
Abstract
Insertion or deletion polymorphism (InDel) is one of the main genetic variations in plant genomes. However, there are few studies on InDels across the whole genome in Populus. In this study, we investigated genome-wide InDels in Populus deltoides and Populus simonii and InDel segregation in their F1 hybrid population with restriction-site associated DNA sequencing (RAD-seq) data. A total of 119,066 InDels were identified in P. deltoides and P. simonii according to the reference genome of Populus trichocarpa, including 58,532 unique InDels in P. deltoides, 54,469 unique InDels in P. simonii, and 6,065 common InDels in both. Meanwhile, the distribution of these InDels was analyzed along chromosomes, indicating that the distribution patterns for both species were largely similar, but the average InDel density was slightly higher in P. deltoides than in P. simonii. GO annotation and enrichment analysis of those genes harboring InDels showed the same patterns between the two poplar species. It is interesting to find that the ratio (~ 46%) of the common InDels within genes to all common InDels was higher than that of the InDels within genes to all InDels in P. deltoides (~ 35%) or in P. simonii (~ 34%), possibly indicating that those InDels are more conservative between poplar species. Moreover, investigation of the InDel segregation patterns demonstrated that a large number of Mendelian InDels could be selected for genetic mapping in the F1 hybrid population. RAD-seq provides genome-wide insights into the InDel distributions in P. deltoides and P. simonii and the segregation patterns in their progeny, providing valuable genomic variation information for genetic and evolutionary studies in Populus.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The genus Populus comprises approximately 30 tree species, naturally distributed in the Northern Hemisphere (Strauss 1994). Some species not only have many attractive biological characteristics but also possess great economic and ecological value (Tong et al. 2016). They are generally diploid plants (2n = 38), and their genome size is close to that of rice, approximately 480 Mb, 4 times that of Arabidopsis thaliana. Due to their small genome, fast growth, long lifespan, and ease of asexual and seed reproduction, this kind of tree has become a model system among forest trees (Woolbright et al. 2008; Zhang et al. 2019). With advances in sequencing technologies, several species of Populus have been successively obtained, including P. trichocarpa (Tuskan et al. 2006), P. pruinosa (Yang et al. 2017), P. alba (Liu et al. 2019b), P. euphratica (Zhang et al. 2020), P. simonii (Wu et al. 2020) and P. deltoides (Bai et al. 2021). These genomic resources provide a fundamental basis for identifying genetic variations and developing molecular markers between or within Populus species.
Next-generation sequencing (NGS) technology allowed us to obtain a large amount of short read data across many plant individuals in a fast and cost-effective way (Song et al. 2015). With available bioinformatics tools, such as BWA (Li and Durbin 2009) and SAMtools (Li et al. 2009), a large number of single nucleotide polymorphisms (SNPs) can be identified and genotyped from the sequencing data of each individual (Liu et al. 2013). SNPs are the most abundant variations in DNA sequences found in most organisms (Ganal et al. 2009; Hu et al. 2014; McCouch et al. 2010), but another form of DNA variation, i.e., a polymorphism in the length of the DNA sequence caused by insertion or deletion of one or more nucleotides (InDels) at a certain site in the genome (Weber et al. 2002), has received relatively little attention compared to widely studied SNPs (Liu et al. 2013). However, InDels represent the second most abundant form of genetic variation in humans and plants (Pena and Pena 2012; Song et al. 2015), and they offer the advantages of a multiallelic nature, codominant inheritance and extensive genome coverage (Das et al. 2015). Compared with SNPs, InDels can be easily identified based on their size (Song et al. 2015). Indeed, InDels have become powerful molecular markers for species diagnostics (Yamaki et al. 2013), evolutionary studies (Weber et al. 2002), genetic linkage map construction (Song et al. 2015; Li et al. 2015), and marker-assisted selection (MAS) breeding (Liu et al. 2013). Recently, Zhu et al. (2018) performed restriction-site associated DNA sequencing (RAD-seq) to identify SNP and InDel markers for constructing a high-density SNP linkage map in Vitis. Meanwhile, Kizil et al. (2020) used double digested restriction site-associated DNA sequencing (ddRAD-seq) data to develop InDel markers for 95 sesame cultivars.
Although the mechanism for the formation of InDels remains elusive, various studies have been conducted to investigate their distribution and associated sites or regions in the genome as well as their impact on proteins. Tian et al. (2008) indicated that the occurrence of InDels was generally associated with proximal nucleotide divergence. However, more studies have shown that InDels have a greater impact on protein structure and function than SNPs (Ramakrishna et al. 2018). It is well known that InDels can change protein conformation and lead to major trait differences in mitochondrial genes (Lin et al. 2017). Therefore, InDels can be used to develop phylogenetic markers. InDel markers not only have high stability and accuracy but are also easy to amplify through polymerase chain reaction (PCR), so they have been proven to be convenient and effective in molecular breeding (Feng et al. 2020; Weber et al. 2002). Two InDels with the same length are extremely unlikely to appear in the same genomic location, which means that the shared InDels are considered to have the same origin, thus avoiding the difficulties of subsequent analysis caused by complexity and specificity (Shedlock and Okada 2000).
In our previous studies, an F1 hybrid population descended from a female P. deltoides and male P. simonii was established and successively sequenced with restriction-site associated DNA sequencing (RAD-seq) technology (Tong et al. 2016; Mousavi et al. 2016). Many high-quality (HQ) SNPs were extracted across the population to construct high-density genetic linkage maps and to then perform quantitative trait locus (QTL) analysis of growth traits, without considering InDels. In this study, we used RAD-seq data to investigate the distribution of InDels in P. deltoides and P. simonii. Moreover, the segregation patterns of these InDels were also analyzed with progeny from the F1 hybrid population. The results facilitate the understanding of the characteristics of InDels in P. deltoides and P. simonii, and the polymorphic InDel markers that follow Mendelian segregation law provide a valuable resource for constructing InDel genetic maps, conducting marker-assisted selection (MAS) breeding, and locating quantitative trait loci (QTLs).
Results
Mapping reads to the reference genome
A total of 915.5 Gb RAD-seq data containing 3,159,482,930 paired-end (PE) reads were obtained from P. deltoides and P. simonii and 47 progeny in the F1 hybrid population (Table 1). After quality control with the NGS QC toolkit (Patel and Jain 2012), we obtained 846.9 Gb of HQ read data. The female parent P. deltoides yielded 5.90 Gb (32,457,232) of HQ reads, whereas the male parent P. simonii retained 12.2 Gb (68,406,849) of HQ reads. A total of 828.8 Gb of HQ reads were obtained from the 47 progeny. With the short read mapping program BWA, 4.92 Gb (27,101,240) of the HQ reads from the female parent and 9.96 Gb (55,888,722) from the male parent were properly mapped to the reference genome of P. trichocarpa. In the progeny, a total of 687.0 Gb (2,340,191,288) of HQ reads were mapped to the reference genome. The mapped HQ reads of the female and male parents reached 13-fold and 25-fold effective genome coverage depths, respectively. For the progeny, the effective coverage depth ranged from 15- to 47-fold. Table 1 summarizes the raw, HQ, and mapped data of the female and male parents as well as the average for the 47 progeny. More detailed information for each sample data is presented in Table S1.
Distribution of InDels in P. deltoides and P. simonii
A total of 64,597 InDels were obtained in P. deltoides, including 32,862 (50.9%) inserted InDels and 31,735 (49.1%) deleted InDels, while in P. simonii 60,534 InDels were detected, of which 29,828 (49.3%) were inserted InDels and 30,706 (50.7%) were deleted InDels (Table 2). Here, the inserted and deleted InDels refer to those in which one of the alleles is inserted and deleted compared with the reference, respectively. Table 2 lists the number of InDels detected across the 19 chromosomes. As expected the highest number of InDels was detected on chromosome 1 in both species because its length is the longest. In contrast, chromosome 19 possessed the lowest number of InDels in each species. Overall, the number of InDels on chromosomes was largely proportional to chromosome length, with a correlation coefficient of 0.891 for P. deltoides and 0.897 for P. simonii. Furthermore, we calculated the number of InDels per 1 Mb on each chromosome. The average number of InDels was 166 per 1 Mb in P. deltoides, ranging from 138 for chromosome 11 to 192 for chromosome 9. Meanwhile, in P. simonii the average number was 155 with a range from 130 for chromosome 11 to 178 for chromosome 8.
Considering the positions, we found 6,065 (~ 5%) InDels at the same loci in the genomes of the two species. There were 58,532 and 54,469 unique InDels in the two species, accounting for 49% and 46% of all InDels identified, respectively (Table 2). We observed that the number of unique InDels in each species was essentially proportional to the chromosome length, with a correlation coefficient of 0.897 for P. deltoides and 0.900 for P. simonii. Furthermore, chromosome 1 harbored the largest number of unique InDels, with 7,460 unique in P. deltoides, 7,003 unique in P. simonii, and 772 common InDels unique to both. In contrast, chromosome 19 contained the fewest InDels, with 2,005 unique InDels in P. deltoides, 1,956 unique InDels in P. simonii, and 772 common unique InDels in both. Except for chromosome 17, the number of InDels in each chromosome of P. deltoides was greater than that in P. simonii.
The distribution of InDel length was also investigated in the two species (Fig. 1). In P. deltoides, single-nucleotide InDels were the most common type, followed by dinucleotide InDels, and these two types accounted for 46.3% of the total InDels. Among all InDels, 90.3% were less than or equal to 10 bp, 5.8% were between 11 and 15 bp, and 3.9% were greater than 15 bp long. In P. simonii, single-nucleotide and dinucleotide InDels accounted for 45.3% of the total InDels, with 89.9% less than or equal to 10 bp, 5.9% between 11 and 15 bp, and 4.2% more than 15 bp long (Table 3). Apparently, there was a tendency for the number of InDels to gradually decrease in accordance with increasing length.

Distribution of the InDel length in P. deltoides and P. simonii. The x-axis represents the InDel length, where the negative numbers indicate the deletions and positive numbers indicate the insertions. The y-axis represents the number of InDels at each length
Figure 2 shows the distribution of InDel numbers in sliding windows of 1 Mb along each chromosome for the two species. The InDel number was unevenly distributed on a single chromosome. In P. deltoides, the number of InDels per 1 Mb ranged from 11 to 274 (Table S2). Among them, the largest density of InDels was found on chromosome 8, while the lowest density was found on chromosome 6. In P. simonii, the number of InDels per 1 Mb ranged from 4 to 258, with the largest density on chromosome 15 and the lowest on chromosome 8 (Table S3). Moreover, we found 102 high-density regions with > 200 InDels per Mb and 15 low-density regions with < 50 InDels per Mb in P. deltoides. Similarly, 59 high-density and 13 low-density regions were found in P. simonii (Table S4). Most of the chromosomes in both parents were composed of a mixture of high-density and low-density InDel regions that were randomly distributed in chromosomes. It was noticed that in both species, there were 8 chromosomes containing no low-density InDel regions. In addition, in P. simonii, there were no high-density regions on chromosome 11, and neither high-density nor low-density regions were found on chromosome 12.

Circular representation of the distribution of InDels in P. deltoides and P. simonii along the 19 chromosomes. (A) The 19 chromosomes are shown in different colors. (B) The number of InDels is shown in sliding windows of 1 Mb in each chromosome for P. deltoides. (C) The number of InDels is shown in sliding windows of 1 Mb in each chromosome for P. simonii
Functional annotation of InDels
The annotation of the P. trichocarpa reference genome v4.0 was used to uncover the distribution of InDels within distinct genomic regions. According to the gene structure of the reference genome, InDels occurred more frequently in intergenic regions than in genic regions. In P. deltoides, 64.91% (41,931) of InDels were located in intergenic regions, whereas 35.09% (22,666) of InDels were located in genic regions, of which 13,864 InDels were in intron regions, 7,092 were in UTR regions and 1,710 were in CDS regions (Table 4). In P. simonii, 65.68% (39,759) of the InDels were located in intergenic regions, but only 34.32% (20,775) of InDels were located in genic regions, of which 12,729 InDels were in intron regions, 6,394 were in UTR regions and 1,652 were in CDS regions. We also found that there were 2,790 common InDels for both species located in the gene regions, including the CDS, UTR, and intron, accounting for 46.00% of the total number of common InDels. A total of 37,861 unique InDels from the two species were located in the gene regions, of which P. deltoides and P. simonii contained 19,876 and 17,985, accounting for 34% and 33% of the total number of their unique InDels, respectively. Among the 37,861 unique InDels, 11,828 (P. deltoides: 6,232 and P. simonii: 5,565) were located in UTR regions, 23,233 (P. deltoides: 12,184 and P. simonii: 11,049) in intron regions, and 2,800 (P. deltoides: 1,429 and P. simonii: 1,371) in CDS regions. Interestingly, we found that the ratio (~ 46%) of the common InDels within genes to all common InDels was higher than that of the InDels within genes to all InDels in P. deltoides (~ 35%) or in P. simonii (~ 34%), possibly indicating that those InDels are more conservative between poplar species.
It is also interesting to investigate the distribution of the number of InDels in the CDS region per gene because they possibly accumulated during evolution. We identified 1,710 and 1,652 InDels in the CDS regions of P. deltoides and P. simonii, respectively. In P. deltoides, InDels in the CDS region were distributed in 1,543 different genes, with 507 genes harboring two or more InDels. In P. simonii, InDels were distributed in 1,450 different genes, with 510 genes harboring two or more InDels (Table 5). On average, 1.11 InDels per gene were detected in P. deltoides, while 1.14 InDels per gene presented in P. simonii. InDels in the CDS region can result in two different types of variants, frameshift (FS) and nonframeshift (NFS) (Lin et al. 2017). There were more NFS InDels than FS InDels in the CDS region (Table S5). We detected 570 and 545 FS InDels in P. deltoides and P. simonii, respectively. FS InDels are usually considered more deleterious. It is possible that some instances of multiple InDels in the same gene could serve as compensatory InDels, thus restoring the translation frame and resulting in less deleterious mutation (Liu et al. 2015).
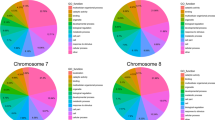
InDels that occur in functionally important regions of genes (typically CDS regions) can affect gene function through frameshifts and structural changes in proteins (Zhang et al. 2016). To better understand the potential functions of InDels within genes, GO term enrichment analysis of genes containing InDels within the CDS region was performed. These genes were classified into three categories: biological processes, cellular components, and molecular functions. We found that these genes in the two species exhibited similar categorizing patterns (Fig. 3). Categories based on biological processes revealed that the mutant genes were related to 22 biological processes; the three most overrepresented GO terms were cellular process, metabolic process and single-organism process, suggesting that these mutated genes were involved in a broad range of physiological functions. In P. deltoides, these genes were ultimately classified into 14 categories based on cellular components, while in P. simonii, they were classified into 17 categories; the three GO terms cell, cell part and organelle were the most abundant. Based on the molecular function category, these genes were ultimately classified into 15 categories; binding and catalytic activity were mainly enriched, while other functions only accounted for a small part.

Gene Ontology (GO) functional annotations of genes containing InDels within the CDS region for P. simonii (A) and P. deltoides (B). The horizontal axis indicates the GO classification types, and the vertical axis represents the number of annotated protein-coding genes
Analysis of InDel segregation in the progeny
At each site of all the InDels in the two species (Table 2), we called genotypes across the two parents and 47 progeny and performed a chi-squared test for the Mendelian segregation ratio. If one of the parental genotypes or 20% of the progeny genotypes were not called at an InDel site, the site was removed from the dataset. Consequently, a total of 11,141 InDel sites were identified for segregation in the progeny and followed Mendelian segregation ratios of 1:1 and 1:2:1 with p ≥ 0.01 (Tables S6-S8). The segregation types of these InDels included ab×aa, aa×ab, and ab×ab, with numbers of 5,811, 5,294, and 36, respectively, where the first two letters represent the female parent genotype and the last two the male parent genotype (Table 6).
Discussion
In this study, by mapping the HQ RAD-seq reads to the reference genome of P. trichocarpa, we found 64,597 and 60,534 InDels in female P. deltoides and male P. simonii, respectively. Because there were 6,065 common InDels in the two species, these InDels amounted to a total of 119,066 unique variant sites (Table 2). The number of InDels in P. simonii was slightly less than that in P. deltoides, which indicated that the heterozygosity of P. simonii was slightly lower than that of P. deltoides. Interestingly, the number of SNPs in P. simonii was also slightly less than that in P. deltoides, which were called with the same RAD-seq data and reference genome as in our previous study (Tong et al. 2016), resulting in a total of 836,895 SNP sites in the same two parents. We noticed that the total number of SNPs found in the two species was approximately 6-fold greater than that of InDels. Such a phenomenon, that the number of SNPs was much greater than that of InDels, could be expected because SNPs are the most abundant genomic variants in most species (Ganal et al. 2009; McCouch et al. 2010; Hu et al. 2014). For example, Liu et al. (2019a) identified 7,511,731 SNPs and 255,218 InDels between two tea cultivars, Camellia sinensis var. sinensis and Camellia sinensis var. assamica, where the number of SNPs is approximately 28-fold larger than the InDels.
The average densities of InDels in P. deltoides and P. simonii were approximately 166 and 155 InDels/Mb, respectively, which were higher than that of pepper (71 InDels/Mb) (Qin et al. 2014) but much lower than those of cucumber (916 InDels/Mb) (Qi et al. 2013) and tomato (1,448 InDels/Mb) (Lin et al. 2014). The main reason for such significant difference may be attributed to the unique genome composition or structure of different plant species (Liu et al. 2019a). Simultaneously, we found that the average density of InDels on chromosome 19 of P. deltoides and P. simonii was slightly lower than that on the other chromosomes except for chromosome 11. This may be related to the implication that chromosome 19 was considered to be responsible for sex determination through a ZW system in Populus (Yin et al. 2008).
It is important to understand the positions of genetic variations in the genome. In P. deltoides, the majority of InDels (64.91%) were more frequently located in intergenic regions, which may be related to the lower pressure of natural selection and/or domestication in these regions (Barreiro et al. 2008), while the rest (22,666) were found to be in genetic regions, of which only 1,710 InDels were within CDS regions (Table 4). A similar situation was also observed in P. simonii. The results showed that a small number of InDels were distributed in the CDS region, which could be explained by the fact that the CDS region only accounts for a small part of the whole genome in Populus and is more conserved than other regions (Liu et al. 2019a). InDels occurring in the CDS region often have a greater impact on genes. FS InDels change the coding sequence of the reading frame starting from the locus of insertion/deletion, producing different protein sequences or premature termination of protein sequences (Lin et al. 2017). These effects are generally considered deleterious, so these InDels may be removed from the population through purification selection (de la Chaux et al. 2007; Taylor et al. 2004). However, it is possible that multiple InDels in the same gene can also be used to restore the coding sequence of the reading frame to ameliorate the deleterious effects caused by FS InDels.
Since Populus belongs to outbred forest trees with a long generation time and high heterozygosity, it is almost impossible to obtain a genetic mapping population like in inbred lines such as the traditional BC and F2 populations (Wu et al. 2000; Zhang et al. 2009). An F1 hybrid population is usually derived by crossing two individuals for linkage mapping in outbred species, especially in forest trees (Grattapaglia and Sederoff 1994; Tong et al. 2020). Maliepaard et al. (1997) summarized that the molecular makers in such an F1 population possibly segregate in various types, such as ab×aa, aa×ab, and ab×ab. Subsequently, great efforts have been made to develop statistical methods for genetic linkage mapping with these different segregation types of markers (Wu et al. 2002; Tong et al. 2010). In our previous studies (Tong et al. 2016, 2020; Mousavi et al. 2016), we only used SNPs to construct the linkage maps of P. deltoides and P. simonii. However, it is interesting to use InDels as molecular markers for genetic linkage mapping (Song et al. 2015; Li et al. 2015). We therefore investigated the segregation patterns at all detected InDel sites across the 47 progeny in the current study. As a result, only 9.36% (11,141) of the total InDel loci were found to follow the Mendelian segregation ratio with p ≥ 0.01. Other InDel loci were excluded because they either had more missing genotypes (≥ 10) or presented distorted segregation (p < 0.01) in the progeny. This result was similar to the situation for calling SNPs in our previous study (Tong et al. 2016), in which 836,895 SNPs were identified but only 2,545 (0.30%) were used for linkage mapping due to uncalled genotypes or distorted segregation. As expected, and similar to the result in SNP calling (Tong et al. 2016), the majority of Mendelian InDels were segregated in the ratio of 1:1 with segregation types of ab×aa and aa×ab (Table 6). This can be attributed to the fact that the two parents belong to different species, and each has high heterozygosity in Populus. Our results demonstrated that abundant InDels could be selected as Mendelian markers with RAD-seq data for genetic linkage mapping studies in an F1 hybrid population. Unlike the traditional PCR-based method (Song et al. 2015; Li et al. 2015), each sample in the mapping population was genotyped by mapping its short reads to a reference genome sequence. However, for such a purpose, the sample size should be increased to much larger than the current study to allow more precise linkage analysis.
Materials and methods
Plant materials and sequence data
The plant materials came from an F1 hybrid population of P. deltoides and P. simonii, which was generated from 2009 to 2011 (Tong et al. 2016). The female P. deltoides ‘I-69’ was chosen from Siyang Forest Farm (SFF), Jiangsu Province, China, while the male P. simonii ‘L-3’ was collected from forestland in Luoning County, Henan Province, China. Approximately 500 progeny were planted in Xiashu Forest Farm of Nanjing Forest University, Jurong County, Jiangsu Province, China. In previous studies, we used the enzyme EcoRI to digest the genomic DNA and performed RAD sequencing of the two parents and their 418 progeny (Tong et al. 2016; Mousavi et al. 2016). The PE read data for each individual are available based on the SRR accession numbers as listed in Tong et al. (2020). Because most individuals have lower genome coverage data, we chose the two parents and 47 progeny with the highest coverage data for identifying InDel markers and performing the segregation analysis in this study. The accession numbers for these selected individual data are also listed in Table S1.
Identification of InDels
The procedure of calling InDel genotypes for each individual was as follows. (1) The command “mem” of BWA (Burrows–Wheeler Aligner) software (Li and Durbin 2009) was used to align the RAD-seq PE reads to the reference genome of P. trichocarpa (v4.0; http://www.phytozome.net), generating a SAM (sequence alignment/map) format file for each sample. We chose P. trichocarpa as the reference sequence because it is the first genome sequence in Populus and has been updated many times since its publication. Simulating the digestion of this reference genome by EcoRI showed that there are a total of 110,418 enzyme sites, of which 35,990 are within 18,660 genes. (2) Each SAM file was converted to a BAM file and then sorted and indexed with SAMtools software (Li et al. 2009). (3) With the BAM files, BCFtools software was used to generate BCF files. (4) Two parental VCF (variant call format) (Danecek et al. 2011) files were generated from the BCF files with the command “bcftools call -m –v”. (5) The InDel sites were extracted from the parental VCF files and saved as a list site file for calling InDel genotypes. (6) For each individual, a VCF file was generated with its BCF file and the list site file generated above. (7) The InDel genotype of each individual at each site was extracted from its VCF file such that the read depth of an allele was at least 3 and the genotype quality was greater than 30.
Location and functional annotation of InDels
The InDel sites were determined according to the reference genome of P. trichocarpa (http://www.phytozome.net). The InDels on chromosomes were annotated as genic or intergenic. The genic InDels were classified as CDS, UTR, and intron according to their localization. Next, the genes containing InDels were annotated by first aligning their coding sequences (CDSs) to the nonredundant protein database (NR) with BLAST and then mapping the blast hits to Gene Ontology (GO) terms with Blast2GO (https://www.blast2go.com).
InDel segregation analysis
At each InDel site, the genotypes of the two parents and 47 progeny were tabulated for analysis of segregation in the population. Then, a chi-square test was performed to check whether each InDel followed Mendelian segregation ratios, such as 1:1 and 1:2:1, in the progeny. If its p value was greater than 0.01 and the number of missing genotypes in the progeny was less than 10 (20%), the InDel site was considered to follow the Mendelian segregation ratio.
References
Bai SJ, Wu HN, Zhang JP, Pan ZL, Zhao W et al (2021) Genome assembly of Salicaceae Populus deltoides (Eastern Cottonwood) I-69 based on nanopore sequencing and Hi-C technologies. J Hered 112(3):303–310
Barreiro LB, Laval G, Quach H, Patin E, Quintana-Murci L (2008) Natural selection has driven population differentiation in modern humans. Nat Genet 40(3):340–345
Danecek P, Auton A, Abecasis G, Albers CA, Banks E et al (2011) The variant call format and VCFtools. Bioinformatics 27(15):2156–2158
Das S, Upadhyaya HD, Srivastava R, Bajaj D, Gowda CLL et al (2015) Genome-wide insertion-deletion (InDel) marker discovery and genotyping for genomics-assisted breeding applications in chickpea. DNA Res 22(5):377–386
de la Chaux N, Messer PW, Arndt PF (2007) DNA indels in coding regions reveal selective constraints on protein evolution in the human lineage. BMC Evol Biol 7:191
Feng JJ, Zhu HY, Zhang M, Zhang XX, Guo LP et al (2020) Development and utilization of an InDel marker linked to the fertility restorer genes of CMS-D8 and CMS-D2 in cotton. Mol Biol Rep 47(2):1275–1282
Ganal MW, Altmann T, Roder MS (2009) SNP identification in crop plants. Curr Opin Plant Biol 12(2):211–217
Grattapaglia D, Sederoff R (1994) Genetic linkage maps of Eucalyptus grandis and Eucalyptus urophylla using a pseudo-testcross: mapping strategy and RAPD markers. Genetics 137:1121–1137
Hu YY, Mao BG, Peng Y, Sun YD, Pan YL et al (2014) Deep re-sequencing of a widely used maintainer line of hybrid rice for discovery of DNA polymorphisms and evaluation of genetic diversity. Mol Genet Genomics 289(3):303–315
Kizil S, Basak M, Guden B, Tosun HS, Uzun B et al (2020) Genome-wide discovery of InDel markers in sesame (Sesamum indicum L.) using ddRADSeq. Plants 9:1262
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J et al (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079
Li W, Cheng J, Wu Z, Qin C, Tan S et al (2015) An InDel-based linkage map of hot pepper (Capsicum annuum). Mol Breed 35(1):32
Lin T, Zhu GT, Zhang JH, Xu XY, Yu QH et al (2014) Genomic analyses provide insights into the history of tomato breeding. Nat Genet 46(11):1220–1226
Lin MX, Whitmire S, Chen J, Farrel A, Shi XH et al (2017) Effects of short indels on protein structure and function in human genomes. Sci Rep 7:9313
Liu B, Wang Y, Zhai W, Deng J, Wang H et al (2013) Development of InDel markers for Brassica rapa based on whole-genome re-sequencing. Theor Appl Genet 126(1):231–239
Liu MM, Watson LT, Zhang LQ (2015) Predicting the combined effect of multiple genetic variants. Hum Genomics 9(1):18
Liu SR, An YL, Tong W, Qin XJ, Samarina L et al (2019a) Characterization of genome-wide genetic variations between two varieties of tea plant (Camellia sinensis) and development of InDel markers for genetic research. BMC Genomics 20(1):935
Liu YJ, Wang XR, Zeng QY (2019b) De novo assembly of white poplar genome and genetic diversity of white poplar population in Irtysh River basin in China. Sci China-Life Sci 62(5):609–618
McCouch SR, Zhao KY, Wright M, Tung CW, Ebana K et al (2010) Development of genome-wide SNP assays for rice. Breed Sci 60(5):524–535
Mousavi M, Tong C, Liu F, Tao S, Wu J et al (2016) De novo SNP discovery and genetic linkage mapping in poplar using restriction site associated DNA and whole-genome sequencing technologies. BMC Genomics 17:656
Patel RK, Jain M (2012) NGS QC Toolkit: A toolkit for quality control of next generation sequencing data. PLoS ONE 7(2):e30619
Pena HB, Pena SDJ (2012) Automated Genotyping of a Highly Informative Panel of 40 Short Insertion-Deletion Polymorphisms Resolved in Polyacrylamide Gels for Forensic Identification and Kinship Analysis. Transfus Med Hemotherapy 39(3):211–216
Qi JJ, Liu X, Shen D, Miao H, Xie BY et al (2013) A genomic variation map provides insights into the genetic basis of cucumber domestication and diversity. Nat Genet 45(12):1510–1515
Qin C, Yu CS, Shen YO, Fang XD, Chen L et al (2014) Whole-genome sequencing of cultivated and wild peppers provides insights into Capsicum domestication and specialization. Proc Natl Acad Sci USA 111(14):5135–5140
Ramakrishna G, Kaur P, Nigam D, Chaduvula PK, Yadav S et al (2018) Genome-wide identification and characterization of InDels and SNPs in Glycine max and Glycine soja for contrasting seed permeability traits. BMC Plant Biol 18:141
Shedlock AM, Okada N (2000) SINE insertions: powerful tools for molecular systematics. BioEssays 22(2):148–160
Song X, Wei H, Cheng W, Yang S, Zhao Y et al (2015) Development of INDEL markers for genetic mapping based on whole genome resequencing in soybean. G3 5:2793–279912
Strauss BSH (1994) Floral phenology and morphology of black cottonwood, Populus trichocarpa (Salicaceae). Am J Bot 81(5):562–567
Taylor MS, Ponting CP, Copley RR (2004) Occurrence and consequences of coding sequence insertions and deletions in mammalian genomes. Genome Res 14(4):555–566
Tian D, Wang Q, Zhang P, Araki H, Yang S et al (2008) Single-nucleotide mutation rate increases close to insertions/deletions in eukaryotes. Nature 455(7209):105–108
Tong C, Zhang B, Shi J (2010) A hidden Markov model approach to multilocus linkage analysis in a full-sib family. Tree Genet Genomes 6:651–662
Tong C, Li H, Wang Y, Li X, Ou J et al (2016) Construction of high-density linkage maps of Populus deltoides × P. simonii using restriction-site associated DNA sequencing. PLoS ONE 11(3):e0150692
Tong C, Yao D, Wu H, Chen Y, Yang W et al (2020) High-quality SNP linkage maps improved QTL mapping and genome assembly in Populus. J Hered 111(6):515–530
Tuskan GA, Difazio S, Jansson S, Bohlmann J, Grigoriev I et al (2006) The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313:1596–1604
Weber JL, David D, Heil J, Fan Y, Zhao CF et al (2002) Human diallelic insertion/deletion polymorphisms. Am J Hum Genet 71(4):854–862
Woolbright SA, DiFazio S, Yin T, Martinsen GD, Zhang X et al (2008) A dense linkage map of hybrid cottonwood (Populus fremontii × P. angustifolia) contributes to long-term ecological research and comparison mapping in a model forest tree. Heredity 100:59–70
Wu RL, Han YF, Hu JJ, Fang JJ, Li L et al (2000) An integrated genetic map of Populus deltoides based on amplified fragment length polymorphisms. Theor Appl Genet 100:1249–1256
Wu RL, Ma CX, Painter I, Zeng ZB (2002) Simultaneous maximum likelihood estimation of linkage and linkage phases in outcrossing species. Theor Popul Biol 61:349–363
Wu HN, Yao D, Chen YH, Yang WG, Zhao W et al (2020) De novo genome assembly of Populus simonii further supports that Populus simonii and Populus trichocarpa belong to different sections. Genes Genomes Genetics 10(2):455–466
Yamaki S, Ohyanagi H, Yamasaki M, Eiguchi M, Miyabayashi T et al (2013) Development of INDEL markers to discriminate all genome types rapidly in the genus Oryza. Breed Sci 63(3):246–254
Yang W, Wang K, Zhang J, Ma J, Liu J et al (2017) The draft genome sequence of a desert tree Populus pruinosa. Gigascience 6(9):1–7
Yin T, DiFazio SP, Gunter LE, Zhang X, Sewell MM et al (2008) Genome structure and emerging evidence of an incipient sex chromosome in Populus (Article). Genome Res 18(3):422–430
Zhang B, Tong CF, Yin TM, Zhang XY, Zhuge QQ et al (2009) Detection of quantitative trait loci influencing growth trajectories of adventitious roots in Populus using functional mapping. Tree Genet Genomes 5:539–552
Zhang JZ, Liu SR, Hu CG (2016) Identifying the genome-wide genetic variation between precocious trifoliate orange and its wild type and developing new markers for genetics research. DNA Res 23(4):403–414
Zhang BY, Zhu WX, Diao S, Wu XJ, Lu JQ et al (2019) The poplar pangenome provides insights into the evolutionary history of the genus. Commun Biology 2:215
Zhang ZY, Chen Y, Zhang JL, Ma XZ, Li YL et al (2020) Improved genome assembly provides new insights into genome evolution in a desert poplar (Populus euphratica). Mol Ecol Resour 20(3):781–794
Zhu JC, Guo YS, Su K, Liu ZD, Ren ZH et al (2018) Construction of a highly saturated Genetic Map for Vitis by Next-generation Restriction Site-associated DNA Sequencing. BMC Plant Biol 18(1):347
Funding
This research was funded by the National Natural Science Foundation of China, grant number 31870654, and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Author information
Authors and Affiliations
Contributions
Conceptualization, C.T. and Z.P.; methodology, Z.P. and Z.L.; formal analysis, Z.P., Z.L. and J.Z.; investigation, Z.P., Z.L., J.Z., S.B. and W.Z.; writing-original draft preparation, Z.P.; writing-review and editing, C.T.; supervision, C.T.; project administration, C.T.; funding acquisition, C.T. All authors have read and agreed to the published version of the manu-script.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Communicated by Yann-Rong Lin.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pan, Z., Li, Z., Zhang, J. et al. Investigation of genome-wide InDel distribution and segregation in Populus with restriction-site associated DNA sequencing data. Tropical Plant Biol. 15, 171–180 (2022). https://doi.org/10.1007/s12042-022-09312-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12042-022-09312-y