
Abstract
Background
High-saturate molecular linkage maps are an important tool in studies on plant molecular biology and assisted breeding. Development of a large set of single nucleotide polymorphisms (SNPs) via next-generation sequencing (NGS)-based methods, restriction-site associated DNA sequencing (RAD-seq), and the generation of a highly saturated genetic map help improve fine mapping of quantitative trait loci (QTL).
Results
We generated a highly saturated genetic map to identify significant traits in two elite grape cultivars and 176 F1 plants. In total, 1,426,967 high-quality restriction site-associated DNA tags were detected; 51,365, 23,683, and 70,061 markers were assessed in 19 linkage groups (LGs) for the maternal, paternal, and integrated maps, respectively. Our map was highly saturated in terms of marker density and average “Gap ≤ 5 cM” percentage.
Conclusions
In this study, RAD-seq of 176 F1 plants and their parents yielded 8,481,484 SNPs and 1,646,131 InDel markers, of which 65,229 and 4832, respectively, were used to construct a highly saturated genetic map for grapevine. This map is expected to facilitate genetic studies on grapevine, including an evaluation of grapevine and deciphering the genetic basis of economically and agronomically important traits. Our findings provide basic essential genetic data the grapevine genetic research community, which will lead to improvements in grapevine breeding.
Similar content being viewed by others

Background
Grapevine is a widely cultivated fruit crop worldwide, with high nutritional value. In 2016, 77 million tons were produced over a total area of 7 million ha (Food and Agriculture Organization). Several studies suggest that consumption of table grapes, grape products, and/or wine has many benefits for human health, and the requirement for high-quality grapes, including seedless and aromatic varieties, has increased considerably over the last several years [1,2,3,4].
Grapevine is a perennial woody plant species with a long juvenile period and is highly heterozygous; grapevine growth is negatively affected under various stressed conditions including natural disasters, disease, and pests. Identifying genes for desirable traits in grapevine cultivars via conventional cross-breeding techniques is challenging for cultivators and breeders. Therefore, alternative methods are necessary for large-scale production of cultivars with these traits. One method of achieving this is via construction of a map of molecular markers on a chromosome based on segregation data from a population resulting from a specific hybridization cross [5]. This approach has been used for numerous woody plants, for instance, to map quantitative trait loci (QTL) for plant quality traits and disease resistance [6].
Considerable progress had been made in the identification of molecular markers and the construction of molecular linkage maps in grapevine. The first molecular map for the 60 F1 progeny and the parental plants (F0) generation “Cayuga White” × “Aurore” was generated using 422 randomly amplified polymorphic DNAs (RAPDs), 16 restriction fragment length polymorphisms (RFLPs), and few isoenzyme markers [7]. Subsequent studies also used RAPDs, amplified fragment length polymorphisms (AFLPs), and sequence-related amplified polymorphisms (SRAPs) in F1 populations [8,9,10,11,12]. However, RAPDs, AFLPs, and SRAPs yield reportedly less stable results owing to uncontrollable experimental conditions [13] and have limited utility owing to their dominant pattern and low transferability. In contrast, simple sequence repeats (SSRs) have the advantages of co-dominance, high polymorphism, distribution throughout the genome, and good transferability with previously reported or annotated primer sequences [14,15,16,17,18,19,20,21,22,23]. (NCBI UniSTShttp://www.ncbi.nlm.nih.gov; Greek Vitis database, http://gvd.biology.uoc.gr/gvd/). Thus, reference genetic maps for the International Grape Genome Program (IGGP) have been constructed using 152 SSR markers and one polymorphic expressed sequence tag spanning 1728 cM [24]; 245 SSR markers spanning 1406.1 cM [14]; and 502 SSRs and 13 other types of PCR-based markers spanning 1647 cM [15]. However, in most cases, the total number of markers in the LGs is < 1000, with some lacking sequence-related information. In addition, there are some inconsistencies in LG number owing to the inefficiency and high cost of marker genotyping, which have prevented the fine mapping of target traits for breeding purposes. Therefore, to date, there are few high-density, high-quality genetic maps for grapevine, which encompass numerous molecular markers with detailed marker-related information.
The rapid development of NGS technologies and the publication of the grapevine reference genome sequence have assisted the identification of single nucleotide polymorphisms (SNPs), which have become the most widely used markers in genetic studies owing to their genomic abundance and stability [25, 26]. SNPs reportedly have revolutionary effects on high-quality genetic map construction [27, 28]. Several NGS-based methods have been used for simultaneous identification and scoring SNPs, including type IIB endonuclease restriction-site associated DNA (2b-RAD), double-digest (dd) RAD, genotyping-by-sequencing (GBS), specific length amplified fragment sequencing (SLAF-seq), and RAD sequencing (RAD-seq) [29,30,31,32,33]. RAD-seq is an NGS-based high-throughput sequencing technique, which simplifies the construction of highly multiplexed, low-representation libraries even in species with large genomes [31]. This method is a technology of reduced-representation genome sequencing (RRGS), with the advantages of simple operation, low experimental cost, and high throughput. RAD-seq is widely used in molecular biology, evolutionary genomics, population genetics, etc. For example, RAD markers were used to construct a high-density, high-quality genetic map in grapevine, which was subsequently applied for the detection of QTLs for sugar and acid production [34].
In this study, we used the F1 population derived from a cross between “Red Globe” (V. vinifera L.) and “Venus seedless” (V. vinifera × V. labrusca.). They were two table grapevine varieties that had significant differences in fruit size, ripening stage, disease resistance, fruit aroma, and number of seeds. Thus, the F1 generation is expected to segregate for labrusca aroma and disease resistance traits of these two elite grapevine cultivars. We performed RAD-seq to identify SNPs and insertion/deletion (InDel) markers to construct a highly saturated SNP-based molecular linkage map for grapevine, which can facilitate studies on grapevine ecology and evolution and facilitate the identification of QTLs for specific traits (grapevine aroma, white rot resistance, and downy mildew resistance), which will aid marker-assisted selection and accelerate genetic improvement of this important crop.
Results
Analysis of RAD-seq data for 176 F1 individuals and two parents
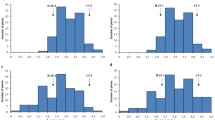
After treating the genomic DNA of F1 individuals and their parents with Taq I, samples were genotyped using high-throughput sequencing. After preprocessing, 388 Gb of raw data were obtained. To prevent sequencing errors, only reads showing < 5 bases with a Q score > 20 were further analyzed, yielding 206,411,693 clean reads ~ 150 bp in length; 94.39% were of a high quality, with quality scores of at least 30 (Q30, indicating a 0.1% chance of an error—i.e., 99.9% confidence). The guanine/cytosine content was 41.02% on average. A total of 1,426,967 RAD tags were detected; average sequencing depths were 52.7 for female parents “Red Globe,” 44.77 for male parents “Venus seedless,” and 7.25 for the progeny (Fig. 1a).

a, b Average read sequencing depth (fold) (a) and number of clean reads (b) expressed as genome equivalents of the 176 F1 individuals (shown in the X axis)
Of these high-quality data, 85,503,839 clean reads were obtained for Red Globe; Venus seedless, 79,625,497 reads (Fig. 1b); per the criteria of segregation distortion (P < 0.05), 70,061 genome-wide DNA markers were used to construct a genetic map. The markers were classified into the following five segregation patterns: ab × cd, ef × eg, hk × hk, lm × ll, and nn × np (Fig. 2).

Number of markers in each of the five segregation patterns
Characteristics of the genetic maps
Mapped markers formed 19 linkage groups numbered in accordance with the chromosome number. LOD values ranged from 4 to 20 depending on the LG. There were 51,265 markers in the female map “Red Globe” (V. vinifera L.) and the total length was 3172.33 cM (Additional file 1: Figure S1). The length of each LG ranged from 125.56 cM for LG1 to 210.21 cM for LG9; mean length, 166.96 cM. LG1 contained the most markers (5325) with an average marker interval of 0.02 cM, whereas LG10 contained the fewest (1361) markers with an average marker interval of 0.11 cM. The “Gap ≤ 5 cM” percentage (gaps where the distance between adjacent markers was < 5 cM) for each LG ranged from 99.72% (LG8) to 100% (LG1, LG4, LG12, LG13, LG14, LG15, LG17, and LG19).
The map of the male parent “Venus seedless (V. vinifera × V. labrusca)” contained 23,683 markers spanning 3221.4 cM (Additional file 2: Figure S2). LG1(132.89 cM) and LG15 (210.12 cM) as the shortest and the longest linkage groups, respectively; mean length, 166.55 cM. LG1 contained 1488 markers with an average genetic interval of 0.09 cM, whereas LG15 contained 758 markers and an average genetic interval of 0.28 cM. The “Gap ≤ 5 cM” percentage for each LG ranged from 99.34% (LG3) to 100.00% (LG1, LG2, LG5, LG8, LG9, LG11, LG17, and LG19).
The integrated map contained a set of 70,061 markers spanning 3014.46 cM, with 3687 markers per LG on average and an average inter-marker distance of 0.05 cM (Fig. 3). The genetic length of LGs ranged from 125.17 cM (LG18) to 195.29 cM (LG6), with an average length of 158.66 cM. LG1contained the most markers (6564) spanning 142.42 cM with an average genetic interval of 0.02 cM, whereas LG3 spanned 152.42 cM and contained fewest markers (2031). The size and number of markers for each LG are described in Table 1. The average “Gap ≤ 5 cM” percentage was 99.99%. The ‘Gap > 5’ attribute was observed only in LG2, LG3, LG4, and LG9 (Table 2); two gaps > 10 cM were located in LG4 and one in LG10; however, they were only 12.91 cM (LG4) and 11.28 cM (LG10).

Genetic lengths and marker distribution in 19 linkage groups of the integrated map. Genetic distance is indicated by the vertical scale in centimorgans (cM). Black lines represent mapped markers. LG1–19 represent corresponding linkage groups ID

Collinear analysis of the consensus between genetic and physical maps

Marker density of integrated map.X-axis: physical position on 19 linkage groups. Y-axis: markers number per LG. The marker density on the genome was calculated by sliding windows using window size of 0.5 cM

Distribution of marker density across the chromosome. The x-axis represents the 10 cM map interval and the y-axis represents the number of RAD markers present in the interval
Comparative analysis of high-saturated linkage maps
The correlation between genetic and physical positions on a linkage map defines its quality [35].
To compare genetic and physical maps, we investigated the locations of all 65,229 SNP markers on the reference grapevine genome (Fig. 4). A high degree of collinearity was observed between genetic and physical distances of all SNP markers in the 19 LGs. All consecutive curves generated from the 19 LGs indicated that the reference genome was sufficiently encompassed with SNP markers positioned accurately within each LG. Most parts of these curves showed a declining trend, suggesting that their genetic and physical positions followed the same order.
To better describe the marker densities across the chromosome, we considered a sliding-window interval of 0.5cM and 10 cM across chromosome 19, respectively (Figs. 5 and 6).Physical coverage represents the proportion of chromosome length encompassed by all markers in the reference genome. In the 19 LGs, physical coverage ranged from 99.17% (LG9) to 99.98% (LG13), with an average of 99.83% (Table 3), indicating that most markers showed a good linear agreement between physical and genetic maps on the basic framework. The average Spearman correlation coefficient between the genetic and physical positions was 0.99, suggesting that the LGs exhibit high levels of genetic collinearity.
Heat maps can indicate the recombination frequency between markers within one single LG (Additional file 3: Figure S3); they can hence be used to identify potential markers ordering errors, pair-wise recombination occurring primarily owing to hotspot regions for genomic recombination and sequencing-related genotyping errors to optimize the genetic map. In general, most LGs yielded a good performance.
Discussion
Genetic maps have long been used as a tool to improve grapevine cultivation and are indispensable for studies aimed at elucidating the genetic architecture of quantitative traits. Construction of a high saturated genetic map of grape is valuable for breeders because it potentially facilitates the identification of genomic regions with characteristics of agronomic interest [36].
Similar to other organisms, numerous SNPs have been used to characterize grapevine genomes [37,38,39,40] and construct high-density genetic maps [41,42,43]. A genetic map for V. vinifera was previously constructed with 994 markers (mostly consisting of 483 SNPs) spanning 1245 cM [41]. A consensus map for a grapevine cultivar (V. vinifera L.) derived from three crosses generated on the basis of 283 SSRs and 501 SNP-based markers was also added to the IGGP [42]. Although SNPs are presumed to be more numerous and genetically stable than other marker types, this is difficult for large scale detection.
An essential step in high-density map construction is scoring tens to hundreds of thousands of stable and accurate molecular markers in a cost-efficient manner. The continuously decreasing cost of NGS has resulted in the development of several NGS-based methods for SNP identification. One of these approaches is RAD-seq, which uses rare-cutter restriction enzymes (6- to 8-bp recognition site) for sequencing short DNA fragments surrounding a particular recognition site throughout the genome [33]. This method is adapted from the RAD tag marker technique for NGS platforms [44,44,46]. Several modifications of the original RAD-seq protocol have been reported, including 2b-RAD-seq methods [47], ddRAD-seq [30], and GBS [31]. For instance, in GBS, a frequent cutter enzyme is used to generate low-representation libraries prior to sequencing [31], although this leads to an increased rate of missing data, which is a major limitation of imputation programs [48, 49]. SLAF-seq was recently developed as a simplified sequencing technique potentially useful for large-scale screening of SNPs [32]. To date, several high-density genetic maps for grapevine have been constructed using NGS technology: RAD sequencing yielded 1646 SNP markers spanning 1917.3 cM from 100 F1 progeny and their parents [29] and 1826 SNP-based markers from 249 individuals and their parents [34]. GBS was used to construct genetic maps for Vitis rupestris “B38” (1146 SNPs) and “Chardonnay” (1215 SNPs) spanning 1645 and 1967 cM, respectively [50]. SLAF-seq of 149 F1 plants and their parents identified 7199 polymorphic markers in a map spanning 1929.13 cM [51]. This method also yielded 10,042 SNPs spanning 1969.95 cM from an analysis of 130 individuals and their parents [52].
In this study, RAD-seq of 176 F1 plants and their parents yielded 8,481,484 SNPs and 1,646,131 InDel markers, of which 65,229 and 4832, respectively, were used to construct a highly saturated genetic map for grapevine, spanning 3014 cM with an average coverage of 99.83% in 19 LGs. The average “Gap ≤ 5 cM” percentage of 99.99% indicated good uniform coverage, whereas the density of the linkage maps was highly saturated. Despite these advantages, the integrated map had two large gaps over 10 cM. The markers flanking these two gaps (LG04 and LG10) were aligned to the reference genome by BLAST. The flanking markers were physically located 16 and 32 kb apart, respectively. Recombination hotspots may be responsible for these results.
We developed 51,365, 23,683, and 70,061 markers for 19 linkage groups (LGs) for the maternal, paternal, and integrated maps, respectively. The genetic map requires molecular markers to display linear correlations with the chromosomes. Linkage maps were constructed as described previously [53], the Multipoint maximum likelihood method was enhanced by determining the degree of support of the possible conformational position of each molecule marker. For example, in LG1, marker chr1_320228, chr1_320235, and chr1_337168, these three markers were located at the same genetic position in close physical proximity. Subsequent studies may yield the location of the QTL in this region. We can increase the mapping population and recalculate the location of these markers for local fine mapping.
This genetic map of a cross with a complex parentage [(V. vinifera) × (V. vinifera × V. labrusca)] has the highest saturated compared to those constructed thus far for grapevine; moreover, it provides genomic tools to improve table grapevine cultivars; most characteristics of agronomic and economic importance in grape are quantitative traits, and it is very important in grape breeding to locate quantitative trait loci (QTLs) and estimate their effects. This genetic map successfully lays the foundation of fine mapping, marker-assisted selection, and cloning of QTLs.
Since the traits of hybrid offspring are inherited from the parents, parental selection is critical in constructing genetic maps. In this study, “Red Globe” (V. vinifera L.) was used as the maternal parent. This cultivar is one of the world’s most important table grape varieties and accounts for over 20% of the total grape cultivation area in China. It is characterized by large clusters and a large grape size, low acid content, firm flesh, and high productivity. It is also late-ripening and is well-preserved during storage and transportation. However, Red Globe shows poor cold tolerance and resistance to pathogens such as elsinoe anthracnose and downy mildew [54]. The paternal parent was “Venus seedless (V. vinifera × V. labrusca),” which is also extensively cultivated in China. It has a labrusca flavor, is seedless and highly resistant to disease, exhibits early ripening and strong growth [55]. Thus, the F1 generation is expected to segregate for the favorable traits of these two elite grapevine cultivars.
Conclusion
In this study, RAD-seq of 176 F1 plants and their parents yielded 8,481,484 SNPs and 1,646,131 InDel markers, of which 65,229 and 4832, respectively, were used to construct a highly saturated genetic map for grapevine, spanning 3014 cM with an average coverage of 99.83% in 19 LGs. The average “Gap ≤ 5 cM” percentage of 99.99% indicated good uniform coverage, whereas the density of the linkage maps was highly saturated. This genetic map contains the largest molecular marker number of the grape maps so far reported. The genetic map will facilitate the QTL mapping of important grapevine traits in the future.
Methods
Mapping population and DNA isolation
An F1 grape hybrid of 176 individuals derived from a cross between “Red Globe” (V. vinifera L.) and “Venus seedless” (V. vinifera × V. labrusca.) was generated in May 2009. Stratification was performed between October 2009 and February 2010. Hybrid seeds were sown in a greenhouse in March 2010. A total of 531 crops were randomly harvested, of which 176 of them and each parent were used as the mapping population. The seedlings of the mapping population were sewn in batches in the vineyard of Shenyang Agriculture University, Liaoning Province, P. R. China (E123°24′, N41°50′) from April to June 2010.
Healthy young leaves (second or third leaves from the apex and less than 1 cm2) were harvested from both parents and each individual progeny plant (F1 generation). Samples immediately frozen in liquid nitrogen and quickly in store a − 80 °C refrigerator. Genomic DNA was extracted using the improved CTAB method [56]. Extracted DNA samples were treated with RNase A to eliminate residual RNA. DNA concentration and quality were evaluated using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA), finally the concentration and volume of each DNA sample was 500 ng·μl− 1 and 50 μl, the extracted DNA samples were electrophoresis on a 0.8% agarose gel. The DNA was diluted to a final concentration of 2.5 ng·μl− 1 for use in subsequent polymerase chain reactions (PCR).
Library construction
RAD-seq libraries were constructed as described previously [33], with a few modifications. Briefly, genomic DNA (0.1–1 μg from either sample) was incubated for 5 min at 37 °C with 20 U of Taq I restriction endonuclease (New England Biolabs, Ipswich, MA, USA) in a 50-μl reaction mixture [57,58,59]. Individually barcoded P1 adapters were ligated to the Taq I restriction site for each sample. Thereafter, samples were pooled in proportional amounts for shearing to an average size of 500 bp with a Bioruptor (Diagenode, Liège, Belgium). Sequencing libraries were constructed a total of 24 samples per library. Libraries were size-selected for 450- to 550-bp fragments on a 2% agarose gel. Libraries were blunt end-repaired, and a 3′-adenine overhang was added to each fragment. We added a P2 adapter containing unique Illumina barcodes (San Diego, CA, USA) to each library. Libraries were amplified by via PCR, under the following conditions: 16 cycles (98 °C for 2 min; 16 cycles at 98 °C for 30 s, 60 °C for 30 s, and 72 °C for 15 s; and 72 °C for 5 min) with Phusion high-fidelity DNA polymerase (New England Biolabs) and column-purified. Samples were sequenced using a HiSeq 2500 system (Illumina) using 150-bp paired-end reads.
In silico analysis
Quality trimming is an essential step to generate high-confidence variant calls. Raw reads were assigned to individual samples in accordance with their nucleotide barcode, using Axe package [60]. Raw reads were processed to obtain high-quality clean reads in accordance with three stringent filtering criteria: 1) elimination of reads with ≥ 10% unidentified nucleotides (N); 2) elimination of reads with > 50% bases having Phred quality scores ≤20; and 3) elimination of reads aligned to the barcode adapter. To identify SNPs, the Burrows-Wheeler Aligner (BWA) was used to align clean reads from each sample against the 12X.0 Vitis vinifera reference genome PN40024 (https://www.ncbi.nlm.nih.gov/assembly/GCF_000003745.3/) with the setting of “mem 4−k 32−M,” where −k is the minimum seed length and − M is an option used to mark shorter split alignment hits as secondary alignments [61]. The markers from a chromosome were used to construct the corresponding LG and the marker order were determined based upon the recombination fractions in the F1 population. Variant calling was performed for all samples, using the Genome Analysis Toolkit (GATK) Unified Genotyper (Broad Institute, Cambridge, MA, USA). Variants were filtered using standard hard filtering parameters in accordance with the GATK Best Practices pipeline. More precisely, SNPs and InDels were obtained on the basis of a mapping quality > 37 and quality depth > 24. Lastly, variants with > 70% call rate and sequence depth > 2-fold were used to construct a linkage map.
Linkage map construction
Variants filtered by quality, as described above, were genotyped in accordance with their heterozygous parents into eight segregation types. After filtering those with no polymorphisms between parents or partial separation based on a P value < 0.01, markers with homozygous parents were used to construct a genetic linkage map for the F1 generation. Genetic marker data were scored in accordance with the criteria of JoinMap v.5.0 with a smooth algorithm. All statistical analyses described below were performed with a cross-pollinating-type population, using the same software designed to analyze data for the F1 outbreeding population containing various genotype configurations. Pairwise analyses were performed; markers were sorted into LGs at a minimum logarithm of odds (LOD) score of 4.0 and modified via genome location. The maximum recombination value was 0.3. Independence LOD scores were used as the grouping parameter, with the maximum likelihood (ML) mapping algorithm. Print map: each round only. The “locus genotype frequency” function was used to calculate chi-square values for each marker to test for the expected Mendelian segregation ratio. Markers deviating significantly from the expected ratio (P < 0.05) were excluded. Linkage distances were estimated for each LG, assuming the Kosambi mapping function [62]. The smooth algorithm was used to detect low-quality genotypes and impute the missing value. A consensus map was constructed using the “Join-combine groups for map integration” command.
Abbreviations
- CTAB:
-
cetyltrimethylammonium bromide
- IGGP:
-
international grape genome program
- LOD:
-
logarithm of odds
- NGS:
-
next-generation sequencing
- QTL:
-
quantitative trait loci
- RAD-seq:
-
restriction-site associated DNA sequencing
- RRGS:
-
reduced-representation genome sequencing
- SNPs:
-
single nucleotide polymorphisms
References
Bertelli AA, Das DK. Grapes, wines, resveratrol, and heart health. J Cardiovasc Pharmacol. 2009;54(6):468–76.
Dohadwala MM, Vita JA. Grapes and cardiovascular disease. J Nutr. 2009;139(9):1788S–93S.
Pezzuto JM, Venkatasubramanian V, Hamad M, Morris KR. Unraveling the relationship between grapes and health. J Nutr. 2009;139(9):1783S–7S.
Wu D. Grape products and oral health. J Nutr. 2009;139(9):1818S–23S.
Collard BC, Jahufer MZ, Brouwer J, Pang EC. An introduction to markers, quantitative trait loci (QTL) mapping and marker-assisted selection for crop improvement: the basic concepts. Euphytica. 2005;142(1–2):169–96.
Longhi S, Giongo L, Buti M, Surbanovski N, Viola R, Velasco R, et al. Molecular genetics and genomics of the Rosoideae: state of the art and future perspectives. Horticulture Research. 2014;1(2–3):381–92.
Lodhi MA, Daly MJ, Ye GN, Weeden NF, Reisch BI. A molecular marker based linkage map of Vitis. Genome. 1995;38(4):786–94.
Dalbó AM, Ye GN, Weeden NF, Steinkellner H, Sefc KM, Reisch BI. A gene controlling sex in grapevines placed on a molecular marker-based genetic map. Genome. 2000;43(2):333–40.
Grando MS, Bellin D, Edwards KJ, Pozzi C, Stefanini M, Velasco R. Molecular linkage maps of Vitis vinifera L. and Vitis riparia Mchx. Theor Appl Genet. 2003;106(7):1213–24.
Doucleff M, Jin Y, Gao F, Riaz S, Krivanek AF, Walker MA. A genetic linkage map of grape, utilizing Vitis rupestris and Vitis arizonica. Theor Appl Genet. 2004;109(6):1178–87.
Liu ZD, Guo XW, Guo YS, Lin H, Zhang PX, Zhao YH, et al. SSR and SRAP markers based linkage map of Vitis Amurensis rupr. Pak J Bot. 2013;45(1):191–5.
Guo YS, Lin H, Liu ZD, Zhao YH, Guo XW, Li K. SSR and SRAP marker-based linkage map of Vitis vinifera L. Biotechnol Biotechnol Equip. 2014;28(2):221–9.
Liu J, Huang S, Sun M, Liu S, Liu Y, Wang W, et al. An improved allele-specific PCR primer design method for SNP marker analysis and its application. Plant Methods. 2012;8(1):34.
Adam-Blondon AF, Roux C, Claux D, Butterlin G, Merdinoglu D, This P. Mapping 245 SSR markers on the Vitis vinifera genome: a tool for grape genetics. Theor Appl Genet. 2004;109(5):1017–27.
Doligez A, Adam-Blondon AF, Cipriani G, Di Gaspero G, Laucou V, Merdinoqlu D, et al. An integrated SSR map of grapevine based on five mapping populations. Theor Appl Genet. 2006;113(3):369–82.
Moreira FM, Madini A, Marino R, Zulini L, Stefanini M, Velasco R, et al. Genetic linkage maps of two interspecific grape crosses (Vitis spp.) used to localize quantitative trait loci for downy mildew resistance. Tree Genet Genomes. 2011;7(1):153–67.
Pap D, Riaz S, Dry IB, Jermakow A, Tenscher AC, Cantu D, et al. Identification of two novel powdery mildew resistance loci, Ren6 and Ren7, from the wild Chinese grape species Vitis piasezkii. BMC Plant Biol. 2016;16(1):170.
Scott KD, Eggler P, Seaton G, Rossetto M, Ablett EM, Lee LS, et al. Analysis of SSRs derived from grape ESTs. Theor Appl Genet. 2000;100(5):723–6.
Decroocq V, Favé MG, Hagen L, Bordenave L, Decroocq S. Development and transferability of aplicot and grape EST mocrosatellite markers across taxa. Theor Appl Genet. 2003;106(5):912–22.
Thomas MR, Scott NS. Microsatellite repeats in grapevine reveal DNA polymorphisms when analysed as sequence-tagged sites (STSs). Theor Appl Genet. 1993;86(8):985–90.
Bowers JE, Dangl GS, Vignani R, Meredith CP. Isolation and characterization of new polymorphic simple sequence repeat loci in grape (Vitis vinifera L.). Genome. 1996;39:628–33.
Bowers JE, Dangl GS, Meredith CP. Development and characterization of additional microsatellite DNA markers for grape. Am J Enol Vitic. 1999;50(3):243–6.
Crespan M. The parentage of Muscat of Hamburg. Vitis. 2003;42(4):193–7.
Riaz S, Dangl GS, Edwards KJ, Meredith CP. A microsatellite marker based framework linkage map of Vitis vinifera L. Theor Appl Genet. 2004;108(5):864–72.
Jaillon O, Aury JM, Noel B, Policriti A, Clepet C, Casagrande A, et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007;449(7161):463–7.
Varshney RK, Nayak SN, May GD, Jackson SA. Next-generation sequencing technologies and their implications for crop genetics and breeding. Trends Biotechnol. 2009;27(9):522–30.
Zhang N, Zhang LN, Tao Y, GUO L, Sun J, Li X, et al. Construction of a high density SNP linkage map of kelp (Saccharina japonica) by sequencing Taq I site associated DNA and mapping of a sex determining locus. BMC Genomics. 2015;16(1):1–11.
Ward JA, Bhangoo J, FernaÂndez-FernaÂndez F, Moore P, Swanson JD, Viola R, et al. Saturated linkage map construction in Rubus idaeus using genotyping by sequencing and genome-independent imputation. BMC Genomics. 2013;14(1):2.
Wang N, Fang LC, Xin HP, Wang LJ, Li SH. Construction of a high-density genetic map for grape using next generation restriction-site associated DNA sequencing. BMC Plant Biol. 2012;12(1):148.
Dacosta JM, Sorenson MD. Amplification biases and consistent recovery of loci in a double-digest RAD-seq protocol. PLoS One. 2014;9(9):e106713.
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One. 2011;6(5):e19379.
Sun X, Liu D, Zhang X, Li W, Liu H, Hong W, et al. SLAF-seq: an efficient method of large-scale Denovo SNP discovery and genotyping using high-throughput sequencing. PLoS One. 2013;8(3):e58700.
Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, et al. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One. 2008;3(10):e3376.
Chen J, Wang N, Fang L, Liang Z, Li S, Wu B. Construction of a high-density genetic map and QTLs mapping for sugars and acids in grape berries. BMC Plant Biol. 2015;15(1):28.
Sim S-C, Durstewitz G, Plieske J, Wieseke R, Ganal MW, Van DA, et al. Development of a large SNP genotyping array and generation of high-density genetic maps in tomato. PLoS One. 2012;7(7):e40563.
Troggio M, Vezzulli S, Pindo M, Malacarne G, Fontana P, Moreira FM, et al. Beyond the genome, opportunities for a modern viticulture: a research overview. Am J Enol Vitic. 2008;59(2):117–27.
Lijavetzky D, Cabezas JA, Ibáñez A, Rodríguez V, Martínez-Zapater JM. High throughput SNP discovery and genotyping in grapevine (Vitis vinifera L.) by combining a re-sequencing approach and SNPlex technology. BioMed Central Genomics. 2007;8(1):424.
Velasco R, Zharkikh A, Troggio M, Cartwright DA, Cestaro A, Pruss D, et al. High quality draft consensus sequence of the genome of a heterozygous grapevine variety. PLoS One. 2007;2(12):e1326.
Salmaso M, Faes G, Segala C, Stefanini M, Salakhutdinov I, Zyprian E, et al. Genome diversity and gene haplotype in the grapevine (Vitis vinifera L. ), as revealed by single nucleotide polymorphisms. Mol Breed. 2004;14(4):385–95.
Pindo M, Vezzulli S, Coppola G, Cartwright DA, Zharkikh A, Velasco R, et al. SNP high-throughput screening in grapevine using the SNPlex™ genotyping system. BioMed Central Plant Biology. 2008;8(1):12.
Troggio M, Malacarne G, Coppola G, Segala C, Cartwright DA, Pindo M, et al. A dense single-nucleotide polymorphyism-based genetic linkage map of grapevine (Vitis vinifera L.) anchoring pinot noir bacterial artificial chromosome contigs. Genetics. 2007;176(4):2637–50.
Vezzulli S, Troggio M, Coppola G, Jermakow A, Cartwright D, Zharkikh A, et al. A reference integrated map for cultivated grapevine (Vitis vinifera L. ) from three crosses, based on 283 SSR and 501 SNP-based markers. Theor Appl Genet. 2008;117(4):499–511.
Salmaso M, Malacarne G, Troggio M, Faes G, Stefanini M, Grando MS, et al. A grapevine (Vitis vinifera L.) genetic map integrating the position of 139 expressed genes. Theor Appl Genet. 2008;116(8):1129–43.
Miller M, Dunham J, Amores A, Cresko W, Johnson E. Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Res. 2007;17(2):240–8.
Davey JL, Blaxter MW. RAD seq: next-generation population genetics. Brief Funct Genomics. 2010;9(5–6):416–23.
Nelson JC, Wang S, Wu Y, Li X, Antony G, White FF, et al. Single-nucleotide polymorphism discovery by high-throughput sequencing in sorghum. BMC Genomics. 2011;12(1):352.
Wang S, Meyer E, Mckay JK, Matz MV. 2b-RAD: a simple and flexible method for genome-wide genotyping. Nat Methods. 2012;9(8):808–10.
Money D, Gardner K, Migicovsky Z, Schwaninger H, Zhong GY, Myles S. LinkImpute: Fast and accurate genotype imputation for non-model organisms. G3 Genes Genomes Genetics. 2015;5(11):2382–90.
Browning SR, Browning BL. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet. 2007;81(5):1084–97.
Barba P, Cadle-Davidson L, Harriman J, Glaubitz JC, Brooks S, Hyma K, et al. Grapevine powdery mildew resistance and susceptibility loci identified on a high-resolution SNP map. Theor Appl Genet. 2014;127(1):73–84.
Guo YS, Shi GL, Liu ZD, Zhao YH, Yang XX, Zhu JC, et al. Using specific length amplified fragment sequencing to construct the high-density genetic map for Vitis (Vitis vinifera L.×Vitis amurensis Rupr.). Front Plant Sci. 2015;6(393):393.
Wang JH, Su K, Guo YS, Xing HY, Zhao YH, Liu ZD, et al. Construction of a high-density genetic map for grape using specific length amplified fragment (SLAF) sequencing. PLoS One. 2017;12(7):e0181728.
Vanooijen JW. Multipoint maximum likelihood mapping in a full-sib family of an outbreeding species. Genet Res Camb. 2011;93(pp):343–9.
Liu CH, Ma XH, Wu G. Grape varieties in China. Beijing: China agriculture press; 2014.
Kong QS, Zhu L, Li SC, Yang CS, Wu DL, Xiu DR, et al. Chinese grapes. Beijing: China Agriculture Science and technology Press; 2004.
Hanania U, Velcheva M, Sahar N, Perl A. An improved method for isolating high-quality DNA from vinifera nuclei. Plant Mol Biol Rptr. 2004;22(2):173–7.
Pfender WF, Saha MC, Johnson EA, Slabaugh MB. Mapping with RAD (restriction-site associated DNA) markers to rapidly identify QTL for stem rust resistance in Lolium perenne. Theor Appl Genet. 2011;122(8):1467–80.
Chutimanitsakun Y, Nipper RW, Cuesta-Marcos A, Cistue L, Corey A, Filichkina T, et al. Construction and application for QTL analysis of a restriction site associated DNA (RAD) linkage map in barley. BMC Genomics. 2011;12(1):4.
Huang X, Feng Q, Qian Q, Zhao Q, Wang L, Wang A, et al. High-throughput genotyping by whole-genome resequencing. Genome Res. 2009;19(6):1068–76.
Murray D, Borevitz, O. Axe: rapid, competitive sequence read demultiplexing using a trie. Bioinformatics.2018;34(22):3924-5.
Li H, Durbin R. Fast and accurate short read alignment with burrows–wheeler transform. Bioinformatics. 2009;25(14):1754–60.
Kosambi DD. The estimation of map distances from recombination values. Ann Hum Genet. 1943;12(1):172–5.
Acknowledgments
The authors are grateful to editage for improving the English in this paper.
Funding
This work was supported by the National Key Research and Development Program(Grant No. 2018YFD1000200); the National Natural Science Foundation of China (Grant No. 31372021, 31572085); the China Agriculture Research System (Grant No.CARS-29-yc-6); the Shenyang Science and Technology Development Funds (Grant No. 18–013–0-35); the Research project in Liaoning Province Science and Technology Department (Grant No. LNKTP2016).
Availability of data and materials
All data generated or analysed during this study are included in this published article (and its Additional file 4).
Author information
Authors and Affiliations
Contributions
HZ and YG organized the entire project and formal analysis. XG funding acquisition and supervision. KS, DL, HR and LK investigation. HZ wrote this manuscript, and YG and XG edited it. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Figure S1. Genetic map of the male parent ‘Venus seedless (V. vinifera × V. labrusca)’. Genetic distance is centimorgans (cM) Kosambi. Black lines represent mapped markers. LG1–19 represent corresponding linkage groups ID. (DOCX 127 kb)
Additional file 2:
Figure S2. Genetic map of the female parent ‘Red Globe’ (V. vinifera L.). Genetic distance is centimorgans (cM) Kosambi. Black lines represent mapped markers. LG1–19 represent corresponding linkage groups ID. (DOCX 89 kb)
Additional file 3:
Figure S3. Heat map of the genetic linkage map. (ZIP 3270 kb)
Additional file 4:
Table S1. Markers used for mapping. (XLS 3199 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhu, J., Guo, Y., Su, K. et al. Construction of a highly saturated Genetic Map for Vitis by Next-generation Restriction Site-associated DNA Sequencing. BMC Plant Biol 18, 347 (2018). https://doi.org/10.1186/s12870-018-1575-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-018-1575-z