% and 74.9%, respectively (p = 0.167). The proportions of patients requiring ICU admission up to Day 28 when deaths were also considered as events were 15.8% and 21.7% (0.56; p = 0.168). In a post-hoc analysis based on the logistic regression model that excluded patients who died, 28-day ICU admission occurred in 20 (12.1%) patients in the reparixin group versus 18 (21.7%) with placebo [0.40 (0.17, 0.95); p = 0.038].
In a post-hoc analysis based on a Poisson regression model, over follow-up durations of 168.46 days in the reparixin group and 80.07 days in the placebo group, the adjusted mean rates of ICU admission days over 4 weeks were 0.0014 in the reparixin group and 0.0034 in the placebo group, equating to a rate ratio of 0.43 (95% CI 0.25, 0.73; p = 0.002). Additional secondary and exploratory endpoints are listed in Table 3.
Safety
A total of 205 treatment-emergent AEs (TEAEs) were reported in 83 patients (45.6%) in the reparixin group compared with 116 TEAEs in 48 patients (54.5%) in the placebo group (Table 4). Serious TEAEs were reported in 20 patients (11.0%) in the reparixin group (23 TEAEs) and 13 patients (14.8%) in the placebo group (16 TEAEs), none of which was related to treatment. The most common TEAEs by system organ class were: gastrointestinal disorders [21 patients (11.5%) in the reparixin group and 12 (13.6%) in the placebo group] and infections and infestations [13 patients (7.1%) in the reparixin group and 14 (15.9%) in the placebo group].
TEAEs leading to treatment discontinuation were reported in 10.4% of patients receiving reparixin versus 12.5% receiving placebo, whereas TEAEs leading to death were reported in 10 patients (5.5%) in the reparixin group and 7 (8.0%) in the placebo group, again none treatment related. Respiratory failure was the most common TEAE leading to death, in 8 patients (4.4%) in the reparixin group and 4 (4.5%) in the placebo group (Table 4). There were no substantial changes from baseline in hematology or laboratory parameters in either group (Table 5), and no significant vital sign or ECG changes.
Discussion
This Phase 3 study did not demonstrate efficacy of reparixin as compared with placebo in adults for severe COVID-19 pneumonia. Nonetheless, there was a positive trend in favor of reparixin in the primary and the secondary efficacy endpoints. More patients in the reparixin group were alive and free of respiratory failure at Days 28 and 60, fewer patients died by Day 28, fewer needed to be transferred to ICU for deterioration of respiratory status, and more had a meaningful recovery. This was consistently supported by the other endpoints; for example, reparixin was associated with statistically significant improvements in the exploratory analyses of serious events, i.e., invasive mechanical ventilation use or ECMO followed by death up to Days 28 and 60, indicating that reparixin may prevent the most invasive rescue treatments that are frequently linked to death. Furthermore, patients in the reparixin group started improving earlier, with significant differences in clinical status and oxygenation status on Day 3 (the first post-dose assessment).
The COVID-19 pandemic has led to close to 7 million deaths worldwide, with 1 million deaths being reported in the USA alone [20], and millions more affected directly and indirectly through impacts on their livelihood, employment opportunities, education, and long-term health-related quality of life. However, the impact of COVID-19 on respiratory pathology, along with the efforts taken to manage the disease, have not only reshaped our awareness of viral pneumonias, but has helped to advance the way we manage patients with infection-related acute hypoxemic respiratory failure.
Pharmacologic management of COVID-19, other than anti-virals, has focused on therapies that modulate the hyperinflammatory state effected by the excessive production of cytokines by a deregulated immune system, which is increasingly recognized as a key feature of severe COVID-19 [21]. Such immunomodulators include steroids, IL-6 inhibitors, and Janus kinase (JAK) inhibitors. The large RECOVERY study demonstrated a significant drop in mortality following early low-dose dexamethasone in patients with COVID-19, with the greatest benefit seen in mechanically ventilated patients whereas there was no benefit for those not requiring supplemental oxygen [22]. Unfortunately, this benefit is associated with significant drawbacks, especially when the use of steroids is protracted, in particular beyond the currently advised 10 days. Indeed, a meta-analysis of 21,350 patients with COVID-19 concluded that the overall effect of steroids on COVID-19-related mortality is less certain than anticipated, with the caveat that there was great heterogeneity among the studies reviewed [23]. When added to steroids, tocilizumab, the most widely used IL-6 receptor inhibitor, led to an improvement of in-hospital mortality—although when optimal SoC was applied this benefit was observed only among the sickest patients [24]. Furthermore, the risk of late-onset infectious complications due to tocilizumab exposure remains a concern [25]. This becomes even more of an issue with inhibition of the JAK–STAT pathway through the use of JAK inhibitors, since the involvement of JAKs in the immune response, especially via interferon-gamma, means that their potential adverse events include immunosuppression with reactivation of latent infection or development of new secondary infections, compounded by thrombotic events, cardiotoxicity, and hepatotoxicity [26].
IL-8 is one of the main neutrophil chemoattractors in the lung. Neutrophils abound in the lungs of patients with fatal COVID-19 [2, 3] and their activation-related functions such as the production of reactive oxygen species, proteases, inflammatory mediators, and release of NETs can result in local tissue injury. NETs are a constant histopathologic feature in the lungs of patients with fatal COVID-19, where their spatial distribution correlates closely with local IL-8 levels [27]. In fact, IL-8 together with other ELR+CXCL (i.e., those with the amino acid sequence Glu-Leu-Arg present) chemokines acting on CXCR1 and CXCR2, are among the most effective NET promoters [28, 29], both persistent and out of proportion to tissue viral load [27]. IL-8 pathway activation is a marker of disease status with serum levels increasing in correlation with progression of disease severity in patients with COVID-19 [30]. Our previous experience with reparixin supported further exploration of the role of IL-8 inhibition in severe COVID-19.
The current study was designed on assumptions informed by the first wave of COVID-19 that approximately 60% of patients in the placebo group would be alive and free of respiratory failure at Day 28, with a difference ≥ 20% in favor of reparixin. These assumptions were not confirmed, given the rapidly evolving natural history and treatment options for COVID-19. In our previous Phase 2 study in a similar patient population, 42.1% of patients in the placebo group experienced the composite endpoint of need for supplemental oxygen or mechanical ventilation use, ICU admission, or requirement of rescue medication by Day 28 (compared with 16.7% in the reparixin group) [17]. In addition, 15.8% of patients in this group died, whereas in the current study mortality by Day 28 was 8.6% in the placebo group and 6.0% in the reparixin group. Given that the patients enrolled in the two studies had comparable COVID-19 severity (as assessed using WHO-OS), the difference in outcomes is most likely attributable to the adaption of new therapies for COVID-19 that became SoC in the meantime, as well as changes in the proportion of vaccinated patients and changes in disease epidemiology and natural history. As disease-related mortality decreases, therapies that have demonstrated a positive effect on survival during the first wave of the disease are likely to lose this effect in subsequent studies. As an example, in the latest tocilizumab study, REMDACTA [24], which failed to demonstrate a survival benefit, mortality in both groups (18% versus 20% with tocilizumab versus placebo, respectively) was much lower than that in the RECOVERY and REMAP-CAP studies that established a mortality benefit (REMAP-CAP, 28% versus 36%; RECOVERY 31% versus 35% [31, 32]).
Lack of mortality benefit, however, does not equate to lack of efficacy, and our results demonstrate a beneficial effect of reparixin on important patient-centered outcomes such as transfer to ICU for deterioration of respiratory status. Even 1 day away from the ICU or not intubated translates to considerable gains in psychological strain for patients and their families and financial cost [33,34,35]. Reparixin appears to assist in achieving this goal, thus adding to the COVID-19 armamentarium in a meaningful way. Furthermore, and in contrast to other COVID-19 therapies [23, 36,37,38], reparixin was not associated with any safety signals, and was very well tolerated. Compared with those receiving placebo, fewer patients in the reparixin group had TEAEs, overall, serious, treatment-related, or severe TEAEs, or TEAEs leading to treatment discontinuation or death. Given that the most commonly reported TEAEs were related to the underlying disease (e.g., respiratory failure or respiratory distress), the fact that patients receiving reparixin experienced fewer AEs is a further indication of efficacy. Consistently with the previous trials [17, 39], there was a very low incidence of secondary infection despite the widespread use of glucocorticoids.
The main limitation of the study is that, because it was conducted during a surge of the pandemic in Italy, study conduct was at times difficult, leading to a high amount of missing clinical and pharmacokinetic data, especially at later time points. Furthermore, the identity and prevalence of SARS-CoV-2 variants in patients participating in the study are unknown, as the majority of the participating centers did not have access to variant screening methods. However, during the study period, the SARS-CoV-2 alpha variant was predominant, corresponding to an increase in infections during the second wave of the epidemic, whereas the omicron variant and its sublineages, which emerged in South Africa in November 2021, were not included in the study [40]. Moreover, there was a rapid change in the prevailing SARS-CoV-2 variant in Italy during the conduction of the study, with prevalence of the alpha variant increasing from 3.5% in December 2020 to 86.7% by March 2021 [40]. Given that evolving viral strains differ in their ability to evade host immunity, it is unclear whether an immune modulator that acts primarily through regulation of neutrophil activation would have the same efficacy across variants. In addition, the attempt to collect inflammatory markers to investigate the link between inflammatory status and drug efficacy was not successful. Finally, although the proportion of patients with concomitant glucocorticoid use was collected, the length of administration was not captured in the study database.
Conclusions
This Phase 3 study demonstrated the safety and tolerability of reparixin in a group of patients with hypoxemic respiratory failure due to COVID-19 pneumonia and indicated that when reparixin is used in addition to optimal therapy, it may still offer an additional advantage in preventing disease deterioration leading to ICU admission and mechanical ventilation or death. Reparixin is free of significant side effects and is not associated with increased risk for secondary infections, which is a significant advantage over currently used immunomodulators. Given the decrease in hospitalizations for COVID-19, our upcoming studies will build on these encouraging findings by expanding the study population to hospitalized patients with acute respiratory failure in pneumonia including COVID-19.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72 314 cases from the Chinese Center For Disease Control and Prevention. JAMA. 2020;323:1239–42. https://doi.org/10.1001/JAMA.2020.2648.
Liu J, Liu Y, Xiang P, Pu L, Xiong H, Li C, et al. Neutrophil-to-lymphocyte ratio predicts critical illness patients with 2019 coronavirus disease in the early stage. J Transl Med. 2020;18:206. https://doi.org/10.1186/s12967-020-02374-0.
Zeng Z-Y, Feng S-D, Chen G-P, Wu J-N. Predictive value of the neutrophil to lymphocyte ratio for disease deterioration and serious adverse outcomes in patients with COVID-19: a prospective cohort study. BMC Infect Dis. 2021;21:80. https://doi.org/10.1186/s12879-021-05796-3.
Masso-Silva JA, Moshensky A, Lam MTY, Odish MF, Patel A, Xu L, et al. Increased peripheral blood neutrophil activation phenotypes and neutrophil extracellular trap formation in critically ill coronavirus disease 2019 (COVID-19) patients: a case series and review of the literature. Clin Infect Dis. 2022;74:479–89. https://doi.org/10.1093/cid/ciab437.
Haick AK, Rzepka JP, Brandon E, Balemba OB, Miura TA. Neutrophils are needed for an effective immune response against pulmonary rat coronavirus infection, but also contribute to pathology. J Gen Virol. 2014;95:578–90. https://doi.org/10.1099/VIR.0.061986-0/CITE/REFWORKS.
Zhao X, Zhou L, Kou Y, Kou J. Activated neutrophils in the initiation and progression of COVID-19: hyperinflammation and immunothrombosis in COVID-19. Am J Transl Res. 2022;14:1454–68.
Feldstein LR, Rose EB, Horwitz SM, Collins JP, Newhams MM, Son MBF, et al. Multisystem inflammatory syndrome in U.S. children and adolescents. N Engl J Med. 2020;383:334–46. https://doi.org/10.1056/NEJMoa2021680.
Malgaj Vrecko M, Veceric-Haler Z. Coronavirus disease 2019-associated thrombotic microangiopathy. J Hematol. 2022;11:148–53.
Ma A, Zhang L, Ye X, Chen J, Yu J, Zhuang L, et al. High levels of circulating IL-8 and soluble IL-2r are associated with prolonged illness in patients with severe COVID-19. Front Immunol. 2021;12:12. https://doi.org/10.3389/FIMMU.2021.626235/BIBTEX.
Cavalcante-Silva LHA, Carvalho DCM, de Lima ÉA, Galvão JGFM, da Silva JSDF, de Sales-Neto JM, et al. Neutrophils and COVID-19: the road so far. Int Immunopharmacol. 2021;90:107233. https://doi.org/10.1016/J.INTIMP.2020.107233.
Bertini R, Allegretti M, Bizzarri C, Moriconi A, Locati M, Zampella G, et al. Noncompetitive allosteric inhibitors of the inflammatory chemokine receptors CXCR1 and CXCR2: prevention of reperfusion injury. Proc Natl Acad Sci USA. 2004;101:11791–6. https://doi.org/10.1073/PNAS.0402090101.
Alsabani M, Abrams ST, Cheng Z, Morton B, Lane S, Alosaimi S, et al. Reduction of NETosis by targeting CXCR1/2 reduces thrombosis, lung injury, and mortality in experimental human and murine sepsis. Br J Anaesth. 2022;128:283–93. https://doi.org/10.1016/J.BJA.2021.10.039.
Zarbock A, Allegretti M, Ley K. Therapeutic inhibition of CXCR2 by reparixin attenuates acute lung injury in mice. Br J Pharmacol. 2008;155:357–64. https://doi.org/10.1038/bjp.2008.270.
Tavares LP, Garcia CC, Machado MG, Queiroz-Junior CM, Barthelemy A, Trottein F, et al. CXCR1/2 antagonism is protective during influenza and post-influenza pneumococcal infection. Front Immunol. 2017. https://doi.org/10.3389/FIMMU.2017.01799.
Cheng IY, Liu CC, Lin JH, Hsu TW, Hsu JW, Li AFY, et al. Particulate matter increases the severity of bleomycin-induced pulmonary fibrosis through KC-mediated neutrophil chemotaxis. Int J Mol Sci. 2019;21:227. https://doi.org/10.3390/IJMS21010227.
Meyers BF, Keshavjee S, Zamora MR, Davis RD, Smith MA, McFadden PM, et al. 405: A multicenter prospective, randomized, placebo-controlled trial of a CXCL8 Inhibitor (reparixin) to prevent primary graft dysfunction after lung transplantation. J Hear Lung Transplant. 2008;27:S206–7. https://doi.org/10.1016/j.healun.2007.11.417.
Landoni G, Zangrillo A, Piersanti G, Scquizzato T, Piemonti L. The effect of reparixin on survival in patients at high risk for in-hospital mortality: a meta-analysis of randomized trials. Front Immunol. 2022;13:932251. https://doi.org/10.3389/fimmu.2022.932251.
National Institutes of Health. Clinical spectrum of SARS-CoV-2 infection [Internet]. 2023 [cited 2023 Jul 25]. Available from: https://www.covid19treatmentguidelines.nih.gov/overview/clinical-spectrum/
World Health Organization. WHO R&D Blueprint: Novel coronavirus [Internet]. 2020 [cited 2023 Jul 25]. Available from: https://www.who.int/publications/i/item/covid-19-therapeutic-trial-synopsis
World Health Organization. WHO Coronavirus (COVID-19) dashboard [Internet]. [cited 2023 Jul 25]. Available from: https://covid19.who.int/?mapFilter=deaths
Gustine JN, Jones D. Immunopathology of hyperinflammation in COVID-19. Am J Pathol. 2021;191:4–17. https://doi.org/10.1016/J.AJPATH.2020.08.009.
The RECOVERY Collaborative Group. Dexamethasone in hospitalized patients with Covid-19. N Engl J Med. 2020;384:693–704. https://doi.org/10.1056/NEJMoa2021436.
Cano EJ, Fonseca Fuentes X, Corsini Campioli C, O’Horo JC, Abu Saleh O, Odeyemi Y, et al. Impact of corticosteroids in coronavirus disease 2019 outcomes: systematic review and meta-analysis. Chest. 2021;159:1019–40. https://doi.org/10.1016/J.CHEST.2020.10.054.
Rosas IO, Diaz G, Gottlieb RL, Lobo SM, Robinson P, Hunter BD, et al. Tocilizumab and remdesivir in hospitalized patients with severe COVID-19 pneumonia: a randomized clinical trial. Intensive Care Med. 2021;47:1258–70. https://doi.org/10.1007/S00134-021-06507-X.
Pettit NN, Nguyen CT, Mutlu GM, Wu D, Kimmig L, Pitrak D, et al. Late onset infectious complications and safety of tocilizumab in the management of COVID-19. J Med Virol. 2021;93:1459–64. https://doi.org/10.1002/JMV.26429.
Levy G, Guglielmelli P, Langmuir P, Constantinescu S. JAK inhibitors and COVID-19. J Immunother Cancer. 2022;10: e002838. https://doi.org/10.1136/JITC-2021-002838.
Melero I, Villalba-Esparza M, Recalde-Zamacona B, Jiménez-Sánchez D, Teijeira Á, Argueta A, et al. Neutrophil extracellular traps, local IL-8 expression, and cytotoxic T-lymphocyte response in the lungs of patients with fatal COVID-19. Chest. 2022;162:1006–16. https://doi.org/10.1016/J.CHEST.2022.06.007.
Teijeira Á, Garasa S, Gato M, Alfaro C, Migueliz I, Cirella A, et al. CXCR1 and CXCR2 chemokine receptor agonists produced by tumors induce neutrophil extracellular traps that interfere with immune cytotoxicity. Immunity. 2020;52:856-871.e8. https://doi.org/10.1016/J.IMMUNI.2020.03.001.
Alfaro C, Sanmamed MF, Rodríguez-Ruiz ME, Teijeira Á, Oñate C, González Á, et al. Interleukin-8 in cancer pathogenesis, treatment and follow-up. Cancer Treat Rev. 2017;60:24–31. https://doi.org/10.1016/J.CTRV.2017.08.004.
Li L, Li J, Gao M, Fan H, Wang Y, Xu X, et al. Interleukin-8 as a biomarker for disease prognosis of coronavirus disease-2019 patients. Front Immunol. 2021;11:602395. https://doi.org/10.3389/FIMMU.2020.602395.
Gordon A, Mouncey P, Al-Beidh F, Rowan K, Nichol A, Arabi Y, et al. Interleukin-6 receptor antagonists in critically ill patients with Covid-19. N Engl J Med. 2021;384:1491–502. https://doi.org/10.1056/NEJMOA2100433.
Abani O, Abbas A, Abbas F, Abbas M, Abbasi S, Abbass H, et al. Tocilizumab in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial. Lancet (London, England). 2021;397:1637–45. https://doi.org/10.1016/S0140-6736(21)00676-0.
Kaier K, Heister T, Wolff J, Wolkewitz M. Mechanical ventilation and the daily cost of ICU care. BMC Health Serv Res. 2020;20:267. https://doi.org/10.1186/S12913-020-05133-5.
Halvorsen K, Jensen JF, Collet MO, Olausson S, Lindahl B, Sætre Hansen B, et al. Patients’ experiences of well-being when being cared for in the intensive care unit—an integrative review. J Clin Nurs. 2022;31:3–19. https://doi.org/10.1111/JOCN.15910.
Topçu S, Alpar ŞE, Gülseven B, Kebapçı A. Patient experiences in intensive care units: a systematic review. Patient Exp J. 2017;4:115–27. https://doi.org/10.35680/2372-0247.1137.
Brotman DJ, Girod JP, Posch A, Jani JT, Patel JV, Gupta M, et al. Effects of short-term glucocorticoids on hemostatic factors in healthy volunteers. Thromb Res. 2006;118:247–52. https://doi.org/10.1016/J.THROMRES.2005.06.006.
Sandhu G, Piraino ST, Piticaru J. Secondary infection risk in patients with severe COVID-19 pneumonia treated with tocilizumab. Am J Ther. 2022;29:E275–8. https://doi.org/10.1097/MJT.0000000000001487.
Charan J, Dutta S, Kaur R, Bhardwaj P, Sharma P, Ambwani S, et al. Tocilizumab in COVID-19: a study of adverse drug events reported in the WHO database. Expert Opin Drug Saf. 2021;20:1125–36. https://doi.org/10.1080/14740338.2021.1946513.
Schott AF, Goldstein LJ, Cristofanilli M, Ruffini PA, McCanna S, Reuben JM, et al. Phase Ib pilot study to evaluate reparixin in combination with weekly paclitaxel in patients with HER-2-negative metastatic breast cancer. Clin Cancer Res. 2017;23:5358–65. https://doi.org/10.1158/1078-0432.CCR-16-2748.
Lai A, Bergna A, Menzo S, Zehender G, Caucci S, Ghisetti V, et al. Circulating SARS-CoV-2 variants in Italy, October 2020-March 2021. Virol J. 2021;18:168. https://doi.org/10.1186/S12985-021-01638-5.
Acknowledgements
The authors would like to thank the investigators and patients at the investigative sites for their support of this study.
Editorial Assistance
Editorial support in the preparation of this manuscript was provided by David Young of Young Medical Communications and Consulting Ltd. This support was funded by Dompé Farmaceutici SpA.
Funding
This study and the manuscript’s Rapid Service fee were funded by Dompé Farmaceutici SpA.
Author information
Authors and Affiliations
Contributions
The study was conceived and designed by Elisabetta Perrotta, Giovanni Goisis, Elizabeth M. Gavioli, Sophie Toya, Maria De Pizzol, Flavio Mantelli, Marcello Allegretti, and Enrico Maria Minnella. Data were collected by Lorenzo Piemonti, Giovanni Landoni, Antonio Voza, Massimo Puoti, Ivan Gentile, Nicola Coppola, Stefano Nava, Alessia Mattei, Franco Marinangeli, Giulia Marchetti, Paolo Bonfanti, Claudio Maria Mastroianni, Matteo Bassetti, Ernesto Crisafulli, Paolo Antonio Grossi, Alberto Zangrillo, Antonio Desai, Marco Merli, Maria Foggia, Marco Carpano, Lorenzo Schiavoni, Antonella D’Arminio Monforte, Luca Bisi, Gianluca Russo, Fabiana Busti, and Cristina Rovelli. All authors were involved in the interpretation of the data, reviewed the manuscript, and approved the submitted version.
Corresponding author
Ethics declarations
Conflict of Interest
Lorenzo Piemonti has no other conflicts of interest to disclose. Giovanni Landoni has no other conflicts of interest to disclose. Antonio Voza declares consulting fees from Sildeha Pharma Swisse; payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events from Minerva Medica and Viatris; payment for expert testimony from GlaxoSmithKline, Alfa Sigma (to his institution), and Aurora Biopharma; and unpaid roles as guest editor of special issue on COVID-19: Prognosis and Long-term Sequelae’ on Viruses, and an advisory board for Critical Reviews in Oncology/Hematology, all outside the scope of the current manuscript. Massimo Puoti declares payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events from Gilead Sciences, GlaxoSmithKline, and AstraZeneca, all outside the scope of the current manuscript. Ivan Gentile declares departmental grants from Gilead; personal fees and payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing, or educational events from MSD, AbbVie, Gilead, Pfizer, GlaxoSmithKline, SOBI, Nordic/InfectoPharm, Angelini, Abbott, and Basilea; support for attending a meeting from Janssen and Gilead; and participation on a data safety monitoring board or advisory board for MSD, AbbVie, Gilead, Pfizer, GlaxoSmithKline, SOBI, Nordic/InfectoPharm, Angelini, Abbott, and Basilea, all outside the scope of the current manuscript. Nicola Coppola has no other conflicts of interest to disclose. Stefano Nava has no other conflicts of interest to disclose. Alessia Mattei has no other conflicts of interest to disclose. Franco Marinangeli declares payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing, or educational events from Grunenthal, Mundipharma, and MSD, all outside the scope of the current manuscript. Giulia Marchetti declares payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing, or educational events from Gilead, ViiV, MSD, Shionogi, and Angelini; support for attending meetings and/or travel from Jannsen and ViiV; and participation on a data safety monitoring board or advisory board for Viiv and Gilead, all outside the scope of the current manuscript. Paolo Bonfanti declares consulting fees and payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing, or educational events from Viiv, Gilead, Merck, Jannsen, and Pfizer, all outside the scope of the current manuscript. Claudio Maria Mastroianni declares consulting fees from Gilead and VIIV; payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing, or educational events from MSD, VIIV, Gilead, Menarini, AstraZeneca, and Pfizer; and support for attending meetings and/or travel from Menarini, all outside the scope of the current manuscript. Matteo Bassetti declares payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing, or educational events from Angelini, Astellas, Bayer, Biomerieux, Cidara, Gilead, Menarini, MSD, Nabriva, and Shionogi, and participation on a data safety monitoring board or advisory board for Angelini, Astellas, Bayer, Biomerieux, Cidara, Gilead, Menarini, MSD, Nabriva, and Shionogi, all outside the scope of the current manuscript. Ernesto Crisafulli declares a grant from GlaxoSmithKline; honoraria for lecturing for AstraZeneca, Boehringer Ingelheim, Chiesi, GlaxoSmithKline, Menarini, Qbgroup, and Sanofi; honoraria for scientific advisory boards for Chiesi and GlaxoSmithKline; and support for attending meetings and/or travel from Chiesi and GlaxoSmithKline, all outside the scope of the current manuscript. Paolo Antonio Grossi declares consulting fees from Takeda, MSD, and Gilead; payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events from Biotest and Gilead; and participation on a data safety monitoring board or advisory board for Reithera, all outside the scope of the current manuscript. Alberto Zangrillo has no other conflicts of interest to disclose. Antonio Desai has no other conflicts of interest to disclose. Marco Merli declares grants or contracts from Gilead, and support for attending meetings and/or travel from Pfizer, all outside the scope of the current manuscript. Maria Foggia has no other conflicts of interest to disclose. Marco Carpano has no other conflicts of interest to disclose. Lorenzo Schiavoni has no other conflicts of interest to disclose. Antonella D’Arminio Monforte has no other conflicts of interest to disclose. Luca Bisi has no other conflicts of interest to disclose. Gianluca Russo declares support for attending a meeting from Gilead, outside the scope of the current manuscript. Fabiana Busti has no other conflicts of interest to disclose. Cristina Rovelli has no other conflicts of interest to disclose. Elisabetta Perrotta, Giovanni Goisis, Elizabeth M. Gavioli, Sophie Toya, Maria De Pizzol, Flavio Mantelli, Marcello Allegretti, and Enrico Maria Minnella are employees of Dompé Farmaceutici SpA, the sponsor of the study.
Ethical Approval
The study was approved by all institutions and was performed in accordance with the Helsinki Declaration and the International Conference of Harmonization Tripartite Guidelines for Good Clinical Practice (ICH/CPMP/135/95), and was registered at EudraCT (2020-005919-51, 19 January 2021) and ClinicalTrials.gov (NCT04878055, 7 May 2021). All patients provided informed consent to participate in the study.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Piemonti, L., Landoni, G., Voza, A. et al. Efficacy and Safety of Reparixin in Patients with Severe COVID-19 Pneumonia: A Phase 3, Randomized, Double-Blind Placebo-Controlled Study. Infect Dis Ther 12, 2437–2456 (2023). https://doi.org/10.1007/s40121-023-00871-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40121-023-00871-5