
Abstract
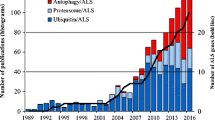
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease affecting upper and lower motor neurons (MNs). Since the identification of the first ALS mutation in 1993, more than 40 genes have been associated with the disorder. The most frequent genetic causes of ALS are represented by mutated genes whose products challenge proteostasis, becoming unable to properly fold and consequently aggregating into inclusions that impose proteotoxic stress on affected cells. In this context, increasing evidence supports the central role played by autophagy dysfunctions in the pathogenesis of ALS. Indeed, in early stages of disease, high levels of proteins involved in autophagy are present in ALS MNs; but at the same time, with neurodegeneration progression, autophagy-mediated degradation decreases, often as a result of the accumulation of toxic protein aggregates in affected cells. Autophagy is a complex multistep pathway that has a central role in maintaining cellular homeostasis. Several proteins are involved in its tight regulation, and importantly a relevant fraction of ALS-related genes encodes products that directly take part in autophagy, further underlining the relevance of this key protein degradation system in disease onset and progression. In this review, we report the most relevant findings concerning ALS genes whose products are involved in the several steps of the autophagic pathway, from phagophore formation to autophagosome maturation and transport and finally to substrate degradation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS) is a rare adult motor neuron disease (MND) characterized by degeneration of upper and lower motor neurons (MNs), leading to progressive muscle atrophy and death within 3–5 years after symptom onset. Besides MNs, skeletal muscle (Dobrowolny et al. 2008; Onesto et al. 2011; Cicardi et al. 2018; Meroni et al. 2019) and glial cells (Trotti et al. 1999; Lobsiger et al. 2009; Philips et al. 2013) can be targeted by ALS. Neurons residing in the frontotemporal cortex might be affected, too, which may lead to the development of a mixed form of MND and frontotemporal dementia (FTD) referred to as ALS/FTD (Robberecht and Philips 2013). No drugs are currently available to treat or cure ALS (Taylor et al. 2016).
Most ALS cases (90%) are sporadic (sALS), while only 10% are characterized by familial inheritance (fALS). Clinical symptoms fully overlap between the two forms of the disease, but fALS tends to be more severe compared to sALS. Inherited ALS cases are associated to both loss-of-function (LOF) and aberrant gain-of-function (GOF) mutations, as well as to mixed LOF and GOF, that can be found in more than 40 genes. Nonetheless, around 30% of fALS genetic causes still need to be identified, indicating that ALS is a disease with a widely heterogeneous genetic background (Cristofani et al. 2020). Noteworthily, the wild-type forms of the gene products causing fALS display an aberrant behavior in sALS too, suggesting that the two forms of the disease probably share some pathogenetic mechanisms (Neumann et al. 2006).
ALS is caused by a combination of genetic, epigenetic, and environmental factors which result in different pathogenetic mechanisms capable of triggering neuronal damage (Morgan and Orrell 2016). To date, the precise etiology of ALS is still unknown, as well as the exact molecular mechanisms involved in the degeneration of motor neurons (Shaw 2005). The analysis of ALS-related genes outlined the main pathophysiological mechanisms involved in neurodegeneration in sALS and fALS: oxidative stress, mitochondrial dysfunction, impairment of axonal transport, excitotoxicity, protein aggregation, endoplasmic reticulum (ER) stress, abnormal RNA processing, and neuroinflammation (Kiernan et al. 2011; Taylor et al. 2016). One of the main disease mechanisms is the alteration in protein quality control (PQC). In this respect, a distinctive hallmark of both fALS and sALS is the formation of aberrant aggregates of TAR DNA-binding protein 43 (TDP-43), often together with a mixture of other proteins, into target cells. The only exceptions are fALS forms linked superoxide dismutase 1 (SOD1) mutation, being the only devoid of TDP-43 inclusions. When mutated, most of the proteins characterizing the aggregates misfold and partition into initially small structures which later degenerate into aggresomes and insoluble inclusions if they are not rapidly degraded (Patel et al. 2015; Boeynaems et al. 2017). The accumulating aggregates end up tampering with the PQC system, that fails in keeping under control protein misfolding and undergoes saturation, leading inclusions to become toxic for the affected cells.
In this review, we will focus on the connections between ALS and autophagy, one of the main branches of the PQC system. To highlight the strict relations between ALS and autophagy and to better clarify the role of ALS-associated autophagic genes, we will describe the function and report the most relevant findings on the pathogenetic mechanisms lying behind the mutations of these genes in ALS (Table 1).
Autophagic Pathway
Autophagy is an evolutionarily conserved cellular mechanism that disassembles old, unnecessary, or dysfunctional cytosolic components and allows their degradation by lysosomal hydrolases to fuel bioenergetic metabolism and repair processes (Klionsky et al. 2021a). Autophagy contributes to the maintenance of proteostasis by avoiding the accumulation of potentially dangerous protein species (Li et al. 2012; Cuervo and Wong 2014; Sica et al. 2015). An active and functional autophagy in neurons is more relevant than in other cell types. Indeed, neurons are cells with no mitotic activity and a very low capacity to regenerate. Moreover, neurons present a particular morphology composed by long axons that makes necessary a characteristic organization for autophagic degradation of synaptic substrates (Cai and Ganesan 2022). Disruption of the autophagic flux occurs in several ALS forms and leads to the accumulation of toxic, ubiquitin-positive inclusion bodies and aberrant stress granules that eventually cause neuronal death (Buchan et al. 2013; Iguchi et al. 2016; Chitiprolu et al. 2018).
Autophagy includes three distinct pathways that promote and regulate the degradation of substrates via lysosomes, microautophagy (Schuck 2020), chaperone-mediated autophagy (CMA) (Kaushik and Cuervo 2018), and macroautophagy, that includes the chaperone assisted–selective autophagy or CASA (Cristofani et al. 2020). Here we will focus on macroautophagy (hereafter autophagy).
Autophagy is characterized by different steps: initiation, elongation, maturation, and degradation (Klionsky et al. 2021b). The initiation consists in the nucleation of the autophagic membrane (phagophore) and is regulated by different complexes. The first complex involved is the preinitiation complex, that is negatively regulated by the mammalian target of rapamycin (mTOR) pathway and is positively regulated by the AMP-activated protein kinase (AMPK) pathway. It is composed by autophagy-related proteins 13 and 101 (ATG13, ATG101), unc-51 like kinase 1/2 (ULK1/2), and FAK family-interacting protein of 200 kDa (FIP200). Another complex is the phosphoinositide 3-kinase (PI3K) complex composed by ATG14, vacuolar protein sorting-associated proteins 34 and 15 (VPS34, VPS15), and Beclin1 (BECN1) (Matsunaga et al. 2010; Cicchini et al. 2015). The contribution of ALS-related genes in this step is represented in Fig. 1.

Schematic representation of the ALS-related gene products involved in autophagy initiation (green) in their conventional intracellular functions
On the forming phagophore are recruited other complexes that regulate its elongation and expansion into the autophagosome. These complexes are the ATG7-ATG3-microtubule-associated protein 1A/1B light chain 3B (MAP1LC3B, or simply LC3) complex that promotes the formation of the lipidated active form of LC3 (LC3-II), and the ATG12-ATG5-ATG16L1 complex (Itakura and Mizushima 2010). Autophagic substrate recognition, targeting, and engulfment are highly regulated. Ubiquitinated misfolded proteins and aggregates are specifically recognized by a complex of chaperones and co-chaperones known as the CASA complex. The components of this complex are heat shock protein B8 (HSPB8), Bcl2-associated athanogene 3 (BAG3), heat shock protein 70 (HSP70), and C-terminus of HSC70-interacting protein (CHIP). The substrates are recognized by HSPB8 and BAG3, and subsequently ubiquitinated by CHIP. The CASA complex is then routed to the forming autophagosome through a dynein-mediated process regulated by BAG3-dynein interaction (Carra et al. 2008). Ubiquitinated substrates are recognized by autophagy receptors such as sequestosome 1 (SQSTM1/p62) or optineurin (OPTN) (Kraft et al. 2010). These proteins act as shuttles mediating substrates to phagophores. Indeed, autophagy receptors, thanks to the specific motifs present in their sequence such as LC3-interacting regions (LIRs), bind components of the autophagic machinery present in the forming autophagosome, such as LC3-II (Rogov et al. 2014). Subsequently to substrate engulfment, the constituted autophagosome maturation may include fusion with an endosome and always terminates in fusion with a lysosome (Zhong et al. 2009). The importance of various ALS-associated genes in these steps has been highlighted in Figs. 2 and 3.

Schematic representation of the ALS-related gene products involved in autophagosome elongation and maturation (green) in their conventional intracellular functions

Schematic representation of the ALS-related gene products involved in autophagosome-lysosome fusion and degradation (green) in their conventional intracellular functions
Strictly correlated to autophagy is the endocytosis pathway where the ALS-related genes described in Fig. 4 exert their function. Indeed, the formed endosome as previously described fuses with autophagosomes contributing to its maturation. The ALS-related genes contribution will be better explained in the dedicated sections.

Schematic representation of the ALS-related gene products involved in endocytosis (green) in their conventional intracellular functions
In neurons, axonal transport plays a relevant role in promoting autophagosome maturation by guiding membrane fusion between autophagy intermediates and components of the endolysosomal pathway (Hollenbeck 2014). Fusion with lysosomes eventually results in the degradation of substrates, thanks to hydrolytic enzymes present in the lysosome lumen (Xu and Ren 2015).
Autophagic Genes Involved in ALS
ALS2
Alsin (ALS2) is a guanine-nucleotide exchange factor (GEF) encoded by the gene ALS2 (chromosome 2q33). It is found in all tissues but particularly in neurons (Hadano et al. 2001; Otomo et al. 2003; Yamanaka et al. 2003). ALS2 comprises three GEF-specific domains: an N-terminal regulator of chromatin condensation–like domain (RCC1-like domain, or RLD), a central Db1-pleckstrin homology (DH/PH) domain, and a C-terminal vacuolar protein sorting 9 (VPS9) domain (Hadano et al. 2001; Yang et al. 2001).
ALS2 VPS9 domain mediates its homo-oligomerization and endosomal localization. ALS2 was indeed demonstrated to localize to structures positive for the early endosomal antigen 1 (EEA1) protein, a marker of early endosomes (Otomo et al. 2003; Topp et al. 2004; Kunita et al. 2004). The VPS9 domain is also essential for ALS2 to act as GEF for the small GTPase RAB5, a key player in endosomal membrane fusion (Fig. 4) (Otomo et al. 2003, 2011; Chandran et al. 2007). Transient ALS2 overexpression was shown to promote endosome enlargement and fusion through a RAB5-dependent mechanism in neuronal and non-neuronal cells, confirming ALS2 role in endosomal maturation (Kunita et al. 2004). Furthermore, ALS2 mutations were demonstrated to be associated with alterations in amphisome formation, and ALS2 downregulation was shown to cause a decrease in the autophagic clearance of substrates, suggesting that ALS2 is also involved in autophagosome maturation (Hadano et al. 2010; Otomo et al. 2011, 2012). Additionally, ALS2 was demonstrated to protect cultured murine MNs from the neurotoxic activity exerted by the ALS-related SOD1 mutants A4T, G85R, and G93R. It has been shown that ALS2 exerts such neuroprotective activity by directly binding to mutant SOD1 while contemporarily acting as GEF for the small GTPase Rac1 through its DH/PH domain. Indeed, Rac1 activation upon ALS2 binding triggers an antiapoptotic response mediated by the PI3K Akt3 that preserves neurons from death. Both Rac1 downregulation through RNA interference and deletion of ALS2 DH/PH domain fully abolish such neuroprotective function. Interestingly, ALS2 does not display the same activity against any other proteins implicated in neurodegeneration, such as Alzheimer’s disease-related amyloid-β and presenilin 1/2 mutants (Kanekura et al. 2004, 2005).
ALS2 mutations causing fALS are characterized by a recessive pattern of inheritance and produce premature stop codons in ALS2 sequence that abolish its VPS9 domain (Hadano et al. 2001; Yang et al. 2001; Kress et al. 2005; Sheerin et al. 2014). Truncated ALS2 isoforms are unstable and get rapidly eliminated, so that ALS2 pool is depleted in neurons harboring ALS2 mutations (Yamanaka et al. 2003). Such loss of ALS2 function is expected to reduce active RAB5 levels, since ALS2-deprived neurons display decreased motility of RAB5-positive endosomes (Lai et al. 2009). Consistently, neurons isolated from Als2 knock-out mice show defective RAB5-dependent endosomal maturation leading to the accumulation of large, EEA1-positive vacuoles. The impairments in the early phases of endosomal trafficking and fusion caused by ALS2 loss increase MN susceptibility to oxidative stress and promote astrogliosis in the brain and in the spinal cord of ALS2-null mice (Cai et al. 2005; Hadano et al. 2006; Devon et al. 2006). Nonetheless, Als2 knock-out mice do not display an overt ALS phenotype but are only characterized by subtle impairments in motor function and learning (Cai et al. 2005; Hadano et al. 2006; Devon et al. 2006; Yamanaka et al. 2006). Taken together, these data suggest that ALS2 loss is per se insufficient to cause neurodegeneration but that it might be responsible for an increased MN vulnerability.
Regarding ALS2 neuroprotective activity, loss of Als2 was shown to worsen the accumulation of protein aggregates in H46R SOD1 mice MNs. Indeed, Als2 knock-out correlated with a reduction in the autophagic clearance of mutant SOD1, as a consequence of impaired autophagosome maturation, and disrupted endosomal trafficking (Hadano et al. 2010). Additionally, ALS2 depletion was reported to exacerbate the effect of SQSTM1/p62 loss in H46R SOD1 mice, enforcing the hypothesis that ALS2 role in endosomal dynamics is essential to support autophagy (Hadano et al. 2016).
C9ORF72
C9ORF72 protein is encoded by chromosome 9 open reading frame 72 (C9ORF72), which presents 10 coding exons and two non-coding exons (Smeyers et al. 2021). C9ORF72 products are 3 transcript variants: a short transcript V1 and two transcripts V2 and V3 that share the coding of exons 2–11 (DeJesus-Hernandez et al. 2011; Smeyers et al. 2021). Interestingly for the development of animal models, the C9ORF72 human gene is conserved in primates but has a very low similarity in nematode and no orthologue in Drosophila (Therrien et al. 2013; Chen et al. 2017). C9ORF72 expression is found in most tissues including all brain regions, spinal cord, and immune system. At cellular level, C9ORF72 is localized in the cytoplasm or in organelles such as Golgi, mitochondria, lysosome, and other components of the endolysosome pathway. The V1 short-isoform was also found localized in the nuclear membrane (Xiao et al. 2015; Atkinson et al. 2015; Aoki et al. 2017; Chitiprolu et al. 2018; Wang et al. 2021). To date, C9ORF72 has been found implicated in different pathways, including nucleocytoplasmic import, stress granule (SG) formation and degradation or recovery, endosomal trafficking, axon growth, and autophagy regulation (Farg et al. 2014; Zhang et al. 2015; Yang et al. 2016; Amick et al. 2016; Sivadasan et al. 2016; Maharjan et al. 2017).
C9ORF72 regulates autophagy at different steps of its pathway as presented in Figs. 1 and 3. Its role in autophagy is supported by two cofactors, Smith–Magenis chromosome region 8 (SMCR8) and WD repeat domain 41 (WDR41), forming the C9ORF72-complex (Tang et al. 2020; Su et al. 2020). Both C9ORF72 and SMC38 present a differentially expressed in normal and neoplastic cells (DENN) domain which functions as a GEF for RAB GTPases (Levine et al. 2013; Iyer et al. 2018). In autophagy initiation, its role is difficult to discriminate as C9ORF72 interacts with different complexes involved in this step. Firstly, C9ORF72 and SMCR8 interact with different members of the ULK1-complex (Yang et al. 2016). During starvation C9ORF72-complex is recruited by solute carrier family 66 member 1 (SLC66A1) to lysosomes, increasing the interaction with ULK1-complex (Amick et al. 2020). Moreover, the C9ORF72 complex promotes the interaction between RAB1 and ULK1-complex, enhancing its recruitment to forming autophagosomes (Webster et al. 2016). Another sign of C9ORF72-positive regulation of autophagy initiation stands in its interaction with RAB5 which promotes the delivery of the PI3K and the ATG7-ATG5-LC3 complexs to autophagosome (Shi et al. 2018; Bingol 2018). The localization of C9ORF72 in forming autophagosomes was suggested to be mediated by SQSTM1/p62 or OPTN, which bind to C9ORF72-complex and RAB39b, another substrate of the complex (Sellier et al. 2016). While these interactions refer to a positive regulation of C9ORF72 complex in autophagy initiation, another mediates a negative regulation of autophagy. Indeed, signs of the interaction of C9ORF72 with mTOR complex 1 (mTORC1) were detected, which suggests a supporting role in mTORC1 function. C9ORF72 silencing decreases mTORC1 activity, promoting autophagy transcriptional factor regulators activation (Ji et al. 2017; Wang et al. 2020). C9ORF72 is also implicated in autophagosome maturation mainly by cooperating with RAB proteins, such as RAB7 and RAB11, which regulate microtubular transport of autophagosomes, late endosomes, multivesicular bodies, and the fusion with lysosomes (Hyttinen et al. 2013; Ao et al. 2014; Aoki et al. 2017; Tang et al. 2020).
C9ORF72 gene mutation consisting in a hexanucleotide expansion of the sequence GGGGCC (G4C2) present in the first intron has been associated to ALS and FTD in 2011 (Renton et al. 2011; DeJesus-Hernandez et al. 2011). The physiological size of the expansion can reach 24 repeats, whereas above 30 repeats it is considered pathological. The pathological length of expansion is very heterogenous; indeed, patients generally present hundreds or even thousands of repeats (DeJesus-Hernandez et al. 2011; Gijselinck et al. 2016). Still, the length of the expansion can differ in brain and blood tissues of the same patient. This mosaicism is due to the instability of the repeat number (van Mossevelde et al. 2017). The mutation has an autosomal-dominant transmission and is associated to 40% of fALS, 80% of ALS/FTD, and 25% of familial FTD cases (Gijselinck et al. 2016; van Mossevelde et al. 2017). The mutation leads to both GOF and LOF that together concur in the onset of the pathology. The LOF is due to protein haploinsufficiency that is triggered by the decrease in gene transcription caused by altered RNA structures and by hypermethylation of the promoter (Haeusler et al. 2014; Gijselinck et al. 2016; Esanov et al. 2017). The GOF is associated to the toxicity of the RNA repeats and the formation of abnormal dipeptide-repeat proteins (DRPs). The RNA repeats are transcribed bidirectionally forming sense and antisense transcripts. These transcripts are instable and form nuclear RNA foci that sequester RNA-binding proteins preventing their functionality (Gendron et al. 2013; Barker et al. 2017). In addition, guanosine-rich RNA repeats tend to fold in a stable secondary structure, known as G-quadruplex, that deleteriously interacts with splicing factors causing splicing errors (Reddy et al. 2013; Conlon et al. 2016). The translation of DPR sense and antisense transcripts occurs in an unconventional ATG-independent mechanism and results in the formation of five DRPs constituted of glycine-alanine (GA), glycine-proline (GP), glycine-arginine (GR), proline-arginine (PR), and proline-alanine (PA) (Zu et al. 2013). All DPRs are found in the brain tissue and spinal cord of patients and exert their toxicity by sequestering proteins, impairing ribosome biogenesis, and altering translation. Specifically, the DPRs poly-PR and poly-GR alter nuclear transport (Freibaum and Taylor 2017; Hayes et al. 2020). In addition, poly-GR also interferes with SGs formation and degradation (Chew et al. 2019). Both GOF and LOF concur in alteration of cellular proteostasis. A sign of altered proteostasis stands in TDP-43 mislocalization and aggregation, which is a hallmark of ALS and FTD C9ORF72 patients (Cook et al. 2020). Another sign of altered proteostasis in C9ORF72-patients is the presence of SQSTM1/p62 and ubiquitin-positive inclusions that frequently also contain DPRs (Mann et al. 2013).
C9ORF72 animal models were developed to mimic LOF and GOF. For what concerns C9ORF72 role in autophagy, transient reduction of C9orf72 expression in conditional knock-out mice in neurons does not cause alterations in behavior or motor phenotype (Koppers et al. 2015), whereas constitutive C9orf72 knock-out animals present dysregulation of the immune system probably associated to autophagy dysfunction, reduced survival, and mild motor and cognitive phenotypes (Jiang et al. 2016; Atanasio et al. 2016). Overall, data on animals support the theory that loss of C9ORF72 function alone is not sufficient to develop ALS. On the other hand, models of GOF present neuronal alterations.
CHMP2B
Charged multivesicular body protein 2B (CHMP2B) is encoded by the gene CHMP2B (chromosome 3p11.2). It is an evolutionarily conserved protein playing a key role in the assembly of the endosomal sorting complex required for transport III (ESCRT-III; Fig. 4). The ESCRT-III machinery is implicated in the generation of multivesicular bodies (MVBs), endosomal sorting, and autophagy in several tissues, including all brain regions (Skibinski et al. 2005; Rusten and Stenmark 2009; Hurley and Hanson 2010; Henne et al. 2013).
When inactive, CHMP2B is found in an autoinhibited conformation masking the N-terminal sites required to recruit the ESCRT-III complex. Activation occurs through the interaction of CHMP2B C-terminal domain with the ATPase vacuolar protein sorting-associated protein 4 (VPS4) and triggers the assembly of the ESCRT-III and VPS4 complexes on the endosomal membrane. VPS4 ATPase activity is then required to let intraluminal vesicles bud from endosomal membranes, hence generating MVBs, and to finally dissociate ESCRT-III proteins from one another for MVB generation (Fig. 4). (Schmidt and Teis, 2012).
Apart from being involved in MVB maturation, CHMP2B also participates to lysosomal membrane repair and to autophagy together with the other components of the ESCRT-III machinery (Krasniak and Ahmad 2016; Radulovic et al. 2018). Loss of ESCRT genes was demonstrated to correlate with autophagosome accumulation in yeast (Roudier et al. 2005), Drosophila (Rusten et al. 2007), and mammals (Komatsu et al. 2006), implying a key role played by ESCRT proteins in autophagosome-lysosome fusion. Even though the mechanism through which CHMP2B is involved in autophagy is still not clearly defined, CHMP2B mutations are associated to important signs of autophagy impairment.
The first CHMP2B mutation linked to neurodegeneration was found in 1995 in a Danish family affected by autosomal-dominant FTD and was characterized as a substitution occurring at exon 6 splice acceptor site (Brown et al. 1995; Gydesen et al. 2002; Skibinski et al. 2005). Such mutation determines the production of two mutant CHMP2B forms, one including intron 5 in the protein sequence and producing a truncated CHMP2B isoform with a valine residue replacing the last 36 amino acids of the wild-type protein (CHMP2BIntron5) and the other generating a 29-amino acid nonsense sequence (CHMP2BΔ10) (Skibinski et al. 2005). Additional CHMP2B mutations were later identified also in fALS and ALS/FTD (Parkinson et al. 2006; Cox et al. 2010; van Blitterswijk et al. 2012c; Narain et al. 2018).
Truncated CHMP2B mutants are incapable of autoinhibition and tend to associate to the rest of the ESCRT-III machinery at a higher frequency compared to wild-type CHMP2B. This abnormal interaction leads to the formation of aberrant complexes that remain associated to endosomal membranes, thus preventing autophagosome-lysosome fusion (Lee et al. 2007; Han et al. 2012). Indeed, CHMP2BIntron5 was shown to induce the accumulation of ubiquitin-positive puncta and/or SQSTM1/p62- and LC3-positive vacuoles in cells (Lee et al. 2007; Filimonenko et al. 2007; West et al. 2020). Autophagosome accumulation was also observed in murine and Drosophila CHMP2BIntron5 models (Ghazi-Noori et al. 2012; Vernay et al. 2016; West et al. 2020) and even in patient-derived fibroblasts and cortical tissue (Urwin et al. 2010), supporting the involvement of autophagic flux blockage in CHMP2BIntron5-related pathogenesis. Additionally, CHMP2BIntron5 was evidenced to localize on accumulating endosomes positive for the small GTPases RAB4, RAB5, and RAB7 in both Drosophila and rat primary neurons (West et al. 2020). Similar observations were made in postmortem brains of fALS patients bearing CHMP2B mutations (T104N, I29V, Q206H), which displayed an accumulation of autophagosomes positive for SQSTM1/p62 and LC3-II (Parkinson et al. 2006; Cox et al. 2010), and the T140N CHMP2B mutant was also shown to localize with other ESCRT-III subunits in RAB5- and RAB7-positive endosomes in cortical neurons (Han et al. 2012). These data support the involvement of endolysosomal and autophagosomal activity dysfunction in CHMP2B-related neurodegeneration.
DCTN1
Dynactin subunit 1 (DCTN1, or p150Glued) is the largest subunit of the dynactin complex and is encoded by the gene DCTN1 (chromosome 2p13.1). The dynactin machinery is composed of at least 11 proteins organized into more than 20 subunits (Urnavicius et al. 2015). The dynactin complex interacts with dynein to increase its processivity in microtubule-based retrograde transport and acts as a multifunctional adaptor between dynein and specific cargos, including vesicles, organelles, cytosolic proteins, and mRNA (Karki and Holzbaur 1999; Culver-Hanlon et al. 2006). Inside the dynactin complex, DCTN1 forms dimers that interact with dynein intermediate chains and bind to microtubules through its N-terminal cytoskeletal-associated protein/glycine-rich (CAP-Gly) domain (Vaughan and Vallee 1995; Karki and Holzbaur 1995; Culver-Hanlon et al. 2006).
Considering autophagy, DCTN1 plays a central role in dynein-mediated retrograde transport of autophagosomes and lysosomes to the perinuclear region (Fig. 3). DCTN1 was indeed shown to interact with Hermansky-Pudlak syndrome 6 (HSP6), a protein that is strictly required to drive lysosomes to the microtubule organizing center for degradation (Li et al. 2014).
Based on the current knowledge, it is still unclear whether DCTN1 mutations promote ALS development through GOF or LOF mechanisms. The first pathogenetic mutation ever identified in DCTN1 sequence was an autosomal dominant missense associated to a form of hereditary motor neuropathy that primarily affectes lower MNs (HMN7B) (Puls et al. 2003). Such mutation produces a G59S substitution in DCTN1 CAP-Gly domain that alters the folding of DCTN1 microtubule-binding domain, reducing the affinity of the mutant protein for microtubules. G59S DCTN1 tends to misfold and form neurotoxic aggregates that were shown to sequester dynein, mitochondria, and TDP-43 (Puls et al. 2003, 2005; Levy et al. 2006; Deshimaru et al. 2021). Additional DCTN1 mutations lying both in DCTN1 CAP-Gly domain and in its dynein-binding domain have then been identified in ALS (Münch et al. 2004; Stockmann et al. 2013; Liu et al. 2014, 2017), including those forms with an FTD component (Münch et al. 2005). Several in vitro studies were performed to characterize the impact of ALS-related DCTN1 mutations on the protein. While some DCTN1 mutants did not evidence any alterations in protein conformation or distribution (Münch et al. 2004; Dixit et al. 2008; Stockmann et al. 2013), other variants identified in sALS patients were shown to form aggregates or abnormal filamentous structures upon overexpression in primary rat MNs (Stockmann et al. 2013). However, most of the DCTN1 mutations causing an aberrant protein behavior are characterized by uncertain patterns of inheritance and can be found also in control populations, suggesting that they might represent risk factors predisposing to ALS, rather than overt pathogenetic variants (Stockmann et al. 2013).
Analyses performed by Jiang and colleagues on postmortem samples revealed that both upper and lower MNs of sALS patients display lower DCTN1 mRNA levels compared to non-ALS controls starting from very early stages of disease progression (Jiang et al. 2005, 2007). Similarly, Kuźma-Kozakiewicz and colleagues evidenced increased DCTN1 mRNA levels but reduced DCTN1 protein levels in the motor cortex of sALS patients compared to the sensory cortex, suggesting that in sALS MNs DCTN1 depletion occurs and cannot be restored by gene expression upregulation, probably due to inefficient translation (Kuźma-Kozakiewicz et al. 2013). In contrast, while homozygous Dctn1 knock-out or G59S DCTN1 knock-in was proven to be embryonic lethal in mice, animals deprived of one Dctn1 allele did not develop MN degeneration (Lai et al. 2007), and neuron-specific Dctn1 knock-out mice only began manifesting neurodegeneration symptoms at 18 months of age (Yu et al. 2018). On the other hand, two heterozygous G59S DCTN1 knock-in strains generated by distinct research groups displayed halved DCTN1 protein levels and no sign of mutant protein aggregation in brain and spinal cord, suggesting that the G59S substitution might lead to rapid protein degradation in vivo, but at the same time, mice started manifesting muscle atrophy due to spinal MN loss at 10 months of age. These symptoms were accompanied by astrogliosis and by the accumulation of cytoskeletal and synaptic vesicle proteins at neuromuscular junctions, probably as a consequence of disrupted retrograde transport (Levy et al. 2006; Lai et al. 2007). Moreover, a third heterozygous G59S DCTN1 murine strain evidenced fast lower MN loss paralleled by impaired ER-to-Golgi vesicular trafficking and by the accumulation of LC3-II- and ubiquitin-positive DCTN1 aggregates in MN cell bodies hinting at an involvement of dysfunctional retrograde transport of autophagosomes in neuronal death (Laird et al. 2008). In line with these observations, knockout of the DCTN1 ortholog in C. elegans, dnc-1, was associated to immature autophagosome accumulation in adult MNs, fostering their degeneration and leading to severe motor deficits that recapitulate ALS symptomatology (Ikenaka et al. 2013). These conflicting data suggest that further studies are required to gain better insight into the pathogenetic mechanisms associated to DCTN1 mutations in neurodegeneration.
FIG4
Factor-induced gene 4 (FIG4) is a magnesium-dependent phosphatase encoded by the gene FIG4 (chromosome 6q21). FIG4 mediates phosphatidylinositol-3,5-bisphosphate (PI(3,5)P2) conversion to phosphatidylinositol-3-phosphate (PI(3)P) on the cytosolic side of endosomal membranes, where FIG4 localizes through the interaction with the scaffolding protein VAC14 (Rudge et al. 2004; Sbrissa et al. 2007; Jin et al. 2008). PI(3)P and its derivatives are central players in endosomal transport and autophagy, with different PI3-phosphate pools generated by distinct kinases and phosphatases localized to different endosomal compartments (Di Paolo and De Camilli 2006). Specifically, PI(3,5)P2 is produced by the kinase 1-phosphatidylinositol 3-phosphate 5-kinase (PIKfyve), which joins the complex formed by FIG4 and VAC14 on endosomal membranes, and this modified phospholipid acts as a key signal for the retrograde trafficking of endosomes (Sbrissa et al. 2007; Ferguson et al. 2009). Conversion of PI(3)P to PI(3,5)P2 is also essential for endosomal maturation since PI(3,5)P2 modulates cargo degradation in late endosomes/lysosomes (Odorizzi et al. 1998; Rutherford et al. 2006; Zhang et al. 2007). Therefore, the PIKfyve-FIG4-VAC14 ternary complex plays a central role in regulating endosomal metabolism (Fig. 4).
The first mutation identified in FIG4 is an insertion of the 5.5-kb retrotransposon ETn2β into intron 18 of Fig4 murine gene, which caused alterations in Fig4 mRNA processing, therefore resulting in a substantial reduction in Fig4 protein levels. Animals affected by this autosomal recessive mutation displayed resting tremor accompanied by diluted pigmentation and were thus referred to as “pale tremor” mice. This phenotype is consistent with Charcot-Marie-Tooth (CMT) axonal neuropathies; notably, recessively inherited FIG4 mutations were subsequently identified in CMT4J patients (Chow et al. 2007). ALS-associated FIG4 mutations include truncating mutations, missenses, and mutations in splice sites and are all predicted to cause loss of protein function, similarly to that observed in pale tremor mice and CMT4J patients (Chow et al. 2007, 2009; Osmanovic et al. 2017). While FIG4 mutations are reported to be responsible for 1–3% of ALS cases among European patients, no deleterious FIG4 variants have been found in larger ALS groups, and some of the variants isolated in the European cohort display reduced penetrance (Osmanovic et al. 2017), so that further investigation is required.
Animal models have been exploited to better characterize the role of FIG4 mutations in endolysosomal trafficking. Concerning pale tremor mice, the relevant reduction in Fig4 protein levels dependent on ETn2β retrotransposition determined a strong decrease in PI(3,5)P2 levels accompanied by the accumulation of large vacuoles harboring ubiquitinated proteins and positive for SQSTM1/p62, LC3-II, and the late-stage endosomal markers lysosomal-associated membrane proteins 1 and 2 (LAMP-1 and LAMP-2) in murine neurons and astrocytes (Ferguson et al. 2009; Lenk et al. 2011). Comparable accumulation of enlarged lysosomes causing the rapid development of severe brain degeneration was observed in both constitutive and neuron-specific Fig4 knock-out murine models (Ferguson et al. 2012), while in Drosophila Fig4 loss correlated with motility impairments and shorter lifespan (Bharadwaj et al. 2016; Kyotani et al. 2016). The decrease in PI(3,5)P2 levels associated to Fig4 loss is hypothesized to depend on reduced PIKfyve activity, since FIG4 is required to stabilize the interaction between PIKfyve and VAC14 (Botelho et al. 2008).
KIF5A
Kinesin 5A (KIF5A) is a neuron-specific kinesin heavy chain encoded by the gene KIF5A (chromosome 12q13.3) (Aizawa et al. 1992). Kinesins are ATP-dependent molecular motors that transport cargo along microtubule tracks in the anterograde direction, from the center of the cell to its periphery (Vale et al. 1985; Brady 1985). KIF5A comprises an N-terminal motor domain involved in microtubule binding and ATP hydrolysis, a stalk domain required for homodimerization and interaction with kinesin light chains, and a C-terminal tail domain which mediates interaction with cargos and adaptor proteins (Miki et al. 2005; Hirokawa and Noda 2008). Among KIF5A cargos, lysosomes can be found, conferring KIF5A a role in the autophagic pathway (Fig. 3) (Liu et al. 2021).
KIF5A was found mutated in ALS in 2018, with genome-wide analyses identifying both low- and high-risk mutations (Brenner et al. 2018; Nicolas et al. 2018). Mutations in KIF5A had been previously linked to other neurodegenerative or neurodevelopmental disorders. Interestingly, mutations targeting different KIF5A domains give rise to distinct phenotypes, with minimal overlapping. Specifically, mutations targeting KIF5A motor or stalk domains cause hereditary spastic paraplegia and CMT2 (Reid et al. 2002; Fichera et al. 2004; Crimella et al. 2012), while mutations falling in its C-terminal tail are associated to neonatal intractable myoclonus as well as ALS (Rydzanicz et al. 2017; Brenner et al. 2018; Nicolas et al. 2018).
Recently, de novo frameshift mutations causing exon 27 skipping (ΔExon27) and downstream elongation of the KIF5A tail have been reported to abolish KIF5A ability to perform autoinhibition, leading to aberrant mitochondrial transport along axons, and to enhance mutant KIF5A interaction with SQSTM1/p62 (Baron et al. 2022). Therefore, neurodegeneration caused by ΔExon27 KIF5A mutations seems to be linked to a toxic GOF disrupting neuronal trafficking and homeostasis. Since lysosomes are part of KIF5A cargos (Liu et al. 2021), the axonal transport alterations implicated in KIF5A-related ALS pathogenesis might tamper with the autophagic flux, too. Further studies are required to better elucidate the contribution of KIF5A mutations to ALS.
OPTN
OPTN is a multifunctional adaptor protein encoded by the OPTN gene (chromosome 10p13). It is highly expressed in brain and skeletal muscle (Rezaie et al. 2002; De Marco et al. 2006) and takes part in a wide variety of cellular processes, such as NF-κB activation, viral sensing, Golgi maintenance, and autophagy (Markovinovic et al. 2017). OPTN acts as an autophagy receptor by binding substrates, including damaged mitochondria, through its C-terminal ubiquitin-binding region of ABIN proteins and NEMO (UBAN) domain and delivers them to phagophores by interacting with LC3, thanks to its LIR domain (Figs. 1 and 2) (Wild et al. 2011; Wong and Holzbaur 2014). Subsequently, it links to myosin VI to promote autophagosome fusion with lysosomes (Tumbarello et al. 2012).
OPTN role as an autophagy receptor makes it a neuroprotective factor. Indeed, OPTN is sequestered into aggregates formed by mutant proteins in several neurodegenerative disorders (Maruyama et al. 2010; Hortobágyi et al. 2011; Osawa et al. 2011), and its deletion promotes aggregates accumulation (Korac et al. 2013). Additionally, OPTN interacts with SOD1 aggregates through its C-terminal coiled-coil domain, therefore in an ubiquitin-independent fashion (Korac et al. 2013).
OPTN mutations were initially associated to primary open-angle glaucoma (Rezaie et al. 2002; Minegishi et al. 2016) and were found to impair the autophagic flux. For example, retinal cells harboring OPTN E50K were shown to be characterized by defects in phagophore formation and by autophagy inhibition upon amino acid starvation (Chalasani et al. 2014). For what concerns ALS, more than 40 OPTN variants have been reported both in sALS and in fALS, with a limited number of mutations being shared between populations of different ethnicity (Maruyama et al. 2010; Del Bo et al. 2011; Tümer et al. 2012; van Blitterswijk et al. 2012b; Iida et al. 2012; Beeldman et al. 2015; Gotkine et al. 2021). ALS-related OPTN mutations are mainly missenses, truncations, and exon 5 deletions. A LOF mechanism is hypothesized for truncating mutations since mRNA decay and loss of OPTN immunoreactivity were reported in patient spinal cord (Maruyama et al. 2010; Iida et al. 2012; Gotkine et al. 2021). Regarding missense variants, they tend to cluster around OPTN UBAN domain (Maruyama et al. 2010). The best characterized one is E478G OPTN, which is defective in ubiquitin binding (Wild et al. 2011) and was indeed reported to lose the ability to associate with polyubiquitinated inclusions and mitochondria to drive them to autophagy (Korac et al. 2013; Wong and Holzbaur 2014). The E478G mutant also sequesters wild-type OPTN into oligomers upon overexpression, limiting the OPTN pool available to guide phagophore formation (Shen et al. 2015) and forming inclusion bodies in sALS neurons (Maruyama et al. 2010).
Furthermore, E478G and G398X OPTN were shown to be unable to bind to myosin VI, determining the accumulation of immature autophagosomes triggering ER and Golgi stress (Sundaramoorthy et al. 2015).
Animal models recapitulate most observations on ALS-associated OPTN mutations made in vitro. OPTN ortholog knock-down in zebrafish is associated to motor axonopathy symptoms resembling models of SOD1 pathology (Korac et al. 2013). Moreover, 470T Optn mice mimic human OPTN truncations and reduced protein levels following mRNA decay are reported for such animal model (Munitic et al. 2013). Finally, the murine equivalent of the human E478G mutation (D477N Optn) is similarly unable to bind ubiquitin (Gleason et al. 2011). Despite the wide variety of data collected about OPTN mutations in ALS, the pathogenetic mechanisms causing autophagy impairments lying behind them are still to be fully characterized.
SQSTM1/p62
SQSTM1/p62 is a multifunctional adaptor protein encoded by the gene SQSTM1 (chromosome 5q35.3). It is highly expressed in spinal cord MNs (Keller et al. 2012) and is implicated in several cellular processes, including NF-κB activation, apoptosis, and proteostasis maintenance (Rea et al. 2013). Concerning the latter process, SQSTM1/p62 acts as an autophagy receptor similarly to OPTN, therefore by sequestering ubiquitinated proteins via its C-terminal ubiquitin-binding domain (UBD), named ubiquitin-associated (UBA) domain, and contemporarily interacting with LC3 through its LIR to deliver substrates to autophagosomes (Figs. 1 and 2) (Bjørkøy et al. 2005; Pankiv et al. 2007; Ichimura et al. 2008). The N-terminal Phox and Bem1p-1 (PB1) ubiquitin-like domain allows SQSTM1/p62 to oligomerize, and both the PB1 and the UBA domains are involved in the interaction with other autophagy receptors, like OPTN and neighbor of BRCA1 gene 1 (NBR1), required to better coordinate autophagosome formation (Johansen and Lamark 2011). SQSTM1/p62 is also able to drive ubiquitinated substrates to proteasomal degradation, thanks to its ubiquitin-like domains (Seibenhener et al. 2004; Babu et al. 2005; Geetha et al. 2008). Evidence supports the hypothesis that SQSTM1/p62 might be a neuroprotective factor, once again similarly to OPTN. Firstly, it is upregulated at the onset of neuronal apoptosis, when ubiquitinated proteins accumulate in response to cell damage (Kuusisto et al. 2001). Moreover, it is found within ubiquitin-positive inclusions formed by mutant proteins in several neurodegenerative disorders (Zatloukal et al. 2002; Arai et al. 2003; Mizuno et al. 2006; King et al. 2013), underlining the central role this autophagy receptor plays in PQC. SQSTM1/p62 is also responsible for driving mutant SOD1 to autophagosomes through an ubiquitin-independent interaction mediated by its SOD1 mutant interaction region (SMIR) (Gal et al. 2009). Lastly, mice deprived of Sqstm1 display tau hyperphosphorylation and memory impairments (Babu et al. 2008), both hallmarks of Alzheimer’s disease, while Sqstm1 knock-down in zebrafish determines the development of locomotory impairments associated with autophagy defects and MN axon shortening (Lattante et al. 2015).
SQSTM1 mutations were firstly identified in 2002 in patients suffering from Paget disease of bone (PDB), a chronic progressive skeletal disorder. PDB-causing SQSTM1 mutations mainly target SQSTM1/p62 UBA domain, with the P392L substitution being the most frequent among them (Laurin et al. 2002). Mutations spanning the whole SQSTM1 sequence, including its promoter, have thereafter been related to fALS, sALS, and ALS/FTD (Rubino et al. 2012; Teyssou et al. 2013; Hirano et al. 2013; Shimizu et al. 2013; Le Ber et al. 2013; Chen et al. 2014; Kwok et al. 2014), and in most cases they have been associated with TDP-43 pathology (van der Zee et al. 2014). Since many ALS-related SQSTM1 variants were also found at relatively high frequencies in control populations, it is still unclear whether they are actually causative of the phenotype, as it remains to be determined how mutations in the same gene give rise to such distinct phenotypes as ALS and PDB (Rea et al. 2014). Nonetheless, the body of evidence connecting ALS-associated SQSTM1 mutations to specific impairments of SQSTM1/p62 functions in autophagy is growing. For example, the L341V substitution in SQSTM1/p62 LIR sequence was shown to cause a strong decrease in its interaction with LC3-II, thus tampering with the ability of the autophagy receptor to deliver cargo to forming phagophores (Goode et al. 2016). Additionally, some studies have confirmed the presence of SQSTM1/p62-positive inclusions along with increased levels of the protein in spinal MNs of SQSTM1 mutation carriers and found frontal cortical atrophy associated with these aggregates, thus providing further evidence of pathologic overlap between ALS and FTLD (Teyssou et al. 2013; Le Ber et al. 2013).
Until now, mechanistic studies on SQSTM1 mutations have mainly concentrated on the P392L substitution. Overexpression of P392L SQSTM1/p62 was shown to enhance autophagosome formation in the osteoclasts of mice harboring the murine equivalent of the P392L mutation (Daroszewska et al. 2011). Additionally, rescue of the locomotor defects displayed by zebrafish upon Sqstm1 downregulation could be achieved through overexpression of human wild-type SQSTM1/p62, but not of the P392L mutant. Amelioration of the phenotype was also evidenced upon administration of the autophagy inductor rapamycin, which further supports the hypothesis of a LOF being connected to the P392L substitution (Lattante et al. 2015). Interestingly, the P392L mutation was later found in ALS patients (Teyssou et al. 2013; Le Ber et al. 2013; Kwok et al. 2014), but its effect on MNs has not been characterized yet.
Finally, Sqstm1 depletion was demonstrated to exacerbate H46R SOD1 mice ALS phenotype, with shorter lifespan and accelerated MN degeneration and weight loss (Hadano et al. 2016).
TBK1
TANK-binding kinase 1 (TBK1) is a serine/threonine kinase encoded by the gene TBK1 (chromosome 12q14.2). TBK1 is highly expressed in neuronal cells belonging to the cerebral cortex, the hippocampus, and the lateral ventricle (Uhlén et al. 2015).
TBK1 was first isolated as OPTN binding partner through yeast two-hybrid screening (Morton et al. 2008). In fact, it phosphorylates OPTN in its UBAN domain, promoting its binding to LC3-II and to ubiquitinated substrates to enhance the autophagic flux (Oakes et al. 2017). OPTN phosphorylation also enforces its role in TBK1 activation, which generates a positive feedback loop between TBK1 and OPTN activation (Lazarou et al. 2015; Heo et al. 2015). TBK1 exerts a similar modulatory activity on nuclear domain 10 protein 52 (NDP52) and SQSTM1/p62, two other autophagy adaptors, increasing their affinity for the ubiquitin chains of their UBDs through phosphorylation (Thurston et al. 2009; Pilli et al. 2012; Li et al. 2018a). TBK1-mediated OPTN, NDP52, and SQSTM1/p62 phosphorylation also promotes their association with damaged mitochondria to support mitophagy (Lazarou et al. 2015; Heo et al. 2015; Richter et al. 2016; Moore and Holzbaur 2016). Additionally, this kinase targets the C9ORF72-coupled GEF SMCR8, leading to C9ORF72 activation and therefore promoting autophagy initiation (Fig. 1) (Sellier et al. 2016). Moreover, TBK1 plays a role in autophagosome maturation, probably by interacting with the small GTPase RAB8b. TBK1 silencing was indeed shown not to interfere with autophagosome formation but to prevent autophagosome-lysosome fusion (Wang et al. 2018). Finally, microtubule-binding proteins required to trigger retrograde transport of autophagosomes and autolysosome formation are among TBK1 substrates, too (Oakes et al. 2017).
TBK1 mutations were firstly identified in ALS in 2015 through whole-exome sequencing of large patient cohorts of European ethnicity. Subsequently, additional studies confirmed the role of TBK1 in ALS pathogenesis, including ALS/FTD (Le Ber et al. 2015; Williams et al. 2015; Shu et al. 2016; Tsai et al. 2016; Borghero et al. 2016; van Rheenen et al. 2016). To date, more than 90 TBK1 mutations have been reported (Abramzon et al. 2020), and while they are rare in sALS patients, they account for around 3–4% of fALS and ALS/FTD cases (Cirulli et al. 2015; Freischmidt et al. 2015; Gijselinck et al. 2015). Most identified TBK1 variants are missense mutations with uncharacterized pathogenic effects (Oakes et al. 2017). On the other hand, nonsense and frameshift TBK1 mutations associated to the deletion of its second coiled-coil domain (CCD2) are reported to alter TBK1 ability to interact with adaptor proteins that regulate its localization and activation of downstream signaling pathways (Ryzhakov and Randow 2007). Nonsense and frameshift TBK1 mutations also correlate with reduced TBK1 expression as a consequence of mRNA decay (Freischmidt et al. 2015; Brenner et al. 2019). Thus, the pathogenetic mechanisms underlying TBK1-related ALS cases are considered to depend on the loss of TBK1 function.
Abolishment of the interaction between TBK1 and OPTN by TBK1 mutations seems to play a prominent role in ALS pathogenesis. Indeed, an ALS-related TBK1 mutant deprived of amino acids 690–713 in the CCD2 domain was shown to be unable to bind OPTN when transiently overexpressed in HEK293T cells while preserving interaction with other TBK1 protein partners (Freischmidt et al. 2015). The same was demonstrated for additional TBK1 mutants (Richter et al. 2016; Moore and Holzbaur 2016; Li et al. 2018a; de Majo et al. 2018). Moreover, E696K TBK1 was shown to correlate with decreased recruitment of OTPN and LC3 to damaged mitochondria, leading to mitochondrial dysfunction and accumulation (Moore and Holzbaur 2016). Finally, TBK1 mutations were shown to correlate with the formation of inclusions positive for TDP-43, ubiquitin, and SQSTM1/p62 in MNs and glial cells of ALS and FTD patients (Van Mossevelde et al. 2016), further reflecting autophagy disruption. Taken together, these data support the hypothesis that the concerted activity of TBK1 and OPTN in autophagy and mitophagy is essential to prevent neurodegeneration.
Animal models have been generated to better investigate TBK1 role in ALS. While heterozygous Tbk1 knock-out mice did not develop MN degeneration or autophagy impairments (Brenner et al. 2019), loss of murine Tbk1 resulted embryonic lethal (Bonnard et al. 2000), and animals undergoing neuron-specific Tbk1 deletion displayed loss of cortical synapses, dendrite dysmorphism, and accumulation of neurofibrillary tangles correlating with motor and cognitive defects reminiscent of ALS/FTD (Duan et al. 2019). Additionally, loss of one Tbk1 allele in a G93A SOD1 murine model accelerated autophagy dysfunction and muscle denervation during the early stage of ALS, but at the same time, it extended mouse survival by reducing neuroinflammation at later stages (Brenner et al. 2018). A comparable disease course was observed in G93A SOD1 mice harboring Tbk1 mutations that impair its kinase activity (Gerbino et al. 2020). Therefore, these findings seem to indicate that loss of TBK1 function does not only lead to ALS development but can also modify the course of the pathology.
TUBA4A
α-tubulin isoform 4a (TUBA4A) is encoded by the gene TUBA4A (chromosome 2q35). It is ubiquitously expressed but particularly enriched in neurons (Rustici et al. 2013; Smith et al. 2014). α-and β-tubulins heterodimerize to form microtubules, cytoskeletal scaffolds along which kinesin- and dynein-mediated transport occurs in cells (Fig. 3).
TUBA4A mutations were firstly identified in familial ALS and ALS/FTD in 2014 based on exome sequencing performed on a large cohort of European and American patients (Smith et al. 2014). In the following years, additional TUBA4A mutations were found in other European and Asian cohorts (Dols-Icardo et al. 2016; Perrone et al. 2017; Li et al. 2018b) and even among sporadic ALS and ALS/FTD patients (Pensato et al. 2015). Differently from other tubulin genes that are highly expressed during brain development and that are therefore found mutated in neurodevelopmental disorders, TUBA4A expression increases with aging (Tischfield et al. 2011; Hersheson et al. 2013), which might explain why its mutations are associated to late-onset neurodegeneration (Smith et al. 2014; Clark et al. 2016) and may account for TUBA4A contributions to ALS progression. The frequency of TUBA4A mutations is anyway low in all tested populations so far, making them a rare cause of ALS (Chia et al. 2018).
Most TUBA4A mutations found in ALS cluster in the protein domain involved in the interaction with other tubulins and with the molecular motors kinesin and dynein (Howes et al. 2014) and are therefore predicted to tamper with microtubule stability and microtubule-based transport in cells. To date, the best characterized ALS-associated TUBA4A mutant is the truncated W407X variant, which displays impaired α/β-tubulin dimer formation and incorporation into microtubules accompanied by aggregation propensity when expressed in primary MNs (Smith et al. 2014).
Concerning autophagy, disruption of microtubule dynamics related to TUBA4A mutations might negatively reflect on autolysosome formation, which is dependent on dynein-mediated retrograde transport (Rademakers and van Blitterswijk 2014). Mutant TUBA4A aggregation might further worsen autophagy impairment; however, up to now, no data are available to confirm this hypothesis except for the fALS-associated W407X mutant (Smith et al. 2014). The generation of animal models could help in characterizing the pathogenetic mechanisms associated to TUBA4A mutations in ALS.
UBQLN2
Ubiquilin-2 (UBQLN2) is one of the four members of the ubiquilin (UBQLN) family in the human genome encoded by the UBQLN2 gene located on chromosome Xp11.21 (Kaye and Shows 2000). It is expressed in various tissues, but the highest expression levels are found in muscles and brain (Lin et al. 2021). UBQLN2 localizes mainly in the cytoplasm where it functions in the maintenance of proteostasis. UBQLN2 presents an ubiquitin-like (UBL) domain at the N-terminus, which interacts with the regulatory cap of the proteasome, and a UBA domain at the C-terminus, which recognizes polyubiquitin chains present on substrates; the UBQLN2 central domain is characterized by the presence of four stress-induced protein 1 (STI-1)-like motifs, which bind to heat shock proteins through a still unclear mechanism, and a proline-rich repeat domain containing 12 PXX repeats whose function is unknown (Kaye et al. 2000; Walters et al. 2002; Ko et al. 2004; Deng et al. 2011).
UBQLN2 activity mainly consists in promoting the disposal of misfolded or redundant proteins through the UPS or autophagy. The UBL and UBA domains permit the shuttling of ubiquitinated proteins to the UPS. UBQLN2 substrates are cytosolic and ER proteins. Indeed, through its binding with ER membrane proteins as ubiquitin regulatory X domain-containing protein 8 protein (Ubxd8) and homocysteine-induced ER protein (Herp), UBQLN2 specifically promotes the disposal of altered ER proteins. UBQLN2 interaction with autophagy is more complex. In the first place, UBQLN2 is implicated in autophagy initiation; indeed, it regulates mTORC1 activity. Loss of UBQLN2 activity prevents mTORC1-mediated autophagy inhibition resulting in an increased autophagy induction (Şentürk et al. 2019). Furthermore, UBQLN2 interacts indirectly with LC3 through its UBA domain promoting autophagosome formation (N’Diaye et al. 2009), and it was found also to interact with OPTN, confirming a role of UBQLN2 in the first steps of the autophagic pathway (Figs. 1 and 2) (Osaka et al. 2015). Recent data showed a role of UBQLN2 also in lysosomal activity by regulating its acidification. Indeed, different groups presented two parallel mechanisms through which UBQLN2 interacts with subunits of v-ATPase, promoting its formation (Fig. 3) (Şentürk et al. 2019; Wu et al. 2020). Thus, silencing or mutations of UBQLN2 gene are associated to an increase in autophagy induction that is paralleled with a blockage of autophagosome disposal due to a loss of lysosome activity (Şentürk et al. 2019).
UBQLN2 mutations were associated in 2011 to a dominantly inherited, X-linked form of ALS and ALS/FTD (Deng et al. 2011). To date, more than 20 mutations have been detected mainly in the PXX domain, while fewer are present in STI-1-like motifs or between domains [as reviewed in (Renaud et al. 2019)]. UBQLN2 mutations impair cellular homeostasis with different mechanisms: by triggering proteasome impairment, preventing the delivery to the proteasome of polyubiquitinated substrates, which accumulate leading to further cellular alterations (Chang and Monteiro 2015); by altering different steps of autophagy as previously described; by interacting with TDP-43 and its C-terminal fragments, triggering their aggregation and formation of cytoplasmatic inclusions (Cassel and Reitz 2013; Picher-Martel et al. 2015); and by promoting neuroinflammation through direct interaction with NF-κB, an inflammation transcriptional factor regulator, or through a TDP-43-dependent process (Swarup et al. 2011). Different pathological mechanisms associated with UBQLN2 mutations result in colocalization of UBQLN2 and TDP-43 cytoplasmic inclusions in the brain and the spinal cord of patients. Of interest, UBQLN2 colocalization with TDP-43 was also present in the spinal cord of sALS patients, underling a central role for UBQLN2 in ALS pathology (Fecto and Siddique 2011).
In vivo models were developed to better understand UBQLN2 pathology. The first model generated was a mouse strain harboring the P497H UBQLN2 substitution under the human UBQLN2 promoter which expressed low protein levels (Gorrie et al. 2014). Because of the limited mutant UBQLN2 expression, this model presented cognitive impairments and hippocampal inclusions but did not show MNs loss. Subsequently, two rat models harboring the same mutation but expressed at higher levels were developed. These models presented impaired autophagy and endocytosis associated with neuronal loss (Wu et al. 2015; Chen et al. 2018). Also, transgenic mice expressing the P520T substitution, which is highly aggressive in humans causing an early ALS/FTD onset (Deng et al. 2011), failed to present neurodegeneration, but only displayed alterations in neuronal proteostasis (Sharkey et al. 2020). Furthermore, mice and rat knock-out models showed late or no neurodegenerative phenotype. Together, these data suggest that UBQLN2 pathology is triggered by both GOF and LOF.
VAPB
VAMPs-associated protein B (VAPB) is a member of the evolutionarily conserved VAP protein family (Skehel et al. 1995) encoded by the gene VAPB (chromosome 20q13.32). VAPs localize on the membrane of the ER and of ER-Golgi intermediates, in close association with vesicle-associated membrane proteins (VAMPs). There, VAPs act as adaptors for the recruitment to the ER surface of cytosolic proteins mainly identified by the two phenylalanines in an acidic tract (FFAT) motif (Loewen et al. 2003). VAP proteins comprise an N-terminal major sperm protein (MSP) domain (Kaiser et al. 2005) involved in target proteins recruitment, a central coiled-coil domain, and a C-terminal hydrophobic sequence anchoring VAPs to the ER membrane. Among VAP interactors, many proteins allowing to put the ER in contact with other organelles can be found. The interaction sites that form through this process are known as membrane contact sites (MCS) and represent regions of communication between organelles achieved in the absence of membrane fusion (Prinz et al. 2020).
Based on these features, VAPB takes part in several cellular processes, among which autophagy can be found. For example, VAPB triggers mitophagy by interacting with the mitochondrial protein tyrosine phosphatase interacting protein 51 (PTPIP51), thus promoting autophagosome formation at the ER-mitochondria interface (Fig. 3) (Gomez-Suaga et al. 2017). Additionally, VABP regulates autophagy through its interaction with RAB3 GTPase-activating protein 1 (RAB3GAP1), which plays a role in autophagy initiation (Spang et al. 2014).
The autosomal dominant P56S substitution was the first and best characterized VAPB mutation ever associated to ALS (Nishimura et al. 2004; Funke et al. 2010; Di et al. 2016; Guber et al. 2018). Other VAPB mutations were thereafter associated to fALS (Chen et al. 2010; van Blitterswijk et al. 2012a; Sun et al. 2017). The observations made in cellular and animal models of P56S VAPB pathology suggest that both LOF and GOF mechanisms might be implicated in neurodegeneration. Nevertheless, data collected in ALS patient-derived cell models are in contrast with the GOF hypothesis. The P56 VAPB residue is evolutionarily conserved and lies in the MSP domain, in close proximity to the FFAT binding site (Nishimura et al. 2004). The P56S substitution almost entirely prevents VAPB binding to its FFAT-containing cytosolic partners; this contemporarily induces mutant VAPB aggregation both in cellular (Nishimura et al. 2004; Kanekura et al. 2006; Teuling et al. 2007; Suzuki et al. 2009) and in murine ALS models (Tudor et al. 2010; Qiu et al. 2013; Kuijpers et al. 2013a; Aliaga et al. 2013). Aggregating P56S VAPB also sequesters into its inclusions both wild-type VAPB (Kanekura et al. 2006; Teuling et al. 2007; Suzuki et al. 2009) and other proteins involved in membrane trafficking and MCS formation (De Vos et al. 2012; Kuijpers et al. 2013b; Hua et al. 2017). Moreover, increased SQSTM1/p62 and LC3 levels were detected in the spinal cord of P56S VAPB mice, possibly indicating alterations in the autophagic flux (Larroquette et al. 2015). This observation, coupled with the fact that P56S VAPB accumulation was shown to determine profound rearrangement of the affected ER sites in cells (Fasana et al. 2010; Papiani et al. 2012), might provide a link between disrupted VAPB function, ER stress, and autophagy impairments. Indeed, the reduction in ER-mitochondria MCS observed upon VAPB depletion resulted in an enhancement of the autophagic flux in HEK293 cells, probably due to impaired ER-to-mitochondria calcium transfer triggering mitophagy (Gomez-Suaga et al. 2017). At the same time, reduced VAPB protein levels and no signs of VAPB aggregation were detected in sALS spinal cord MNs (Teuling et al. 2007; Anagnostou et al. 2010) and in P56S VAPB fibroblasts, iPSCs, and iPSC-derived MNs (Mitne-Neto et al. 2011). Additionally, despite harboring MN inclusions, most P56S VAPB murine models (Tudor et al. 2010; Qiu et al. 2013; Kuijpers et al. 2013a) did not develop ALS-like motor symptoms, except for the ones overexpressing the highest levels of the transgenic protein (Aliaga et al. 2013). The same occurred for Drosophila models harboring the P56S substitution (Sanhueza et al. 2014; Moustaqim-Barrette et al. 2014). At the same time, though, VAPB-null mice develop mild motor dysfunction (Kabashi et al. 2013), suggesting that loss of VAPB function alone might not be sufficient to justify the onset of ALS symptoms in patients. Overall, the contrasting evidence coming from cellular and animal models indicates that further investigation is required to better elucidate the role of VAPB mutations in ALS pathogenesis.
VCP
Valosin-containing protein (VCP) is an ATPase associated with diverse cellular activities (AAA+) encoded by VCP gene (chromosome 9p13.3). VCP is ubiquitously expressed in tissues (Koller and Brownstein 1987). At cellular level, VCP localizes mainly found in the cytoplasm, while a smaller fraction binds to organelles or localizes in the nucleus (Acharya et al. 1995; Latterich et al. 1995; Madeo et al. 1998; Xu et al. 2011; Ramanathan and Ye 2012). VCP functions assembling in a homo-hexamer. Each monomer presents an N-terminal domain that interacts with adaptors and cofactors; two ATPase domains, D1 and D2, that function respectively in the formation of the homo-hexamer and in accomplishing VCP activity; and a C-terminal domain that cooperates with D2 activity and binds to a small subset of VCP partners (Huyton et al. 2003; Niwa et al. 2012; Chou et al. 2014).
VCP mechanism of action is to identify and segregate ubiquitinated proteins from different cellular compartment and to enhance their degradation through the UPS or the autophagic pathway. Thanks to the binding of various cofactors and adaptors, VCP has a key role in the maintenance of cellular homeostasis, regulating different cellular pathways as endoplasmic reticulum-associated degradation (Stein et al. 2014), organelle degradation (Tanaka et al. 2010; Papadopoulos et al. 2017), ribosome-associated degradation (Verma et al. 2013), regulation of autophagy (Hill et al. 2021), chaperone activity (Hirabayashi et al. 2001), chromatin-associated degradation (Meerang et al. 2011), NF-κB activation (Dai et al. 1998), and membrane fusion (Amenta et al. 1978). Most of VCP activity is aimed at regulating and preserving cellular proteostasis. To contribute to the maintenance of proteostasis, VCP is involved in the routing of misfolded proteins and aggregates to the UPS or autophagy for their disposal (Figs. 1 and 2).
VCP role in addressing substrates to autophagy is still not completely clear. However, it was established that VCP interacts with different partners involved in aggresome formation and promotes substrates routing to autophagosomes (Iwata et al. 2005; Boyault et al. 2006). Besides this, VCP regulates different steps of the autophagic pathway [as reviewed in (Ferrari et al. 2022)]. VCP participates in the activation of transcriptional regulators of autophagy, such as NF-κB and TFEB. In particular, VCP promotes the degradation of IκBα that releases NF-κB permitting its activation (Dai et al. 1998). On the other hand, VCP mechanism in the regulation of TFEB is unknown, but when lysosomal damage is induced, the silencing or inhibition of VCP activity stabilizes TFEB activation, suggesting that VCP modulates TFEB action (Arhzaouy et al. 2019). In addition, VCP is involved in autophagy initiation and maturation (Figs. 1 and 2). Recent data showed that VCP promotes the formation of the initiation complex ATG14-VPS34-VPS15-BECN1 through two distinct pathways (Hill et al. 2021). Different studies show that VCP mutation impacts on autophagosome maturation; indeed, VCP silencing or mutations are correlated with an increase in LC3-II and SQSTM1 protein levels and accumulation of autophagosomes with increased size (Ju et al. 2009; Tresse et al. 2010). Finally, VCP has a key role in regulating lysosome stability and disposal. Studies on muscle tubular lysosomes show that VCP, supported by its cofactor small VCP interacting protein (SVIP), preserves lysosome stability (Fig. 3). Indeed, the inhibition or downregulation of VCP causes lysosomal fragmentation (Johnson et al. 2015). VCP, together with its cofactors phospholipase A2 activating protein (PLAA), UBX domain-containing protein 6 (UBXD1), and ubiquitin thioesterase OTU1 (YOD1), is also essential for damaged lysosome degradation. Indeed, they promote the removal of ubiquitinated proteins exposed on damaged lysosome membrane, an essential step in lysosome disposal (Papadopoulos et al. 2017).
The essential involvement of VCP in proteostasis is also demonstrated by cellular alterations of VCP-patients affected tissue. VCP mutations have been mainly associated in 2004 to inclusion bodies myopathy Paget disease and frontotemporal dementia (IBMPFD), a multisystem proteinopathy, and in 2010 to fALS (Watts et al. 2004; Johnson et al. 2010). To date, more than 40 missense mutations, localized at the interface between N-terminal and D1 domains, were associated to IBMPFD, and almost 20 mutations, localized at N-terminal and D1 interface and in D2, were associated to ALS (Johnson et al. 2010; Mehta et al. 2013; Kenna et al. 2013). VCP-patients present different phenotypes, yet they display similar alterations at cellular level. In skeletal muscles of VCP-patients inclusions positive to ubiquitin and VCP, damaged lysosomes and rimmed vacuoles can be found (Watts et al. 2004; Kimonis and Watts 2005; Ritson et al. 2010). Similarly, in affected neurons cytoplasmatic inclusions positive to VCP and ubiquitin are also found (Kimonis and Watts 2005). Moreover, in both muscles and neurons, TDP-43 mislocalizes and forms inclusions (Ritson et al. 2010).
Animal models recapitulate most observations made on VCP mutations in vitro and in VCP-patients. VCP mouse models with VCP mutations or conditional knockout (Watts et al. 2004; Johnson et al. 2010) present lower survival with TDP-43 pathology in spinal cord and/or in skeletal muscle, muscle weakness, increase in fibers size that harbor ubiquitinated protein inclusions, and increased autophagy activity (Yin et al. 2012; Nalbandian et al. 2012). VCP mutants-induced neurodegeneration is also visible in Drosophila VCP models that overexpress mutations of TER94 (VCP ortholog in Drosophila), such as R152H, R188Q, and A229E (corresponding to human VCP mutations R155H, R191Q, and A232E). These models present disruption in nervous system and in muscle tissue that additionally shows specific lysosomal alterations (Chang et al. 2011; Johnson et al. 2015). Together, data obtained from animal models show an impairment in the autophagic pathway, reinforcing VCP essential role in this pathway.
Conclusions
Research over the past decades has highlighted the central role played by the autophagic pathway in ALS. Autophagy has a determinant role in the disposal of toxic proteins, including the aberrant aggregates formed by mutated proteins in ALS, and damaged organelles, and its failure in keeping under control protein misfolding upon saturating conditions is strictly associated to ALS progression. For this reason, the modulation of the autophagic pathway has been studied by different groups and has been outlined as a target to ameliorate ALS-related pathological conditions. Moreover, as this review summarizes, several ALS-associated mutations are found in genes implicated in all different steps of the autophagic pathway, from transport to protein degradation. Despite the efforts in studying ALS, to date no cure is available, and no therapies that drastically ameliorate life-span have been discovered yet. Thus, by analyzing the physiological role of ALS-related genes involved in autophagy and by evaluating the main experimental data linking mutations in these genes to ALS, it is clear that not all the implications of autophagy dysfunction in ALS have been completely unraveled yet. Indeed, in several cases, the molecular mechanisms explaining the pathogenicity of autophagy-related gene mutations found in ALS are still to be fully deciphered. In some cases, in vitro and cellular analyses might contrast with observations made in patients or in animal models, indicating that further studies are required to fully elucidate the connection between genes involved in the various steps of autophagy and ALS. In addition, this review highlights that alterations in autophagy are directly linked with ALS progression and suggests that a better functionality of autophagy could ameliorate the phenotype. Indeed, various studies on modulation of autophagy have been carried out and are ongoing with promising results. Compounds that positively modulate autophagy, such as trehalose, rapamycin, and colchicine, have been tested in animals or are already in clinical trial (Castillo et al. 2013; Mandrioli et al. 2018, 2019). These compounds stimulate autophagy through different mechanisms that have still to be completely clarified. Potentiation of the autophagic flux in ALS is surely positive in presence of a LOF of autophagy or in presence of impairment of organelles or accumulation of protein aggregates. Contrary, the implication of a potentiation of autophagy in presence of alterations derived by mutations of autophagic components has to be carefully analyzed. In this context, a better understanding of ALS-related genes function and contribution in each step of autophagy is fundamental to fully comprehend the pathological mechanisms and to display novel rescue strategies.
References
Abramzon YA, Fratta P, Traynor BJ, Chia R (2020) The overlapping genetics of amyotrophic lateral sclerosis and frontotemporal dementia. Front Neurosci 14:42. https://doi.org/10.3389/fnins.2020.00042
Acharya U, Jacobs R, Peters JM et al (1995) The formation of Golgi stacks from vesiculated Golgi membranes requires two distinct fusion events. Cell 82:895–904. https://doi.org/10.1016/0092-8674(95)90269-4
Aizawa H, Sekine Y, Takemura R et al (1992) Kinesin family in murine central nervous system. J Cell Biol 119:1287–1296. https://doi.org/10.1083/jcb.119.5.1287
Aliaga L, Lai C, Yu J et al (2013) Amyotrophic lateral sclerosis-related VAPB P56S mutation differentially affects the function and survival of corticospinal and spinal motor neurons. Hum Mol Genet 22:4293–4305. https://doi.org/10.1093/hmg/ddt279
Amenta JS, Hlivko TJ, McBee AG et al (1978) Specific inhibition by NH4CL of autophagy-associated proteloysis in cultured fibroblasts. Exp Cell Res 115:357–366. https://doi.org/10.1016/0014-4827(78)90289-6
Amick J, Roczniak-Ferguson A, Ferguson SM (2016) C9orf72 binds SMCR8, localizes to lysosomes, and regulates mTORC1 signaling. Mol Biol Cell 27:3040–3051. https://doi.org/10.1091/mbc.e16-01-0003
Amick J, Tharkeshwar AK, Talaia G, Ferguson SM (2020) PQLC2 recruits the C9orf72 complex to lysosomes in response to cationic amino acid starvation. J Cell Biol. https://doi.org/10.1083/jcb.201906076
Anagnostou G, Akbar MT, Paul P et al (2010) Vesicle associated membrane protein B (VAPB) is decreased in ALS spinal cord. Neurobiol Aging 31:969–985. https://doi.org/10.1016/j.neurobiolaging.2008.07.005
Ao X, Zou L, Wu Y (2014) Regulation of autophagy by the Rab GTPase network. Cell Death Differ 21:348–358. https://doi.org/10.1038/cdd.2013.187
Aoki Y, Manzano R, Lee Y et al (2017) C9orf72 and RAB7L1 regulate vesicle trafficking in amyotrophic lateral sclerosis and frontotemporal dementia. Brain 140:887–897. https://doi.org/10.1093/brain/awx024
Arai T, Nonaka T, Hasegawa M et al (2003) Neuronal and glial inclusions in frontotemporal dementia with or without motor neuron disease are immunopositive for p62. Neurosci Lett 342:41–44. https://doi.org/10.1016/s0304-3940(03)00216-7
Arhzaouy K, Papadopoulos C, Schulze N et al (2019) VCP maintains lysosomal homeostasis and TFEB activity in differentiated skeletal muscle. Autophagy 15:1082–1099. https://doi.org/10.1080/15548627.2019.1569933
Atanasio A, Decman V, White D et al (2016) C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production and glomerulonephropathy in mice. Sci Rep 6:23204. https://doi.org/10.1038/srep23204
Atkinson RAK, Fernandez-Martos CM, Atkin JD et al (2015) C9ORF72 expression and cellular localization over mouse development. Acta Neuropathol Commun 3:59. https://doi.org/10.1186/s40478-015-0238-7
Babu J, Lamar Seibenhener M, Peng J et al (2008) Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J Neurochem 106:107–120. https://doi.org/10.1111/j.1471-4159.2008.05340.x
Babu JR, Geetha T, Wooten MW (2005) Sequestosome 1/p62 shuttles polyubiquitinated tau for proteasomal degradation. J Neurochem 94:192–203. https://doi.org/10.1111/j.1471-4159.2005.03181.x
Barker H, v., Niblock M, Lee Y-B et al (2017) RNA misprocessing in C9orf72-linked neurodegeneration. Front Cell Neurosci. https://doi.org/10.3389/fncel.2017.00195
Baron DM, Fenton AR, Saez-Atienzar S et al (2022) ALS-associated KIF5A mutations abolish autoinhibition resulting in a toxic gain of function. Cell Rep 39:110598. https://doi.org/10.1016/j.celrep.2022.110598
Beeldman E, van der Kooi AJ, de Visser M et al (2015) A Dutch family with autosomal recessively inherited lower motor neuron predominant motor neuron disease due to optineurin mutations. Amyotroph Lateral Scler Frontotemporal Degener 16:410–411. https://doi.org/10.3109/21678421.2015.1066821
Bharadwaj R, Cunningham KM, Zhang K, Lloyd TE (2016) FIG4 regulates lysosome membrane homeostasis independent of phosphatase function. Hum Mol Genet 25:681–692. https://doi.org/10.1093/hmg/ddv505
Bingol B (2018) Autophagy and lysosomal pathways in nervous system disorders. Mol Cell Neurosci 91:167–208. https://doi.org/10.1016/j.mcn.2018.04.009
Bjørkøy G, Lamark T, Brech A et al (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 171:603–614. https://doi.org/10.1083/jcb.200507002
Boeynaems S, Bogaert E, Kovacs D et al (2017) Phase separation of C9orf72 dipeptide repeats perturbs stress granule dynamics. Mol Cell 65:1044-1055.e5. https://doi.org/10.1016/j.molcel.2017.02.013
Bonnard M, Mirtsos C, Suzuki S et al (2000) Deficiency of T2K leads to apoptotic liver degeneration and impaired NF-kappaB-dependent gene transcription. EMBO J 19:4976–4985. https://doi.org/10.1093/emboj/19.18.4976
Borghero G, Pugliatti M, Marrosu F et al (2016) TBK1 is associated with ALS and ALS-FTD in Sardinian patients. Neurobiol Aging 43:180.e1–5. https://doi.org/10.1016/j.neurobiolaging.2016.03.028
Botelho RJ, Efe JA, Teis D, Emr SD (2008) Assembly of a Fab1 phosphoinositide kinase signaling complex requires the Fig4 phosphoinositide phosphatase. Mol Biol Cell 19:4273–4286. https://doi.org/10.1091/mbc.e08-04-0405
Boyault C, Gilquin B, Zhang Y et al (2006) HDAC6-p97/VCP controlled polyubiquitin chain turnover. EMBO J 25:3357–3366. https://doi.org/10.1038/sj.emboj.7601210
Brady ST (1985) A novel brain ATPase with properties expected for the fast axonal transport motor. Nature 317:73–75. https://doi.org/10.1038/317073a0
Brenner D, Sieverding K, Bruno C et al (2019) Heterozygous Tbk1 loss has opposing effects in early and late stages of ALS in mice. J Exp Med 216:267–278. https://doi.org/10.1084/jem.20180729
Brenner D, Yilmaz R, Müller K et al (2018) Hot-spot KIF5A mutations cause familial ALS. Brain 141:688–697. https://doi.org/10.1093/brain/awx370
Brown J, Ashworth A, Gydesen S et al (1995) Familial non-specific dementia maps to chromosome 3. Hum Mol Genet 4:1625–1628. https://doi.org/10.1093/hmg/4.9.1625
Buchan JR, Kolaitis R-M, Taylor JP, Parker R (2013) Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 153:1461–1474. https://doi.org/10.1016/j.cell.2013.05.037
Cai H, Lin X, Xie C et al (2005) Loss of ALS2 function is insufficient to trigger motor neuron degeneration in knock-out mice but predisposes neurons to oxidative stress. J Neurosci 25:7567 LP – 7574. https://doi.org/10.1523/JNEUROSCI.1645-05.2005
Cai Q, Ganesan D (2022) Regulation of neuronal autophagy and the implications in neurodegenerative diseases. Neurobiol Dis 162:105582. https://doi.org/10.1016/j.nbd.2021.105582
Carra S, Seguin SJ, Landry J (2008) HspB8 and Bag3: a new chaperone complex targeting misfolded proteins to macroautophagy. Autophagy 4:237–239. https://doi.org/10.4161/auto.5407
Cassel JA, Reitz AB (2013) Ubiquilin-2 (UBQLN2) binds with high affinity to the C-terminal region of TDP-43 and modulates TDP-43 levels in H4 cells: characterization of inhibition by nucleic acids and 4-aminoquinolines. Biochim Biophys Acta 1834:964–971. https://doi.org/10.1016/j.bbapap.2013.03.020
Castillo K, Nassif M, Valenzuela V et al (2013) Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy 9:1308–1320. https://doi.org/10.4161/auto.25188
Chalasani MLS, Kumari A, Radha V, Swarup G (2014) E50K-OPTN-induced retinal cell death involves the Rab GTPase-activating protein, TBC1D17 mediated block in autophagy. PLoS ONE 9:e95758. https://doi.org/10.1371/journal.pone.0095758
Chandran J, Ding J, Cai H (2007) Alsin and the molecular pathways of amyotrophic lateral sclerosis. Mol Neurobiol 36:224–231. https://doi.org/10.1007/s12035-007-0034-x
Chang L, Monteiro MJ (2015) Defective proteasome delivery of polyubiquitinated proteins by ubiquilin-2 proteins containing ALS mutations. PLoS ONE 10:e0130162. https://doi.org/10.1371/journal.pone.0130162
Chang Y-C, Hung W-T, Chang Y-C et al (2011) Pathogenic VCP/TER94 alleles are dominant actives and contribute to neurodegeneration by altering cellular ATP level in a drosophila IBMPFD model. PLoS Genet 7:e1001288. https://doi.org/10.1371/journal.pgen.1001288
Chen H-J, Anagnostou G, Chai A et al (2010) Characterization of the properties of a novel mutation in VAPB in familial amyotrophic lateral sclerosis. J Biol Chem 285:40266–40281. https://doi.org/10.1074/jbc.M110.161398
Chen T, Huang B, Shi X et al (2018) Mutant UBQLN2P497H in motor neurons leads to ALS-like phenotypes and defective autophagy in rats. Acta Neuropathol Commun 6:122. https://doi.org/10.1186/s40478-018-0627-9
Chen Y, Zheng Z-Z, Chen X et al (2014) SQSTM1 mutations in Han Chinese populations with sporadic amyotrophic lateral sclerosis. Neurobiol Aging 35:726.e7–9. https://doi.org/10.1016/j.neurobiolaging.2013.09.008
Chen Z, Lin K, Macklis JD, Al-Chalabi A (2017) Proposed association between the hexanucleotide repeat of C9orf72 and opposability index of the thumb. Amyotroph Lateral Scler Frontotemporal Degener 18:175–181. https://doi.org/10.1080/21678421.2016.1257024
Chew J, Cook C, Gendron TF et al (2019) Aberrant deposition of stress granule-resident proteins linked to C9orf72-associated TDP-43 proteinopathy. Mol Neurodegener 14:9. https://doi.org/10.1186/s13024-019-0310-z
Chia R, Chiò A, Traynor BJ (2018) Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. The Lancet Neurology 17:94–102. https://doi.org/10.1016/S1474-4422(17)30401-5
Chitiprolu M, Jagow C, Tremblay V et al (2018) A complex of C9ORF72 and p62 uses arginine methylation to eliminate stress granules by autophagy. Nat Commun 9:2794. https://doi.org/10.1038/s41467-018-05273-7
Chou T-F, Bulfer SL, Weihl CC et al (2014) Specific inhibition of p97/VCP ATPase and kinetic analysis demonstrate interaction between D1 and D2 ATPase domains. J Mol Biol 426:2886–2899. https://doi.org/10.1016/j.jmb.2014.05.022
Chow CY, Landers JE, Bergren SK et al (2009) Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am J Hum Genet 84:85–88. https://doi.org/10.1016/j.ajhg.2008.12.010
Chow CY, Zhang Y, Dowling JJ et al (2007) Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature 448:68–72. https://doi.org/10.1038/nature05876
Cicardi M, Cristofani R, Rusmini P et al (2018) Tdp-25 routing to autophagy and proteasome ameliorates its aggregation in amyotrophic lateral sclerosis target cells. Sci Rep. https://doi.org/10.1038/s41598-018-29658-2
Cicchini M, Karantza V, Xia B (2015) Molecular pathways: autophagy in cancer—a matter of timing and context. Clin Cancer Res 21:498–504. https://doi.org/10.1158/1078-0432.CCR-13-2438
Cirulli ET, Lasseigne BN, Petrovski S et al (2015) Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347:1436–1441. https://doi.org/10.1126/science.aaa3650
Clark JA, Yeaman EJ, Blizzard CA et al (2016) A case for microtubule vulnerability in amyotrophic lateral sclerosis: altered dynamics during disease. Front Cell Neurosci 10:204. https://doi.org/10.3389/fncel.2016.00204
Conlon EG, Lu L, Sharma A et al (2016) The C9ORF72 GGGGCC expansion forms RNA G-quadruplex inclusions and sequesters hnRNP H to disrupt splicing in ALS brains. Elife. https://doi.org/10.7554/eLife.17820
Cook CN, Wu Y, Odeh HM et al (2020) C9orf72 poly(GR) aggregation induces TDP-43 proteinopathy. Science Translational Medicine. https://doi.org/10.1126/scitranslmed.abb3774
Cox LE, Ferraiuolo L, Goodall EF et al (2010) Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS). PLoS ONE 5:e9872. https://doi.org/10.1371/journal.pone.0009872
Crimella C, Baschirotto C, Arnoldi A et al (2012) Mutations in the motor and stalk domains of KIF5A in spastic paraplegia type 10 and in axonal Charcot-Marie-Tooth type 2. Clin Genet 82:157–164. https://doi.org/10.1111/j.1399-0004.2011.01717.x
Cristofani R, Crippa V, Cicardi ME et al (2020) A crucial role for the protein quality control system in motor neuron diseases. Frontiers in Aging Neuroscience. https://doi.org/10.3389/fnagi.2020.00191
Cuervo AM, Wong E (2014) Chaperone-mediated autophagy: roles in disease and aging. Cell Res 24:92–104. https://doi.org/10.1038/cr.2013.153
Culver-Hanlon TL, Lex SA, Stephens AD et al (2006) A microtubule-binding domain in dynactin increases dynein processivity by skating along microtubules. Nat Cell Biol 8:264–270. https://doi.org/10.1038/ncb1370
Dai RM, Chen E, Longo DL et al (1998) Involvement of valosin-containing protein, an ATPase co-purified with IκBalpha and 26 S proteasome, in ubiquitin-proteasome-mediated degradation of IkBalpha. J Biol Chem 273:3562–3573. https://doi.org/10.1074/jbc.273.6.3562
Daroszewska A, van ’t Hof RJ, Rojas JA, et al (2011) A point mutation in the ubiquitin-associated domain of SQSMT1 is sufficient to cause a Paget’s disease-like disorder in mice. Hum Mol Genet 20:2734–2744. https://doi.org/10.1093/hmg/ddr172
de Majo M, Topp SD, Smith BN et al (2018) ALS-associated missense and nonsense TBK1 mutations can both cause loss of kinase function. Neurobiol Aging 71:266.e1-266.e10. https://doi.org/10.1016/j.neurobiolaging.2018.06.015
De Marco N, Buono M, Troise F, Diez-Roux G (2006) Optineurin increases cell survival and translocates to the nucleus in a Rab8-dependent manner upon an apoptotic stimulus. J Biol Chem 281:16147–16156. https://doi.org/10.1074/jbc.M601467200
De Vos KJ, Mórotz GM, Stoica R et al (2012) VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum Mol Genet 21:1299–1311. https://doi.org/10.1093/hmg/ddr559
DeJesus-Hernandez M, Mackenzie IR, Boeve BF et al (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72:245–256. https://doi.org/10.1016/j.neuron.2011.09.011
Del Bo R, Tiloca C, Pensato V et al (2011) Novel optineurin mutations in patients with familial and sporadic amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 82:1239–1243. https://doi.org/10.1136/jnnp.2011.242313
Deng H-X, Chen W, Hong S-T et al (2011) Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 477:211–215. https://doi.org/10.1038/nature10353
Deshimaru M, Kinoshita-Kawada M, Kubota K et al (2021) DCTN1 binds to TDP-43 and regulates TDP-43 aggregation. Int J Mol Sci. https://doi.org/10.3390/ijms22083985
Devon RS, Orban PC, Gerrow K, et al (2006) Als2-deficient mice exhibit disturbances in endosome trafficking associated with motor behavioral abnormalities. Proc Natl Acad Sci USA 103:9595 LP – 9600. https://doi.org/10.1073/pnas.0510197103
Di L, Chen H, Da Y et al (2016) Atypical familial amyotrophic lateral sclerosis with initial symptoms of pain or tremor in a Chinese family harboring VAPB-P56S mutation. J Neurol 263:263–268. https://doi.org/10.1007/s00415-015-7965-3
Di Paolo G, De Camilli P (2006) Phosphoinositides in cell regulation and membrane dynamics. Nature 443:651–657. https://doi.org/10.1038/nature05185
Dixit R, Levy JR, Tokito M et al (2008) Regulation of dynactin through the differential expression of p150Glued isoforms. J Biol Chem 283:33611–33619. https://doi.org/10.1074/jbc.M804840200
Dobrowolny G, Aucello M, Molinaro M, Musarò A (2008) Local expression of mIgf-1 modulates ubiquitin, caspase and CDK5 expression in skeletal muscle of an ALS mouse model. Neurol Res 30:131–136. https://doi.org/10.1179/174313208X281235
Dols-Icardo O, Iborra O, Valdivia J et al (2016) Assessing the role of TUBA4A gene in frontotemporal degeneration. Neurobiol Aging 38:215.e13-215.e14. https://doi.org/10.1016/j.neurobiolaging.2015.10.030
Duan W, Guo M, Yi L, et al (2019) Deletion of Tbk1 disrupts autophagy and reproduces behavioral and locomotor symptoms of FTD-ALS in mice. Aging 11:2457–2476. https://doi.org/10.18632/aging.101936
Esanov R, Cabrera GT, Andrade NS et al (2017) A C9ORF72 BAC mouse model recapitulates key epigenetic perturbations of ALS/FTD. Mol Neurodegener 12:46. https://doi.org/10.1186/s13024-017-0185-9
Farg MA, Sundaramoorthy V, Sultana JM et al (2014) C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet 23:3579–3595. https://doi.org/10.1093/hmg/ddu068
Fasana E, Fossati M, Ruggiano A et al (2010) A VAPB mutant linked to amyotrophic lateral sclerosis generates a novel form of organized smooth endoplasmic reticulum. FASEB J 24:1419–1430. https://doi.org/10.1096/fj.09-147850
Fecto F, Siddique T (2011) Making connections: pathology and genetics link amyotrophic lateral sclerosis with frontotemporal lobe dementia. J Mol Neurosci 45:663–675. https://doi.org/10.1007/s12031-011-9637-9
Ferguson CJ, Lenk GM, Jones JM et al (2012) Neuronal expression of Fig4 is both necessary and sufficient to prevent spongiform neurodegeneration. Hum Mol Genet 21:3525–3534. https://doi.org/10.1093/hmg/dds179
Ferguson CJ, Lenk GM, Meisler MH (2009) Defective autophagy in neurons and astrocytes from mice deficient in PI(3,5)P2. Hum Mol Genet 18:4868–4878. https://doi.org/10.1093/hmg/ddp460
Ferrari V, Cristofani R, Tedesco B et al (2022) Valosin containing protein (VCP): a multistep regulator of autophagy. Int J Mole Sci 23
Fichera M, lo Giudice M, Falco M et al (2004) Evidence of kinesin heavy chain (KIF5A) involvement in pure hereditary spastic paraplegia. Neurology 63:1108–1110. https://doi.org/10.1212/01.wnl.0000138731.60693.d2
Filimonenko M, Stuffers S, Raiborg C et al (2007) Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J Cell Biol 179:485–500. https://doi.org/10.1083/jcb.200702115
Freibaum BD, Taylor JP (2017) The role of dipeptide repeats in C9ORF72-related ALS-FTD. Front Mol Neurosci. https://doi.org/10.3389/fnmol.2017.00035
Freischmidt A, Wieland T, Richter B et al (2015) Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci 18:631–636. https://doi.org/10.1038/nn.4000
Funke AD, Esser M, Krüttgen A et al (2010) The p. P56S mutation in the VAPB gene is not due to a single founder: the first European case. Clin Genet 77:302–303
Gal J, Ström A-L, Kwinter DM et al (2009) Sequestosome 1/p62 links familial ALS mutant SOD1 to LC3 via an ubiquitin-independent mechanism. J Neurochem 111:1062–1073. https://doi.org/10.1111/j.1471-4159.2009.06388.x
Geetha T, Seibenhener ML, Chen L et al (2008) p62 serves as a shuttling factor for TrkA interaction with the proteasome. Biochem Biophys Res Commun 374:33–37. https://doi.org/10.1016/j.bbrc.2008.06.082
Gendron TF, Bieniek KF, Zhang Y-J et al (2013) Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol 126:829–844. https://doi.org/10.1007/s00401-013-1192-8
Gerbino V, Kaunga E, Ye J et al (2020) The loss of TBK1 kinase activity in motor neurons or in all cell types differentially impacts ALS disease progression in SOD1 mice. Neuron 106:789-805.e5. https://doi.org/10.1016/j.neuron.2020.03.005
Ghazi-Noori S, Froud KE, Mizielinska S et al (2012) Progressive neuronal inclusion formation and axonal degeneration in CHMP2B mutant transgenic mice. Brain 135:819–832. https://doi.org/10.1093/brain/aws006
Gijselinck I, van Mossevelde S, van der Zee J et al (2016) The C9orf72 repeat size correlates with onset age of disease, DNA methylation and transcriptional downregulation of the promoter. Mol Psychiatry 21:1112–1124. https://doi.org/10.1038/mp.2015.159
Gijselinck I, Van Mossevelde S, van der Zee J et al (2015) Loss of TBK1 is a frequent cause of frontotemporal dementia in a Belgian cohort. Neurology 85:2116–2125. https://doi.org/10.1212/WNL.0000000000002220
Gleason CE, Ordureau A, Gourlay R et al (2011) Polyubiquitin binding to optineurin is required for optimal activation of TANK-binding kinase 1 and production of interferon β. J Biol Chem 286:35663–35674. https://doi.org/10.1074/jbc.M111.267567
Gomez-Suaga P, Paillusson S, Stoica R et al (2017) The ER-mitochondria tethering complex VAPB-PTPIP51 regulates autophagy. Curr Biol 27:371–385. https://doi.org/10.1016/j.cub.2016.12.038
Goode A, Butler K, Long J et al (2016) Defective recognition of LC3B by mutant SQSTM1/p62 implicates impairment of autophagy as a pathogenic mechanism in ALS-FTLD. Autophagy 12:1094–1104. https://doi.org/10.1080/15548627.2016.1170257
Gorrie GH, Fecto F, Radzicki D et al (2014) Dendritic spinopathy in transgenic mice expressing ALS/dementia-linked mutant UBQLN2. Proc Natl Acad Sci 111:14524–14529. https://doi.org/10.1073/pnas.1405741111
Gotkine M, de Majo M, Wong CH et al (2021) A recessive S174X mutation in Optineurin causes amyotrophic lateral sclerosis through a loss of function via allele-specific nonsense-mediated decay. Neurobiol Aging 106:351.e1-351.e6. https://doi.org/10.1016/j.neurobiolaging.2021.05.009
Guber RD, Schindler AB, Budron MS et al (2018) Nucleocytoplasmic transport defect in a North American patient with ALS8. Ann Clin Transl Neurol 5:369–375. https://doi.org/10.1002/acn3.515
Gydesen S, Brown JM, Brun A et al (2002) Chromosome 3 linked frontotemporal dementia (FTD-3). Neurology 59:1585–1594. https://doi.org/10.1212/01.wnl.0000034763.54161.1f
Hadano S, Benn SC, Kakuta S et al (2006) Mice deficient in the Rab5 guanine nucleotide exchange factor ALS2/alsin exhibit age-dependent neurological deficits and altered endosome trafficking. Hum Mol Genet 15:233–250. https://doi.org/10.1093/hmg/ddi440
Hadano S, Hand CK, Osuga H et al (2001) A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat Genet 29:166–173. https://doi.org/10.1038/ng1001-166
Hadano S, Mitsui S, Pan L et al (2016) Functional links between SQSTM1 and ALS2 in the pathogenesis of ALS: cumulative impact on the protection against mutant SOD1-mediated motor dysfunction in mice. Hum Mol Genet 25:3321–3340. https://doi.org/10.1093/hmg/ddw180
Hadano S, Otomo A, Kunita R et al (2010) Loss of ALS2/Alsin exacerbates motor dysfunction in a SOD1-expressing mouse ALS model by disturbing endolysosomal trafficking. PLoS ONE 5:e9805. https://doi.org/10.1371/journal.pone.0009805
Haeusler AR, Donnelly CJ, Periz G et al (2014) C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 507:195–200. https://doi.org/10.1038/nature13124
Han J-H, Ryu H-H, Jun M-H et al (2012) The functional analysis of the CHMP2B missense mutation associated with neurodegenerative diseases in the endo-lysosomal pathway. Biochem Biophys Res Commun 421:544–549. https://doi.org/10.1016/j.bbrc.2012.04.041
Hayes LR, Duan L, Bowen K et al (2020) C9orf72 arginine-rich dipeptide repeat proteins disrupt karyopherin-mediated nuclear import. Elife. https://doi.org/10.7554/eLife.51685
Henne WM, Stenmark H, Emr SD (2013) Molecular mechanisms of the membrane sculpting ESCRT pathway. Cold Spring Harb Perspect Biol. https://doi.org/10.1101/cshperspect.a016766
Heo J-M, Ordureau A, Paulo JA et al (2015) The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 Activation to Promote Mitophagy. Mol Cell 60:7–20. https://doi.org/10.1016/j.molcel.2015.08.016
Hersheson J, Mencacci NE, Davis M et al (2013) Mutations in the autoregulatory domain of β-tubulin 4a cause hereditary dystonia. Ann Neurol 73:546–553. https://doi.org/10.1002/ana.23832
Hill SM, Wrobel L, Ashkenazi A et al (2021) VCP/p97 regulates beclin-1-dependent autophagy initiation. Nat Chem Biol 17:448–455. https://doi.org/10.1038/s41589-020-00726-x
Hirabayashi M, Inoue K, Tanaka K et al (2001) VCP/p97 in abnormal protein aggregates, cytoplasmic vacuoles, and cell death, phenotypes relevant to neurodegeneration. Cell Death Differ 8:977–984. https://doi.org/10.1038/sj.cdd.4400907
Hirano M, Nakamura Y, Saigoh K et al (2013) Mutations in the gene encoding p62 in Japanese patients with amyotrophic lateral sclerosis. Neurology 80:458–463. https://doi.org/10.1212/WNL.0b013e31827f0fe5
Hirokawa N, Noda Y (2008) Intracellular transport and kinesin superfamily proteins, KIFs: structure, function, and dynamics. Physiol Rev 88:1089–1118. https://doi.org/10.1152/physrev.00023.2007
Hollenbeck PJ (2014) Directing traffic and autophagy in axonal transport. Dev Cell 29:505–506. https://doi.org/10.1016/j.devcel.2014.05.016
Hortobágyi T, Troakes C, Nishimura AL et al (2011) Optineurin inclusions occur in a minority of TDP-43 positive ALS and FTLD-TDP cases and are rarely observed in other neurodegenerative disorders. Acta Neuropathol 121:519–527. https://doi.org/10.1007/s00401-011-0813-3
Howes SC, Alushin GM, Shida T et al (2014) Effects of tubulin acetylation and tubulin acetyltransferase binding on microtubule structure. Mol Biol Cell 25:257–266. https://doi.org/10.1091/mbc.E13-07-0387
Hua R, Cheng D, Coyaud É et al (2017) VAPs and ACBD5 tether peroxisomes to the ER for peroxisome maintenance and lipid homeostasis. J Cell Biol 216:367–377. https://doi.org/10.1083/jcb.201608128
Hurley JH, Hanson PI (2010) Membrane budding and scission by the ESCRT machinery: it’s all in the neck. Nat Rev Mol Cell Biol 11:556–566. https://doi.org/10.1038/nrm2937
Huyton T, Pye VE, Briggs LC et al (2003) The crystal structure of murine p97/VCP at 3.6A. J Struct Biol 144:337–348. https://doi.org/10.1016/j.jsb.2003.10.007
Hyttinen JMT, Niittykoski M, Salminen A, Kaarniranta K (2013) Maturation of autophagosomes and endosomes: a key role for Rab7. Biochimica et Biophysica Acta (BBA) - Mole Cell Res 1833:503–510. https://doi.org/10.1016/j.bbamcr.2012.11.018
Ichimura Y, Kumanomidou T, Sou Y et al (2008) Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem 283:22847–22857. https://doi.org/10.1074/jbc.M802182200
Iguchi Y, Eid L, Parent M et al (2016) Exosome secretion is a key pathway for clearance of pathological TDP-43. Brain 139:3187–3201. https://doi.org/10.1093/brain/aww237
Iida A, Hosono N, Sano M et al (2012) Novel deletion mutations of OPTN in amyotrophic lateral sclerosis in Japanese. Neurobiol Aging 33:1843.e19–24. https://doi.org/10.1016/j.neurobiolaging.2011.12.037
Ikenaka K, Kawai K, Katsuno M et al (2013) dnc-1/dynactin 1 knockdown disrupts transport of autophagosomes and induces motor neuron degeneration. PLoS ONE 8:e54511. https://doi.org/10.1371/journal.pone.0054511
Itakura E, Mizushima N (2010) Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 6:764–776. https://doi.org/10.4161/auto.6.6.12709
Iwata A, Riley BE, Johnston JA, Kopito RR (2005) HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem 280:40282–40292. https://doi.org/10.1074/jbc.M508786200
Iyer S, Subramanian V, Acharya KR (2018) C9orf72, a protein associated with amyotrophic lateral sclerosis (ALS) is a guanine nucleotide exchange factor. PeerJ 6:e5815. https://doi.org/10.7717/peerj.5815
Ji YJ, Ugolino J, Brady NR et al (2017) Systemic deregulation of autophagy upon loss of ALS- and FTD-linked C9orf72. Autophagy 13:1254–1255. https://doi.org/10.1080/15548627.2017.1299312
Jiang J, Zhu Q, Gendron TF et al (2016) Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron 90:535–550. https://doi.org/10.1016/j.neuron.2016.04.006
Jiang Y-M, Yamamoto M, Kobayashi Y et al (2005) Gene expression profile of spinal motor neurons in sporadic amyotrophic lateral sclerosis. Ann Neurol 57:236–251. https://doi.org/10.1002/ana.20379
Jiang Y-M, Yamamoto M, Tanaka F et al (2007) Gene expressions specifically detected in motor neurons (dynactin 1, early growth response 3, acetyl-CoA transporter, death receptor 5, and cyclin C) differentially correlate to pathologic markers in sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 66:617–627. https://doi.org/10.1097/nen.0b013e318093ece3
Jin N, Chow CY, Liu L et al (2008) VAC14 nucleates a protein complex essential for the acute interconversion of PI3P and PI(3,5)P(2) in yeast and mouse. EMBO J 27:3221–3234. https://doi.org/10.1038/emboj.2008.248
Johansen T, Lamark T (2011) Selective autophagy mediated by autophagic adapter proteins. Autophagy 7:279–296. https://doi.org/10.4161/auto.7.3.14487
Johnson AE, Shu H, Hauswirth AG et al (2015) VCP-dependent muscle degeneration is linked to defects in a dynamic tubular lysosomal network in vivo. Elife. https://doi.org/10.7554/eLife.07366
Johnson JO, Mandrioli J, Benatar M et al (2010) Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68:857–864. https://doi.org/10.1016/j.neuron.2010.11.036
Ju J-S, Fuentealba RA, Miller SE et al (2009) Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J Cell Biol 187:875–888. https://doi.org/10.1083/jcb.200908115
Kabashi E, El Oussini H, Bercier V et al (2013) Investigating the contribution of VAPB/ALS8 loss of function in amyotrophic lateral sclerosis. Hum Mol Genet 22:2350–2360. https://doi.org/10.1093/hmg/ddt080
Kaiser SE, Brickner JH, Reilein AR et al (2005) Structural basis of FFAT motif-mediated ER targeting. Structure 13:1035–1045. https://doi.org/10.1016/j.str.2005.04.010
Kanekura K, Hashimoto Y, Kita Y et al (2005) A Rac1/phosphatidylinositol 3-kinase/Akt3 anti-apoptotic pathway, triggered by AlsinLF, the product of the ALS2 gene, antagonizes Cu/Zn-superoxide dismutase (SOD1) mutant-induced motoneuronal cell death. J Biol Chem 280:4532–4543. https://doi.org/10.1074/jbc.M410508200
Kanekura K, Hashimoto Y, Niikura T et al (2004) Alsin, the product of ALS2 gene, suppresses SOD1 mutant neurotoxicity through RhoGEF domain by interacting with SOD1 mutants. J Biol Chem 279:19247–19256. https://doi.org/10.1074/jbc.M313236200
Kanekura K, Nishimoto I, Aiso S, Matsuoka M (2006) Characterization of amyotrophic lateral sclerosis-linked P56S mutation of vesicle-associated membrane protein-associated protein B (VAPB/ALS8). J Biol Chem 281:30223–30233. https://doi.org/10.1074/jbc.M605049200
Karki S, Holzbaur ELF (1999) Cytoplasmic dynein and dynactin in cell division and intracellular transport. Curr Opin Cell Biol 11:45–53. https://doi.org/10.1016/S0955-0674(99)80006-4
Karki S, Holzbaur ELF (1995) Affinity chromatography demonstrates a direct binding between cytoplasmic dynein and the dynactin complex. J Biol Chem 270:28806–28811. https://doi.org/10.1074/jbc.270.48.28806
Kaushik S, Cuervo AM (2018) The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol 19:365–381. https://doi.org/10.1038/s41580-018-0001-6
Kaye FJ, Modi S, Ivanovska I et al (2000) A family of ubiquitin-like proteins binds the ATPase domain of Hsp70-like Stch. FEBS Lett 467:348–355. https://doi.org/10.1016/s0014-5793(00)01135-2
Kaye FJ, Shows TB (2000) Assignment of ubiquilin2 (UBQLN2) to human chromosome xp11. 23–>p11.1 by GeneBridge radiation hybrids. Cytogenet Cell Genet 89:116–117. https://doi.org/10.1159/000015588
Keller BA, Volkening K, Droppelmann CA et al (2012) Co-aggregation of RNA binding proteins in ALS spinal motor neurons: evidence of a common pathogenic mechanism. Acta Neuropathol 124:733–747. https://doi.org/10.1007/s00401-012-1035-z
Kenna KP, McLaughlin RL, Byrne S et al (2013) Delineating the genetic heterogeneity of ALS using targeted high-throughput sequencing. J Med Genet 50:776–783. https://doi.org/10.1136/jmedgenet-2013-101795
Kiernan MC, Vucic S, Cheah BC et al (2011) Amyotrophic lateral sclerosis. The Lancet 377:942–955. https://doi.org/10.1016/S0140-6736(10)61156-7
Kimonis VE, Watts GDJ (2005) Autosomal dominant inclusion body myopathy, Paget disease of bone, and frontotemporal dementia. Alzheimer Dis Assoc Disord 19(Suppl 1):S44–S47. https://doi.org/10.1097/01.wad.0000183081.76820.5a
King A, Al-Sarraj S, Troakes C et al (2013) Mixed tau, TDP-43 and p62 pathology in FTLD associated with a C9ORF72 repeat expansion and p.Ala239Thr MAPT (tau) variant. Acta Neuropathol 125:303–310. https://doi.org/10.1007/s00401-012-1050-0
Klionsky DJ, Abdel-Aziz AK, Abdelfatah S et al (2021a) Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1). Autophagy 17:1–382. https://doi.org/10.1080/15548627.2020.1797280
Klionsky DJ, Petroni G, Amaravadi RK et al (2021b) Autophagy in major human diseases. EMBO J 40. https://doi.org/10.15252/embj.2021b108863
Ko HS, Uehara T, Tsuruma K, Nomura Y (2004) Ubiquilin interacts with ubiquitylated proteins and proteasome through its ubiquitin-associated and ubiquitin-like domains. FEBS Lett 566:110–114. https://doi.org/10.1016/j.febslet.2004.04.031
Koller KJ, Brownstein MJ (1987) Use of a cDNA clone to identify a supposed precursor protein containing valosin. Nature 325:542–545. https://doi.org/10.1038/325542a0
Komatsu M, Waguri S, Chiba T et al (2006) Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441:880–884. https://doi.org/10.1038/nature04723
Koppers M, Blokhuis AM, Westeneng H et al (2015) C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann Neurol 78:426–438. https://doi.org/10.1002/ana.24453
Korac J, Schaeffer V, Kovacevic I et al (2013) Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J Cell Sci 126:580–592. https://doi.org/10.1242/jcs.114926
Kraft C, Peter M, Hofmann K (2010) Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol 12:836–841. https://doi.org/10.1038/ncb0910-836
Krasniak CS, Ahmad ST (2016) The role of CHMP2B(Intron5) in autophagy and frontotemporal dementia. Brain Res 1649:151–157. https://doi.org/10.1016/j.brainres.2016.02.051
Kress JA, Kühnlein P, Winter P et al (2005) Novel mutation in the ALS2 gene in juvenile amyotrophic lateral sclerosis. Annals Neurol 58:800–803. https://doi.org/10.1002/ana.20665
Kuijpers M, van Dis V, Haasdijk ED et al (2013a) Amyotrophic lateral sclerosis (ALS)-associated VAPB-P56S inclusions represent an ER quality control compartment. Acta Neuropathol Commun 1:24. https://doi.org/10.1186/2051-5960-1-24
Kuijpers M, Lou YuK, Teuling E et al (2013b) The ALS8 protein VAPB interacts with the ER-Golgi recycling protein YIF1A and regulates membrane delivery into dendrites. EMBO J 32:2056–2072. https://doi.org/10.1038/emboj.2013.131
Kunita R, Otomo A, Mizumura H et al (2004) Homo-oligomerization of ALS2 through its unique carboxyl-terminal regions is essential for the ALS2-associated Rab5 guanine nucleotide exchange activity and its regulatory function on endosome trafficking. J Biol Chem 279:38626–38635. https://doi.org/10.1074/jbc.M406120200
Kuusisto E, Suuronen T, Salminen A (2001) Ubiquitin-binding protein p62 expression is induced during apoptosis and proteasomal inhibition in neuronal cells. Biochem Biophys Res Commun 280:223–228. https://doi.org/10.1006/bbrc.2000.4107
Kuźma-Kozakiewicz M, Chudy A, Kaźmierczak B et al (2013) Dynactin deficiency in the CNS of humans with sporadic ALS and mice with genetically determined motor neuron degeneration. Neurochem Res 38:2463–2473. https://doi.org/10.1007/s11064-013-1160-7
Kwok CT, Morris A, de Belleroche JS (2014) Sequestosome-1 (SQSTM1) sequence variants in ALS cases in the UK: prevalence and coexistence of SQSTM1 mutations in ALS kindred with PDB. Eur J Hum Genet 22:492–496. https://doi.org/10.1038/ejhg.2013.184
Kyotani A, Azuma Y, Yamamoto I et al (2016) Knockdown of the Drosophila FIG4 induces deficient locomotive behavior, shortening of motor neuron, axonal targeting aberration, reduction of life span and defects in eye development. Exp Neurol 277:86–95. https://doi.org/10.1016/j.expneurol.2015.12.011
Lai C, Lin X, Chandran J et al (2007) The G59S mutation in p150(glued) causes dysfunction of dynactin in mice. J Neurosci 27:13982–13990. https://doi.org/10.1523/JNEUROSCI.4226-07.2007
Lai C, Xie C, Shim H et al (2009) Regulation of endosomal motility and degradation by amyotrophic lateral sclerosis 2/alsin. Mol Brain 2:23. https://doi.org/10.1186/1756-6606-2-23
Laird FM, Farah MH, Ackerley S et al (2008) Motor neuron disease occurring in a mutant dynactin mouse model is characterized by defects in vesicular trafficking. J Neurosci 28:1997–2005. https://doi.org/10.1523/JNEUROSCI.4231-07.2008
Larroquette F, Seto L, Gaub PL et al (2015) Vapb/Amyotrophic lateral sclerosis 8 knock-in mice display slowly progressive motor behavior defects accompanying ER stress and autophagic response. Hum Mol Genet 24:6515–6529. https://doi.org/10.1093/hmg/ddv360
Lattante S, de Calbiac H, Le Ber I et al (2015) Sqstm1 knock-down causes a locomotor phenotype ameliorated by rapamycin in a zebrafish model of ALS/FTLD. Hum Mol Genet 24:1682–1690. https://doi.org/10.1093/hmg/ddu580
Latterich M, Fröhlich K-U, Schekman R (1995) Membrane fusion and the cell cycle: Cdc48p participates in the fusion of ER membranes. Cell 82:885–893. https://doi.org/10.1016/0092-8674(95)90268-6
Laurin N, Brown JP, Morissette J, Raymond V (2002) Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am J Hum Genet 70:1582–1588. https://doi.org/10.1086/340731
Lazarou M, Sliter DA, Kane LA et al (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524:309–314. https://doi.org/10.1038/nature14893
Le Ber I, Camuzat A, Guerreiro R et al (2013) SQSTM1 mutations in French patients with frontotemporal dementia or frontotemporal dementia with amyotrophic lateral sclerosis. JAMA Neurol 70:1403–1410. https://doi.org/10.1001/jamaneurol.2013.3849
Le Ber I, De Septenville A, Millecamps S et al (2015) TBK1 mutation frequencies in French frontotemporal dementia and amyotrophic lateral sclerosis cohorts. Neurobiol Aging 36:3116.e5–3116.e8. https://doi.org/10.1016/j.neurobiolaging.2015.08.009
Lee J-A, Beigneux A, Ahmad ST et al (2007) ESCRT-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr Biol 17:1561–1567. https://doi.org/10.1016/j.cub.2007.07.029
Lenk GM, Ferguson CJ, Chow CY et al (2011) Pathogenic mechanism of the FIG4 mutation responsible for Charcot-Marie-Tooth disease CMT4J. PLoS Genet 7:e1002104. https://doi.org/10.1371/journal.pgen.1002104
Levine TP, Daniels RD, Gatta AT et al (2013) The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics 29:499–503. https://doi.org/10.1093/bioinformatics/bts725
Levy JR, Sumner CJ, Caviston JP et al (2006) A motor neuron disease–associated mutation in p150Glued perturbs dynactin function and induces protein aggregation. J Cell Biol 172:733–745. https://doi.org/10.1083/jcb.200511068
Li F, Xu D, Wang Y et al (2018a) Structural insights into the ubiquitin recognition by OPTN (optineurin) and its regulation by TBK1-mediated phosphorylation. Autophagy 14:66–79. https://doi.org/10.1080/15548627.2017.1391970
Li J, He J, Tang L et al (2018b) Screening for TUBA4A mutations in a large Chinese cohort of patients with ALS: re-evaluating the pathogenesis of TUBA4A in ALS. J Neurol Neurosurg Psychiatry 89:1350–1352
Li K, Yang L, Zhang C et al (2014) HPS6 interacts with dynactin p150Glued to mediate retrograde trafficking and maturation of lysosomes. J Cell Sci 127:4574–4588. https://doi.org/10.1242/jcs.141978
Li W, Li J, Bao J (2012) Microautophagy: lesser-known self-eating. Cell Mol Life Sci 69:1125–1136. https://doi.org/10.1007/s00018-011-0865-5
Lin BC, Higgins NR, Phung TH, Monteiro MJ (2021) UBQLN proteins in health and disease with a focus on UBQLN2 in ALS/FTD. FEBS J. https://doi.org/10.1111/febs.16129
Liu M, Pi H, Xi Y et al (2021) KIF5A-dependent axonal transport deficiency disrupts autophagic flux in trimethyltin chloride-induced neurotoxicity. Autophagy 17:903–924. https://doi.org/10.1080/15548627.2020.1739444
Liu X, Yang L, Tang L et al (2017) DCTN1 gene analysis in Chinese patients with sporadic amyotrophic lateral sclerosis. PLoS ONE 12:e0182572. https://doi.org/10.1371/journal.pone.0182572
Liu Z-J, Li H-F, Tan G-H et al (2014) Identify mutation in amyotrophic lateral sclerosis cases using HaloPlex target enrichment system. Neurobiol Aging 35:2881.e11-2881.e15. https://doi.org/10.1016/j.neurobiolaging.2014.07.003
Lobsiger CS, Boillee S, McAlonis-Downes M et al (2009) Schwann cells expressing dismutase active mutant SOD1 unexpectedly slow disease progression in ALS mice. Proc Natl Acad Sci 106:4465–4470. https://doi.org/10.1073/pnas.0813339106
Loewen CJR, Roy A, Levine TP (2003) A conserved ER targeting motif in three families of lipid binding proteins and in Opi1p binds VAP. EMBO J 22:2025–2035. https://doi.org/10.1093/emboj/cdg201
Madeo F, Schlauer J, Zischka H et al (1998) Tyrosine phosphorylation regulates cell cycle-dependent nuclear localization of Cdc48p. Mol Biol Cell 9:131–141. https://doi.org/10.1091/mbc.9.1.131
Maharjan N, Künzli C, Buthey K, Saxena S (2017) C9ORF72 regulates stress granule formation and its deficiency impairs stress granule assembly, hypersensitizing cells to stress. Mol Neurobiol 54:3062–3077. https://doi.org/10.1007/s12035-016-9850-1
Mandrioli J, Crippa V, Cereda C et al (2019) Proteostasis and ALS: protocol for a phase II, randomised, double-blind, placebo-controlled, multicentre clinical trial for colchicine in ALS (Co-ALS). BMJ Open 9:e028486. https://doi.org/10.1136/bmjopen-2018-028486
Mandrioli J, D’Amico R, Zucchi E et al (2018) Rapamycin treatment for amyotrophic lateral sclerosis. Medicine 97:e11119. https://doi.org/10.1097/MD.0000000000011119
Mann DM, Rollinson S, Robinson A et al (2013) Dipeptide repeat proteins are present in the p62 positive inclusions in patients with frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol Commun 1:68. https://doi.org/10.1186/2051-5960-1-68
Markovinovic A, Cimbro R, Ljutic T et al (2017) Optineurin in amyotrophic lateral sclerosis: multifunctional adaptor protein at the crossroads of different neuroprotective mechanisms. Prog Neurobiol 154:1–20. https://doi.org/10.1016/j.pneurobio.2017.04.005
Maruyama H, Morino H, Ito H et al (2010) Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465:223–226. https://doi.org/10.1038/nature08971
Matsunaga K, Morita E, Saitoh T et al (2010) Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J Cell Biol 190:511–521. https://doi.org/10.1083/jcb.200911141
Meerang M, Ritz D, Paliwal S et al (2011) The ubiquitin-selective segregase VCP/p97 orchestrates the response to DNA double-strand breaks. Nat Cell Biol 13:1376–1382. https://doi.org/10.1038/ncb2367
Mehta SG, Khare M, Ramani R et al (2013) Genotype-phenotype studies of VCP-associated inclusion body myopathy with Paget disease of bone and/or frontotemporal dementia. Clin Genet 83:422–431. https://doi.org/10.1111/cge.12000
Meroni M, Crippa V, Cristofani R et al (2019) Transforming growth factor beta 1 signaling is altered in the spinal cord and muscle of amyotrophic lateral sclerosis mice and patients. Neurobiol Aging 82:48–59. https://doi.org/10.1016/j.neurobiolaging.2019.07.001
Miki H, Okada Y, Hirokawa N (2005) Analysis of the kinesin superfamily: insights into structure and function. Trends Cell Biol 15:467–476. https://doi.org/10.1016/j.tcb.2005.07.006
Minegishi Y, Nakayama M, Iejima D et al (2016) Significance of optineurin mutations in glaucoma and other diseases. Prog Retin Eye Res 55:149–181. https://doi.org/10.1016/j.preteyeres.2016.08.002
Mitne-Neto M, Machado-Costa M, Marchetto MCN et al (2011) Downregulation of VAPB expression in motor neurons derived from induced pluripotent stem cells of ALS8 patients. Hum Mol Genet 20:3642–3652. https://doi.org/10.1093/hmg/ddr284
Mizuno Y, Amari M, Takatama M et al (2006) Immunoreactivities of p62, an ubiqutin-binding protein, in the spinal anterior horn cells of patients with amyotrophic lateral sclerosis. J Neurol Sci 249:13–18. https://doi.org/10.1016/j.jns.2006.05.060
Moore AS, Holzbaur ELF (2016) Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc Natl Acad Sci U S A 113:E3349–E3358. https://doi.org/10.1073/pnas.1523810113
Morgan S, Orrell RW (2016) Pathogenesis of amyotrophic lateral sclerosis. Br Med Bull 119(1):87-98. https://doi.org/10.1093/bmb/ldw026
Morton S, Hesson L, Peggie M, Cohen P (2008) Enhanced binding of TBK1 by an optineurin mutant that causes a familial form of primary open angle glaucoma. FEBS Lett 582:997–1002. https://doi.org/10.1016/j.febslet.2008.02.047
Moustaqim-Barrette A, Lin YQ, Pradhan S et al (2014) The amyotrophic lateral sclerosis 8 protein, VAP, is required for ER protein quality control. Hum Mol Genet 23:1975–1989. https://doi.org/10.1093/hmg/ddt594
Münch C, Rosenbohm A, Sperfeld A-D et al (2005) Heterozygous R1101K mutation of the DCTN1 gene in a family with ALS and FTD. Ann Neurol 58:777–780. https://doi.org/10.1002/ana.20631
Münch C, Sedlmeier R, Meyer T et al (2004) Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurol 63:724 LP – 726. https://doi.org/10.1212/01.WNL.0000134608.83927.B1
Munitic I, Giardino Torchia ML, Meena NP et al (2013) Optineurin insufficiency impairs IRF3 but not NF-κB activation in immune cells. J Immunol 191:6231–6240. https://doi.org/10.4049/jimmunol.1301696
Nalbandian A, Llewellyn KJ, Kitazawa M et al (2012) The homozygote VCPR155H/R155H mouse model exhibits accelerated human VCP-associated disease pathology. PLoS ONE 7:e46308. https://doi.org/10.1371/journal.pone.0046308
Narain P, Pandey A, Gupta S et al (2018) Targeted next-generation sequencing reveals novel and rare variants in Indian patients with amyotrophic lateral sclerosis. Neurobiol Aging 71:265.e9-265.e14. https://doi.org/10.1016/j.neurobiolaging.2018.05.012
N’Diaye E-N, Kajihara KK, Hsieh I et al (2009) PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. EMBO Rep 10:173–179. https://doi.org/10.1038/embor.2008.238
Neumann M, Sampathu DM, Kwong LK et al (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Sci 314:130 LP – 133. https://doi.org/10.1126/science.1134108
Nicolas A, Kenna KP, Renton AE et al (2018) Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron 97:1268-1283.e6. https://doi.org/10.1016/j.neuron.2018.02.027
Nishimura AL, Mitne-Neto M, Silva HCA et al (2004) A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet 75:822–831. https://doi.org/10.1086/425287
Niwa H, Ewens CA, Tsang C et al (2012) The role of the N-domain in the ATPase activity of the mammalian AAA ATPase p97/VCP. J Biol Chem 287:8561–8570. https://doi.org/10.1074/jbc.M111.302778
Oakes JA, Davies MC, Collins MO (2017) TBK1: a new player in ALS linking autophagy and neuroinflammation. Mol Brain 10:5. https://doi.org/10.1186/s13041-017-0287-x
Odorizzi G, Babst M, Emr SD (1998) Fab1p PtdIns(3)P 5-kinase function essential for protein sorting in the multivesicular body. Cell 95:847–858. https://doi.org/10.1016/s0092-8674(00)81707-9
Onesto E, Rusmini P, Crippa V et al (2011) Muscle cells and motoneurons differentially remove mutant SOD1 causing familial amyotrophic lateral sclerosis. J Neurochem 118:266–280. https://doi.org/10.1111/j.1471-4159.2011.07298.x
Osaka M, Ito D, Yagi T et al (2015) Evidence of a link between ubiquilin 2 and optineurin in amyotrophic lateral sclerosis. Hum Mol Genet 24:1617–1629. https://doi.org/10.1093/hmg/ddu575
Osawa T, Mizuno Y, Fujita Y et al (2011) Optineurin in neurodegenerative diseases. Neuropathology 31:569–574. https://doi.org/10.1111/j.1440-1789.2011.01199.x
Osmanovic A, Rangnau I, Kosfeld A et al (2017) FIG4 variants in central European patients with amyotrophic lateral sclerosis: a whole-exome and targeted sequencing study. Eur J Hum Genet 25:324–331. https://doi.org/10.1038/ejhg.2016.186
Otomo A, Hadano S, Okada T et al (2003) ALS2, a novel guanine nucleotide exchange factor for the small GTPase Rab5, is implicated in endosomal dynamics. Hum Mol Genet 12:1671–1687. https://doi.org/10.1093/hmg/ddg184
Otomo A, Kunita R, Suzuki-Utsunomiya K et al (2011) Defective relocalization of ALS2/alsin missense mutants to Rac1-induced macropinosomes accounts for loss of their cellular function and leads to disturbed amphisome formation. FEBS Lett 585:730–736. https://doi.org/10.1016/j.febslet.2011.01.045
Otomo A, Pan L, Hadano S (2012) Dysregulation of the autophagy-endolysosomal system in amyotrophic lateral sclerosis and related motor neuron diseases. Neurol Res Int 2012:1–12. https://doi.org/10.1155/2012/498428
Pankiv S, Clausen TH, Lamark T et al (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282:24131–24145. https://doi.org/10.1074/jbc.M702824200
Papadopoulos C, Kirchner P, Bug M et al (2017) VCP/p97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy. EMBO J 36:135–150. https://doi.org/10.15252/embj.201695148
Papiani G, Ruggiano A, Fossati M et al (2012) Restructured endoplasmic reticulum generated by mutant amyotrophic lateral sclerosis-linked VAPB is cleared by the proteasome. J Cell Sci 125:3601–3611. https://doi.org/10.1242/jcs.102137
Parkinson N, Ince PG, Smith MO et al (2006) ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology 67:1074–1077. https://doi.org/10.1212/01.wnl.0000231510.89311.8b
Patel A, Lee HO, Jawerth L et al (2015) A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162:1066–1077. https://doi.org/10.1016/j.cell.2015.07.047
Pensato V, Tiloca C, Corrado L et al (2015) TUBA4A gene analysis in sporadic amyotrophic lateral sclerosis: identification of novel mutations. J Neurol 262:1376–1378
Perrone F, Nguyen HP, Van Mossevelde S et al (2017) Investigating the role of ALS genes CHCHD10 and TUBA4A in Belgian FTD-ALS spectrum patients. Neurobiol Aging 51:177.e9-177.e16. https://doi.org/10.1016/j.neurobiolaging.2016.12.008
Philips T, Bento-Abreu A, Nonneman A et al (2013) Oligodendrocyte dysfunction in the pathogenesis of amyotrophic lateral sclerosis. Brain 136:471–482. https://doi.org/10.1093/brain/aws339
Picher-Martel V, Dutta K, Phaneuf D et al (2015) Ubiquilin-2 drives NF-κB activity and cytosolic TDP-43 aggregation in neuronal cells. Mol Brain 8:71. https://doi.org/10.1186/s13041-015-0162-6
Pilli M, Arko-Mensah J, Ponpuak M et al (2012) TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 37:223–234. https://doi.org/10.1016/j.immuni.2012.04.015
Prinz WA, Toulmay A, Balla T (2020) The functional universe of membrane contact sites. Nat Rev Mol Cell Biol 21:7–24. https://doi.org/10.1038/s41580-019-0180-9
Puls I, Jonnakuty C, LaMonte BH et al (2003) Mutant dynactin in motor neuron disease. Nat Genet 33:455–456. https://doi.org/10.1038/ng1123
Puls I, Oh SJ, Sumner CJ et al (2005) Distal spinal and bulbar muscular atrophy caused by dynactin mutation. Ann Neurol 57:687–694. https://doi.org/10.1002/ana.20468
Qiu L, Qiao T, Beers M et al (2013) Widespread aggregation of mutant VAPB associated with ALS does not cause motor neuron degeneration or modulate mutant SOD1 aggregation and toxicity in mice. Mol Neurodegener 8:1. https://doi.org/10.1186/1750-1326-8-1
Rademakers R, van Blitterswijk M (2014) Excess of rare damaging TUBA4A variants suggests cytoskeletal defects in ALS. Neuron 84:241–243. https://doi.org/10.1016/j.neuron.2014.10.002
Radulovic M, Schink KO, Wenzel EM et al (2018) ESCRT-mediated lysosome repair precedes lysophagy and promotes cell survival. EMBO J 37:. https://doi.org/10.15252/embj.201899753
Ramanathan HN, Ye Y (2012) The p97 ATPase associates with EEA1 to regulate the size of early endosomes. Cell Res 22:346–359. https://doi.org/10.1038/cr.2011.80
Rea SL, Majcher V, Searle MS, Layfield R (2014) SQSTM1 mutations - bridging Paget disease of bone and ALS/FTLD. Exp Cell Res 325:27–37. https://doi.org/10.1016/j.yexcr.2014.01.020
Rea SL, Walsh JP, Layfield R et al (2013) New insights into the role of sequestosome 1/p62 mutant proteins in the pathogenesis of Paget’s disease of bone. Endocr Rev 34:501–524. https://doi.org/10.1210/er.2012-1034
Reddy K, Zamiri B, Stanley SYR et al (2013) The disease-associated r(GGGGCC) repeat from the C9orf72 gene forms tract length-dependent uni- and multimolecular RNA G-quadruplex Structures. J Biol Chem 288:9860–9866. https://doi.org/10.1074/jbc.C113.452532
Reid E, Kloos M, Ashley-Koch A et al (2002) A kinesin heavy chain (KIF5A) mutation in hereditary spastic paraplegia (SPG10). Am J Hum Genet 71:1189–1194. https://doi.org/10.1086/344210
Renaud L, Picher-Martel V, Codron P, Julien J-P (2019) Key role of UBQLN2 in pathogenesis of amyotrophic lateral sclerosis and frontotemporal dementia. Acta Neuropathol Commun 7:103. https://doi.org/10.1186/s40478-019-0758-7
Renton AE, Majounie E, Waite A et al (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72:257–268. https://doi.org/10.1016/j.neuron.2011.09.010
Rezaie T, Child A, Hitchings R et al (2002) Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science 295:1077–1079. https://doi.org/10.1126/science.1066901
Richter B, Sliter DA, Herhaus L et al (2016) Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci U S A 113:4039–4044. https://doi.org/10.1073/pnas.1523926113
Ritson GP, Custer SK, Freibaum BD et al (2010) TDP-43 mediates degeneration in a novel Drosophila model of disease caused by mutations in VCP/p97. J Neurosci 30:7729–7739. https://doi.org/10.1523/JNEUROSCI.5894-09.2010
Robberecht W, Philips T (2013) The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci 14:248–264. https://doi.org/10.1038/nrn3430
Rogov V, Dötsch V, Johansen T, Kirkin V (2014) Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol Cell 53:167–178. https://doi.org/10.1016/j.molcel.2013.12.014
Roudier N, Lefebvre C, Legouis R (2005) CeVPS-27 is an endosomal protein required for the molting and the endocytic trafficking of the low-density lipoprotein receptor-related protein 1 in Caenorhabditis elegans. Traffic 6:695–705. https://doi.org/10.1111/j.1600-0854.2005.00309.x
Rubino E, Rainero I, Chiò A et al (2012) SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology 79:1556–1562. https://doi.org/10.1212/WNL.0b013e31826e25df
Rudge SA, Anderson DM, Emr SD (2004) Vacuole size control: regulation of PtdIns(3,5)P2 levels by the vacuole-associated Vac14-Fig4 complex, a PtdIns(3,5)P2-specific phosphatase. Mol Biol Cell 15:24–36. https://doi.org/10.1091/mbc.e03-05-0297
Rusten TE, Stenmark H (2009) How do ESCRT proteins control autophagy? J Cell Sci 122:2179–2183. https://doi.org/10.1242/jcs.050021
Rusten TE, Vaccari T, Lindmo K et al (2007) ESCRTs and Fab1 regulate distinct steps of autophagy. Curr Biol 17:1817–1825. https://doi.org/10.1016/j.cub.2007.09.032
Rustici G, Kolesnikov N, Brandizi M et al (2013) ArrayExpress update–trends in database growth and links to data analysis tools. Nucleic Acids Res 41:D987–D990. https://doi.org/10.1093/nar/gks1174
Rutherford AC, Traer C, Wassmer T et al (2006) The mammalian phosphatidylinositol 3-phosphate 5-kinase (PIKfyve) regulates endosome-to-TGN retrograde transport. J Cell Sci 119:3944–3957. https://doi.org/10.1242/jcs.03153
Rydzanicz M, Jagła M, Kosinska J et al (2017) KIF5A de novo mutation associated with myoclonic seizures and neonatal onset progressive leukoencephalopathy. Clin Genet 91:769–773. https://doi.org/10.1111/cge.12831
Ryzhakov G, Randow F (2007) SINTBAD, a novel component of innate antiviral immunity, shares a TBK1-binding domain with NAP1 and TANK. EMBO J 26:3180–3190. https://doi.org/10.1038/sj.emboj.7601743
Sanhueza M, Zechini L, Gillespie T, Pennetta G (2014) Gain-of-function mutations in the ALS8 causative gene VAPB have detrimental effects on neurons and muscles. Biol Open 3:59–71. https://doi.org/10.1242/bio.20137070
Sbrissa D, Ikonomov OC, Fu Z et al (2007) Core protein machinery for mammalian phosphatidylinositol 3,5-bisphosphate synthesis and turnover that regulates the progression of endosomal transport. Novel Sac phosphatase joins the ArPIKfyve-PIKfyve complex. J Biol Chem 282:23878–23891. https://doi.org/10.1074/jbc.M611678200
Schmidt O, Teis D (2012) The ESCRT machinery. Curr Biol 22:R116–R120. https://doi.org/10.1016/j.cub.2012.01.028
Schuck S (2020) Microautophagy – distinct molecular mechanisms handle cargoes of many sizes. J Cell Sci. https://doi.org/10.1242/jcs.246322
Seibenhener ML, Babu JR, Geetha T et al (2004) Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol 24:8055–8068. https://doi.org/10.1128/MCB.24.18.8055-8068.2004
Sellier C, Campanari M, Julie Corbier C, et al (2016) Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin‐2 to induce motor neuron dysfunction and cell death. EMBO J 35:1276–1297. https://doi.org/10.15252/embj.201593350
Şentürk M, Lin G, Zuo Z et al (2019) Ubiquilins regulate autophagic flux through mTOR signalling and lysosomal acidification. Nat Cell Biol 21:384–396. https://doi.org/10.1038/s41556-019-0281-x
Sharkey LM, Sandoval-Pistorius SS, Moore SJ et al (2020) Modeling UBQLN2-mediated neurodegenerative disease in mice: shared and divergent properties of wild type and mutant UBQLN2 in phase separation, subcellular localization, altered proteostasis pathways, and selective cytotoxicity. Neurobiol Dis 143:105016. https://doi.org/10.1016/j.nbd.2020.105016
Shaw PJ (2005) Molecular and cellular pathways of neurodegeneration in motor neurone disease. J Neurol Neurosurg Psychiatry 76:1046–1057. https://doi.org/10.1136/jnnp.2004.048652
Sheerin U-M, Schneider SA, Carr L et al (2014) ALS2 mutations. Neurol 82:1065 LP – 1067. https://doi.org/10.1212/WNL.0000000000000254
Shen W-C, Li H-Y, Chen G-C et al (2015) Mutations in the ubiquitin-binding domain of OPTN/optineurin interfere with autophagy-mediated degradation of misfolded proteins by a dominant-negative mechanism. Autophagy 11:685–700. https://doi.org/10.4161/auto.36098
Shi Y, Lin S, Staats KA et al (2018) Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat Med 24:313–325. https://doi.org/10.1038/nm.4490
Shimizu H, Toyoshima Y, Shiga A et al (2013) Sporadic ALS with compound heterozygous mutations in the SQSTM1 gene. Acta Neuropathol 126:453–459. https://doi.org/10.1007/s00401-013-1150-5
Shu S, Li XL, Liu Q et al (2016) Screening of the TBK1 gene in familial and sporadic amyotrophic lateral sclerosis patients of Chinese origin. Amyotroph Lateral Scler Frontotemporal Degener 17:605–607. https://doi.org/10.1080/21678421.2016.1183681
Sica V, Galluzzi L, Bravo-San Pedro JM et al (2015) Organelle-specific initiation of autophagy. Mol Cell 59:522–539. https://doi.org/10.1016/j.molcel.2015.07.021
Sivadasan R, Hornburg D, Drepper C et al (2016) C9ORF72 interaction with cofilin modulates actin dynamics in motor neurons. Nat Neurosci 19:1610–1618. https://doi.org/10.1038/nn.4407
Skehel PA, Martin KC, Kandel ER, Bartsch D (1995) A VAMP-binding protein from Aplysia required for neurotransmitter release. Science 269:1580–1583. https://doi.org/10.1126/science.7667638
Skibinski G, Parkinson NJ, Brown JM et al (2005) Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet 37:806–808. https://doi.org/10.1038/ng1609
Smeyers J, Banchi E-G, Latouche M (2021) C9ORF72: what it is, what it does, and why it matters. Front Cell Neurosci 15:661447. https://doi.org/10.3389/fncel.2021.661447
Smith BN, Ticozzi N, Fallini C et al (2014) Exome-wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron 84:324–331. https://doi.org/10.1016/j.neuron.2014.09.027
Spang N, Feldmann A, Huesmann H et al (2014) RAB3GAP1 and RAB3GAP2 modulate basal and rapamycin-induced autophagy. Autophagy 10:2297–2309. https://doi.org/10.4161/15548627.2014.994359
Stein A, Ruggiano A, Carvalho P, Rapoport TA (2014) Key steps in ERAD of luminal ER proteins reconstituted with purified components. Cell 158:1375–1388. https://doi.org/10.1016/j.cell.2014.07.050
Stockmann M, Meyer-Ohlendorf M, Achberger K et al (2013) The dynactin p150 subunit: cell biology studies of sequence changes found in ALS/MND and Parkinsonian syndromes. J Neural Transm (vienna) 120:785–798. https://doi.org/10.1007/s00702-012-0910-z
Su M-Y, Fromm SA, Zoncu R, Hurley JH (2020) Structure of the C9orf72 ARF GAP complex that is haploinsufficient in ALS and FTD. Nature 585:251–255. https://doi.org/10.1038/s41586-020-2633-x
Sun Y-M, Dong Y, Wang J et al (2017) A novel mutation of VAPB in one Chinese familial amyotrophic lateral sclerosis pedigree and its clinical characteristics. J Neurol 264:2387–2393. https://doi.org/10.1007/s00415-017-8628-3
Sundaramoorthy V, Walker AK, Tan V et al (2015) Defects in optineurin- and myosin VI-mediated cellular trafficking in amyotrophic lateral sclerosis. Hum Mol Genet 24:3830–3846. https://doi.org/10.1093/hmg/ddv126
Suzuki H, Kanekura K, Levine TP et al (2009) ALS-linked P56S-VAPB, an aggregated loss-of-function mutant of VAPB, predisposes motor neurons to ER stress-related death by inducing aggregation of co-expressed wild-type VAPB. J Neurochem 108:973–985. https://doi.org/10.1111/j.1471-4159.2008.05857.x
Swarup V, Phaneuf D, Dupré N et al (2011) Deregulation of TDP-43 in amyotrophic lateral sclerosis triggers nuclear factor κB-mediated pathogenic pathways. J Exp Med 208:2429–2447. https://doi.org/10.1084/jem.20111313
Tanaka A, Cleland MM, Xu S et al (2010) Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 191:1367–1380. https://doi.org/10.1083/jcb.201007013
Tang D, Sheng J, Xu L et al (2020) Cryo-EM structure of C9ORF72–SMCR8–WDR41 reveals the role as a GAP for Rab8a and Rab11a. Proc Natl Acad Sci 117:9876–9883. https://doi.org/10.1073/pnas.2002110117
Taylor JP, Brown RH, Cleveland DW (2016) Decoding ALS: from genes to mechanism. Nature 539:197–206. https://doi.org/10.1038/nature20413
Teuling E, Ahmed S, Haasdijk E et al (2007) Motor neuron disease-associated mutant vesicle-associated membrane protein-associated protein (VAP) B recruits wild-type VAPs into endoplasmic reticulum-derived tubular aggregates. J Neurosci 27:9801–9815. https://doi.org/10.1523/JNEUROSCI.2661-07.2007
Teyssou E, Takeda T, Lebon V et al (2013) Mutations in SQSTM1 encoding p62 in amyotrophic lateral sclerosis: genetics and neuropathology. Acta Neuropathol 125:511–522. https://doi.org/10.1007/s00401-013-1090-0
Therrien M, Rouleau GA, Dion PA, Parker JA (2013) Deletion of C9ORF72 results in motor neuron degeneration and stress sensitivity in C. elegans. PLoS One 8:e83450. https://doi.org/10.1371/journal.pone.0083450
Thurston TLM, Ryzhakov G, Bloor S et al (2009) The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol 10:1215–1221. https://doi.org/10.1038/ni.1800
Tischfield MA, Cederquist GY, Gupta MLJ, Engle EC (2011) Phenotypic spectrum of the tubulin-related disorders and functional implications of disease-causing mutations. Curr Opin Genet Dev 21:286–294. https://doi.org/10.1016/j.gde.2011.01.003
Topp JD, Gray NW, Gerard RD, Horazdovsky BF (2004) Alsin is a Rab5 and Rac1 guanine nucleotide exchange factor. J Biol Chem 279:24612–24623. https://doi.org/10.1074/jbc.M313504200
Tresse E, Salomons FA, Vesa J et al (2010) VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy 6:217–227. https://doi.org/10.4161/auto.6.2.11014
Trotti D, Rolfs A, Danbolt NC et al (1999) Erratum: SOD1 mutants linked to amyotrophic lateral sclerosis selectively inactivate a glial glutamate transporter. Nat Neurosci 2:848–848. https://doi.org/10.1038/12227
Tsai P-C, Liu Y-C, Lin K-P et al (2016) Mutational analysis of TBK1 in Taiwanese patients with amyotrophic lateral sclerosis. Neurobiol Aging 40:191.e11-191.e16. https://doi.org/10.1016/j.neurobiolaging.2015.12.022
Tudor EL, Galtrey CM, Perkinton MS et al (2010) Amyotrophic lateral sclerosis mutant vesicle-associated membrane protein-associated protein-B transgenic mice develop TAR-DNA-binding protein-43 pathology. Neuroscience 167:774–785. https://doi.org/10.1016/j.neuroscience.2010.02.035
Tumbarello DA, Waxse BJ, Arden SD et al (2012) Autophagy receptors link myosin VI to autophagosomes to mediate Tom1-dependent autophagosome maturation and fusion with the lysosome. Nat Cell Biol 14:1024–1035. https://doi.org/10.1038/ncb2589
Tümer Z, Bertelsen B, Gredal O et al (2012) Novel heterozygous nonsense mutation of the OPTN gene segregating in a Danish family with ALS. Neurobiol Aging 33:208.e1–5. https://doi.org/10.1016/j.neurobiolaging.2011.07.001
Uhlén M, Fagerberg L, Hallström BM, et al (2015) Proteomics. tissue-based map of the human proteome. Sci 347:1260419. https://doi.org/10.1126/science.1260419
Urnavicius L, Zhang K, Diamant AG et al (2015) The structure of the dynactin complex and its interaction with dynein. Science 347:1441–1446. https://doi.org/10.1126/science.aaa4080
Urwin H, Authier A, Nielsen JE et al (2010) Disruption of endocytic trafficking in frontotemporal dementia with CHMP2B mutations. Hum Mol Genet 19:2228–2238. https://doi.org/10.1093/hmg/ddq100
Vale RD, Reese TS, Sheetz MP (1985) Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell 42:39–50. https://doi.org/10.1016/s0092-8674(85)80099-4
van Blitterswijk M, van Es MA, Koppers M et al (2012a) VAPB and C9orf72 mutations in 1 familial amyotrophic lateral sclerosis patient. Neurobiol Aging 33:2950.e1–4. https://doi.org/10.1016/j.neurobiolaging.2012.07.004
van Blitterswijk M, van Vught PWJ, van Es MA et al (2012b) Novel optineurin mutations in sporadic amyotrophic lateral sclerosis patients. Neurobiol Aging 33:1016.e1–7. https://doi.org/10.1016/j.neurobiolaging.2011.05.019
van Blitterswijk M, Vlam L, van Es MA et al (2012c) Genetic overlap between apparently sporadic motor neuron diseases. PLoS ONE 7:e48983. https://doi.org/10.1371/journal.pone.0048983
van der Zee J, Van Langenhove T, Kovacs GG et al (2014) Rare mutations in SQSTM1 modify susceptibility to frontotemporal lobar degeneration. Acta Neuropathol 128:397–410. https://doi.org/10.1007/s00401-014-1298-7
van Mossevelde S, van der Zee J, Cruts M, van Broeckhoven C (2017) Relationship between C9orf72 repeat size and clinical phenotype. Curr Opin Genet Dev 44:117–124. https://doi.org/10.1016/j.gde.2017.02.008
Van Mossevelde S, van der Zee J, Gijselinck I et al (2016) Clinical features of TBK1 carriers compared with C9orf72, GRN and non-mutation carriers in a Belgian cohort. Brain 139:452–467. https://doi.org/10.1093/brain/awv358
van Rheenen W, Shatunov A, Dekker AM et al (2016) Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat Genet 48:1043–1048. https://doi.org/10.1038/ng.3622
Vaughan KT, Vallee RB (1995) Cytoplasmic dynein binds dynactin through a direct interaction between the intermediate chains and p150Glued. J Cell Biol 131:1507–1516. https://doi.org/10.1083/jcb.131.6.1507
Verma R, Oania RS, Kolawa NJ, Deshaies RJ (2013) Cdc48/p97 promotes degradation of aberrant nascent polypeptides bound to the ribosome. Elife 2:e00308. https://doi.org/10.7554/eLife.00308
Vernay A, Therreau L, Blot B et al (2016) A transgenic mouse expressing CHMP2Bintron5 mutant in neurons develops histological and behavioural features of amyotrophic lateral sclerosis and frontotemporal dementia. Hum Mol Genet 25:3341–3360. https://doi.org/10.1093/hmg/ddw182
Walters KJ, Kleijnen MF, Goh AM et al (2002) Structural studies of the interaction between ubiquitin family proteins and proteasome subunit S5a. Biochemistry 41:1767–1777. https://doi.org/10.1021/bi011892y
Wang J, Hussain T, Yue R et al (2018) MicroRNA-199a inhibits cellular autophagy and downregulates IFN-β expression by targeting TBK1 in Mycobacterium bovis infected cells. Front Cell Infect Microbiol 8:238. https://doi.org/10.3389/fcimb.2018.00238
Wang M, Wang H, Tao Z et al (2020) C9orf72 associates with inactive Rag GTPases and regulates mTORC1-mediated autophagosomal and lysosomal biogenesis. Aging Cell. https://doi.org/10.1111/acel.13126
Wang T, Liu H, Itoh K et al (2021) C9orf72 regulates energy homeostasis by stabilizing mitochondrial complex I assembly. Cell Metab 33:531-546.e9. https://doi.org/10.1016/j.cmet.2021.01.005
Watts GDJ, Wymer J, Kovach MJ et al (2004) Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet 36:377–381. https://doi.org/10.1038/ng1332
Webster CP, Smith EF, Bauer CS et al (2016) The C9orf72 protein interacts with Rab1a and the ULK 1 complex to regulate initiation of autophagy. EMBO J 35:1656–1676. https://doi.org/10.15252/embj.201694401
West RJH, Ugbode C, Fort-Aznar L, Sweeney ST (2020) Neuroprotective activity of ursodeoxycholic acid in CHMP2B(Intron5) models of frontotemporal dementia. Neurobiol Dis 144:105047. https://doi.org/10.1016/j.nbd.2020.105047
Wild P, Farhan H, McEwan DG et al (2011) Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 333:228–233. https://doi.org/10.1126/science.1205405
Williams KL, McCann EP, Fifita JA et al (2015) Novel TBK1 truncating mutation in a familial amyotrophic lateral sclerosis patient of Chinese origin. Neurobiol Aging 36:3334.e1–3334.e5. https://doi.org/10.1016/j.neurobiolaging.2015.08.013
Wong YC, Holzbaur ELF (2014) Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci U S A 111:E4439–48. https://doi.org/10.1073/pnas.1405752111
Wu JJ, Cai A, Greenslade JE et al (2020) ALS/FTD mutations in UBQLN2 impede autophagy by reducing autophagosome acidification through loss of function. Proc Natl Acad Sci U S A 117:15230–15241. https://doi.org/10.1073/pnas.1917371117
Wu Q, Liu M, Huang C et al (2015) Pathogenic Ubqln2 gains toxic properties to induce neuron death. Acta Neuropathol 129:417–428. https://doi.org/10.1007/s00401-014-1367-y
Xiao S, MacNair L, McGoldrick P et al (2015) Isoform-specific antibodies reveal distinct subcellular localizations of C9orf72 in amyotrophic lateral sclerosis. Ann Neurol 78:568–583. https://doi.org/10.1002/ana.24469
Xu H, Ren D (2015) Lysosomal physiology. Annu Rev Physiol 77:57–80. https://doi.org/10.1146/annurev-physiol-021014-071649
Xu S, Peng G, Wang Y et al (2011) The AAA-ATPase p97 is essential for outer mitochondrial membrane protein turnover. Mol Biol Cell 22:291–300. https://doi.org/10.1091/mbc.E10-09-0748
Yamanaka K, Miller TM, McAlonis-Downes M et al (2006) Progressive spinal axonal degeneration and slowness in ALS2-deficient mice. Ann Neurol 60:95–104. https://doi.org/10.1002/ana.20888
Yamanaka K, Vande Velde C, Eymard-Pierre E et al (2003) Unstable mutants in the peripheral endosomal membrane component ALS2 cause early-onset motor neuron disease. Proc Natl Acad Sci U S A 100:16041 LP – 16046. https://doi.org/10.1073/pnas.2635267100
Yang M, Liang C, Swaminathan K et al (2016) A C9ORF72/SMCR8-containing complex regulates ULK1 and plays a dual role in autophagy. Sci Adv. https://doi.org/10.1126/sciadv.1601167
Yang Y, Hentati A, Deng H-X et al (2001) The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat Genet 29:160–165. https://doi.org/10.1038/ng1001-160
Yin HZ, Nalbandian A, Hsu C-I et al (2012) Slow development of ALS-like spinal cord pathology in mutant valosin-containing protein gene knock-in mice. Cell Death Dis 3:e374–e374. https://doi.org/10.1038/cddis.2012.115
Yu J, Lai C, Shim H et al (2018) Genetic ablation of dynactin p150(Glued) in postnatal neurons causes preferential degeneration of spinal motor neurons in aged mice. Mol Neurodegener 13:10. https://doi.org/10.1186/s13024-018-0242-z
Zatloukal K, Stumptner C, Fuchsbichler A et al (2002) p62 Is a common component of cytoplasmic inclusions in protein aggregation diseases. Am J Pathol 160:255–263. https://doi.org/10.1016/S0002-9440(10)64369-6
Zhang K, Donnelly CJ, Haeusler AR et al (2015) The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525:56–61. https://doi.org/10.1038/nature14973
Zhang Y, Zolov SN, Chow CY et al (2007) Loss of Vac14, a regulator of the signaling lipid phosphatidylinositol 3,5-bisphosphate, results in neurodegeneration in mice. Proc Natl Acad Sci U S A 104:17518–17523. https://doi.org/10.1073/pnas.0702275104
Zhong Y, Wang QJ, Li X et al (2009) Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1–phosphatidylinositol-3-kinase complex. Nat Cell Biol 11:468–476. https://doi.org/10.1038/ncb1854
Zu T, Liu Y, Bañez-Coronel M et al (2013) RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. PNAS 110:E4968 LP-E4977. https://doi.org/10.1073/pnas.1315438110
Acknowledgements
The authors are grateful to Prof. Angelo Poletti (Dipartimento di Scienze Farmacologiche e Biomolecolari, Università degli Studi di Milano, 20133, Milan, Italy) for the useful and inspiring discussion and the critical reading of the manuscript. The authors thank the European Society for Neurochemistry (ESN) for inviting them to participate to the special issue of the Journal of Molecular Neuroscience.
Funding
Open access funding provided by Università degli Studi di Milano within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Contributions
Marta Cozzi and Veronica Ferrari designed and wrote the manuscript and critically discussed all sections of the review. In addition, Marta Cozzi and Veronica Ferrari prepared the figures.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cozzi, M., Ferrari, V. Autophagy Dysfunction in ALS: from Transport to Protein Degradation. J Mol Neurosci 72, 1456–1481 (2022). https://doi.org/10.1007/s12031-022-02029-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-022-02029-3