
Abstract
Macrophages are one of the first innate immune cells to reach the site of infection or injury. Diverse functions from the uptake of pathogen or antigen, its killing, and presentation, the release of pro- or anti-inflammatory cytokines, activation of adaptive immune cells, clearing off tissue debris, tissue repair, and maintenance of tissue homeostasis have been attributed to macrophages. Besides tissue-resident macrophages, the circulating macrophages are recruited to different tissues to get activated. These are highly plastic cells, showing a spectrum of phenotypes depending on the stimulus received from their immediate environment. The macrophage differentiation requires colony-stimulating factor-1 (CSF-1) or macrophage colony-stimulating factor (M-CSF), colony-stimulating factor-2 (CSF-2), or granulocyte–macrophage colony-stimulating factor (GM-CSF) and different stimuli activate them to different phenotypes. The richness of tissue macrophages is precisely controlled via the CSF-1 and CSF-1R axis. In this review, we have given an overview of macrophage origin via hematopoiesis/myelopoiesis, different phenotypes associated with macrophages, their clinical significance, and how they are altered in various diseases. We have specifically focused on the function of CSF-1/CSF-1R signaling in deciding macrophage fate and the outcome of aberrant CSF-1R signaling in relation to macrophage phenotype in different diseases. We further extend the review to briefly discuss the possible strategies to manipulate CSF-1R and its signaling with the recent updates.
Graphical Abstract

Similar content being viewed by others


Introduction
Macrophages (macro, big; phage (from Greek phageîn), to devour/eat) are the phagocytes or the big guzzlers (eaters) of our immune system. They are part of the mononuclear phagocyte system. The mononuclear phagocyte system consists of cells that originate from a common precursor in the bone marrow, circulate through the bloodstream, and then mature and activate in distinct organs or yolk-sac derived (brain, microglia; liver, Kupffer cells; skin, Langerhans cells; lung, alveolar macrophages; peritoneum, peritoneal macrophages) [1]. They are one of the first responders of the innate immune system to any infection or injury, to mediate pathogen engulfment [2]. Macrophages are highly malleable cells. Their activities, properties, nature, and functions change according to their immediate environment [3]. It is fascinating to observe that macrophages change their nature based on the ambiance they are present in, the signals they receive, need of the surrounding tissue, i.e., to kill or to repair. However, a plethora of phenotypes are displayed by macrophages (explained ahead in Sect. 3), and the two major activated polarized states shown by them are classically activated, pro-inflammatory M1 macrophages and alternatively activated anti-inflammatory M2 macrophages [4]. In healthy conditions, the ratio of M1/M2 macrophages is greatly controlled and regulated in tissues. Generally, the M1-dominant phenotype is required in clearing off infections or tumor cells in the initial phases, and the M2-dominant phenotype is required in the latter phases for maintaining tissue repair and homeostasis [5,6,7,8]. The imbalance of inflammatory and anti-inflammatory macrophage phenotypes is the major culprit in pathology associated with many diseases and persistent infections. In Alzheimer’s disease (AD), the chronic and pre-dominant, M1-mediated inflammatory response can lead to neuronal loss-mediated disease progression [9]. While in cancer, the chronic M2 phenotype or the TAM-like phenotype aids in disease progression [10]. Similarly, the fungi-induced M2 subset participates in mediating lung pathology [11]. Various lab groups across the globe including ours have been exploring the effect of different environments like tumors, acute or chronic infections, and antibiotics on macrophage activation and subsequent polarization to different phenotypes [12,13,14,15].
Historically, macrophage-like phagocytic cells, found in invertebrates, are phylogenetically ancient innate immune mediators [16]. For instance, when Drosophila is infected with bacteria, it surrounds the invading bacteria with cells similar to macrophages called hemocytes [17]. Hemocytes which phagocytize the microbes and promote coagulation of the surrounding hemolymph thus could be called the phylogenetic ancestors of macrophages. Macrophages were first discovered by Russian scientist Élie Metchnikoff in the late 1990s [18].
Metchnikoff was recognized for his early discovery of phagocytosis in starfish larvae in 1883 [19]. In the decades after the 1920s, significant progress was made in describing macrophage biology, particularly in the murine cell culture models pioneered by Zanvil Cohn and co-workers [20]. The macrophages can be derived from murine bone marrow (BM) cells by culturing the BM-derived hematopoietic stem cells (HSCs) in the presence of colony proliferator cytokines like macrophage colony-stimulating factor (M-CSF/CSF-1) or granulocyte–macrophage colony-stimulating factor (GM-CSF/CSF-2) [10, 21] (Table 1) (explained in Sect. 2.). The lushness of tissue macrophages is precisely controlled via the CSF-1 or M-CSF, IL-34, and CSF-1R axis. However, the role of CSF-1R in dictating the fate of macrophage activation to M1 or M2 is a grey area in macrophage biology.
Henceforth, it is interesting for us to sum up available literature to spawn a better understanding of macrophage polarization in health and diseases and how it concerns with CSF-1R axis. In this review, we have tried to sum up the available literature in the context of macrophage polarization in general and in relation to CSF-1R and its clinical implications. We have first explained the generation of macrophages via myelopoiesis in normal and stress/disease conditions, briefed about macrophage phenotypes, and their clinical significance, and then discussed CSF-1R and its possible connotation with macrophage polarization and clinical implications of dysregulated macrophage polarization and CSF-1R axis.
Myelopoiesis and macrophage generation
The formation of myeloid cells is called myelopoiesis. The center of the bones (bone marrow) produces ample amounts of white blood cells (monocytes, lymphocytes, neutrophils, eosinophils, basophils, and macrophages), red blood cells (erythrocytes), and platelets [22]. These blood cells are produced via proliferation and differentiation of pluripotent hematopoietic stem cells (HSCs) (hematopoiesis). Hematopoietic cells are “self-renewing” and generate both lymphoid and myeloid progenitors. HSCs form a hierarchy of proliferative progenitor populations (PPPs) which get committed into lymphoid, myeloid, or erythroid lineages. HSCs generate multipotent progenitor precursors (MPPs) (Fig. 1). MPPs differentiate into immature lineage-biased progenitors—lymphoid-primed multipotent progenitors (LMPPs)/multilymphoid progenitors (MLPs) which differentiate into common myeloid progenitors (CMPs). CMPs form the granulocyte monocyte progenitors (GMPs) [23]. The differentiation of MPPs into CMPs and then to GMPs is regulated by a transcription factor PU.1 [24] (ETS domain transcription factor encoded by Spi-1 proto-oncogene that activates gene expression during myeloid and B-lymphoid cell development).

Normal and demand adapted or emergency myelopoiesis. The quiescent bone marrow-HSCs maintain the normal hematopoiesis/myelopoiesis. Any systemic inflammation/infection/stress leading to the release of inflammatory cytokines, PAMPs, and DAMPs is sensed by HSCs. HSCs activate and differentiate to myeloid progenitors. Under inflammation/infection/stress conditions, the GMPs are recruited to organs like the spleen in a CCR-2-dependent manner to carry out extramedullary myelopoiesis to meet the demand adapted emergency myelopoiesis. HSCs, hematopoietic stem cells; CMPs, common myeloid progenitors; GMPs, granulocyte monocyte progenitors
Another transcription factor IRF8 or ICSBP1 (interferon regulatory factor 8 or interferon consensus sequence-binding protein 1) binds to the IFN-stimulated response element (ISRE) and regulates the expression of genes stimulated by type I IFNs-IFN-α/β and is involved in directing GMPs to form granulocyte progenitors (GPs) and monocyte progenitors (MPs) [25]. MPs differentiate into mononuclear phagocytes, i.e., monocytes, macrophages, and myeloid DCs. GPs generate polymorphonuclear phagocytes, i.e., neutrophils, basophils, and eosinophils.
In mice, the monocyte subsets show differential expression of chemokine receptors CCR2 and inflammatory monocyte marker Ly6C/Gr1 [26]. Mouse monocytes are grouped in Ly6C+/Ly6Chigh/Ly6Cmiddle and Ly6C−/Ly6Clow subsets. The Ly6C+ monocytes could further be CD11b+CD115+ and CCR2highCX3CR1low, and Ly6C− monocytes could be CD11b+CD115+ and CCR2lowCX3CR1high [26, 27]. The CCR2+/high monocyte subset has a greater migratory/infiltration ability than the CCR2−/low subset. Ly-6C+ and Ly-6C− monocytes enter the blood from the bone marrow. Thus, CD115 (CSF-1R) plays an important role in the generation of both monocyte subsets [27,28,29] (Fig. 1). Macrophage colony-stimulating factor (M-CSF/CSF-1), one of the ligands to CSF-1R or CD115, is one of the key cytokines for macrophage generation.
CD115/CSF-1R is expressed on monocytes, macrophages, and mononuclear phagocyte precursors [30]. Mouse Ly6C + monocytes leave the bone marrow in a CCR2-dependent manner. They could differentiate into Ly6C− monocytes in circulation which further develop into tissue-resident macrophages/DCs. They are drafted to normal tissue via a LAF/ICAM1-dependent CX3CR1/CCL3 interaction and become tissue-resident macrophages/DCs. Ly6C− monocytes in tissue differentiate into M2-like anti-inflammatory macrophages, which secrete anti-inflammatory cytokines like IL-10, and contribute to tissue repair/remodeling and maintenance of tissue homeostasis [31], whereas Ly6C + monocytes demonstrate a higher phagocytic ability and better generation of reactive oxidative stress (ROS), higher secretion of inflammatory cytokines like TNFα, and IL-1β, and better antimicrobial competence as compared to the Ly6C− monocytes. During inflammatory episodes, Ly6C + monocytes invade tissue via a VLA-1/VCAM1-dependent CCR2/CCL2 (MPC-1) interaction and mature into inflammatory M1-like macrophages. M1 macrophages secrete pro-inflammatory cytokines like TNFα, IL-6, IL-12, etc., and cause T cell activation [14, 26, 32].
Emergency or demand adapted myelopoiesis
In response to an inflammatory event like an infection, allergy, and tumor, there is an increased demand for myeloid cells, i.e., monocytes, macrophages, neutrophils, and DCs. The adaptive immune cells (T and B cells) robustly multiply/proliferate when specific antigens are sensed by them; innate immune cells have the low proliferative ability and thus are required to be replenished from the HSCs and progenitors in the bone marrow. This is called stress-induced or emergency or demand-adapted myelopoiesis (Fig. 1).
During inflammatory episodes, the monocytes, macrophages, and neutrophils are deployed from bone marrow to the site of infection which is essential for containing the pathogens [33]. The molecular mechanism dictating emergency myelopoiesis is not completely understood. Emergency or demand-adapted myelopoiesis is facilitated by activation of HSCs and progenitor cells in the bone marrow [34] showing a myeloid-biased differentiation during such conditions.
This is principally mediated by cytokines, like TNF-α, IL-1β, IL-6, IL-12, IFN-α, IFN-γ, M-CSF, and GM-CSF, and pathogen-derived factors, e.g., lipopolysaccharide (LPS), which are sensed by HSCs. The elevated inflammatory cytokines, PAMPs/DAMPs, lead to the expansion in myeloid cell turnover within the bone marrow and extramedullary tissues like the spleen (Fig. 1). Recently, one of the IL-6 family cytokines IL-27 has been reported to play a crucial role in emergency myelopoiesis [35]. IL-27 is reported to expand lineage (Lin)− Sca-1+c-Kit+ (LSK) cells. LSK cells are enriched in bone marrow HSCs in cooperation with stem cell factor c-Kit ligand. The HSCs differentiate into myeloid progenitors via signal transducer and activator of transcription 1 (STAT1) and STAT3. Infections induce cytokines like IFN-γ to boost IL-27 production. IL-27, as discussed above, promotes the expansion and mobilization of LSK cells, resulting in heightened myelopoiesis in response to infections [36].
Macrophage phenotypes
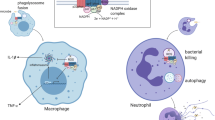
Macrophages perform several important functions in innate and adaptive immunity like ingestion and killing of microorganisms, generation of reactive oxygen and nitrogen species, proteolytic digestion of ingested pathogens, and presenting the antigens to adaptive immune cells. Macrophages gobble up the dead host cells to clean up the tissue microenvironment after infections or tissue injuries and maintain tissue homeostasis (Fig. 2). Macrophages can divide at an inflammatory site and thus are the imperative effector cells of innate immune response even after several days of infection. Other than immunological activities, macrophages promote tissue repair by inducing angiogenesis and fibrosis (formation of collagen-rich extracellular matrix (ECM)). Owing to the vast functions associated with macrophages, they are known to possess a spectrum of phenotypes. Depending upon the nature of activating stimuli, macrophages show different functional capabilities.

A balance of inflammatory (M1) and anti-inflammatory (M2) macrophages is required for a healthy immune response. Macrophage phenotypes play an important role in maintaining a healthy immune response and resolving infections and other diseases. Various factors induce the M1 or M2 macrophage phenotype, which is tightly regulated by transcriptional factors as depicted in the image. Any dis-regulation in the activation of macrophage phenotypes may lead to chronic infections, inflammatory disorders, cytokine storm, or tumor escape and development to cancer
As macrophages are extremely plastic cells, they can change their functional profile through macrophage polarization (Fig. 2). Macrophage polarization is the process by which macrophages respond to stimuli coming from the local microenvironment and acquire a specific functional phenotype. Based on specific programs of gene expression leading to the gain of different markers on the cellular surface, the secretion of certain cytokines (Table 2), and metabolic adaptations, macrophages are generally classified into classically activated, pro-inflammatory, or M1 macrophages and alternatively activated, anti-inflammatory, or M2 macrophages. Below we have briefly described the major macrophage phenotypes:
M1 macrophages
M1 phenotype in macrophages is induced by pathogen-associated molecular pattern molecules (PAMPs) and cytokines secreted by T helper 1 (TH1) lymphocytes like IFN-γ and TNF-α[7].
PAMPs are derived from microorganisms, e.g., LPS, and are recognized by pattern recognition receptors (PRRs) of different kinds like toll-like receptors (TLRs), nucleotide oligomerization domain (NOD)-like receptors (NLRs), retinoic acid-inducible gene-I (RIG-I)-like receptors (RLRs), C-type lectin receptors (CLRs), and absent in melanoma-2 (AIM2)-like receptors (ALRs) present in APCs like macrophages[46]. From the functional point of view, M1 macrophages are characterized by their ability to kill pathogens and present antigens to T lymphocytes for initiation of adaptive responses. M1 has higher expression of CD80, CD86, class II trans activator (CIITA), major histocompatibility complex class II receptor (MHC-II), cyclooxygenase 2 (COX-2), and inducible nitric oxide synthase (iNOS) and produces high levels of pro-inflammatory cytokines, such as TNF-α, IL1-β, IL-6, IL-12, and IL-23, and promotes TH1 responses[47]. The expression of these cytokines is mainly controlled by the activation and nuclear translocation of the transcription factor nuclear factor kappa-light-chain enhancer of B cell (NF-κB), with STAT1, STAT3, IFN-γ regulatory factor 4 (IRF4), hypoxia-induced factor 1 alpha (HIF1α), and activator protein 1 (AP1)[4, 37, 47, 48] (Fig. 2).
M2a macrophages
M2a is induced by IL-4 and/or IL-13 secreted by innate and adaptive immune cells, mast cells, basophils, and TH2 lymphocytes[47]. Alternatively, activated macrophages are characterized by an anti-inflammatory profile, which permits the resolution of inflammation and tissue repair. They express high levels of mannose receptor (CD206) and the decoy receptor IL-1R as well as the IL-1R antagonist and produce pro-fibrotic factors such as the transforming growth factor-beta (TGF-β) and insulin-like growth factor-1 (IGF-1), thus actively suppressing inflammation and promoting repair. Markers and effectors associated with M2 polarization include STAT6, GATA-binding protein 3 (GATA3), suppressor of cytokine signaling 1 (SOCS1), peroxisome proliferator-activated receptor gamma (PPARγ), CD163, and CD36, found in inflammatory zone 1 (FIZZ1), matrix metallo-proteinases (MMPs), and arginase 1 [4, 47, 49]. The increased arginase activity results in the production of polyamines and collagen and favors tissue remodeling and wound healing[50]. M2 macrophages induce angiogenesis and lymphangiogenesis by producing vascular endothelial growth factor-A (VEGF-A), endothelial growth factor (EGF), platelet-derived growth factor (PDGF), and IL-8[51].
M2b or regulatory macrophages
M2b macrophages are induced by stimulation with immune complexes (ICs) and TLR ligands or by IL-1R agonists. These ICs and LPS induce the signaling pathways mediated by NF‐κB, MAPK, PI3K/Akt, and IRF3 to actuate the M2b phenotype. Moreover, triggers like radiation induce miR‐222/GAS5 signaling to promote M2b polarization[58]. They produce both pro- and anti-inflammatory cytokines, such as IL-10, IL-1β, and TNF-α, and regulate both immune and inflammatory reactions.
M2c macrophages
M2c macrophages are activated by glucocorticoids or IL10 and exhibit a strong anti-inflammatory profile by releasing IL-10 and TGF-β. M2c expresses high IL-10 and TGF-β and does not express surface FIZZ-1; instead, M2c expresses a regulatory surface molecule B7-H4. This macrophage phenotype has HIF-1α and STAT3 expression and induces Tregs, impeding macrophage activation and T cell proliferation [56].
M2d macrophages
M2d macrophages are one of the tumor-associated macrophages (TAMs) [49]. These are induced by TLR ligands and A2 adenosine receptor (A2R) agonists, or by IL-6; they secrete high levels of IL-10, TGF-β, VEGF, and low IL-12, TNF-α, and IL-1β and contribute to tumor angiogenesis, growth, and metastasis. M2d macrophages originate from circulating monocytes drafted to the tumor site via CCL2, M-CSF, and VEGF. IL-6 along with M-CSF is known to induce M2d phenotype in tumor microenvironment (TME). Recently, it has been reported that Fra-1 upstream to IL-6 binds to IL-6 promoter and augments IL-6 expression and thus causes the generation of M2d macrophages [59].
Macrophage phenotypes: their clinical significance
In the initial phases of infections, pathogenic PAMPs are recognized by PRRs such as TLRs, and macrophages are activated to pro-inflammatory M1 phenotype [46]. They produce a large amount of pro-inflammatory mediators such as TNF-α, IL-1β, and IL-12 and generate ROS and NO to kill invading pathogens and activate adaptive immunity [5, 60].
In the resolving phase of infections to protect the host from excessive harm and facilitate wound healing, M1 macrophages undergo apoptosis or polarize to an anti-inflammatory M2 phenotype and counterbalance the excessive inflammatory response [61] (Fig. 2).
The mechanism by which the host’s immune system is activated plays a decisive role in shaping the consequence of an infection. A suitable activation of cell-mediated immunity eliminates intracellular infections; on the other hand, an appropriately mounted antibody-dependent humoral immune response mediates the elimination of extracellular infections.
Thus, intracellular infections require the M1-mediated cytotoxic T lymphocyte (CTL) response or the M1, TH1-mediated CTL to clear off infection, while extracellular infections are better cleared off via M2-mediated TH2-based humoral response. However, we are not going to discuss T cell-based response here, as it is outside the scope of this review, and we will focus only on macrophages. If a host fails to mount the balanced immune axis (M1 (TH1), M2 (TH2)), the infection persists [61]. This is partly dictated by the subset of macrophages which primarily phagocyte the pathogen and carry their endosomal degradation, subsequently presenting the processed antigens to T cells. As discussed earlier, macrophages can exist in different phenotypic and functional states (M1 and M2), depending on the environmental cues they receive (Fig. 2).
M1, the inflammatory macrophages, mount a TH1-mediated CTL response and participate in clearing off intracellular infection, while M2, the anti-inflammatory macrophages, mount a TH2-mediated humoral response that engages itself in clearing off extracellular pathogen and has a major role in tissue remodeling and homeostasis. Pathogens may utilize this as a strategy to evade the host immune mechanisms for their long-term survival. The balance between pro- and anti-inflammatory macrophages is crucial for mounting a proper immune response and deciding the outcome of the disease (Fig. 2).
We here discuss the involvement and dysregulation of macrophage phenotypes in various infections/diseases (Figs. 2 and 3) and summarize Table 3 for the diseases not covered in the text.

Pathogens and tumor cells cause a dis-regulation in the generation of macrophage phenotypes to escape the immune system. ASFV, African swine flu virus; MTB, mycobacterium tuberculosis; MYXV, myxoma virus; HSV, herpes simplex virus; HIV, human immunodeficiency virus; HCV, hepatitis C virus; TME, tumor microenvironment; DD, death domain; TRADD, TNFR1-associated death domain; FADD, Fas-associated protein with death domain; BID, BH3 interacting domain death; MYD88, myeloid differentiation primary response 88; TRAF6, TNF receptor-associated factor 6; MAPK, mitogen-activated protein kinase, SHP, protein tyrosine phosphatase; vOX2, CD200 viral analogue; vIL-10, viral IL-10 analogue; E2P, E2 viral protein; nef, HIV viral protein; MIF, migration inhibition factor
Macrophage phenotype in viral infection
It requires an M1 response to clear off the virus during the initial phases of infection and an M2-dominant response during the resolving phase of infection, to save the host from inflammatory cytokine storm-induced damage (Figs. 2 and 3). It is observed in highly infectious and pathogenic influenza viruses and the recent coronavirus (SARS CoV-2) outbreak that excessive M1 response can lead to an excessive release of inflammatory cytokines, leading to host tissue damage and eventually multiple organ failure and death [75]. Thus, viral infections generating severe inflammation-related immune-dysregulation are related to severe sickness and mortality in patients. M1 macrophages use strategies like generation of oxidative stress (ROS, NO) and production of antiviral cytokines (IFNs), activating other immune cells to fight off and kill the invading virus.
Oxidative stress (NO, ROS) related to viral control or limitation of viral infection has been reported in the vaccinia virus and SARS-CoV-2. Inflammatory M1 cytokines TNF-α, IL-1, IL-6, IL-8, and IL-12 directly or indirectly contain the viral infection. Influenza A virus, human immunodeficiency virus type 1 (HIV1), reproductive syndrome virus (RSV), and classical swine fever virus are directly reported to be inhibited by TNF-α treatment [70]. IL-6, IL-8, and IL-1β have been reported to limit viral infections directly, whereas IL-12 secreted by M1 macrophages has been reported to contain the viral infections indirectly by activating NK cells and promoting the naïve CD4+ T cell differentiation into TH1 [76, 77]. M1 macrophages boost the cytotoxicity of NK cells via secretion of IL-1β, IFN-β, and IL-15 [78].
However, despite the antiviral activities of M1 macrophages, they also play a prime role in promoting viral infections.
In the initial stages, pathogens like intracellular bacteria and viruses are delivered to permissive tissues by infected macrophages and are disseminated by inflammatory cell death of macrophages along with the release of inflammatory mediators like IL-1β and IL-18 [79].
Viruses have evolved multiple strategies to evade host immune responses and establish an infection. Viruses cause inhibition of macrophage polarization to M1 and can block downstream antiviral response signaling to evade the host immune response (Fig. 3).
Herpes viruses can cause M2 polarization of macrophages to evade host immune surveillance by encoding an IL-10 homolog known as vIL-10 which competes with IL-10 and binds to its receptor IL-10R [80]. vIL-10-IL-10R interaction causes STAT3 activation and ultimately amplifies the M2 response instead of the required M1 [81].
A similar strategy has been employed by the hepatitis C virus (HCV), which encodes an E2 protein. E2 upregulates STAT3 phosphorylation and downregulates SOCS3 expression, leading to enhanced IL-10 and skewing immune response to an immunosuppressive M2 type, favorable for viral replication [82].
African swine fever virus (ASFV) lowers the expression of iNOS causing NO suppression, leading to its survival in the infected macrophage [83]. Myxoma virus (MYXV) employs an interesting strategy of encoding the viral homolog of CD200, i.e., viral OX2 or vOX2 encoded by viral gene M141R [84] and the viral homolog of CD47 encoded by viral M128L, which are inhibitory regulators or suppressors of M1 phenotype and thus suppresses the M1 polarization [85]. Similarly, the herpes simplex virus type 1 (HSV-1) increases the expression of PD-L1/PD-1 and inhibits STAT1 activation, thus inhibiting M1 polarization and increasing viral survival [86, 87]. Other than these, viruses can suppress the inflammatory cytokine production during the initial phases of infection to establish infection. HIV-1 encodes Nef and Gag proteins. Nef enhances MHC-I degradation leading to its reduced expression on cell surface, thus causing impaired antigen presentation [88]. Moreover, if macrophages are treated with IL-4/IL-13 (M2 stimulants), it augments Ebola virus (EV) glycoprotein-dependent infection in murine peritoneal macrophages [89].
Thus, the key macrophage phenotype required for viral clearance is M1 in the initial stages and M2 in the resolving phases. The viruses have evolved the strategies to overcome M1-mediated viral clearance and generation of an adaptive immune response, to successfully establish an infection. If M1 is activated for a prolonged time, the excess of pro-inflammatory cytokines (cytokine storm) may cause multiple organ failures. If M2 is activated and becomes dominant during the initial phases of infection, the virus may persist and cause severe infection. The M1/M2 misbalance created by the invading virus thus costs the host infection and sometimes an eventual death.
Macrophage phenotype in bacterial infection
M. tuberculosis drives the macrophage polarization from M1 to M2 by blocking the NO metabolism, as an escape strategy [90] (Fig. 3). Biswas et al. reported that in the presence of a porin from Shigella spp., macrophages demonstrated increased MHC-II, CD80, and CD40 expression accompanying increased release of TNF-α and IL-12 indicating M1-like macrophage generation [91]. Recognition by PRRs and phagocytosis facilitates consequent degradation of the antigens. Cell wall components of bacteria activate cell surface TLRs and NLRP1, and DNA activates TLR9, while other proteins may activate AIM2, TLR5, and NLRC4 inflammasome leading to secretion of multiple inflammatory cytokines [92].
The inflammasome is a characteristic of M1 macrophages [93] and is authoritative for killing the internalized microbe along with the release of inflammatory mediators like IL-1β/IL-18.
Recognition thus leads to the generation and release of inflammatory mediators like cytokines, fatty acid metabolites, and free radicals. Many PRR activators and mediators also participate in the activation of inflammasome assembly that induces caspase-1, required to cleave pro–IL-1β to secretory IL-1β. Infection with Shigella flexneri is reported to activate NLRP1, NLRP3, and NLRC4 [94]. It is also reported that inflammasome activation following S. flexneri infection induces pyroptotic cell death in macrophages [95].
M1 macrophages, being high in co-stimulatory molecules, particularly CD40 are expected to prime the T cell response more specifically to TH1 and subsequently to CD8+ cytotoxic T cells. However, M2 macrophages prime the T cells to mount TH17, Treg, or TH2 responses. The pro-inflammatory M1 macrophages are associated with host protection during acute infections. It has been observed that mice deficient in IL-12, TNF-α, and IFN-γ and their respective receptors are generally prone to bacterial infections and may die [96].
The pathogens, like Leishmania, Salmonella, Shigella, Mycobacterium, Chlamydia, and Escherichia spp., require macrophages to be M1 polarized during initial phases to limit the infection.
However, the bacteria have also evolved strategies to evade host immune response, and they can modulate the host macrophage gene profile in their favor.
Mycobacterium tuberculosis (MTB) has been reported to modulate the gene expression relating to the M1 program. Tuberculosis patients have been reported to have dominant M2-type patterns [97], which could aid in the occurrence of chronic, latent, or secondary infections. Our lab has been exploring the host–pathogen interactions of chlamydial infection in a mouse model. Processed data from our lab indicates that Chlamydia trachomatis evades the host immune system by altering the host macrophage polarization program, CD40 signaling, and makes it biased towards M2 like during long-term or chronic infections. Similarly, we are exploring the effect of antibiotic treatment during intracellular infections and have found that immunomodulating antibiotics like azithromycin can bias the host immune system towards an anti-inflammatory M2 type program and can cause inefficient bacterial clearance from the host (processed lab data, unpublished). The visible disease symptoms can disappear due to the antibiotic-aided lowering of the bacterial load, but a small persistent population can still be present dormant in the host accounting for later secondary infections as observed in MTB. Nitric oxide (NO) is an important feature of M1 polarization. The reactive nitrogen species is imperative for causing oxidative stress to kill internalized bacteria like Salmonella [98]. Antibiotics like azithromycin have been observed to lower the NO release by murine macrophages and thus could aid in inefficient bacterial clearance (processed lab data, unpublished).
M1 polarization backs resistance to intracellular bacteria and limits the acute phase of infection. However, sustained M1 polarization is lethal for the host (Fig. 2). This is best understood in sepsis. Sepsis demonstrates an uncontrolled systemic inflammatory response as a result of immune dysregulation. This leads to tissue damage and multiple organ failure, as also observed in an advanced stage of bacterial sepsis [99]. Severe septic patients have been reported to have high systemic concentrations of M1 or pro-inflammatory cytokines which co-relate with a high rate of mortality [100]. Thus, to balance the deleterious effects of the M1 phenotype, macrophages are M2 biased during the resolving phases of infection.
However, in chronic infections, the macrophage reprograms towards an M2 profile; e.g., chronic brucellosis demonstrates IL-10-mediated M2 polarization [101], chlamydial infections have also been observed to cause IL-10-mediated M2 polarization, and bacteria are observed to actively replicate in M2-like macrophages (processed lab data, unpublished). Thus, a balance of M1 and M2 phenotypes and their timely activation and dominance is critical for resolving bacterial infections as well as saving the host from inflammatory damage.
Macrophage phenotype in cancer
Like intracellular infections, the balance of M1 and M2 phenotypes is crucial for the elimination or escape of tumor cells in the host (Figs. 2 and 3). Generally, M1 macrophages are required in the initial phase of tumor development to slay the aberrant cells. The M2 macrophages are required to subdue the deleterious effects of the inflammatory storm and to save the host from sustained inflammation. But tumor cells create their microenvironment and evade the host immune response by immunomodulating strategies like the generation of pre-cancerous macrophage phenotype [49]. The macrophage phenotype primarily associated with tumors is known as TAM [49, 102].
TAMs are abundant immune cells in cancer and employ a strong influence on tumor initiation, progression, and metastasis [57] (Fig. 3). TAMs secrete cytokines such as IL-10 and TGF-β to suppress T cell-dependent antitumor functions [41, 49, 57, 103,104,105]. The bone marrow-derived, circulating inflammatory Ly6C+CCR2+ monocytes give rise to TAMs [106]. These monocytes are recruited to the tumor tissue, and there they differentiate into TAMs. Initially, the inflammatory monocytes show an increased expression of CD11c and a decreased expression of Ly6C; i.e., they are CD11chigh and Ly6Clow via recombination signal binding protein for immunoglobulin kappa J region (RBPJ)-mediated notch signaling [107].
In the latter phase of differentiation, RBPJ induced upregulation of MHC-II, and downregulation of CD11b is observed in these monocytes [106]. Vcam1 is also upregulated significantly later on the inflammatory monocytes [108]; eventually, CCR2+ inflammatory monocytes differentiate to Ly6C−CD11c+MHCII+CD11blowVcam1+ TAMs [109]. TAMs can further differentiate tumors according to the local stimuli received. TAMs can be polarized into pro-inflammatory (M1-like) and anti-inflammatory (M2-like) under the stimuli of different tumor microenvironments (TME) [110]. Landry et al. using single-cell RNA sequencing (scRNA seq) data and cell trajectory analysis reported that TAMs evolve towards a pro-inflammatory state in human glioblastoma, while peripheral TAMs develop an anti-inflammatory phenotype [111].TAMs are polarized towards a conventionally activated endotype in colorectal cancer and produce pro-inflammatory cytokines like IFNγ, which trigger cytotoxic CD8+ T cell responses to enhance tumor elimination [112].
The creation of a macrophage migration inhibitor factor (MIF) is another mechanism by which TAMs can kill tumor cells [113]. MIF activates the key tumoricidal mechanisms. MIF is known to induce tumor cell killing via phagocytosis [114] and the release of apoptosis-inducing TNFα and IL1β [115]. In addition to blocking macrophage recruitment, TAMs release IL-18 and IL-22 in tumor milieu, and it has been linked to tumor cell death by boosting cytokine production (especially IFN-γ and IL-2) and by augmenting the cytotoxic activity of NK cells [116]. TAMs have been reported to be significant factors of prognostic response to postsurgical adjuvant chemotherapy (chemotherapy after the primary treatment), usually surgery in pancreatic cancer due to TAM re-education to slow tumor development [117]. However, other than the M1 TAMs, the existence of M2 TAMs in TME contributes to the maintenance of an immunosuppressive environment [118], which could be pro-cancerous. The dominance of M2 TAMs is one of the key causes for the formation of the immunosuppressive-TME. As tumors progress, the immunosuppressive factors like PGE2, IL-10, and TGF-β are secreted by cancer cells [49, 119, 120]. The excessive presence of these cytokine in the TME eventually impairs the M1-mediated immune responses, which are required for killing the tumor cells. The cross-talk between the different cytokines released by tumor cells and macrophages can further aid to the formation of pro-cancerous, immunosuppressive TAMs. This cross talk of macrophage cytokines and dual role of TAMs, leading to tumor escape or elimination, have been beautifully described by Challagundla et al. recently [49]. IL-4 and TGF-β secreted in TME can decrease the IL-12 secretion by macrophages and thus hamper the proliferation of NK cells and cytotoxic T cells, which are imperative to kill tumor cells. Additionally, necrotic tumor cells produce, immunosuppressive factors like IL-10 and S1P, which polarize the macrophages to M2 TAMs with reduced iNOS and NO. These deviations caused by TAMs play a vital role in immunosuppression and eventually tumor progression. Thus, TAMs play a dual that is both anti- and pro-cancerous role in TME, and a balanced, timely activation of required TAM phenotype could cause the elimination of tumors. On the other hand, inhibition of over-dominant M2 TAM generation can slow down tumor progression [110, 112, 121].
CSF-1R is expressed on monocytic cells; thus, blocking CSF-1R could directly inhibit TAMs generation. The humanized anti-CSF-1R antibody emactuzumab (RG7155) reduced TAM infiltration and enhanced CD8+ T cell growth in mice when used as a single agent [122]. Emactuzumab administration to patients resulted in a significant reduction of TAMs in tumor tissue, resulting in a significant therapeutic benefit for patients with diffuse-type giant cell tumors [123]. Moreover, our group has recently reported that berberine can restore the imbalanced, anti-inflammatory, pro-cancerous M2 macrophage phenotype to pro-inflammatory, anticancerous phenotype M1 [10], resulting in decreased tumor progression in berberine-treated mice. Hence, this signifies the importance of macrophage phenotypes in tumor progression or elimination.
Macrophage phenotype in allergies
Histamine is said to be the major culprit of allergic reactions [124]. Mast cells and basophils principally produce histamines. Macrophages are too reported to produce histamines [125]. If we talk about the pathogenesis of allergies and allergy-induced diseases, majorly alternatively activated macrophages or M2 are associated with it [126, 127].
In vivo studies by Naruhito Iwasaki et al. showed that macrophages produce histamine [128]. Macrophages interacted with antigen-specific TH2 cells to produce histamine in the culture supernatant. The authors further demonstrated that macrophages in association with TH2 cells caused allergic rhinitis in the mouse model. Allergic asthma is marked by the occurrence of high IL-4 and IL-13 levels which are known as M2 polarization inducers [128].
The role of M2 macrophages during allergic lung inflammation has been studied in the mouse model. To confirm the role of M2, allergic inflammation-induced mice were treated with an M2 polarization inhibitor during induction of allergy. The M2 inhibitor-treated mice developed less severe eosinophilia (increase in the number of eosinophils), mediated lung inflammation, and less collagen-mediated scarring in airway tissue. However, the M2 inhibition can shift eosinophil-mediated lung airway inflammation towards neutrophilic inflammation and aggravate airway hyper-responsiveness (exaggerated obstructive airway response). Thus, M2 macrophages are found to be associated with the development of eosinophil-mediated lung inflammation as well as prevent the development of neutrophil-mediated lung inflammation. This study also suggested that M2-like phenotype of macrophages contributes to determining the development of eosinophilic or neutrophilic lung inflammation in asthma [129]. Thus, a continued M2-mediated TH2 response can cause allergic inflammation and add to allergy-associated pathogenesis in various allergic diseases.
Macrophage phenotype in tissue injury and repair
For healing of injured, inflamed tissues, macrophages (tissue-resident as well as circulatory) are essential. Inflammation, proliferation, and remodeling are the overlapping stages of tissue repair and healing. Macrophages participate in all these stages and follow a transition of phenotypes from inflammatory to homeostatic as the healing progresses. If there is any dysregulation in macrophage function and phenotype transition, it messes up the healing or fibrosis and leads to pathological conditions, e.g., lung scarring, and chronic no-healing wounds can lead to cancer [130]. Tissue-resident macrophages (TRMs) originate from the yolk sac and fetal liver during embryonic development and persist in many tissues via self-renewal [131].
To maintain tissue homeostasis, TRMs mop up the dying, apoptotic cells. TRMs also respond to toxins, particulates, and pathogens found in the tissue’s local microenvironment [132]. Whenever there is any tissue injury, the BM-derived monocytes are recruited to the site of injury, and there they get differentiated into monocyte-derived macrophages (MoMs) [132].
Through an injury, both the TRMs and MoMs play separate roles. While the resolution phase of inflammation or any injury is going on, both TRMs and MOMs share a fate of death or self-renewal.
TRMs may repopulate the niche there in the healing tissue via self-renewal. With time, the recruited MoMs and the TRMs seem to develop similarities in phenotypes. During tissue injury, the invading pathogens, and the dying cells undergoing necroptosis/pyroptosis/apoptosis, release PAMPs and DAMPs that activate inflammatory signaling pathways in macrophages [132] to clear off any infection or aberrant cell growth or toxins. Thus, again like any infection or aberrant condition, an M1 macrophage phenotype is required in the initial phases, and an M2 phenotype is required in the resolving phases. Our immune system has devised a perfect way to maintain the healthy balance of the killer macrophages (M1) and the healer macrophages (M2), to tackle any injuries or inflammation (sterile or infection mediated). Satoshi Watanabe et al. have proposed two models for tissue repair.
5a. Macrophage phenotype in passive macrophage repair
Satoshi Watanabe et al. proposed that in the passive macrophage repair model, there is observed a progressive differentiation of MoMs. As tissue regeneration progresses, the promotion of the transition of MOMs into TRM-similar phenotypes is restored. These transitioning macrophages are homeostatic, as more transitioning happens, it speeds up the tissue repair. This leads to the generation of a positive feed-forward loop of tissue healing, and homeostatic macrophage generation is formed, which progresses the tissue repair. This restores the tissue homeostasis, and injured tissue is healed [132, 133]. These macrophages develop a capacity for self-renewal and persist in tissue after resolution through downregulation of transcription factor V-MAF musculoaponeurotic fibrosarcoma oncogene homolog B (MAFB, a basic leucine zipper (bZIP) transcription factor, plays role in development and regulation of lineage-specific hematopoiesis) [134, 135]. It includes the upregulation of molecules that interact with the epithelium to promote homeostasis, e.g., include receptor/ligand pairs CD200/CD200R, signal regulatory protein-α (SIRPα)/CD47, and GM-CSF/GM-CSF-R and immune/epithelial E-cadherin interactions.
5b. Macrophage phenotype in active macrophage repair
In the active repair model as proposed by Satoshi Watanabe et al., MoMs respond to the stimuli received from their immediate tissue microenvironment (TME).
They secrete factors to drive active tissue repair. The MoMs interact with TME via the uptake of apoptotic cells, Tregs, pathogens, and epithelial cells. The MoMs might promote inflammation resolution by secreting anti-inflammatory, pro-repair factors like metabolic intermediates, pro-resolution lipid mediators, anti-inflammatory cytokines, and matrix remodeling proteins, e.g., anti-inflammatory molecules (IL-10 and TGF-β), growth factors (VEGF, PDGFA), matrix metallo-proteinases (MMP-8, -10, -28), and osteopontin (osteopontin is a matricellular protein that plays important role in chronic inflammatory and autoimmune diseases) [132, 133].
Colony-stimulating factor-1 and macrophage differentiation
As discussed above for the production of granulocytes and macrophages, colony-stimulating factors (CSFs) or hematopoietic growth factors ally to effectuate the production and maintenance of these demand-driven cells by widely scattered deposits of marrow cells [136, 137].
There are four types of CSFs, i.e., granulocyte–macrophage colony-stimulating factor (GM-CSF), granulocyte colony-stimulating factor (G-CSF), macrophage colony-stimulating factor (M-CSF/CSF-1), and multipotential colony-stimulating factor also termed as IL-3(interleukin-3).
We are going to focus on CSF-1/M-CSF in this review. These hematopoietic growth factors belong to the family of glycoproteins. The CSF-1 or M-CSF is encoded by the CSF-1 gene and is secreted by endothelial cells, osteoblasts, fibroblasts, bone marrow stromal cells, astrocytes, myoblasts, keratinocytes, and mesothelial cells [138].
The human CSF-1 gene is present on chromosome 1 and is of about 21 kilobase pairs. Human CSF-1 mRNA codes for a precursor protein of 522 amino acids, whereas murine gene codes a 520 amino acids precursor having 60% homology to humans [139]. The N-terminal of CSF-1, which is imperative for its biological activity, is conserved in the human and murine CSF-1 genes [140]. CSF-1 has various functions like promoting the proliferation, survival, and differentiation of mononuclear phagocytic cells. It has also been reported to promote cytoskeletal reorganization and osteoclast migration [141] (Fig. 4).

CSF-1R signaling and its inhibitors. M279, rat monoclonal antibody (mAb) targeted to CSF-1R;IMC CS4, mAB targeted to CSF-1R; AFS98, mAB targeted to CSF-1R; RG-7155 (emactuzumab), humanized mAb targeted to CSF-1R; RK-20449 (A 419259), broad-spectrum pyrrolo-pyrimidine Src family inhibitor; idelalisib (IDEL), an oral phosphatidylinositol 3-kinase inhibitor; CYC11645, small molecules targeting the tyrosine kinase domain of CSF-1R; Ki20227, orally active, selective tyrosine kinase inhibitor; BLZ945, a highly selective, brain penetrant, CSF-1R kinase domain inhibitor; GW2580, an orally available, selective inhibitor of the tyrosine kinase activity of CSF1R; PLX3397 (pexidartinib), oral tyrosine kinase inhibitor of CSF1R; PLX5622, a highly selective, brain penetrant, orally active CSF1R inhibitor
The CSF-1 gene is alternatively spliced to give two biologically active forms, membrane-bound, and a secreted form. The ectodomain of membrane-bound CSF-1 could also be cleaved by metalloprotease disintegrin and metalloproteinase domain-containing protein 17/TNF-α-converting enzyme (ADAM 17/TACE), to generate soluble CSF-1. CSF-1 is a ligand to CSF-1R/cFMS which is expressed on a wide variety of cells like multipotent hematopoietic cells, mononuclear phagocyte progenitor cells, monocytes, tissue macrophages, and osteoclasts. It is a homo-dimeric type III receptor tyrosine kinase (RTK) and is encoded by cfms proto-oncogene. CSF-1R has an extracellular domain that binds to the ligand (CSF-1, IL-34), a transmembrane domain, and an intracellular domain having kinase activity [142]. CSF-1 when binds to the CSF-1R the Src and phosphoinositide-3-kinase (PI3-K) interact with the CSF-1R and mitogen-activated protein kinase (MAPK), and Akt pathways are activated [143]. CSF-1R controls the differentiation of myeloid progenitors into monocytes, macrophages, dendritic cells, and bone-resorbing osteoclasts [141] (Fig. 4).
We will focus on the monocyte-macrophage-CSF-1 differentiation axis according to the scope of our review. The known CSF-1R ligands are CSF-1 and IL-34. The normal detected concentration CSF-1 in serum is around 8–10 ng/ml [144]. CSF-1 binding auto-phosphorylates CSF-1R on various tyrosine residues (8 intracellular domain tyrosines: juxtamembrane domain Tyr-559 and Tyr-544, Tyr-697, kinase insert domain Tyr-706, Tyr-721 and carboxy-terminal tail Tyr-807, Tyr-921, Tyr-974) [145].
These phosphorylations create docking sites where different signaling molecules bind and different signaling pathways are activated. Phosphorylation of Tyr-559 activates Src family kinases and MAPK, extracellular signal regulated kinase (ERK) 5 [146] (via Tyr-561(human)/Tyr-559(mouse)). Tyr-721 phosphorylation of CSF-1R activates PI3K. Tyr-807 phosphorylation activates MAPK, ERK1, and ERK2, and recruited Grb2 by phosphorylated Tyr-921/Tyr-697 activates other signaling pathways. The structure of CSF-1R CSF-1R is the only RTK (receptor tyrosine kinase) that is regulated by two different cytokines, colony-stimulating factor 1 (CSF-1) and interleukin-34 (IL-34), both of which have identical four-helix bundle folds despite their different sequences and levels of spatio-temporal expression [147, 148].
The extracellular domain of CSF-1R is separated from the intracellular cytoplasmic region. The extracellular domain contains immunoglobulin (Ig)-like domains, a linker region, and a single-pass transmembrane helix to which ligands bind [141].
The first three N-terminal Ig domains (D1–D3) are involved in ligand recognition, while the next two Ig domains (D4–D5) are involved in ligand-receptor complex stabilization (Fig. 4).
Two kinase domains, a kinase insert, a juxtamembrane domain, and a carboxyl-terminal tail make up the cytoplasmic domain posttranslational changes to CSF-1R include phosphorylation and glycosylation [149]. CSF-1R is in an inactive auto-inhibitory state in the absence of ligands. The juxtamembrane domain transfers from its auto-inhibitory position upon ligand binding, and CSF-1R shifts to an active, elongated shape [141].
The extracellular region of the CSF-1 has 5 Ig-like domains in the extracellular region—D1, D2, D3, D4, and D5. D2 and D3 act as ligand-binding domains. The transmembrane domain, juxtamembrane domain, two-kinase domains, a kinase insert, and cytoplasmic domains comprise the intracellular domains. CSF-1R dimerization and phosphorylation of tyrosine residues are induced by ligand interaction that promotes differentiation and proliferation.
CSF-1-CSF-1R signaling pathways play a significant role in embryonic development, innate immunization, inflammation, tissue repair, and the tumor microenvironment and are associated with its extensive pattern of expression [150].
CSF-1R and macrophage polarization
The role of CSF-1-CSF-1R signaling in macrophage polarization has not been well understood yet. CSF-1 has been considered a potent colony proliferator, but how it can shape the polarization of differentiated macrophages to M1 or M2 phenotype is implicit. During the early stages of myeloid differentiation, CSF-1 works together with other hematopoietic growth factors like stem cell factor (SCF) and IL-3 to generate mononuclear phagocyte progenitor (MPP) cells [151].
The proliferation and differentiation of MPPs to monocytes and macrophages are regulated by CSF-1, and it regulates further macrophage differentiation as well. CSF-1R elicited PI-3 K signaling pathway is known to regulate the M1/M2 polarization axis of macrophages [146].
When CSF-1 binds to the CSF-1R, it starts a signaling cascade of class I PI-3ks [152] that leads to the activation of AKT1 and mTORC2.
The activated AKTs also inhibit the NF-ҡB-mediated M1 signals [153]. The signaling elicited by phosphorylation of Tyr-721 has been reported to downregulate pro-inflammatory genes (IL-1β, IL-12, TNF-α, etc.) associated with the M1 macrophage profile, while it upregulates expression of genes associated with M2 polarization profile (Arginase (Arg1) and IL-10) [48, 153].
CSF-1-dependent pTyr-721 and PI3K pathway eventually generate mi RNA (miR-21) which has a reported role in the regulation of macrophage activation, and if miR-21 is inhibited, it obliterates the CSF-1-mediated downregulation of IL-1β [154]. In vivo peritoneal LPS-induced inflammation model, the miR-21 weakens the recruitment of Ly6Chigh inflammatory monocytes to the peritoneal cavity, further suggesting a definitive role of CSF-1-dependent pTyr-721 and PI3K induced miR-21 in macrophage polarization axis [155].
Nevertheless, it is interesting to note that both M1- and M2-inducing signals, i.e., LPS, IFN-γ, IL-4, and IL-13, respectively, downregulate CSF-1R expression as observed in our lab, too (processed lab data, unpublished).
The endothelial cells that express CSF-1 are reported to support the growth of murine BM HSCs (expressing CSF-1R). It has been reported that when murine endothelial cells are co-cultured with murine BM HSCs along with growth factors like SCF, IL-3, thrombopoietin, and VEGF, differentiated F4/80+ macrophage colonies are formed [156]. The colony macrophages were observed to express high levels of M2 markers (Arg1, CD206/Mrc1, CD36), and no detectable markers for M1 (IL-12, TNFα, IL-1β) were observed suggesting a role of CSF-1R in deciding the M1/M2 fate of developing macrophages [157]. A CSF-1R inhibitor GW2580 was used to confirm the results; in our lab, we have also observed that if BM HSCs are cultured with GW2580 and then polarized with M2 stimulus (IL-4 + IL-10), there is no observed differentiation to M2-like phenotype. We have observed that the expression of CD206 (M2 marker) did not increase in GW2580-treated macrophages even after treatment with M2 stimulus (processed lab data, unpublished). If we compare the differentiation of BMDMs cultured with GM-CSF or CSF-1, the GM-CSF cultured BM HSCs are biased towards an M1-like phenotype, and the CSF-1 cultured BM HSCs are biased towards an M2-like phenotype [158].
However, GM-CSF and CSF-1 do not program BM HSCs towards an M1- and M2-like phenotype exclusively. As the monocytes and macrophages have phenotypic plasticity and their M1 and M2 polarizing spectrum are somewhat overlapping, none of the GM-CSF or CSF-1 can exclusively dictate the fate of a differentiating monocyte to M1 or M2, respectively. So, CSF-1 and GM-CSF-differentiated macrophages may further react to the polarization signals LPS, IFNγ, IL-4, and IL-13 alike. If CSF-1-treated BM HSCs are given polarizing signals with IL-4/IL-10/TGFβ, they give a robust M2 phenotype [159].
But also, CSF-1-differentiated macrophages could undergo a later polarization to the M1-like phenotype, and still higher expressions of M2 markers, i.e., IL-10, CCL14, and CD206, are retained [30]. Furthermore, it has been explored that another cytokine IL-32γ which could induce both M1 and M2 phenotypes if given as co-treatment with CSF-1 preferentially fast-tracks the M2 polarization in differentiating BM HSCs [160].
CSF-1-CSF-1R and macrophage phenotypes: clinical importance
It has been reported in studies that mice that have a non-functional CSF-1 ligand or receptor show deficiency in macrophage populations and display an attenuated response to inflammatory challenges [161, 162]. These studies substantiate the significance of the CSF-1-CSF-1R-receptor pathway in regulating the macrophage lineages [162]. In addition, the CSF-1-CSF-1R pathway is upregulated in a lot of human pathologies that require chronic activation of tissue-resident macrophages, like cancer [163], hence could be a potential drug therapy target. Different studies on the role of CSF-1R signaling in various diseases and pathological conditions have led to our knowledge about CSF-1R signaling in maintaining healthy immune responses or anomalies. CSF-1 has been found to promote osteoclast development and bone degradation in vivo [141]. CSF-1 could favor excessive osteoclast activity during osteoporosis as well as at sites of orthopedic implant failure [164]. In one of the studies, CSF-1was found to be elevated in the synovial fluid of rheumatoid arthritis patients, and synovial fibroblasts from rheumatoid arthritis patients were shown to produce high levels of CSF-1 [165]. An increase in CSF-1 production is also found to be associated with the accumulation of tissue-resident macrophages as seen in inflammatory bowel disease [166], glomerulonephritis [167], allograft rejection [168], and arteriosclerosis [169]. On top, the growth of several tumor types is found to be linked to overexpression of CSF-1 and cFMS receptors in cancer cells and tumor stroma [170].
Different studies have reported the use of antibodies and genetic techniques to evaluate the role of the CSF-1-CSF-1R pathway in animal disease models (Fig. 4) (Table 4). It has been shown that anti-CSF-1R antibodies inhibited the early stages of atherogenesis [171] in mice and decreased macrophage accumulation in mouse models of renal inflammation [167] and allograft rejection [168].
Moreover, it was reported that inhibition of CSF-1 signaling by the use of antisense and small interfering RNAs resulted in the inhibition of tumor growth in mice [172].
Additionally, it has also been reported that tumors in mice with non-functional CSF-1 grow more slowly as compared to control [173]. These findings advocate that the CSF-1/CSF-1R axis might directly encourage oncogenic effects on tumor cells. CSF-1-driven monocyte-to-macrophage differentiation is related to the activation of cell cycle genes, substantiating the underestimated proliferation potential of monocytes (Fig. 4).
As reviewed above, CSF-1 is a key regulator of macrophage homeostasis in vivo and can increase the generation of pure macrophage colonies from bone marrow progenitors in vivo [144]. CSF-1 stimulates monocyte growth and macrophage proliferation under homeostatic conditions, controlled by a negative feedback system.
CSF-1R is upregulated in mature mononuclear phagocytes, which are responsible for CSF-1 removal. In a healthy state, the CSF level is low (8–10 ng/ml in serum), but during inflammation, the amount of CSF in the bloodstream rises dramatically. Lee et al. reported that pexidartinib is a dual CSF-1R and c-KIT inhibitor and was approved by the US FDA in 2019 for the treatment of tenosynovial giant cell tumors (TGCT or PVNS), a rare condition caused by CSF-1R overexpression [174].
CSF-1R in central nervous system (CNS) disorders
The stimulation of CSF-1R and other receptors in the presence of pro-inflammatory cytokines, amyloids, lipopolysaccharides, or myelin debris causes the pro-inflammatory M1 phenotype, which contributes to neurodegeneration [175]. Stimulation of CSF-1R and other receptors in the presence of anti-inflammatory cytokines results in an anti-inflammatory M2 phenotype, which contributes to neuroprotection via phagocytosis, proliferation, and re-myelination.
Microglia polarize to an M1 phenotype in response to acute injury to resist the insult/infection and then transition to a more M2-like phenotype to allow tissue repair [176]. However, more M1 phenotype in microglia is maintained throughout chronic neurodegeneration, and the microglia primarily exacerbates the disease in this way. Several studies have found that using CSF-1R inhibitors lowered the severity of the condition and retarded its course, which was linked to a decrease in the number of microglia and infiltrating markers.
However, there is evidence that suggests the opposite, emphasizing the neuroprotective role of CSF-1/CSF-1R. CSF-1R inhibitor PLX3397 has been reported to significantly reduce the number of microglia which resulted in exacerbated brain infarction and vividly increased the production of inflammatory mediators by astrocytes [177, 178] (Fig. 4) (Table 4).
In a recent study, intranasal injection of recombinant human CSF-1R, 24 h after ischemia, reduced infarcted regions and corrected neurobehavioral impairments in rats with hypoxic-ischemic encephalopathy [179]. CSF-1-CSF-1R signaling pathways have also been shown to have neuroprotective effects in a few experimental autoimmune encephalomyelitis (EAE) and Alzheimer’s disease (AD) models. According to a recent study, administering CSF-1 and IL-34 to the cisterna magna increased the proliferation of CD11c+ microglia, a population thought to be important for primary myelination, and reduced EAE symptoms and demyelination [180].
A few in vitro and in vivo studies support CSF-1’s neuroprotective role in AD models, based on the fact that CSF-1 induced activation of microglia improves their ability to phagocytose-amyloid and reduces memory impairments, while Luo et al. (2013) discovered that the favorable effects of CSF-1 on cognitive performance in hAPP mice (first and most widely used mouse models of AD, based on the transgenic expression of human APP) are most likely independent of Aβ accumulation [181]. Despite the continued debate between research on neurodegenerative and neuroprotective effects of CSF-1R, according to the majority of studies, blocking the CSF-1/CSF-1R system has neuroprotective properties in a variety of neurodegenerative models.
CSF-1R in cancer
CSF-1 enhances the invasion and survival of immunosuppressive tumor-related cells in the tumor microenvironment macrophages, i.e., TAMs. TAMs levels in malignancies are high and inextricably linked to a negative prognosis outcome.
CSF-1 levels in tumors are high, and as discussed earlier in CSF-1 and macrophage phenotype and emergency myelopoiesis section that monocytes differentiate into anti-inflammatory M2-like TAMs by TME, a greater number of M2-like macrophages encourage tumor progression even more by inhibiting effector T cells functions [182, 183]. According to preclinical research, blocking CSF-1R or inhibiting its kinase activity may lower tumor volume by reducing the number of immunosuppressive M2-like TAMs which in turn results in a boost in antitumor response in a variety of tumor types [184] (Fig. 4) (Table 4).
CSF-1R blockade also results in increased immune-stimulatory or pro-inflammatory cytokines including interferon (IFNγ), which boosts antitumor T cell responses. Some cancer cells may upregulate PDL-1 as a result of the decrease in immunosuppressive signals mediated by M2-like TAMs.
Targeting the CSF-1R route in combination with other possibly complementary immune pathways, according to preclinical studies, could be a vital approach for more efficiently activating the antitumor immune response [144, 185]. CSF-1 mRNA is alternatively spliced to generate CSF-1 mRNA 3′UTR variants (var) [186]. According to Ho-Hyung Woo et al., CSF-1 protein translated by var-1 mRNA having long 3′UTR has a swift secretion rate compared to the CSF-1 protein translated by var-4 mRNA having short 3′UTR.
The authors reported that the secretion kinetics showed that HuR (human embryonic lethal abnormal vision-like protein, overexpressed in cancers, especially breast cancer) binds preferentially to the CSF-1 var-1 mRNA, not to var-4 mRNA, and accelerates the secretion of CSF-1 [186]. Thus, HuR overexpression escalates the secretion rate of CSF-1. Both CSF-1 var-1 and -4 mRNA were reported to be involved in increasing the migration and invasion rate of tumor cells in breast cancer [186].
CSF-1R in bone diseases
The usefulness of targeting CSF-1R in treating bone diseases is constantly being researched. In inflammatory arthritis, inflammatory bone deterioration, and osteoporosis, blocking or depleting CSF-1R inhibits the production and activity of osteoclasts and reduces pathological bone resorption. Osteoclasts are monocyte-/macrophage-derived multinucleated, hulks (gigantic size) of immune cells. These specialize in bone resorption (bones are broken down and absorbed, and osteoclasts remove hard bone tissue trailing which osteoblasts lay down new bone cells, to restore them from usual wear-tear) via proteolytic degradation and acid decalcification of bone matrix. Osteoclast-mediated bone resorption is essential for skeletal development and normal bone remodeling. In animal models of arthritis, blocking the activation of CSF-1R with CSF-1R inhibitors like Ki20227 [187], AFS98 [188], and GW2580 [189] slows the progression of joint inflammation and systemic bone erosion (Fig. 4) (Table 4). In serum-induced inflammatory arthritis and TNF-induced inflammatory osteolysis, anti-CSF-1R antibodies decrease osteoclastogenesis and bone resorption [190]. Imatinib, a tyrosine kinase inhibitor, prevents and cures type II collagen antibody-induced arthritis (CAIA) as well as collagen-induced arthritis [191].
Imatinib also inhibits CSF-1R expression and promotes apoptosis in mature osteoclasts. In a rat model, the CSF-1R inhibitor PLX3397 dramatically reduced the bone degradation and biomechanical characteristics caused by LPS [192]. In the human tumor necrosis factor transgenic (hTNFtg) mouse model, downregulation of CSF-1R and receptor activator of NF-kB (RANK) utilizing extracellular binding immunoglobulin protein (BiP) lowered inflammation and bone loss [193].
CSF-1R inhibition as a therapeutic strategy to restore dysregulated M1/M2 balance
CSF-1R inhibition is preventing CSF-1 from binding to its receptor and executing its functions. As discussed above, CSF-1-CSF-1R signaling has a role in biasing the differentiating macrophages towards an anti-inflammatory M2-like phenotype. The inhibition of CSF-1-CSF-1R signaling seems to be a gorgeous strategy to restore dysregulated M1/M2 balance in various diseases. The use of inhibitors targeted against the receptor’s protein tyrosine kinase activity and the use of agents that inhibit CSF-1’s binding to its receptor are two types of techniques to inhibit CSF-1’s action. CSF-1R inhibitors such as CYC10268 [194], imatinib [195], SU11248 [196], and GW2580 [189] represent a strong tool for understanding the role of the CSF-1/CSF-1R signaling system in a variety of biological systems and have therapeutic potential (Fig. 4) (Table 4). Regulating signals which are controlled by CSF-1 such as Akt and ERK-1/2 and expression of genes like toll-like receptor 9, apolipoprotein E, and LPS-inducible cytokine production in BMDM are suppressed by CSF-1R inhibitors [194]. Anujan et al. demonstrated that inhibiting the CSF-1R and MAPK signaling pathways at the same time with dual-kinase inhibitor-loaded supra-molecular nanoparticles (DSNs) improves the repolarization of pro-tumorigenic M2 macrophages to the antitumorigenic M1 phenotype [197].
Macrophage-based combination therapies for cancer treatment give better results when used along with therapeutic dual inhibition of CSF-1R and MAPK pathways using supra-molecular nanoparticles[197, 209]. Edwards et al. suggested that using CSF-1R inhibitors to eliminate tumor supportive cells or TAMs may be an effective treatment for a subset of patients with acute myeloid leukemia (AML) [210, 211]. Moreover, combining the highly specific CSF-1R inhibitors such as DCC-3014 and ARRY-382 with avelumab and pembrolizumab, respectively, has shown significant potential as a technique for inducing tumor suppression by multifactorial immune cell regulation[136, 212]. Blocking CSF-1/CSF-1R signaling in pancreatic tumors depletes CD206Hi TAMs and reprograms residual macrophages to enhance antitumor immunity, according to current data[213]. Its blockade enhances antitumor interferon responses, promotes CTL infiltration, and delays tumor progression when used alone. The induction of T cell checkpoint molecules, such as PDL1 on tumor cells and CTLA4 on T cells, restricts the therapeutic efficacy[214]. The combination of these agents with CSF-1R blockade inhibits the PD1 and CTLA4 checkpoint immunotherapy substantially enhanced efficacy and results in the regression of potently elicited PDAC tumor regressions. These results suggest that CSF-1/CSF-1R signaling can be a beneficial clinical target for reprogramming checkpoint-based immunotherapeutics[215]. In Table 4, we have summarized potential CSF-1R inhibitors and their target signaling molecules.
Concluding remarks
Macrophage phenotypes play a crucial role in fighting off infections and diseases as well as in maintaining a healthy body environment or homeostasis. The hematopoietic growth factors like CSF-1or M-CSF are imperative for the origin of macrophages from HSCs; however, their role in shaping the fate of macrophage phenotypes has not been well understood. The role of CSF-1 in dictating macrophage phenotype is imperial as indicated by the dominance of M1 and M2 balance dysregulation in various disease conditions and co-related abnormal expression of CSF-1R. Still, there is a dearth of studies connecting the dots between CSF-1-CSF-1-R signaling and macrophage polarization.
We have attempted to concise the available literature to relate the CSF-1-CSF-1-R axis with macrophage differentiation and with the clinical significance of this facet. However, more research on this aspect needs to be done to dearly understand the fate of macrophage development and differentiation in presence of CSF-1 and M1/M2 polarizing signals. The clinical applications of CSF-1-CSF-1-R signaling axis and macrophage polarization could be vast in developing therapeutic strategies in diseases like cancer, inflammatory disorders, injuries like spinal cord injuries, neurodegeneration, and bone disorders.
Abbreviations
- A2R :
-
Adenosine receptor 2
- AD :
-
Alzheimer’s disease
- ADAM17 :
-
A disintegrin and metalloproteinase 17
- AIM2 :
-
Absent in melanoma 2
- AKT :
-
Alpha serine/threonine-protein kinase gene
- AML :
-
Acute myeloid leukemia
- AMPK :
-
AMP-activated protein kinase
- AP1 :
-
Activator protein 1
- Arg1 :
-
Arginase 1
- ASFV :
-
African swine fever virus
- BiP :
-
Binding immunoglobulin protein
- BMDMs :
-
Bone marrow-derived macrophages
- bZIP :
-
Basic lucien zipper
- CAIA :
-
Collagen antibody-induced arthritis
- CCR2 :
-
Chemokine receptor 2
- CD :
-
Cluster of differentiation
- CIITA :
-
Class II trans activator
- CMPs :
-
Common myeloid progenitors
- COX-2 :
-
Cyclooxygenase 2
- CSF-1 :
-
Colony-stimulating factor-1
- CSF-1R :
-
CSF-1 receptor
- CTL :
-
Cytotoxic T lymphocyte
- CTLA4 :
-
CTL-associated protein-4
- DAMPs :
-
Damage associated molecular patterns
- DCs :
-
Dendritic cells
- DSNs :
-
Dual-kinase inhibitor loaded supra-molecular nanoparticles
- EAE :
-
Experimental autoimmune encephalomyelitis
- ECM :
-
Extracellular matrix
- EGF :
-
Endothelial growth factor
- ERK :
-
Extracellular signal-regulated kinase
- FIZZ1 :
-
Found in inflammatory zone 1
- GATA3 :
-
GATA-binding protein 3
- G-CSF :
-
Granulocyte colony-stimulating factor
- GM-CSF :
-
Granulocyte-macrophage colony-stimulating factor
- GMCSF-R :
-
GMCSF Receptor
- GMPs :
-
Granulocyte monocyte progenitors
- GPs :
-
Granulocyte progenitors
- Grb2 :
-
Growth factor receptor bound protein 2
- hAPP :
-
Human amyloid precursor protein
- HCV :
-
Hepatitis C virus
- HIF-1a :
-
Hypoxia inducible factor-1 alpha
- HIV-1 :
-
Human immunodeficiency virus-1
- HSCs :
-
Hematopoietic stem cells
- HSV-1 :
-
Herpes simplex virus-1
- hTNFtg :
-
Human tumor necrosis factor transgenic
- IFN :
-
Interferon
- IFR :
-
Interferon regulatory factor
- Ig :
-
Immunoglobulin
- IGF-1 :
-
Insulin-like growth factor-1
- IL :
-
Interleukin
- iNOS :
-
Inducible nitric oxide synthase
- IRF8 :
-
Interferon regulatory factor 8
- JAK :
-
Janus activated kinase
- JNK :
-
C-Jun N-terminal kinase
- KLF4 :
-
Krüppel-like factor 4
- LMPPs :
-
Lymphoid-primed multipotent progenitors
- LPS :
-
Lipopolysaccharide
- Ly6C :
-
Lymphocyte antigen 6 complex
- M128L :
-
Myxoma virus 128L gene
- M141R :
-
Myxoma virus 141 R
- MAFB :
-
Musculo aponeurotic fibrosarcoma oncogene homolog B
- MAPK :
-
Mitogen-activated protein kinase
- M-CSF :
-
Macrophage colony-stimulating factor
- MHC-II :
-
Major histocompatibility complex class-II
- MIF :
-
Migration inhibitory factor
- miR-21 :
-
Micro RNA-21
- MLPs :
-
Multi-lymphoid progenitors
- MMPs :
-
Matrix metallo-proteinase
- MoMs :
-
Monocyte-derived macrophages
- MPPs :
-
Multipotent progenitor precursors
- MPs :
-
Monocyte progenitors
- Mrc1 :
-
Mannose receptor c-type 1
- MTB :
-
Mycobacterium tuberculosis
- MYXV :
-
Myxoma virus
- NF-κB :
-
Nuclear factor-kappa-b
- NK cells :
-
Natural killer cells
- NLRC4 :
-
NLR family CARD domain-containing protein 4
- NLRP :
-
Nucleotide-binding domain and lucien-rich repeat containing protein
- NO :
-
Nitrogen oxide
- P13-K :
-
Phosphoinositide-3-kinase
- PAMPs :
-
Pathogen-associated molecular pattern
- PD-1 :
-
Programmed cell death protein-1
- PDAC :
-
Pancreatic ductal adenocarcinoma
- PDGF :
-
Platelet-derived growth factor
- PD-L1 :
-
PD-ligand 1
- PKCδ :
-
Protein kinase C delta
- PLX3397 :
-
Pexidartinib
- PPARγ :
-
Peroxisome proliferator-activated receptor gamma
- PPPs :
-
Proliferative progenitor populations
- PRRs :
-
Pathogen recognition receptors
- PyK2 :
-
Proline-rich tyrosine kinase 2
- Ras :
-
(Rat sarcoma virus) small GTPase protein
- Raf :
-
Serine/threonine-specific protein kinases (rapidly accelerated fibrosarcoma)
- RBPJ :
-
Recombination signal binding protein for immunoglobulin kappa J region
- RTK :
-
Receptor tyrosine kinase
- SCF :
-
Stem cell factor
- scRNA seq :
-
Single-cell RNA sequencing
- SIRPa :
-
Signal regulatory protein a
- SOCS :
-
Suppressor of cytokine signaling
- SRC :
-
Non-receptor tyrosine kinases
- STAT :
-
Signal transducer and activator of transcription
- TACE :
-
Tumor necrosis factor alpha-converting enzyme
- TAMs :
-
Tumor-associated macrophages
- TGF-β :
-
Transforming growth factor-beta
- TH :
-
T helper
- TLR:
-
Toll-like receptors
- TME :
-
Tumor microenvironment
- TNFα :
-
Tumor necrosis factor alpha
- TRMs :
-
Tissue-resident macrophages
- Tyr :
-
Tyrosine
- Vcam1 :
-
Vascular cell adhesion molecule-1
- VEGF :
-
Vascular endothelial growth factor
References
Mertens, C. et al. The macrophage iron signature in health and disease. Int. J. Mol. Sci. 22, (2021).
Hirayama, D., Iida, T. & Nakase, H. The Phagocytic function of macrophage-enforcing innate immunity and tissue homeostasis. Int. J. Mol. Sci. 19, (2017).
Sica A, Mantovani A. Science in medicine macrophage plasticity and polarization : in vivo veritas. J Clin Invest. 2012;122:787–95.
Italiani, P. & Boraschi, D. From monocytes to M1/M2 macrophages: phenotypical vs. functional differentiation. Frontiers in Immunology vol. 5 (2014).
Benoit M, Desnues B, Mege J-L. Macrophage polarization in bacterial infections. J Immunol. 2008;181:3733–9.
Macrophage polarization in health and disease - PubMed. https://pubmed.ncbi.nlm.nih.gov/22194670/.
Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage M1–M2 polarization balance. Front Immunol. 2014;5:614.
Mosser DM, Hamidzadeh K, Goncalves R. Macrophages and the maintenance of homeostasis. Cell Mol Immunol. 2021;18:579–87.
Wang WY, Tan MS, Yu JT, Tan L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann Transl Med. 2015;3:7–7.
Shah, D. et al. Berberine mediates tumor cell death by skewing tumor-associated immunosuppressive macrophages to inflammatory macrophages. Phytomedicine 99, (2021).
Moreira AP, et al. Serum amyloid P attenuates M2 macrophage activation and protects against fungal spore–induced allergic airway disease. J Allergy Clin Immunol. 2010;126:712-721.e7.
A-Gonzalez, N. et al. Phagocytosis imprints heterogeneity in tissue-resident macrophages. J. Exp. Med. 214, 1281–1296 (2017).
Zhang B, et al. Azithromycin drives alternative macrophage activation and improves recovery and tissue sparing in contusion spinal cord injury. J Neuroinflammation. 2015;12:1–13.
Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49–61.
Challagundla N, Saha B, Agrawal-Rajput R. Insights into inflammasome regulation: cellular, molecular, and pathogenic control of inflammasome activation. Immunol Res. 2022;2022:1–29. https://doi.org/10.1007/S12026-022-09286-9.
Arroyo Portilla, C., Tomas, J., Gorvel, J. P. & Lelouard, H. From species to regional and local specialization of intestinal macrophages. Front. Cell Dev. Biol. 8, (2020).
Gold KS, Brückner K. Macrophages and cellular immunity in Drosophila melanogaster. Semin Immunol. 2015;27:357.
Cavaillon J-M. The historical milestones in the understanding of leukocyte biology initiated by Elie Metchnikoff. J Leukoc Biol. 2011;90:413–24.
Gordon S. Elie Metchnikoff: father of natural immunity. Eur J Immunol. 2008;38:3257–64.
Steinman RM, Moberg CL. Zanvil Alexander Cohn 1926–1993. J Exp Med. 1994;179:1–30.
Gaikwad S, Naveen C, Agrawal-Rajput R. Toll-like receptor-4 antagonism mediates benefits during neuroinflammation. Neural Regen Res. 2016;11:552–3.
Jagannathan-Bogdan M, Zon LI. Hematopoiesis. Development. 2013;140:2463–7.
Hoggatt J, Kfoury Y, Scadden DT. Hematopoietic stem cell niche in health and disease. Annu Rev Pathol. 2016;11:555–81.
Mak, K. S., Funnell, A. P. W., Pearson, R. C. M. & Crossley, M. PU.1 and haematopoietic cell fate: dosage matters. Int. J. Cell Biol. 2011, (2011).
Qiu Q, et al. IRF8 regulates cell cycle of hematopoietic stem cells. Blood. 2015;126:2353–2353.
Ruytinx P, Proost P, Van Damme J, Struyf S. Chemokine-induced macrophage polarization in inflammatory conditions. Front Immunol. 2018;9:1930.
Broxmeyer HE. Chemokines in hematopoiesis. Curr Opin Hematol. 2008;15:49–58.
M, M. et al. Numerous growth factors, cytokines, and chemokines are secreted by human CD34(+) cells, myeloblasts, erythroblasts, and megakaryoblasts and regulate normal hematopoiesis in an autocrine/paracrine manner. Blood 97, 3075–3085 (2001).
Stegelmeier, A. A. et al. Myeloid cells during viral infections and inflammation. Viruses vol. 11 (2019).
Rovida, E. & Sbarba, P. Dello. Colony-stimulating factor-1 receptor in the polarization of macrophages: a target for turning bad to good ones? (2015) doi:https://doi.org/10.4172/2155-9899.1000379.
Yang J, Zhang L, Yu C, Yang XF, Wang H. Monocyte and macrophage differentiation: circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomark Res. 2014;2:1.
Weston CJ, Zimmermann HW, Adams DH. The role of myeloid-derived cells in the progression of liver disease. Front Immunol. 2019;10:893.
Schultze JL, Mass E, Schlitzer A. Emerging principles in myelopoiesis at homeostasis and during infection and inflammation. Immunity. 2019;50:288–301.
Boettcher S, Manz MG. Regulation of inflammation- and infection-driven hematopoiesis. Trends Immunol. 2017;38:345–57.
Furusawa, J. I. et al. Promotion of expansion and differentiation of hematopoietic stem cells by interleukin-27 into myeloid progenitors to control infection in emergency myelopoiesis. PLoS Pathog. 12, (2016).
Kumar R, Fossati V, Israel M, Snoeck H-W. Lin-Sca1+Kit- bone marrow cells contain early lymphoid-committed precursors that are distinct from common lymphoid progenitors. J Immunol. 2008;181:7507.
Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008;13:453–61.
Heideveld E, et al. Methods for macrophage differentiation and in vitro generation of human tumor associated-like macrophages. Methods Enzymol. 2020;632:113–31.
Duan Z, Luo Y. Targeting macrophages in cancer immunotherapy. Signal Transduct Target Ther. 2021;6:127.
Orecchioni, M., Ghosheh, Y., Pramod, A. B. & Ley, K. Macrophage polarization: different gene signatures in M1(Lps+) vs. classically and M2(LPS-) vs. alternatively activated macrophages. Front. Immunol. 10, 1084 (2019).
Zhang F, et al. TGF-β induces M2-like macrophage polarization via SNAIL-mediated suppression of a pro-inflammatory phenotype. Oncotarget. 2016;7:52294.
Makita N, Hizukuri Y, Yamashiro K, Murakawa M, Hayashi Y. IL-10 enhances the phenotype of M2 macrophages induced by IL-4 and confers the ability to increase eosinophil migration. Int Immunol. 2015;27:131–41.
Melton DW, McManus LM, Gelfond JAL, Shireman PK. Temporal phenotypic features distinguish polarized macrophages in vitro. Autoimmunity. 2015;48:161–76.
Zhang M-Z, et al. IL-4/IL-13-mediated polarization of renal macrophages/dendritic cells to an M2a phenotype is essential for recovery from acute kidney injury. Kidney Int. 2017;91:375–86.
Rogers KJ, et al. IL-4/IL-13 polarization of macrophages enhances Ebola virus glycoprotein-dependent infection. PLoS Negl Trop Dis. 2019;13: e0007819.
Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol. 2011;30:16–34.
Viola A, Munari F, Sánchez-Rodríguez R, Scolaro T, Castegna A. The metabolic signature of macrophage responses. Front Immunol. 2019;10:1462.
Caescu, C. I. et al. e-Blood Colony stimulating factor-1 receptor signaling networks inhibit mouse macrophage inflammatory responses by induction of microRNA-21 Key Points. (2015) doi:https://doi.org/10.1182/blood-2014.
Challagundla N, Shah D, Yadav S, Agrawal-Rajput R. Saga of monokines in shaping tumour-immune microenvironment: origin to execution. Cytokine. 2022;157: 155948.
Durante W. Role of arginase in vessel wall remodeling. Front Immunol. 2013;4:111.
Corliss BA, Azimi MS, Munson JM, Peirce SM, Murfee WL. Macrophages: an inflammatory link between angiogenesis and lymphangiogenesis. Microcirculation. 2016;23:95.
Medvedeva GF, Kuzmina DO, Nuzhina J, Shtil AA, Dukhinova MS. How macrophages become transcriptionally dysregulated: a hidden impact of antitumor therapy. Int J Mol Sci. 2021;22:1–27.
Schliefsteiner, C. et al. Human placental Hofbauer cells maintain an anti-inflammatory m2 phenotype despite the presence of gestational diabetes mellitus . Frontiers in Immunology vol. 8 (2017).
Raggi F, et al. Regulation of human macrophage M1–M2 polarization balance by hypoxia and the triggering receptor expressed on myeloid cells-1. Front Immunol. 2017;8:1–18.
Mantovani A, Martinez FO, Gordon S, Locati M. Gene expression polarization: new molecules and patterns of and monocyte-to-macrophage differentiation transcriptional profiling of the human. 2006. https://doi.org/10.4049/jimmunol.177.10.7303.
Lu J, et al. Discrete functions of M2a and M2c macrophage subsets determine their relative efficacy in treating chronic kidney disease. Kidney Int. 2013;84:745–55.
Rhee I. Diverse macrophages polarization in tumor microenvironment. Arch Pharm Res. 2016;39:1588–96.
Wang, L. xun, Zhang, S. xi, Wu, H. juan, Rong, X. lu & Guo, J. M2b macrophage polarization and its roles in diseases. J. Leukoc. Biol. 106, 345 (2019).
Wang, Q. et al. Fra-1 protooncogene regulates IL-6 expression in macrophages and promotes the generation of M2d macrophages. Cell Res. 2010 206 20, 701–712 (2010).
Mills CD. M1 and M2 macrophages: oracles of health and disease. Crit Rev Immunol. 2012;32:463–88.
Atri, C., Guerfali, F. Z. & Laouini, D. Role of human macrophage polarization in inflammation during infectious diseases. Int. J. Mol. Sci. 19, (2018).
Kazankov K, et al. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol. 2019;16:145–59.
Medbury, H. J., Williams, H. & Fletcher, J. P. Clinical significance of macrophage phenotypes in cardiovascular disease. Clin. Transl. Med. 3, (2014).
Huang, J., Hou, F. L., Zhang, A. Y. & Li, Z. L. Protective effect of the polarity of macrophages regulated by IL-37 on atherosclerosis. Genet. Mol. Res. 15, (2016).
Ardura-Fabregat, A. et al. Targeting neuroinflammation to treat Alzheimer’s disease. CNS Drugs 2017 3112 31, 1057–1082 (2017).
Zhang, Q. wen et al. Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PLoS One 7, e50946 (2012).
Gobejishvili, L. et al. Chronic ethanol-mediated decrease in cAMP primes macrophages to enhanced LPS-inducible NF-kappaB activity and TNF expression: relevance to alcoholic liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 291, (2006).
McClain CJ, Cohen DA. Increased tumor necrosis factor production by monocytes in alcoholic hepatitis. Hepatology. 1989;9:349–51.
Ju C, Mandrekar P. Macrophages and alcohol-related liver inflammation. Alcohol Res. 2015;37:251.
Yu S, Ge H, Li S, Qiu HJ. Modulation of macrophage polarization by viruses: turning off/on host antiviral responses. Front Microbiol. 2022;13:130.
Wasson, M., Kapoor, S., Garg, M., Singh, S. & Prakash, H. Macrophage polarization is decisive for chronic bacterial infection-induced carcinogenesis. in Macrophage Activation - Biology and Disease (IntechOpen, 2020). doi:https://doi.org/10.5772/intechopen.88171.
Heung LJ. Monocytes and the host response to fungal pathogens. Front Cell Infect Microbiol. 2020;10:34.
Jimenez MDP, Walls L, Fierer J. High levels of interleukin-10 impair resistance to pulmonary coccidioidomycosis in mice in part through control of nitric oxide synthase 2 expression. Infect Immun. 2006;74:3387–95.
Cosma NC, et al. M1/M2 polarization in major depressive disorder: disentangling state from trait effects in an individualized cell-culture-based approach. Brain Behav Immun. 2021;94:185–95.
Zirui Tay, M., Meng Poh, C., Rénia, L., MacAry, P. A. & P Ng, L. F. The trinity of COVID-19: immunity, inflammation and intervention. Nat. Rev. Immunol. (2019) doi:https://doi.org/10.1038/s41577-020-0311-8.
Velazquez-Salinas L, Verdugo-Rodriguez A, Rodriguez LL, Borca MV. The role of interleukin 6 during viral infections. Front Microbiol. 2019;10:1057.
Komastu T, Ireland DDC, Reiss CS. IL-12 and viral infections. Cytokine Growth Factor Rev. 1998;9:277.
Mattiola I, et al. Priming of human resting NK cells by autologous m1 macrophages via the engagement of IL-1β, IFN-β, and IL-15 pathways. J Immunol. 2015;195:2818–28.
Sahoo M, Ceballos-Olvera I, Del Barrio L, Re F. Role of the inflammasome, IL-1β, and IL-18 in bacterial infections. Sci World J. 2011;11:2037.
Ouyang P, et al. IL-10 encoded by viruses: a remarkable example of independent acquisition of a cellular gene by viruses and its subsequent evolution in the viral genome. J Gen Virol. 2014;95:245–62.
Andres, A., Suarez, R., Van Renne, N., Baumert, T. F. & Lupberger, J. Viral manipulation of STAT3: Evade, exploit, and injure. (2018) doi:https://doi.org/10.1371/journal.ppat.1006839.
Kwon YC, et al. Hepatitis C virus E2 envelope glycoprotein induces an immunoregulatory phenotype in macrophages. Hepatology. 2019;69:1873.
Granja AG, Sabina P, Salas ML, Fresno M, Revilla Y. Regulation of inducible nitric oxide synthase expression by viral A238L-mediated inhibition of p65/RelA acetylation and p300 transactivation. J Virol. 2006;80:10487–96.
Cameron CM, Barrett JW, Liu L, Lucas AR, McFadden G. Myxoma virus M141R expresses a viral CD200 (vOX-2) that is responsible for down-regulation of macrophage and T-cell activation in vivo. J Virol. 2005;79:6052–67.
Cameron CM, Barrett JW, Mann M, Lucas A, McFadden G. Myxoma virus M128L is expressed as a cell surface CD47-like virulence factor that contributes to the downregulation of macrophage activation in vivo. Virology. 2005;337:55–67.
Reichard AC, Cheemarla NR, Bigley NJ. SOCS1/3 expression levels in HSV-1-Infected, cytokine-polarized and -unpolarized macrophages. J Interf Cytokine Res. 2015;35:32.
Yokota SI, et al. Herpes simplex virus type 1 suppresses the interferon signaling pathway by inhibiting phosphorylation of STATs and janus kinases during an early infection stage. Virology. 2001;286:119–24.
Stumptner-Cuvelette P, et al. Human immunodeficiency virus-1 nef expression induces intracellular accumulation of multivesicular bodies and major histocompatibility complex Class II complexes: potential role of phosphatidylinositol 3-kinase. Mol Biol Cell. 2003;14:4857.
Rogers KJ, et al. IL-4/IL-13 polarization of macrophages enhances Ebola virus glycoprotein-dependent infection. PLoS Negl Trop Dis. 2019;13: e0007819.
Miller BH, et al. Mycobacteria inhibit nitric oxide synthase recruitment to phagosomes during macrophage infection. Infect Immun. 2004;72:2872–8.
Biswas T. Role of porin of Shigella dysenteriae type 1 in modulation of lipopolysaccharide mediated nitric oxide and interleukin-1 release by murine peritoneal macrophages. FEMS Immunol Med Microbiol. 2000;29:129–36.
Kieser KJ, Kagan JC. Multi-receptor detection of individual bacterial products by the innate immune system. Nat Rev Immunol. 2017;17:376.
Zhang J, et al. NLRP3 inflammasome mediates M1 macrophage polarization and IL-1β production in inflammatory root resorption. J Clin Periodontol. 2020;47:451–60.
Lupfer C, Kanneganti TD. The role of inflammasome modulation in virulence. Virulence. 2012;3:262–70.
Suzuki T, et al. Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog. 2007;3:1082–91.
Cooper AM, Magram J, Ferrante J, Orme IM. Interleukin 12 (IL-12) Is crucial to the development of protective immunity in mice intravenously infected with mycobacterium tuberculosis. J Exp Med. 1997;186:39–45.
Huang, Z. et al. Mycobacterium tuberculosis-induced polarization of human macrophage orchestrates the formation and development of tuberculous granulomas in vitro. PLoS One 10, (2015).
Fang FC, Vázquez-Torres A. Reactive nitrogen species in host-bacterial interactions. Curr Opin Immunol. 2019;60:96.
Gustot T. Multiple organ failure in sepsis: prognosis and role of systemic inflammatory response. Curr Opin Crit Care. 2011;17:153–9.
Chaudhry, H. et al. Role of cytokines as a double-edged sword in sepsis. in Vivo 27, 669 (2013).
Wang Y, et al. Brucella dysregulates monocytes and inhibits macrophage polarization through LC3-dependent autophagy. Front Immunol. 2017;8:691.
Challagundla, N., Shah, D., Yadav, S. & Agrawal-rajput, R. CO.
Afik R, et al. Tumor macrophages are pivotal constructors of tumor collagenous matrix. J Exp Med. 2016;213:2315–31.
Cassetta L, Cassol E, Poli G. Macrophage polarization in health and disease. Sci World J. 2011;11:2391.
Corthay A, et al. Primary antitumor immune response mediated by CD4+ T cells. Immunity. 2005;22:371–83.
Liu Y, Cao X. The origin and function of tumor-associated macrophages. Cell Mol Immunol. 2015;12:1.
Gamrekelashvili J, et al. Notch and TLR signaling coordinate monocyte cell fate and inflammation. Elife. 2020;9:1–19.
Kong, D. H., Kim, Y. K., Kim, M. R., Jang, J. H. & Lee, S. Emerging roles of vascular cell adhesion molecule-1 (VCAM-1) in immunological disorders and cancer. Int. J. Mol. Sci. 19, (2018).
Kitamura, T. et al. Monocytes differentiate to immune suppressive precursors of metastasis-associated macrophages in mouse models of metastatic breast cancer. Front. Immunol. 8, (2018).
He, Z. & Zhang, S. Tumor-associated macrophages and their functional transformation in the hypoxic tumor microenvironment. Front. Immunol. 12, (2021).
Landry, A. P., Balas, M., Alli, S., Spears, J. & Zador, Z. Distinct regional ontogeny and activation of tumor associated macrophages in human glioblastoma. Sci. Rep. 10, (2020).
Wang, H., Tian, T. & Zhang, J. Tumor-Associated Macrophages (TAMs) in Colorectal Cancer (CRC): from mechanism to therapy and prognosis. Int. J. Mol. Sci. 22, (2021).
Krockenberger M, et al. Macrophage migration inhibitory factor (MIF) contributes to the immune escape of ovarian cancer by downregulating NKG2D. J Immunol. 2008;180:7338.
Onodera S, et al. Macrophage migration inhibitory factor induces phagocytosis of foreign particles by macrophages in autocrine and paracrine fashion. Immunology. 1997;92:131–7.
Pozzi LAM, Weiser WY. Human recombinant migration inhibitory factor activates human macrophages to kill tumor cells. Cell Immunol. 1992;145:372–9.
Mao C, Ding Y, Xu N. A double-edged sword role of cytokines in prostate cancer immunotherapy. Front Oncol. 2021;11:4732.
Di Caro G, et al. Dual prognostic significance of tumour-associated macrophages in human pancreatic adenocarcinoma treated or untreated with chemotherapy. Gut. 2016;65:1710–20.
Rhee I. Diverse macrophages polarization in tumor microenvironment. Arch Pharm Res. 2016;39:1588–96.
Jakowlew SB. Transforming growth factor-beta in cancer and metastasis. Cancer Metastasis Rev. 2006;25:435–57.
Erreni M, Mantovani A, Allavena P. Tumor-associated macrophages (TAM) and inflammation in colorectal cancer. Cancer Microenviron. 2011;4:141–54.
Di Caro G, et al. Dual prognostic significance of tumour-associated macrophages in human pancreatic adenocarcinoma treated or untreated with chemotherapy. Gut. 2015;65:1710–20.
Liu, K. X. & Joshi, S. “Re-educating” tumor associated macrophages as a novel immunotherapy strategy for neuroblastoma. Front. Immunol. 11, (2020).
Gomez-Roca C, et al. Anti-CSF-1R emactuzumab in combination with anti-PD-L1 atezolizumab in advanced solid tumor patients naïve or experienced for immune checkpoint blockade. J Immunother Cancer. 2022;10: e004076.
Thangam EB, et al. The role of histamine and histamine receptors in mast cell-mediated allergy and inflammation: the hunt for new therapeutic targets. Front Immunol. 2018;9:1873.
Nakano K, Takamatsu S. Histamine produced by macrophage and T lymphocyte: a new type of signal transducer. Nihon Yakurigaku Zasshi. 2001;118:15–22.
Suzuki K, Meguro K, Nakagomi D, Nakajima H. Roles of alternatively activated M2 macrophages in allergic contact dermatitis. Allergol Int. 2017;66:392–7.
Saradna A, Do DC, Kumar S, Fu QL, Gao P. Macrophage polarization and allergic asthma. Transl Res. 2018;191:1.
Iwasaki N, et al. Th2 cells and macrophages cooperatively induce allergic inflammation through histamine signaling. PLoS ONE. 2021;16:1–18.
Draijer, C., Robbe, P., Boorsma, C. E., Hylkema, M. N. & Melgert, B. N. Dual role of YM1+ M2 macrophages in allergic lung inflammation. Sci. Reports 2018 81 8, 1–12 (2018).
Day DL, Chakari W, Matzen SH. Malignant transformation of a non-healing traumatic wound on the lower extremity: A case report. Int J Surg Case Rep. 2018;53:468–70.
Hoeffel G, Ginhoux F. Ontogeny of tissue-resident macrophages. Front Immunol. 2015;6:486.
Watanabe S, Alexander M, Misharin AV, Budinger GRS. The role of macrophages in the resolution of inflammation. J Clin Invest. 2019;129:2619–28.
Novak ML, Koh TJ. Phenotypic transitions of macrophages orchestrate tissue repair. Am J Pathol. 2013;183:1352.
Kang K, et al. Interferon-γ represses M2 gene expression in human macrophages by disassembling enhancers bound by the transcription factor MAF. Immunity. 2017;47:235.
Eychène A, Rocques N, Pouponnot C. A new MAFia in cancer. Nat Rev Cancer. 2008;8:683–93.
Achkova D, Maher J. Role of the colony-stimulating factor (CSF)/CSF-1 receptor axis in cancer. Biochem Soc Trans. 2016;44:333–41.
Metcalf D. The colony-stimulating factors and cancer. Cancer Immunol Res. 2013;1:351.
Sinha SK, et al. HHS Public Access. 2022;41:220–33.
Ladner MB, et al. Human CSF-1: gene structure and alternative splicing of mRNA precursors. EMBO J. 1987;6:2693–8.
Ryan GR, et al. Rescue of the colony-stimulating factor 1 (CSF-1)-nullizygous mouse (Csf1op/Csf1op) phenotype with a CSF-1 transgene and identification of sites of local CSF-1 synthesis. Blood. 2001;98:74–84.
Mun SH, Park PSU, Park-Min KH. The M-CSF receptor in osteoclasts and beyond. Exp Mol Med. 2020;52:1239–54.
Horiuchi K, et al. Cell surface colony-stimulating factor 1 can be cleaved by tnf-α converting enzyme or endocytosed in a clathrin-dependent manner. J Immunol. 2007;179:6715–24.
Endele M, et al. CSF-1-induced Src signaling can instruct monocytic lineage choice. Blood. 2017;129:1691–701.
Hume DA, MacDonald KPA. Therapeutic applications of macrophage colony-stimulating factor-1 (CSF-1) and antagonists of CSF-1 receptor (CSF-1R) signaling. Blood. 2012;119:1810–20.
Yu W, et al. Macrophage proliferation is regulated through CSF-1 receptor tyrosines 544, 559, and 807. J Biol Chem. 2012;287:13694–704.
Yu W, et al. Macrophage proliferation is regulated through CSF-1 receptor tyrosines 544, 559, and 807. J Biol Chem. 2012;287:13694–704.
Felix J, et al. Structure and assembly mechanism of the signaling complex mediated by human CSF-1. Structure. 2015;23:1621–31.
Wei 10_Functional overlap but differential expression of CSF-1 and IL-34 in their CSF-1.pdf.
Liu H, et al. The mechanism of shared but distinct CSF-1R signaling by the non-homologous cytokines IL-34 and CSF-1. Biochim Biophys Acta - Proteins Proteomics. 2012;1824:938–45.
Stanley, E. R. & Chitu, V. CSF-1 receptor signaling in myeloid cells. Cold Spring Harb. Perspect. Biol. 6, (2014).
Seita J, Weissman IL. Published in final edited form as: Hematopoietic stem cell: self-renewal versus differentiation. Wiley Interdiscip Rev Syst Biol Med. 2010;2:1–20.
Lee AWM, States DJ. Colony-stimulating factor-1 requires PI3-kinase-mediated metabolism for proliferation and survival in myeloid cells. Cell Death Differ. 2006;13:1900–14.
Vergadi E, Ieronymaki E, Lyroni K, Vaporidi K, Tsatsanis C. Akt signaling pathway in macrophage activation and M1/M2 polarization. J Immunol. 2017;198:1006–14.
Caescu CI, et al. Colony stimulating factor-1 receptor signaling networks inhibit mouse macrophage inflammatory responses by induction of microRNA-21. Blood. 2015;125:e1–13.
Feng J, et al. MiR-21 attenuates lipopolysaccharide-induced lipid accumulation and inflammatory response: Potential role in cerebrovascular disease. Lipids Health Dis. 2014;13:1–9.
He H, et al. Endothelial cells provide an instructive niche for the differentiation and functional polarization of M2-like macrophages. Blood. 2012;120:3152–62.
Hamilton TA, Zhao C, Pavicic PG, Datta S. Myeloid colony-stimulating factors as regulators of macrophage polarization. Front Immunol. 2014;5:1–6.
J Leukocyte Bio - 2009 - Fleetwood - GM‐CSF‐ and M‐CSF‐dependent macrophage phenotypes display differential dependence on.pdf.
Mia S, Warnecke A, Zhang XM, Malmström V, Harris RA. An optimized protocol for human M2 macrophages using M-CSF and IL-4/IL-10/TGF-β yields a dominant immunosuppressive phenotype. Scand J Immunol. 2014;79:305–14.
Osman A, et al. M-CSF inhibits anti–HIV-1 activity of IL-32, but they enhance M2-like phenotypes of macrophages. J Immunol. 2014;192:5083–9.
Erblich, B., Zhu, L., Etgen, A. M., Dobrenis, K. & Pollard, J. W. Absence of colony stimulation factor-1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PLoS One 6, (2011).
Rojo R, et al. Deletion of a Csf1r enhancer selectively impacts CSF1R expression and development of tissue macrophage populations. Nat Commun. 2019;10:1–17.
Cannarile MA, et al. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J Immunother Cancer. 2017;5:1–13.
Journal Orthopaedic Research - 2008 - Sarahrudi - The impact of colony‐stimulating factor‐1 on fracture healing An.pdf.
Takei I, et al. High macrophage-colony stimulating factor levels in synovial fluid of loose artificial hip joints. J Rheumatol. 2000;27:894–9.
Klebl FH, Olsen JE, Jain S, Doe WF. Expression of macrophage-colony stimulating factor in normal and inflammatory bowel disease intestine. J Pathol. 2001;195:609–15.
Isbel, N. M., Nikolic-Paterson, D. J., Hill, P. A., Dowling, J. & Atkins, R. C. Local macrophage proliferation correlates with increased renal M-CSF expression in human glomerulonephritis. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. - Eur. Ren. Assoc. 16, 1638–1647 (2001).
Jose MD, Le Meur Y, Atkins RC, Chadban SJ. Blockade of macrophage colony-stimulating factor reduces macrophage proliferation and accumulation in renal allograft rejection. Am J Transplant Off J Am Soc Transplant Am Soc Transpl Surg. 2003;3:294–300.
Rosenfeld ME, et al. Macrophage colony-stimulating factor mRNA and protein in atherosclerotic lesions of rabbits and humans. Am J Pathol. 1992;140:291–300.
Lin, E. Y., Nguyen, A. V, Russell, R. G. & Pollard, J. W. Colony-Stimulating Factor 1 Promotes Progression of Mammary Tumors to Malignancy. J. Exp. Med. 193, 727–740 (2001).
Beirão BCB, et al. A blocking antibody against canine CSF-1R maturated by limited CDR mutagenesis. Antib Ther. 2020;3:193–204.
Aharinejad S, et al. Colony-Stimulating factor-1 blockade by antisense oligonucleotides and small interfering RNAs suppresses growth of human mammary tumor xenografts in mice. Cancer Res. 2004;64:5378–84.
Strachan DC, et al. CSF1R inhibition delays cervical and mammary tumor growth in murine models by attenuating the turnover of tumor-associated macrophages and enhancing infiltration by CD8+ T cells. Oncoimmunology. 2013;2:1–12.
Lee K-H, et al. Discovery of BPR1R024, an orally active and selective CSF1R inhibitor that exhibits antitumor and immunomodulatory activity in a murine colon tumor model. J Med Chem. 2021;64:14477–97.
Bo L, Bo X. Colony stimulating factor 1: friend or foe of neurons? Neural Regen Res. 2022;17:773–4.
Orihuela R, McPherson CA, Harry GJ. Microglial M1/M2 polarization and metabolic states. Br J Pharmacol. 2016;173:649–65.
Xu Y, Jin M-Z, Yang Z-Y, Jin W-L. Microglia in neurodegenerative diseases. Neural Regen Res. 2021;16:270–80.
Wyatt-Johnson SK, Sommer AL, Shim KY, Brewster AL. Suppression of microgliosis with the colony-stimulating factor 1 receptor inhibitor PLX3397 does not attenuate memory defects during epileptogenesis in the rat. Front Neurol. 2021;12: 651096.
Hu, X. et al. Rh-CSF1 Attenuates oxidative stress and neuronal apoptosis via the CSF1R/PLCG2/PKA/UCP2 signaling pathway in a rat model of neonatal HIE. Oxid. Med. Cell. Longev. 2020, (2020).
Wlodarczyk A, et al. CSF1R stimulation promotes increased neuroprotection by CD11c+ microglia in EAE. Front Cell Neurosci. 2019;12:1–10.
Luo J, et al. Colony-stimulating factor 1 receptor (CSF1R) signaling in injured neurons facilitates protection and survival. J Exp Med. 2013;210:157–72.
Huang, L., Xu, X. & Hao, Y. The possible mechanisms of tumor progression via CSF-1/CSF-1R pathway activation. Rom. J. Morphol. Embryol. = Rev. Roum. Morphol. Embryol. 55, 501–506 (2014).
Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160:48–61.
Holmgaard RB, et al. Timing of CSF-1/CSF-1R signaling blockade is critical to improving responses to CTLA-4 based immunotherapy. Oncoimmunology. 2016;5:e1151595–e1151595.
Kowal J, Kornete M, Joyce JA. Re-education of macrophages as a therapeutic strategy in cancer. Immunotherapy. 2019;11:677–89.
Woo H-H, Chambers SK. The alternative spliced 3’-UTR mediated differential secretion of macrophage colony stimulating factor in breast cancer cells. Biochem Biophys Res Commun. 2020;525:1004–10.
Han J, et al. Inhibition of colony stimulating factor-1 receptor (CSF-1R) as a potential therapeutic strategy for neurodegenerative diseases: opportunities and challenges. Cell Mol Life Sci. 2022;79:219.
MacDonald KPA, et al. An antibody against the colony-stimulating factor 1 receptor depletes the resident subset of monocytes and tissue- and tumor-associated macrophages but does not inhibit inflammation. Blood. 2010;116:3955–63.
Conway JG, et al. Effects of the cFMS kinase inhibitor 5-(3-methoxy-4-((4-methoxybenzyl)oxy)benzyl)pyrimidine-2,4-diamine (GW2580) in normal and arthritic rats. J Pharmacol Exp Ther. 2008;326:41–50.
Toh, M.-L. et al. A CSF-1 receptor monoclonal antibody has potent bone and cartilage protective effects in experimental arthritis. Arthritis Rheumatol. 66, (2014).
Koyama K, et al. Imatinib mesylate both prevents and treats the arthritis induced by type II collagen antibody in mice. Mod Rheumatol. 2007;17:306–10.
Wang X-F, et al. Colony-stimulating factor 1 receptor inhibition prevents against lipopolysaccharide -induced osteoporosis by inhibiting osteoclast formation. Biomed Pharmacother. 2019;115: 108916.
Zaiss, M. et al. Binding immunoglobulin protein (BIP) inhibits TNF‐α–induced osteoclast differentiation and systemic bone loss in an erosive arthritis model. ACR Open Rheumatol. 1, (2019).
Irvine K, et al. A CSF-1 receptor kinase inhibitor targets effector functions and inhibits pro-inflammatory cytokine production from murine macrophage populations. FASEB J. 2006;20:1921–3.
Paniagua RT, et al. C-Fms-mediated differentiation and priming of monocyte lineage cells play a central role in autoimmune arthritis. Arthritis Res Ther. 2010;12:R32.
Murray LJ, et al. SU11248 inhibits tumor growth and CSF-1R-dependent osteolysis in an experimental breast cancer bone metastasis model. Clin Exp Metastasis. 2003;20:757–66.
Ramesh A, Brouillard A, Kumar S, Nandi D, Kulkarni A. Dual inhibition of CSF1R and MAPK pathways using supramolecular nanoparticles enhances macrophage immunotherapy. Biomaterials. 2020;227: 119559.
Rattanaburee T, Tipmanee V, Tedasen A, Thongpanchang T, Graidist P. Inhibition of CSF1R and AKT by (±)-kusunokinin hinders breast cancer cell proliferation. Biomed Pharmacother. 2020;129: 110361.
Gelhorn HL, et al. Patient-reported symptoms of tenosynovial giant cell tumors. Clin Ther. 2016;38:778–93.
Rattanaburee T, Tipmanee V, Tedasen A, Thongpanchang T, Graidist P. Inhibition of CSF1R and AKT by (±)-kusunokinin hinders breast cancer cell proliferation. Biomed Pharmacother. 2020;129: 110361.
Shah RR, Morganroth J, Shah DR. Hepatotoxicity of tyrosine kinase inhibitors: clinical and regulatory perspectives. Drug Saf. 2013;36:491–503.
Kumari A, Silakari O, Singh RK. Recent advances in colony stimulating factor-1 receptor/c-FMS as an emerging target for various therapeutic implications. Biomed Pharmacother. 2018;103:662–79.
Wiktor-Jedrzejczak W, et al. Inhibition of colony-stimulating-factor-1 signaling in vivo with the orally bioavailable cFMS kinase inhibitor GW2580. Proc Natl Acad Sci U S A. 1990;87:4828–32.
Gerngross L, Lehmicke G, Belkadi A, Fischer T. Role for cFMS in maintaining alternative macrophage polarization in SIV infection: implications for HIV neuropathogenesis. J Neuroinflammation. 2015;12:1–15.
Clanchy FIL, Hamilton JA. HUVEC co-culture and haematopoietic growth factors modulate human proliferative monocyte activity. Cytokine. 2012;59:31–4.
Ohno H, et al. A c-fms tyrosine kinase inhibitor, Ki20227, suppresses osteoclast differentiation and osteolytic bone destruction in a bone metastasis model. Mol Cancer Ther. 2006;5:2634–43.
Saleh, R. et al. CSF-1 in inflammatory and arthritic pain development. J. Immunol. 201, 2042 LP – 2053 (2018).
Lim AKH, et al. Antibody blockade of c-fms suppresses the progression of inflammation and injury in early diabetic nephropathy in obese db/db mice. Diabetologia. 2009;52:1669–79.
Tian L, et al. Macrophage-based combination therapies as a new strategy for cancer immunotherapy. Kidney Dis. 2022;8:26–43.
Edwards 5th, D. K. et al. CSF1R inhibitors exhibit antitumor activity in acute myeloid leukemia by blocking paracrine signals from support cells. Blood. 2019;133, 588–599.
Edwards V, D. K. et al. CSF1R inhibitors exhibit antitumor activity in acute myeloid leukemia by blocking paracrine signals from support cells. Blood, 2019; 133, 588–599.
Gross S, Rahal R, Stransky N, Lengauer C, Hoeflich KP. Targeting cancer with kinase inhibitors. J Clin Invest. 2015;125:1780–9.
Zhu Y, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014;74:5057–69.
Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and anti-CTLA-4 therapies in cancer: mechanisms of action, efficacy, and limitations. Front Oncol. 2018;8:86.
Gunturu KS, Rossi GR, Saif MW. Immunotherapy updates in pancreatic cancer: are we there yet? Ther Adv Med Oncol. 2013;5:81–9.
Acknowledgements
SY’s fellowship is funded by the Indian Council of Medical Research-Senior Research Fellowship; DB is supported by the Department of Biotechnology (DBT), India; the work of RAR was supported by the Department of Science and Technology (DST)-SERB (CRG/2019/002802), Gujarat State Biotechnology Mission (GSBTM) (GSBTM/JD (R&D)/610/20-21/345).
Author information
Authors and Affiliations
Contributions
SY drafted, wrote the manuscript, and did the artwork and table preparation; AP contributed to writing the manuscript and helped in preparing artwork; DB contributed to writing the manuscript and helped in preparing tables; RAR conceptualized, drafted, and proofread the manuscript to the final version.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Yadav, S., Priya, A., Borade, D.R. et al. Macrophage subsets and their role: co-relation with colony-stimulating factor-1 receptor and clinical relevance. Immunol Res 71, 130–152 (2023). https://doi.org/10.1007/s12026-022-09330-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12026-022-09330-8